[level-membership-for-basic-science-category]
Chapter 8 General pharmacology
• Qualitative aspects: receptors, enzymes, selectivity.
• Quantitative aspects: dose response, potency, therapeutic efficacy, tolerance.
• Time course of drug concentration: drug passage across cell membranes; order of reaction; plasma half-life and steady-state concentration; therapeutic drug monitoring.
• Individual processes: absorption, distribution, metabolism, elimination.
• Drug dosage: dosing schedules.
• Chronic pharmacology: the consequences of prolonged drug administration and drug discontinuation syndromes.
Pharmacodynamics
Qualitative aspects
Mechanisms
• Ligand-gated ion channels, i.e. receptors coupled directly to membrane ion channels; neurotransmitters act on such receptors in the postsynaptic membrane of a nerve or muscle cell and give a response within milliseconds.
• G-protein-coupled receptor systems, i.e. receptors bound to the cell membrane and coupled to intracellular effector systems by a G-protein. For instance, catecholamines (the first messenger) activate β-adrenoceptors through a coupled G-protein system. This increases the activity of intracellular adenylyl cyclase, increasing the rate of formation of cyclic AMP (the second messenger), a modulator of the activity of several enzyme systems that cause the cell to act. The process takes seconds.
• Protein kinase receptors, so called because the structure incorporates a protein kinase, are targets for peptide hormones involved in the control of cell growth and differentiation, and the release of inflammatory mediators over a course of hours.
• Cytosolic (nuclear) receptors, i.e. within the cell itself, regulate DNA transcription and, thereby, protein synthesis, e.g. by steroid and thyroid hormones, a process that takes hours or days.
Drugs also act on processes within or near the cell by:
• Enzyme inhibition, e.g. platelet cyclo-oxygenase by aspirin, cholinesterase by pyridostigmine, xanthine oxidase by allopurinol.
• Inhibition or induction of transporter processes that carry substances into, across and out of cells, e.g. blockade of anion transport in the renal tubule cell by probenecid is used to protect against the nephrotoxic effects of cidofovir (used for cytomegalovirus retinitis).
• Incorporation into larger molecules, e.g. 5-fluorouracil, an anticancer drug, is incorporated into messenger RNA in place of uracil.
• In the case of successful antimicrobial agents, altering metabolic processes unique to microorganisms, e.g. penicillin interferes with formation of the bacterial cell wall; or by showing enormous quantitative differences in affecting a process common to both humans and microbes, e.g. inhibition of folic acid synthesis by trimethoprim.
Outside the cell drugs act by:
• Direct chemical interaction, e.g. chelating agents, antacids.
• Osmosis, as with purgatives, e.g. magnesium sulphate, and diuretics, e.g. mannitol, which are active because neither they nor the water in which they are dissolved is absorbed by the cells lining the gut and kidney tubules respectively.
Receptors
Radioligand binding studies have shown that the receptor numbers do not remain constant but change according to circumstances. When tissues are continuously exposed to an agonist, the number of receptors decreases (down-regulation) and this may be a cause of tachyphylaxis (loss of efficacy with frequently repeated doses), e.g. in asthmatics who use adrenoceptor agonist bronchodilators excessively. Prolonged contact with an antagonist leads to formation of new receptors (up-regulation). Indeed, one explanation for the worsening of angina pectoris or cardiac ventricular arrhythmia in some patients following abrupt withdrawal of a β-adrenoceptor blocker is that normal concentrations of circulating catecholamines now have access to an increased (up-regulated) population of β-adrenoceptors (see Chronic pharmacology, p. 98).
Enzymes
Irreversible inhibition occurs with organophosphorus insecticides and chemical warfare agents (see Chap. 10), which combine covalently with the active site of acetylcholinesterase; recovery of cholinesterase activity depends on the formation of new enzyme. Covalent binding of aspirin to cyclo-oxygenase (COX) inhibits the enzyme in platelets for their entire lifespan because platelets have no system for synthesising new protein; this is why low doses of aspirin are sufficient for antiplatelet action.
Selectivity
Modification of drug structure
Many drugs have in their design a structural similarity to some natural constituent of the body, e.g. a neurotransmitter, a hormone, a substrate for an enzyme; replacing or competing with that natural constituent achieves selectivity of action. Enormous scientific effort and expertise go into the synthesis and testing of analogues of natural substances in order to create drugs capable of obtaining a specified effect, and that alone (see Therapeutic index, below). The approach is the basis of modern drug design and it has led to the production of adrenoceptor antagonists, histamine receptor antagonists and many other important medicines.
Drug molecules are three-dimensional and many drugs contain one or more asymmetrical or chiral1 centres in their structures, i.e. a single drug can be, in effect, a mixture of two non-identical mirror images (like a mixture of left- and right-handed gloves). The two forms, which are known as enantiomorphs, can exhibit very different pharmacodynamic, pharmacokinetic and toxicological properties.
For example, (1) the S form of warfarin is four times more active than the R form,2 (2) the peak plasma concentration of S fenoprofen is four times that of R fenoprofen after oral administration of RS fenoprofen, and (3) the S, but not the R, enantiomorph of thalidomide is metabolised to primary toxins.
Quantitative aspects
Dose–response relationships
Dose–response curves for wanted and unwanted effects can illustrate and quantify selective and non-selective drug action (see Fig. 8.1).
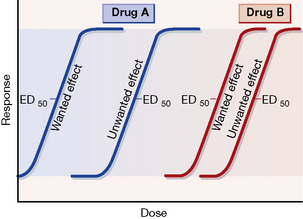
Potency and efficacy
Ehrlich (see p. 162) introduced the concept of the therapeutic index or ratio as the maximum tolerated dose divided by the minimum curative dose, but the index is never calculated thus as such single doses cannot be determined accurately in humans. More realistically, a dose that has some unwanted effect in 50% of humans, e.g. in the case of an adrenoceptor agonist bronchodilator a specified increase in heart rate, is compared with that which is therapeutic in 50% (ED50), e.g. a specified decrease in airways resistance.
In practice, such information is not available for many drugs but the therapeutic index does embody a concept that is fundamental in comparing the usefulness of one drug with another, namely, safety in relation to efficacy. Figure 8.1 expresses the concept diagrammatically.
Tolerance
Continuous or repeated administration of a drug is often accompanied by a gradual diminution of the effect it produces. A state of tolerance exists when it becomes necessary to increase the dose of a drug to get an effect previously obtained with a smaller dose, i.e. reduced sensitivity. By contrast, the term tachyphylaxis describes the phenomenon of progressive lessening of effect (refractoriness) in response to frequently administered doses (see Receptors, p. 75); it tends to develop more rapidly than tolerance.
Accelerated metabolism by enzyme induction (see p. 93) also leads to tolerance, as experience shows with alcohol, taken regularly as opposed to sporadically. There is commonly cross-tolerance between drugs of similar structure.
Failure of certain individuals to respond to normal doses of a drug, e.g. resistance to warfarin, vitamin D, constitutes a form of natural tolerance (see Pharmacogenetics, p. 101).
Pharmacokinetics
Pharmacokinetics3 is concerned with the rate at which drug molecules cross cell membranes to enter the body, to distribute within it and to leave the body, as well as with the structural changes (metabolism) to which they are subject within it.
The discussion covers the following topics:
• Drug passage across cell membranes.
• Order of reaction or process (first and zero order).
• Time course of drug concentration and effect:


• The individual processes: absorption, distribution, metabolism (biotransformation), elimination.
Drug passage across cell membranes
Passive diffusion
This is the most important means by which a drug enters the tissues and distributes through them. It refers simply to the natural tendency of any substance to move passively from an area of high concentration to one of low concentration. In the context of an individual cell, the drug moves at a rate proportional to the concentration difference across the cell membrane, i.e. it shows first-order kinetics (see p. 81); cellular energy is not required, which means that the process does not become saturated and is not inhibited by other substances.
It is useful to classify drugs in a physicochemical sense into:
• Those that are variably ionised according to environmental pH (electrolytes) (lipid soluble or water soluble).
• Those that are incapable of becoming ionised whatever the environmental pH (un-ionised, non-polar substances) (lipid soluble).
• Those that are permanently ionised whatever the environmental pH (ionised, polar substances) (water soluble).
Drugs that ionise according to environmental pH
• Acidic groups become less ionised in an acidic environment.
• Basic groups become less ionised in a basic (alkaline) environment and vice versa.
This in turn influences diffusibility because:
Permanently ionised drugs
The following are particular examples of the relevance of drug passage across membranes.
Maternal blood bathes the chorionic villi, which consist of a layer of trophoblastic cells that enclose fetal capillaries. Their large surface area and the high placental blood flow (500 mL/min) are essential for gas exchange, uptake of nutrients and elimination of waste products. Thus a lipid barrier separates the fetal and maternal bloodstreams, allowing the passage of lipid-soluble substances but excluding water-soluble compounds, especially those with a molecular weight exceeding 600.4
This exclusion is of particular importance with short-term use, e.g. tubocurarine (mol. wt. 772) (lipid insoluble) or gallamine (mol. wt. 891) used as a muscle relaxant during caesarean section do not affect the infant; with prolonged use, however, all compounds will eventually enter the fetus to some extent (see Index).
Carrier-mediated transport
Some carrier-mediated transport processes operate passively, i.e. do not require cellular energy, and this is facilitated diffusion, e.g. vitamin B12 absorption. Other, energy-requiring processes move substrates into or out of cells against a concentration gradient very effectively, i.e. by active transport; they are subject to saturation, inhibition and induction (see p. 91).
The order of reaction or process
• First-order processes by which a constant fraction of drug is transported/metabolised in unit time.
• Zero-order processes by which a constant amount of drug is transported/metabolised in unit time.
Zero-order processes (saturation kinetics)
As the amount of drug in the body rises, metabolic reactions or processes that have limited capacity become saturated. In other words, the rate of the process reaches a maximum amount at which it stays constant, e.g. due to limited activity of an enzyme, and any further increase in rate is impossible despite an increase in the dose of drug. In these circumstances, the rate of reaction is no longer proportional to dose, and exhibits rate-limited or dose-dependent5 or zero-order or saturation kinetics. In practice, enzyme-mediated metabolic reactions are the most likely to show rate limitation because the amount of enzyme present is finite and can become saturated. Passive diffusion does not become saturated. There are some important consequences of zero-order kinetics.
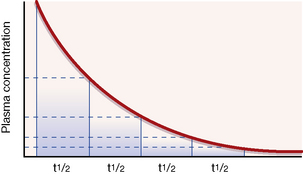
An illustration. Consider a man of average size who drinks about half (375 mL) a standard bottle of whisky (40% alcohol), i.e. 150 mL alcohol, over a short period, absorbs it and goes drunk to bed at midnight with a blood alcohol concentration of about 250 mg/dL. If alcohol metabolism were subject to first-order kinetics, with a t½ of 1 h throughout the whole range of social consumption, the subject would halve his blood alcohol concentration each hour (see Fig. 8.2). It is easy to calculate that, when he drives his car to work at 08.00 hours the next morning, he has a negligible blood alcohol concentration (less than 1 mg/dL) though, no doubt, a hangover might reduce his driving skill.
Time course of drug concentration and effect
Plasma half-life and steady-state concentration
Decrease in plasma concentration after an intravenous bolus injection
Following an intravenous bolus injection (a single dose injected in a period of seconds as distinct from a continuous infusion), plasma concentration rises quickly as drug enters the blood to reach a peak. There is then a sharp drop as the drug distributes round the body (distribution phase), followed by a steady decline as drug is removed from the blood by the liver or kidneys (elimination phase). If the elimination processes are first order, the time taken for any concentration point in the elimination phase to fall to half its value (the t½) is always the same; see Figure 8.2. Note that the drug is virtually eliminated from the plasma in five t½ periods.
Increase in plasma concentration with constant dosing
With a constant rate infusion, the amount of drug in the body and with it the plasma concentration rise until a state is reached at which the rate of administration to the body is exactly equal to the rate of elimination from it: this is called the steady state. The plasma concentration is then on a plateau, and the drug effect is stable. Figure 8.3 depicts the smooth changes in plasma concentration that result from a constant intravenous infusion. Clearly, giving a drug by regularly spaced oral or intravenous doses will result in plasma concentrations that fluctuate between peaks and troughs, but in time all of the peaks will be of equal height and all of the troughs will be of equal depth; this is also called a steady-state concentration, as the mean concentration is constant.6
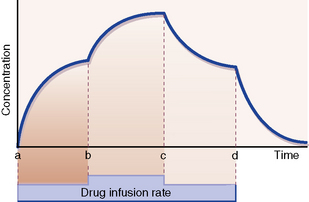
Time to reach steady state
of the ultimate steady state.
Change in plasma concentration with change or cessation of dosing
The same principle holds for change from any steady-state plasma concentration to a new steady state brought about by increase or decrease in the rate of drug administration. Provided the kinetics remain first order, increasing or decreasing the rate of drug administration (b and c in Fig. 8.3) gives rise to a new steady-state concentration in a time equal to 5 × t½ periods.
Similarly, starting at any steady-state plasma concentration (100%), discontinuing the dose (d in Fig. 8.3) will cause the plasma concentration to fall to virtually zero in 5 × t½ periods, as described in Figure 8.2.
Some t½ values appear in Table 8.1 to illustrate their range and implications for dosing in clinical practice.
| Drug | t½ |
|---|---|
| Adenosine | < 2 s |
| Dobutamine | 2 min |
| Benzylpenicillin | 30 min |
| Amoxicillin | 1 h |
| Paracetamol | 2 h |
| Midazolam | 3 h |
| Tolbutamide | 6 h |
| Atenolol | 7 h |
| Dosulepin | 25 h |
| Diazepam | 40 h |
| Piroxicam | 45 h |
| Ethosuximide | 54 h |
Therapeutic drug monitoring
Plasma concentration may correlate well with effect
Plasma concentration monitoring has proved useful:
• As a guide to the effectiveness of therapy, e.g. plasma gentamicin and other antimicrobials against sensitive bacteria, plasma theophylline for asthma, plasma ciclosporin to avoid transplant rejection, lithium for mood disorder.
• To reduce the risk of adverse drug effects when therapeutic doses are close to toxic doses (low therapeutic index), e.g. otic damage with aminoglycoside antibiotics; adverse CNS effects of lithium, nephrotoxicity with ciclosporin.
• When the desired effect is suppression of infrequent sporadic events such as epileptic seizures or episodes of cardiac arrhythmia.
• To check patient compliance on a drug regimen, when there is failure of therapeutic effect at a known effective dose, e.g. antiepilepsy drugs.
• To diagnose and manage drug overdose.
• When lack of therapeutic effect and toxicity may be difficult to distinguish. Digoxin is both a treatment for, and sometimes the cause of, cardiac supraventricular tachycardia; a plasma digoxin measurement will help to distinguish whether an arrhythmia is due to too little or too much digoxin.
Interpreting plasma concentration measurements
• The target therapeutic concentration range for a drug is a guide to optimise dosing together with other clinical indicators of progress.
• Take account of the time needed to reach steady-state dosing conditions (see above). Additionally, some drugs alter their own rates of metabolism by enzyme induction, e.g. carbamazepine and phenytoin, and it is best to allow 2–4 weeks between change in dose and meaningful plasma concentration measurement.
• As a general rule, when a drug has a short t½ it is desirable to know both peak (15 min after an intravenous dose) and trough (just before the next dose) concentrations to provide efficacy without toxicity, as with gentamicin (t½ 2.5 h). For a drug with a long t½, it is usually best to sample just before a dose is due; effective immunosuppression with ciclosporin (t½ 27 h) is obtained with trough concentrations of 50–200 micrograms/L when the drug is given by mouth.
Individual pharmacokinetic processes
Drug absorption into, distribution around, metabolism by and elimination from the body are reviewed.
Absorption
• Enteral: by mouth (swallowed) or by sublingual or buccal absorption; by rectum.
• Parenteral: by intravenous injection or infusion, intramuscular injection, subcutaneous injection or infusion, inhalation, topical application for local (skin, eye, lung) or for systemic (transdermal) effect.
• Other routes, e.g. intrathecal, intradermal, intranasal, intratracheal, intrapleural, are used when appropriate.
The features of the various routes, their advantages and disadvantages are relevant.
Absorption from the gastrointestinal tract
The small intestine is the principal site for absorption of nutrients and it is also where most orally administered drugs enter the body. This part of the gut has an enormous surface area due to the intestinal villi, and an epithelium through which fluid readily filters in response to osmotic differences caused by the presence of food. Disturbed alimentary motility can reduce drug absorption, i.e. if food slows gastric emptying, or gut infection accelerates intestinal transit. Additionally, it is becoming apparent that uptake and efflux transporters in enterocytes (see p. 93) play a substantial role in controlling the absorption of certain drugs, e.g. digoxin, ciclosporin. Many sustained-release formulations probably depend on absorption from the colon.
Systemic availability and bioavailability
Pharmaceutical factors7
Differences in bioavailability are prone to occur with modified-release (m/r) formulations, i.e. where the rate or place of release of the active ingredients has been modified (also called sustained, controlled or delayed release) (see p. 97). Modified-release preparations from different manufacturers may differ in their bioavailability profiles despite containing the same amount of drug, i.e. there is neither bioequivalence nor therapeutic equivalence, and the problem is particularly acute where the therapeutic ratio is narrow. In this case, ‘brand name prescribing’, i.e. using only a particular brand name for a particular patient is justified, e.g. for m/r preparations of theophylline, lithium, nifedipine and diltiazem.
Biological factors
Biological factors related to the gut include limitation of drug absorption by drug transporter systems (see p. 93), destruction of drug by gastric acid, e.g. benzylpenicillin, and impaired absorption due to rapid intestinal transit, which is important for all drugs that are absorbed slowly. Drugs may also bind to food constituents, e.g. tetracyclines to calcium (in milk), and to iron, or to other drugs (e.g. acidic drugs to colestyramine), and the resulting complex is not absorbed.
Advantages and disadvantages of enteral administration
By swallowing
Advantages are convenience and acceptability.
Disadvantages are that absorption may be delayed, reduced or even enhanced after food, or slow or irregular after drugs that inhibit gut motility (antimuscarinic, opioid). Differences in presystemic elimination are a cause of variation in drug effect between patients. Some drugs are not absorbed (gentamicin) and others are destroyed in the gut (insulin, oxytocin, some penicillins). Tablets taken with too small a quantity of liquid and in the supine position, can lodge in the oesophagus with delayed absorption9 and may even cause ulceration (sustained-release potassium chloride and doxycycline tablets), especially in the elderly and those with an enlarged left atrium which impinges on the oesophagus.10
Advantages and disadvantages of parenteral administration
(for systemic and local effect)
Subcutaneous injection
Advantages are that the route is reliable and is acceptable for self-administration.
Disadvantages are poor absorption in peripheral circulatory failure. Repeated injections at one site can cause lipoatrophy, resulting in erratic absorption (see Insulin, Ch. 36).
Topical application
e.g. to skin, eye, lung, anal canal, rectum, vagina.
Advantage is the provision of high local concentration without systemic effect (usually11).
Disadvantage is that absorption can occur, especially when there is tissue destruction so that systemic effects result, e.g. adrenal corticosteroids and neomycin to the skin, atropine to the eye. Ocular administration of a β-adrenoceptor blocker may cause systemic effects (bypassing first-pass elimination) and such eye drops are contraindicated in asthma or chronic lung disease.12 There is extensive literature on this subject characterised by expressions of astonishment that serious effects, even death, can occur.
Distribution
Distribution volume
The pattern of distribution from plasma to other body fluids and tissues is a characteristic of each drug that enters the circulation, and it varies between drugs. Precise information on the concentration of drug attained in various tissues and fluids is usually not available for humans.13 But blood plasma is sampled readily in humans, the drug concentration in which, taking account of the dose given, is a measure of whether a drug tends to remain in the circulation or to distribute from the plasma into the tissues. In other words:
• If a drug remains mostly in the plasma, its distribution volume will be small.
• If a drug is present mainly in other tissues, the distribution volume will be large.
Such information can be useful. In drug overdose, if a major proportion of the total body load is known to be in the plasma, i.e. the distribution volume is small, then haemodialysis/filtration is likely to be a useful option (as is the case with severe salicylate poisoning), but it is an inappropriate treatment for overdose with dosulepin (see Table 8.2).
Table 8.2 Apparent distribution volume of some drugs (values are in litres for a 70-kg person who would displace about 70 L)*

The principle for measuring the distribution volume is essentially that of using a dye to find the volume of a container filled with liquid. The weight of added dye divided by the concentration of dye once mixing is complete gives the distribution volume of the dye, which is the volume of the container. Similarly, the distribution volume of a drug in the body may be determined after a single intravenous bolus dose by dividing the dose given by the concentration achieved in plasma.14
The list in Table 8.2 illustrates a range of apparent distribution volumes. The names of those substances that distribute within (and have been used to measure) physiological spaces are printed in italics.
Plasma protein and tissue binding
may modify protein binding of drugs to an extent that is clinically relevant, as Table 8.3 shows. In chronic renal failure, hypoalbuminaemia and retention of products of metabolism that compete for binding sites on protein are both responsible for the decrease in protein binding of drugs. Most affected are acidic drugs that are highly protein bound, e.g. phenytoin, and initiating or modifying the dose of such drugs for patients with renal failure requires special attention (see also Prescribing in renal disease, p. 462).
Table 8.3 Examples of plasma protein binding of drugs and effects of disease
| Drug | % Unbound (free) |
|---|---|
| Warfarin | 1 |
| Diazepam | 2 (6% in liver disease) |
| Furosemide | 2 (6% in nephrotic syndrome) |
| Tolbutamide | 2 |
| Amitriptyline | 5 |
| Phenytoin | 9 (19% in renal disease) |
| Triamterene | 19 (40% in renal disease) |
| Trimethoprim | 30 |
| Theophylline | 35 (71% in liver disease) |
| Morphine | 65 |
| Digoxin | 75 (82% in renal disease) |
| Amoxicillin | 82 |
| Ethosuximide | 100 |
Chronic liver disease also leads to hypoalbuminaemia and an increase of endogenous substances such as bilirubin that may compete for binding sites on protein. Drugs that are normally extensively protein bound should be used with special caution, for increased free concentration of diazepam, tolbutamide and phenytoin have been demonstrated in patients with this condition (see also Prescribing for patients with liver disease, p. 547).
The free, unbound, and therefore pharmacologically active percentages of some drugs appear in Table 8.3 to illustrate the range and, in some cases, changes recorded in disease.
Some drugs distribute readily to regions of the body other than plasma, as a glance at Table 8.2 will show. These include many lipid-soluble drugs, which may enter fat stores, e.g. most benzodiazepines, verapamil and lidocaine. There is less information about other tissues, e.g. muscle, than about plasma protein binding because solid tissue samples require invasive biopsy. Extensive binding to tissues delays elimination from the body and accounts for the long t½ of chloroquine and amiodarone.
Metabolism
Altering biological activity
1. Conversion of a pharmacologically active to an inactive substance – this applies to most drugs.
2. Conversion of one pharmacologically active to another active substance – this has the effect of prolonging drug action, as shown below.
| Active drug | Active metabolite |
|---|---|
| amitriptyline | nortriptyline |
| codeine | morphine |
| chloroquine | hydroxychloroquine |
| diazepam | oxazepam |
| spironolactone | canrenone |
3. Conversion of a pharmacologically inactive to an active substance (then called a prodrug). The process then follows 1 or 2, above.
| Inactive substance | Active metabolite(s) | Comment |
|---|---|---|
| aciclovir | aciclovir triphosphate | see p. 213 |
| colecalciferol | calcitriol and alfacalcidol | highly active metabolites of vitamin D3; see p. 635 |
| cyclophosphamide | phosphoramide mustard | another metabolite, acrolein, causes the bladder toxicity; see p. 517 |
| perindopril | perindoprilat | less risk of first dose hypotension (applies to all ACE inhibitors except captopril) |
| levodopa | dopamine | levodopa, but not dopamine, can cross the blood–brain barrier |
| sulindac | sulindac sulphide | possibly reduced gastric toxicity |
| sulfasalazine | 5-aminosalicylic acid | see p. 541 |
| zidovudine | zidovudine triphosphate | see p. 217 |
The metabolic processes
The following explanation provides a background to the P450 nomenclature that accompanies accounts of the metabolism of several individual drugs in this book. The many cytochrome P450 isoenzymes15 are indicated by the letters CYP (from cytochrome P450) followed by a number denoting a family group, then a subfamily letter, and then a number for the individual enzyme within the family: for example, CYP2E1 is an isoenzyme that catalyses a reaction involved in the metabolism of alcohol, paracetamol, estradiol and ethinylestradiol.
The enzymes of families CYP1, 2 and 3 metabolise 70–80% of clinically used drugs as well as many other foreign chemicals and, within these, CYP3A, CYP2D and CYP2C are the most important. The very size and variety of the P450 superfamily ensures that we do not need new enzymes for every existing or yet-to-be synthesised drug. Induction and inhibition of P450 enzymes is a fruitful source of drug–drug interactions.16
Each P450 enzyme protein is encoded by a separate gene (57 have been identified in humans), and variation in genes leads to differences between individuals, and sometimes between ethnic groups, in the ability to metabolise drugs. Persons who exhibit polymorphisms (see p. 107) inherit diminished or increased ability to metabolise substrate drugs, predisposing to toxicity or lack of efficacy.
Note that some drug oxidation reactions do not involve the P450 system: several biologically active amines are inactivated by monoamine oxidase (see p. 319) and methylxanthines (see p. 154); mercaptopurine by xanthine oxidase (see p. 250); ethanol by alcohol dehydrogenase (see p. 143).
Most efflux transporters are members of the ATP-binding cassette (ABC) superfamily that utilises energy derived from the hydrolysis of ATP; they include the P-glycoprotein family that expresses multidrug resistance protein 1 (MDR1) (see p. 516).
• Enterocytes of the small intestine, controlling absorption and thus bioavailability, e.g. of ciclosporin, digoxin.
• Liver cells, controlling uptake from the blood and excretion into the bile, e.g. of pravastatin.
• Renal tubular cells, controlling uptake from the blood, secretion into tubular fluid (and thus excretion) of organic anions, e.g. β-lactam antibiotics, diuretics, non-steroidal anti-inflammatory drugs.
• Brain capillary endothelial cells, controlling passage across the blood–brain barrier, e.g. of levodopa (but not dopamine) for benefit in Parkinson’s disease (see p. 361).
In time, it is likely that drug occupancy of transporter processes will provide explanations for some drug-induced toxicities and for a number of drug–drug interactions.
Enzyme induction is relevant to drug therapy because:
• Clinically important drug–drug (and drug–herb18) interactions may result, for example, in failure of oral contraceptives, loss of anticoagulant control, failure of cytotoxic chemotherapy.
• Disease may result. Antiepilepsy drugs accelerate the breakdown of dietary and endogenously formed vitamin D, producing an inactive metabolite – in effect a vitamin D deficiency state, which can result in osteomalacia. The accompanying hypocalcaemia can increase the tendency to fits and a convulsion may lead to fracture of the demineralised bones.
• Tolerance to drug therapy may result in and provide an explanation for suboptimal treatment, e.g. with an antiepilepsy drug.
• Variability in response to drugs is increased. Enzyme induction caused by heavy alcohol drinking or heavy smoking may be an unrecognised cause for failure of an individual to achieve the expected response to a normal dose of a drug, e.g. warfarin, theophylline.
• Drug toxicity may occur. A patient who becomes enzyme induced by taking rifampicin is more likely to develop liver toxicity after paracetamol overdose by increased production of a hepatotoxic metabolite. (Such a patient will also present with a deceptively low plasma concentration of paracetamol due to accelerated metabolism; see p. 246).
Enzyme inhibition
The consequences of inhibiting drug metabolism can be more profound and more selective than enzyme induction because the outcome is prolongation of action of a drug or metabolite. Consequently, enzyme inhibition offers more scope for therapy (Table 8.4). Enzyme inhibition by drugs is also the basis of a number of clinically important drug interactions (see p. 107).
| Drug | Enzyme inhibited | In treatment of |
|---|---|---|
| Acetazolamide | Carbonic anhydrase | Glaucoma |
| Allopurinol | Xanthine oxidase | Gout |
| Benserazide | DOPA decarboxylase | Parkinson’s disease |
| Disulfiram | Aldehyde dehydrogenase | Alcoholism |
| Enalapril | Angiotensin-converting enzyme | Hypertension, cardiac failure |
| Moclobemide | Monoamine oxidase, A type | Depression |
| Non-steroidal anti-inflammatory drugs | cyclo-oxygenase | Pain, inflammation |
| Selegiline | Monoamine oxidase, B type | Parkinson’s disease |
Elimination
Renal elimination
The following mechanisms are involved.
The rate at which a drug enters the glomerular filtrate depends on the concentration of free drug in plasma water and on its molecular weight. Substances having a molecular weight in excess of 50 000 do not cross into the glomerular filtrate, whereas those of molecular weight less than 10 000 (which includes almost all drugs)19 pass easily through the pores of the glomerular membrane.
Uptake and efflux transporters in proximal renal tubule cells transfer organic anions and cations between the plasma and the tubular fluid (see p. 453).
Faecal elimination
When any drug intended for systemic effect is taken by mouth a proportion may remain in the bowel and be excreted in the faeces. Some drugs are intended not be absorbed from the gut, as an objective of therapy, e.g. neomycin. The cells of the intestinal epithelium contain several carrier-mediated transporters that control the absorption of drugs. The efflux transporter MDR1, for example, drives drug from the enterocyte into the gut lumen, limiting its bioavailability (see p. 93). Drug in the blood may also diffuse passively into the gut lumen, depending on its pKa and the pH difference between blood and gut contents. The effectiveness of activated charcoal by mouth for drug overdose depends partly on its adsorption of such diffused drug, and subsequent elimination in the faeces (see p. 125).
Transporters regulate the uptake of organic cations and anions from portal blood to hepatocyte, and thence to the bile (see p. 86). The bile canaliculi tend to reabsorb small molecules and in general, only compounds having a molecular weight greater than 300 pass into bile. (See also Enterohepatic circulation, p. 86.)
Drugs and breast feeding20
• Alimentary tract. Sulfasalazine may cause adverse effects and mesalazine appears preferable.
• Anti-asthma. The neonate eliminates theophylline and diprophylline slowly; observe the infant for irritability or disturbed sleep.
• Anticancer. Regard all as unsafe because of inherent toxicity.
• Antidepressants. Avoid doxepin, a metabolite of which may cause respiratory depression.
• Anti-arrhythmics (cardiac). Amiodarone is present in high and disopyramide in moderate amounts.
• Antiepilepsy. General note of caution: observe the infant for sedation and poor suckling. Primidone, ethosuximide and phenobarbital are present in milk in high amounts; phenytoin and sodium valproate less so.
• Anti-inflammatory. Regard aspirin (salicylates) as unsafe (possible association with Reye’s syndrome).
• Antimicrobials. Metronidazole is present in milk in moderate amounts; avoid prolonged exposure (though no harm recorded). Avoid nalidixic acid and nitrofurantoin where glucose-6-phosphate dehydrogenase deficiency is prevalent. Avoid clindamycin, dapsone, lincomycin, sulphonamides. Regard chloramphenicol as unsafe.
• Antipsychotics. Phenothiazines, butyrophenones and thioxanthenes are best avoided unless the indications are compelling: amounts in milk are small but animal studies suggest adverse effects on the developing nervous system. In particular, moderate amounts of sulpiride enter milk. Avoid lithium if possible.
• Anxiolytics and sedatives. Benzodiazepines are safe if use is brief but prolonged use may cause somnolence or poor suckling.
• β-Adrenoceptor blockers. Neonatal hypoglycaemia may occur. Sotalol and atenolol are present in the highest amounts in this group.
• Hormones. Oestrogens, progestogens and androgens suppress lactation in high dose. Oestrogen–progestogen oral contraceptives are present in amounts too small to be harmful, but may suppress lactation if it is not well established.
• Miscellaneous. Bromocriptine suppresses lactation. Caffeine may cause infant irritability in high doses.
Drug dosage
Drug dosage can be of five main kinds.
Dosing schedules
To specify a maintenance dose:
1. Half-life 6–12 h. In this instance, replacing half the initial dose at intervals equal to the t½ can indeed be a satisfactory solution because dosing every 6–12 h is acceptable.
2. Half-life greater than 24 h. With once-daily dosing (which is desirable for compliance), giving half the priming dose every day means that more drug is entering the body than is leaving it each day, and the drug will accumulate to give unwanted effects. The solution is to replace only the amount of drug that leaves the body in 24 h, calculated from the inital dose, dose interval, and t½.
3. Half-life less than 3 h. Dosing at intervals equal to the t½ would be so frequent as to be unacceptable. The answer is to use continuous intravenous infusion if the t½ is very short, e.g. dopamine t½ 2 min (steady-state plasma concentration will be reached in 5 × t½ = 10 min), or, if the t½ is longer, e.g. lidocaine (t½ 90 min), to use a priming dose as an intravenous bolus followed by a constant intravenous infusion. Intermittent administration of a drug with short t½ is nevertheless reasonable provided large fluctuations in plasma concentration are acceptable, i.e. that the drug has a large therapeutic index. Benzylpenicillin has a t½ of 30 min but is effective in a 6-hourly regimen because the drug is so non-toxic that it is possible safely to give a dose that achieves a plasma concentration many times in excess of the minimum inhibitory concentration for sensitive organisms.
Dose calculation by body-weight and surface area
The relationship between body surface area and weight is curvilinear, but a reasonable approximation is that a 70-kg human has a body surface area of 1.8 m2. A combination of body-weight and height gives a more precise value for surface area (obtained from standard nomograms) and other more sophisticated methods.21
The issue takes on special significance for children, if the only dose known is that for the adult; adjustment is then commonly made by body-weight, or body surface area, among other factors (see p. 104).
Prolongation of drug action
• Vasoconstriction will reduce local blood flow so that distribution of drug away from an injection site is retarded, e.g. combination with adrenaline/epinephrine prolongs local anaesthetic action.
• Slowing of metabolism may usefully extend drug action, as when a dopa decarboxylase inhibitor, e.g. carbidopa, is combined with levodopa (as co-careldopa) for parkinsonism.
• Delayed excretion is seldom practicable, the only important example being the use of probenecid to block renal tubular excretion of penicillin for single-dose treatment of gonorrhoea.
• Altered molecular structure can prolong effect, e.g. the various benzodiazepines.
• Pharmaceutical formulation. Manipulating the form in which a drug is presented by modified-release22 systems can achieve the objective of an even as well as a prolonged effect.
Fixed-dose drug combinations
• Convenience, with improved patient compliance, is appropriate with two drugs used at constant dose, long term, for an asymptomatic condition, e.g. a thiazide plus an ACE inhibitor in mild or moderate hypertension, and other antihypertensive drug combinations.
• Enhanced effect. Single-drug treatment of tuberculosis leads to the emergence of resistant mycobacteria and is prevented or delayed by using two or more drugs simultaneously. Combining isoniazid with rifampicin (Rifinah, Rimactazid) ensures that single-drug treatment cannot occur; treatment has to be two drugs or no drug at all. An oestrogen and progestogen combination provides effective oral contraception, for the same reason.
• Minimisation of unwanted effects. Levodopa combined with benserazide (Madopar) or with carbidopa (Sinemet) slows its metabolism outside the CNS so that smaller amounts of levodopa can be used, reducing its adverse effects.
Chronic pharmacology
Interference with self-regulating systems
The number (density) of receptors on cells (for hormones, autacoids or local hormones, and drugs), the number occupied (receptor occupancy) and the capacity of the receptor to respond (affinity, efficacy) can change in response to the concentration of the specific binding molecule or ligand,23 whether this be agonist or antagonist (blocker). The effects always tend to restore cell function to its normal or usual state. Prolonged high concentrations of agonist (whether administered as a drug or over-produced in the body by a tumour) cause a reduction in the number of receptors available for activation (down-regulation); changes in receptor occupancy and affinity and the prolonged occupation of receptors antagonists lead to an increase in the number of receptors (up-regulation). At least some of this may be achieved by receptors moving inside the cell and out again (internalisation and externalisation).
Down-regulation, and the accompanying receptor changes, may explain the ‘on–off’ phenomenon in Parkinson’s disease (see p. 362) and the action of luteinising hormone releasing hormone (LHRH) super-agonists in reducing follicle stimulating hormone (FSH) concentrations for treating endocrine-sensitive prostate cancer.
Abrupt withdrawal
• Cardiovascular system: β-adrenoceptor blockers, antihypertensives (especially clonidine).
• Nervous system: all depressants (hypnotics, sedatives, alcohol, opioids), antiepileptics, antiparkinsonian agents, tricyclic antidepressants.
Other aspects of chronic drug use
Conclusions
Drugs not only induce their known listed primary actions, but may:
• evoke compensatory responses in the complex interrelated physiological systems they affect, and these systems need time to recover on withdrawal of the drug (gradual withdrawal is sometimes mandatory and never harmful)
• induce metabolic changes that may be trivial in the short term, but serious if they persist for a long time
• produce localised effects in specially susceptible tissues and induce serious cell damage or malfunction
• increase susceptibility to intercurrent illness and to interaction with other drugs that may be taken for new indications.
Pharmacogenomics
Sources of variability
In general, variability in drug response can be due to pharmacokinetic and/or pharmacodynamic factors (Fig. 8.4). Variability in the expression of the cytochrome P450 enzymes, which are responsible for Phase I drug metabolism, has been the focus of most of the work in pharmacokinetics. Cytochrome P450 2D6 (CYP2D6), for example, is one of the most variable P450 enzymes in man, is absent in 8% of the UK population, and is responsible for the metabolism of 25% of drugs, including CNS (antidepressants and antipsychotics) and cardiovascular (β-blockers and anti-arrhythmics) drugs. Much less work has been done on pharmacodynamic factors causing variation in drug response, but as drugs can affect almost any protein in the body, almost every gene may have an effect on how individual drugs vary in their response. It is important to note, however, that for most drugs variability in response is due to a combination of both pharmacokinetic and pharmacodynamic factors, both of which can be affected by environmental or genetic factors. Specific examples are provided below.
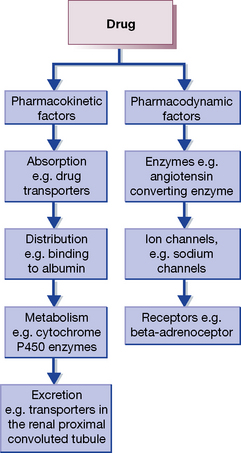
Examples of pharmacogenomic variation
Drug toxicity
Immune-mediated adverse drug reactions
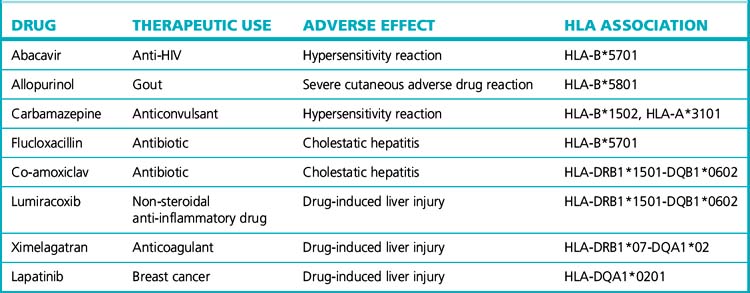
Many type B or idiosyncratic adverse drug reactions are immunologically mediated. Immune response to antigens, including those derived from drugs, is partly under the control of the HLA genes on chromosome 6, which is the polymorphic region of the human genome. Not surprisingly, HLA genes are now being found to be important determinants of susceptibility to these immune-mediated adverse reactions. The best example of this is with abacavir, a drug used to treat HIV, which causes hypersensitivity (skin rash, fever, gastrointestinal and respiratory manifestations) in 5% of patients. A strong association of abacavir hypersensitivity with the HLA allele, HLA-B*5701, has now been shown in several populations. Furthermore, genotyping for HLA-B*5701 before prescribing abacavir has been shown to reduce the frequency of hypersensitivity, and is a cost-effective approach. In Europe, it is now mandatory to undertake HLA-B*5701 testing before the prescription of abacavir. Very strong genetic associations between the HLA genes and different forms of hypersensitivity, including those affecting the skin and liver, occur with a number of drugs (Table 8.5).
Summary
There are many genetic variations identified to be risk factors for lack of efficacy or predisposition to toxicity (Table 8.6). As the technologies to interrogate the human genome improve, it is likely that more genetic tests will be introduced which will need to be used before the prescription of the drug. The net effect will be prediction of individual responses and thereby reduction in variability through better drug choices and/or drug doses.
Table 8.6 Drugs which contain pharmacogenetic information in their product labels
| Drug | Drug class | Genomic variation |
|---|---|---|
| Maraviroc | Antiretroviral, antagonist of the CC chemokine receptor 5 (CCR5) | CCR5 promoter and coding sequence polymorphisms |
| Trastuzumab (Herceptin) | Anticancer drug, anti-HER-2/neu monoclonal antibody used where there is over-expression of the human epidermal growth factor receptor-2 (HER2) | HER2/neu |
| Abacavir | Antiretroviral, nucleoside reverse transcriptase inhibitor | Human leucocyte antigen HLA-B*5701 allele |
| Carbamazepine | Antiepileptic | HLA-B*1502 in patients of Asian ancestry |
| Warfarin | Anticoagulans, vitamin K epoxide reductase inhibitor | CYP 2 C9*2 and 2 C9*3 and VKORC1 variants |
| Azathioprine | Antiproliferative immunosuppressant | Thiopurine methyltransferase (TPMT) deficiency |
| Valproic acid | Antiepileptic and antimanic drug | Urea cycle disorder (UCD) deficiency |
| Hydralazine | Vasodilator antihypertensive drug | N-acetyl transferase (NAT) |
| Rifampicin Isoniazid |
Antituberculous drug | N-acetyl transferase (NAT) |
| Voriconazole | Antifungal | CYP 2 C19 variants poor and extensive metabolisers |
| Diazepam | Anxiolytic | CYP 2 C19 variants poor and extensive metabolisers |
| Fluoxetine | Selective serotonin reuptake inhibitor | Cytochrome P450 CYP 2D6, substrate and inhibitor |
| Tramadol | Analgesic | CYP 2D6 |
| Propranolol | β-Adrenoceptor blocking drug | CYP 2D6 |
| Tamoxifen | Oestrogen-receptor antagonist | CYP 2D6 |
| Tretinoin | Acid form of vitamin A used in acute promyelocytic leukaemia | Presence of the t(15;17) translocation and/or PML/RARα gene fusion |
| Celecoxib | Non-steroidal anti-inflamatory drug, selective COX-2 inhibitor | CYP 2 C9 variants with poor metaboliser status |
| Primaquine Chloroquine |
Antimalarials | Glucose-6-phosphate dehydrogenase (G6PD) deficiency |
| Suxamethonium | Anaesthetics | Butyrylcholinesterase deficiency |
Environmental and host influences
Age
The neonate, infant and child24
• Rectal absorption is efficient with an appropriate formulation, e.g. of diazepam and theophyllines; this route may be preferred with an uncooperative infant.
• The intramuscular or subcutaneous routes tend to give unpredictable plasma concentrations, e.g. of digoxin or gentamicin, because of the relatively low proportion of skeletal muscle and fat. Intravenous administration is preferred in the seriously ill newborn.
• Drugs or other substances that come in contact with the skin are readily absorbed as the skin is well hydrated and the stratum corneum is thin; overdose toxicity may result, e.g. with hexachlorophene used in dusting powders and emulsions to prevent infection.
An understandable reluctance to test drugs extensively in children means that reliable information is often lacking. Many drugs do not have a licence to be used for children, and their prescription must be ‘off-licence’, a practice that is recognised as necessary, if not actually promoted, by the UK drug regulatory authorities.25 Attempts to correct this are underway across Europe.
Distribution
• Weight-related loading doses of aminoglycosides, aminophylline, digoxin and furosemide need to be larger for neonates than for older children.
• Less extensive binding of drugs to plasma proteins is generally without clinical importance but there is a risk of kernicterus in the jaundiced neonate following displacement of bilirubin from protein-binding sites by vitamin K, X-ray contrast media or indometacin.
Pharmacodynamic responses
There is scant information about developmental effects of interaction between drugs and receptors. Other sources suggest possible effects: e.g. thalidomide causes phocomelia only in the forming limb (see Index); tetracyclines stain only developing enamel; young children are particularly sensitive to liver toxicity from valproate.
Dosage in the young
No single rule or formula is suitable for all cases. Computation by body-weight may overdose an obese child, for whom calculation of ideal weight from age and height is preferred. Doses based on body surface area are generally more accurate, and preferably should take into account both body-weight and height.26 The fact that the surface area of a 70-kg adult human is 1.8 m2 (see p. 97) then allows adjustment, as follows:

General guidance is available from formularies, e.g. the British National Formulary, and specialist publications.27,28
The elderly
• The increasing number of drugs that the elderly need because they tend to have multiple diseases.
• Poor compliance with dosing regimens.
• Bodily changes of ageing that require modified dosage regimens.
Distribution
reflects the following changes:
• Lean body mass is less and standard adult doses provide a greater amount of drug per kilogram.
• Body fat increases and may act as a reservoir for lipid-soluble drugs.
• Total body water is less and, in general, water-soluble drugs have a lower distribution volume. Standard doses of drugs, especially the loading doses of those that are water soluble, may thus exceed the requirement.
• Plasma albumin concentration is well maintained in the healthy elderly but may fall with chronic disease, giving scope for a greater proportion of unbound (free) drug, which may be important when priming doses are given.
Metabolism
reduces as liver mass and liver blood flow decline. Consequently:
• Metabolic inactivation of drugs is slower, mostly for Phase I (oxidation) reactions; the capacity for Phase II (conjugation) is better preserved.
• Drugs normally extensively eliminated in first pass through the liver appear in higher concentration in the systemic circulation and persist in it for longer. There is, therefore, particular need initially to use lower doses of most neuroleptics, tricyclic antidepressants and cardiac anti-arrhythmic agents.
Pharmacodynamic
• Drugs that act on the CNS appear to produce an exaggerated response in relation to that expected from the plasma concentration, and sedatives and hypnotics may have a pronounced hangover effect. These drugs are also more likely to depress respiration because of reduced vital capacity and maximum breathing capacity in the elderly.
• Response to β-adrenoceptor agonists and antagonists may diminish in old age, possibly through reduced affinity for adrenoceptors, or smaller number of receptors.
• Baroreceptor sensitivity reduces, leading to greater potential for orthostatic hypotension with drugs that reduce blood pressure.
These pharmacokinetic and pharmacodynamic differences, together with broader issues particular to the elderly, influence the choice and use of drugs for this age group, as follows:
Rules of prescribing for the elderly29
1. Think about the necessity for drugs. Is the diagnosis correct and complete? Is the drug really necessary? Is there a better alternative?
2. Do not prescribe drugs that are not useful. Think carefully before giving an old person a drug that may have major side-effects, and consider alternatives, including prescribing nothing.
3. Think about the dose. Is it appropriate to possible alterations in the patient’s physiological state? Is it appropriate to the patient’s renal and hepatic function at the time?
4. Think about drug formulation. Is a tablet the most appropriate form of drug or would an injection, a suppository or a syrup be better? Is the drug suitably packaged for the elderly patient, bearing in mind any disabilities?
5. Assume any new symptoms may be due to drug side-effects or, more rarely, to drug withdrawal, unless shown to be otherwise. Rarely (if ever) treat a side-effect of one drug with another.
6. Take a careful drug history. Bear in mind the possibility of interaction with substances the patient may be taking without your knowledge, such as herbal or other non-prescribed remedies, old drugs taken from the medicine cabinet or drugs obtained from friends.
7. Use fixed combinations of drugs only when they are logical and well studied, and they either aid compliance or improve tolerance or efficacy. Few fixed combinations meet this standard.
8. When adding a new drug to the therapeutic regimen, see whether another can be withdrawn.
9. Attempt to check whether the patient’s compliance is adequate, e.g. by counting remaining tablets. Has the patient (or relatives) been properly instructed?
10. Remember that stopping a drug is as important as starting it.
The old (80 + years) are particularly intolerant of neuroleptics (given for confusion) and of diuretics (given for ankle swelling that is postural and not due to heart failure), which cause adverse electrolyte changes. Both classes of drug may result in admission to hospital of semi-comatose ‘senior citizens’ who deserve better treatment from their juniors.
Pregnancy
As pregnancy evolves, profound changes occur in physiology, including fluid and tissue composition.
Disease
Pharmacokinetic changes
Metabolism
Acute inflammatory disease of the liver (viral, alcoholic) and cirrhosis affect both the functioning of the hepatocytes and blood flow through the liver. Reduced extraction from the plasma of drugs that are normally highly cleared in first pass through the liver results in increased systemic availability of drugs such as metoprolol, labetalol and clomethiazole. Many other drugs exhibit prolonged t½ and reduced clearance in patients with chronic liver disease, e.g. diazepam, tolbutamide, rifampicin (see p. 87). Thyroid disease has the expected effects, i.e. drug metabolism accelerates in hyperthyroidism and decelerates in hypothyroidism.
Pharmacodynamic changes
• Asthmatic attacks can be precipitated by β-adrenoceptor blockers.
• Malfunctioning of the respiratory centre (raised intracranial pressure, severe pulmonary insufficiency) causes patients to be intolerant of opioids, and indeed any sedative may precipitate respiratory failure.
• Myocardial infarction predisposes to cardiac arrhythmia with digitalis glycosides or sympathomimetics.
• Myasthenia gravis is aggravated by quinine and quinidine, and myasthenics are intolerant of competitive neuromuscular blocking agents and aminoglycoside antibiotics.
Food
• The presence of food in the stomach, especially if it is fatty, delays gastric emptying and the absorption of certain drugs, e.g. ampicillin and rifampicin. More specifically, calcium, for instance in milk, interferes with absorption of tetracyclines and iron (by chelation).
• Substituting protein for fat or carbohydrate in the diet is associated with an increase in drug oxidation rates. Some specific dietary factors induce drug metabolising enzymes, e.g. alcohol, charcoal grilled (broiled) beef, cabbage and Brussels sprouts.
Protein malnutrition causes changes that are likely to influence pharmacokinetics, e.g. loss of body-weight, reduced hepatic metabolising capacity, hypoproteinaemia.
Drug interactions
For completeness, alterations in drug action caused by diet (above) are termed drug–food interactions, and those by herbs drug–herb interactions.30
Clinical importance of drug interactions
• Drugs that have a steep dose–response curve and a small therapeutic index (see p. 77) because small quantitative changes at the target site, e.g. receptor or enzyme, lead to substantial changes in effect, e.g. digoxin or lithium.
• Drugs that are known enzyme inducers or inhibitors (see pp. 93–94).
• Drugs that exhibit saturable metabolism (zero-order kinetics), when small interference with kinetics may lead to large alteration of plasma concentration, e.g. phenytoin, theophylline.
• Drugs that are used long term, where precise plasma concentrations are required, e.g. oral contraceptives, antiepilepsy drugs, cardiac anti-arrhythmia drugs, lithium.
• Severely ill patients, for they may be receiving several drugs; signs of iatrogenic disease may be difficult to distinguish from those of existing disease and the patient’s condition may be such that they cannot tolerate further adversity.
• Patients who have significantly impaired liver or kidney function, for these are the principal organs that terminate drug action.
• The elderly, for they tend to have multiple pathology, and may receive several drugs concurrently (see p. 105).
Pharmacological basis of drug interactions
Drug interactions are of two principal kinds:
• Pharmacodynamic interaction: both drugs act on the target site of clinical effect, exerting synergism (below) or antagonism. The drugs may act on the same or different receptors or processes, mediating similar biological consequences. Examples include: alcohol + benzodiazepine (to produce enhanced sedation), atropine + β-adrenoceptor blocker (to indirectly reverse β-adrenoceptor blocker overdose).
• Pharmacokinetic interaction: the drugs interact remotely from the target site to alter plasma (and other tissue) concentrations so that the amount of the drug at the target site of clinical effect is altered, e.g. enzyme induction by rifampicin reduces the plasma concentration of warfarin; enzyme inhibition by ciprofloxacin increases the concentration of theophylline.
Interaction may result in antagonism or synergism.
Synergism31
1. Summation or addition occurs when the effects of two drugs having the same action are additive, i.e. 2 + 2 = 4 (a β-adrenoceptor blocker plus a thiazide diuretic have an additive antihypertensive effect).
2. Potentiation (to make more powerful) occurs when one drug increases the action of another, i.e. 2 + 2 = 5. Sometimes the two drugs both have the action concerned (trimethoprim plus sulfonamide), and sometimes one drug lacks the action concerned (benserazide plus levodopa), i.e. 0 + 2 = 3.
In broad terms, it is useful to distinguish the drug–drug interactions that occur:
During distribution
Carrier-mediated transporters control processes such as bioavailability, passage into the CNS, hepatic uptake and entry into bile, and renal tubular excretion (see Index). Inhibitors and inducers of drug transporters can profoundly influence the disposition of drugs. The transporter MDR1 controls the entry of digoxin into cells; quinidine, verapamil and ciclosporin inhibit this transporter and increase the plasma concentration of digoxin (with potentially toxic effects). Probenecid inhibits the organic anion renal transporter, which decreases the renal clearance of penicillin (usefully prolonging its effect) but also that of methotrexate (with danger of toxicity). Elucidation of the location and function of transport systems will give the explanation for, and allow the prediction of, many more drug–drug interactions.
During metabolism
Enzyme induction (see p. 93) and, even more powerfully, enzyme inhibition (see p. 94) are important sources of drug–drug interaction.
Alfirevic A., Pirmohamed M. Drug-induced hypersensitivity reactions and pharmacogenomics: past, present and future. Pharmacogenomics. 2010;11:497–499.
Callellini M.D., Fiorelli G. Glucose-6-phosphate dehydrogenase. deficiency. Lancet. 2008;371:64–72.
Daly A.K. Genome-wide association studies in pharmacogenomics. Nat. Rev. Genet.. 2010;11:241–246.
Han P.Y., Duffull S.B., Kirkpatrick C.M.J., Green B. Dosing in obesity: a simple answer to a big problem. Clin. Pharmacol. Ther.. 2007;82:505–508.
Ito S. Drug therapy for breast-feeding women. N. Engl. J. Med.. 2000;343:118–126.
Link E., Parish S., Armitage J., et al. SLCO1B1 variants and statin-induced myopathy – a genomewide study. N. Engl. J. Med.. 2008;359:789–799.
Peck C.C., Cross J.T. ‘Getting the dose right’: facts, a blueprint, and encouragements. Clin. Pharmacol. Ther.. 2007;82:12–14.
Phillips E.J., Mallal S.A. Pharmacogenetics of drug hypersensitivity. Pharmacogenomics. 2010;11:973–987.
Ping P. Getting to the heart of proteomics. N. Engl. J. Med.. 2009;360:532–534.
Pirmohamed M., James S., Meakin S., et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. Br. Med. J.. 2004;329:15–19.
Sim S.C., Ingelman-Sundberg M. Pharmacogenomic biomarkers: new tools in current and future drug therapy. Trends Pharmacol. Sci.. 2011;32:72–81.
Strauss S.E. Geriatric medicine. Br. Med. J.. 2001;322:86–88.
Tucker G.T. Chiral switches. Lancet. 2000;355:1085–1087.
Wadelius M., Pirmohamed M. Pharmacogenetics of warfarin: current status and future challenges. Pharmacogenomics J.. 2007;7:99–111.
2 R (rectus) and S (sinister) refer to the sequential arrangement of the constituent parts of the molecule around the chiral centre.
3 Greek: pharmacon = drug; kinein = to move.
4 Most drugs have a molecular weight of less than 600 (e.g. diazepam 284, morphine 303) but some have more (erythromycin 733, digoxin 780).
5 We quote all of these terms since they appear in the relevant literature. Note: because the rate of a reaction is constant when it is zero order, it is dose independent, but as zero order is approached, with increasing dose the kinetics alter, and thus are called dose dependent.
6 The peaks and troughs can be of practical importance with drugs of low therapeutic index, e.g. aminoglycoside antibiotics; it may be necessary to monitor for both safe and effective therapy.
7 Some definitions of enteral dose forms. Tablet: a solid dose form in which the drug is compressed or moulded with pharmacologically inert substances (excipients); variants include sustained-release and coated tablets. Capsule: the drug is provided in a gelatin shell or container. Mixture: a liquid formulation of a drug for oral administration. Suppository: a solid dose form shaped for insertion into rectum (or vagina, when it may be called a pessary); it may be designed to dissolve or it may melt at body temperature (in which case there is a storage problem in countries where the environmental temperature may exceed 37°C); the vehicle in which the drug is carried may be fat, glycerol with gelatin, or macrogols (polycondensation products of ethylene oxide) with gelatin. Syrup: the drug is provided in a concentrated sugar (fructose or other) solution. Linctus: a viscous liquid formulation, traditional for cough.
8 For a more detailed list, see Wilkinson G R 2005 Drug metabolism and variability among patients in drug response. New England Journal of Medicine 352:2211–2221.
9 A woman’s failure to respond to antihypertensive medication was explained when she was observed to choke on drinking. Investigation revealed a large pharyngeal pouch that was full of tablets and capsules. Her blood pressure became easy to control when the pouch was removed. Birch D J, Dehn T C B 1993 British Medical Journal 306:1012.
10 Ideally solid dose forms should be taken while standing up, and washed down with 150 mL (a teacup) of water; even sitting (higher intra-abdominal pressure) impairs passage. At least, patients should be told to sit and take three or four mouthfuls of water (a mouthful = 30 mL) or a cupful. Some patients do not even know they should take water.
11 A cautionary tale. A 70-year-old man reported left breast enlargement and underwent mastectomy; histological examination revealed benign gynaecomastia. Ten months later the right breast enlarged. Tests of endocrine function were normal but the patient himself was struck by the fact that his wife had been using a vaginal cream (containing 0.01% dienestrol), initially for atrophic vaginitis but latterly the cream had been used to facilitate sexual intercourse which took place two or three times a week. On the assumption that penile absorption of oestrogen was responsible for the disorder, exposure to the cream was terminated. The gynaecomastia in the remaining breast then resolved (DiRaimondo C V, Roach A C, Meador C K 1980 Gynecomastia from exposure to vaginal estrogen cream. New England Journal of Medicine 302:1089–1090).
12 Two drops of 0.5% timolol solution, one to each eye, can equate to 10 mg by mouth.
13 But positive emission tomography (PET) offers a prospect of obtaining similar information. With PET, a positron emitting isotope, e.g.15O, is substituted for a stable atom without altering the chemical behaviour of the molecule. The radiation dose is very low but can be imaged tomographically using photomultiplier–scintillator detectors. PET can be used to monitor effects of drugs on metabolism in the brain, e.g. ‘on’ and ‘off’ phases in parkinsonism. There are many other applications.
14 Clearly a problem arises in that the plasma concentration is not constant but falls after the bolus has been injected. To get round this, use is made of the fact that the relation between the logarithm of plasma concentration and the time after a single intravenous dose is a straight line. The log concentration–timeline extended back to zero time gives the theoretical plasma concentration at the time the drug was given. In effect, the assumption is made that drug distributes instantaneously and uniformly through a single compartment, the distribution volume. This mechanism, although rather theoretical, does usefully characterise drugs according to the extent to which they remain in or distribute out from the circulation.
15 An isoenzyme is one of a group of enzymes that catalyse the same reaction but differ in protein structure.
16 In this expanding field, useful lists of substrate drugs for P450 enzymes with inducers and inhibitors can be found in reviews, e.g. Wilkinson G R 2005 Drug metabolism and variability among patients in drug response. New England Journal of Medicine 352:2211–2221, already cited.
17 Parts of this section are based on the review by Ho R H, Kim R B 2005 Transporters and drug therapy: implications for drug disposition and disease. Clinical Pharmacology and Therapeutics 78:260–277.
18 Tirona R G, Bailey D G 2006 Herbal product–drug interactions mediated by induction. British Journal of Clinical Pharmacology 61:677–681.
19 Most drugs have a molecular weight of less than 1000.
20 Bennett P N (ed) 1996 Drugs and Human Lactation. Elsevier, Amsterdam.
21 For example, Livingston E H, Lee S 2001 Body surface area prediction in normal-weight and obese patients. American Journal of Physiology Endocrinology and Metabolism 281:586–591.
22 The term modified covers several drug delivery systems. Delayed release: available other than immediately after administration (mesalazine in the colon); sustained release: slow release as governed by the delivery system (iron, potassium); controlled release: at a constant rate to maintain unvarying plasma concentration (nitrate, hormone replacement therapy).
24 A neonate is under 1 month and an infant is 1–12 months of age.
25 Stephenson T 2006 The medicines for children agenda in the UK. British Journal of Clinical Pharmacology 61:716–719.
26 For example, Insley J 1996 A Paediatric Vade-Mecum, 13th edn. Arnold, London.
27 Neonatal and Paediatric Pharmacists Group, Royal College of Paediatrics and Child Health 2001 Pocket Medicines for Children. Royal College of Paediatrics and Child Health Publications, London.
28 For practical advice, see World Health Organization 2005 Pocket Book of Hospital Care for Children. WHO, Geneva.
29 By permission from Caird F I (ed) 1985 Drugs for the elderly. WHO (Europe), Copenhagen.
30 Hu Z, Yang X, Ho P C et al 2005 Herb–drug interactions: a literature review. Drugs 65:1239–1282.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 8 General pharmacology
• Qualitative aspects: receptors, enzymes, selectivity.
• Quantitative aspects: dose response, potency, therapeutic efficacy, tolerance.
• Time course of drug concentration: drug passage across cell membranes; order of reaction; plasma half-life and steady-state concentration; therapeutic drug monitoring.
• Individual processes: absorption, distribution, metabolism, elimination.
• Drug dosage: dosing schedules.
• Chronic pharmacology: the consequences of prolonged drug administration and drug discontinuation syndromes.
Pharmacodynamics
Qualitative aspects
Mechanisms
• Ligand-gated ion channels, i.e. receptors coupled directly to membrane ion channels; neurotransmitters act on such receptors in the postsynaptic membrane of a nerve or muscle cell and give a response within milliseconds.
• G-protein-coupled receptor systems, i.e. receptors bound to the cell membrane and coupled to intracellular effector systems by a G-protein. For instance, catecholamines (the first messenger) activate β-adrenoceptors through a coupled G-protein system. This increases the activity of intracellular adenylyl cyclase, increasing the rate of formation of cyclic AMP (the second messenger), a modulator of the activity of several enzyme systems that cause the cell to act. The process takes seconds.
• Protein kinase receptors, so called because the structure incorporates a protein kinase, are targets for peptide hormones involved in the control of cell growth and differentiation, and the release of inflammatory mediators over a course of hours.
• Cytosolic (nuclear) receptors, i.e. within the cell itself, regulate DNA transcription and, thereby, protein synthesis, e.g. by steroid and thyroid hormones, a process that takes hours or days.
Drugs also act on processes within or near the cell by:
• Enzyme inhibition, e.g. platelet cyclo-oxygenase by aspirin, cholinesterase by pyridostigmine, xanthine oxidase by allopurinol.
• Inhibition or induction of transporter processes that carry substances into, across and out of cells, e.g. blockade of anion transport in the renal tubule cell by probenecid is used to protect against the nephrotoxic effects of cidofovir (used for cytomegalovirus retinitis).
• Incorporation into larger molecules, e.g. 5-fluorouracil, an anticancer drug, is incorporated into messenger RNA in place of uracil.
• In the case of successful antimicrobial agents, altering metabolic processes unique to microorganisms, e.g. penicillin interferes with formation of the bacterial cell wall; or by showing enormous quantitative differences in affecting a process common to both humans and microbes, e.g. inhibition of folic acid synthesis by trimethoprim.
Outside the cell drugs act by:
• Direct chemical interaction, e.g. chelating agents, antacids.
• Osmosis, as with purgatives, e.g. magnesium sulphate, and diuretics, e.g. mannitol, which are active because neither they nor the water in which they are dissolved is absorbed by the cells lining the gut and kidney tubules respectively.
Receptors
Radioligand binding studies have shown that the receptor numbers do not remain constant but change according to circumstances. When tissues are continuously exposed to an agonist, the number of receptors decreases (down-regulation) and this may be a cause of tachyphylaxis (loss of efficacy with frequently repeated doses), e.g. in asthmatics who use adrenoceptor agonist bronchodilators excessively. Prolonged contact with an antagonist leads to formation of new receptors (up-regulation). Indeed, one explanation for the worsening of angina pectoris or cardiac ventricular arrhythmia in some patients following abrupt withdrawal of a β-adrenoceptor blocker is that normal concentrations of circulating catecholamines now have access to an increased (up-regulated) population of β-adrenoceptors (see Chronic pharmacology, p. 98).
Enzymes
Irreversible inhibition occurs with organophosphorus insecticides and chemical warfare agents (see Chap. 10), which combine covalently with the active site of acetylcholinesterase; recovery of cholinesterase activity depends on the formation of new enzyme. Covalent binding of aspirin to cyclo-oxygenase (COX) inhibits the enzyme in platelets for their entire lifespan because platelets have no system for synthesising new protein; this is why low doses of aspirin are sufficient for antiplatelet action.
Selectivity
Modification of drug structure
Many drugs have in their design a structural similarity to some natural constituent of the body, e.g. a neurotransmitter, a hormone, a substrate for an enzyme; replacing or competing with that natural constituent achieves selectivity of action. Enormous scientific effort and expertise go into the synthesis and testing of analogues of natural substances in order to create drugs capable of obtaining a specified effect, and that alone (see Therapeutic index, below). The approach is the basis of modern drug design and it has led to the production of adrenoceptor antagonists, histamine receptor antagonists and many other important medicines.
Drug molecules are three-dimensional and many drugs contain one or more asymmetrical or chiral1 centres in their structures, i.e. a single drug can be, in effect, a mixture of two non-identical mirror images (like a mixture of left- and right-handed gloves). The two forms, which are known as enantiomorphs, can exhibit very different pharmacodynamic, pharmacokinetic and toxicological properties.
For example, (1) the S form of warfarin is four times more active than the R form,2 (2) the peak plasma concentration of S fenoprofen is four times that of R fenoprofen after oral administration of RS fenoprofen, and (3) the S, but not the R, enantiomorph of thalidomide is metabolised to primary toxins.
Quantitative aspects
Dose–response relationships
Dose–response curves for wanted and unwanted effects can illustrate and quantify selective and non-selective drug action (see Fig. 8.1).
Potency and efficacy
Ehrlich (see p. 162) introduced the concept of the therapeutic index or ratio as the maximum tolerated dose divided by the minimum curative dose, but the index is never calculated thus as such single doses cannot be determined accurately in humans. More realistically, a dose that has some unwanted effect in 50% of humans, e.g. in the case of an adrenoceptor agonist bronchodilator a specified increase in heart rate, is compared with that which is therapeutic in 50% (ED50), e.g. a specified decrease in airways resistance.
In practice, such information is not available for many drugs but the therapeutic index does embody a concept that is fundamental in comparing the usefulness of one drug with another, namely, safety in relation to efficacy. Figure 8.1 expresses the concept diagrammatically.
Tolerance
Continuous or repeated administration of a drug is often accompanied by a gradual diminution of the effect it produces. A state of tolerance exists when it becomes necessary to increase the dose of a drug to get an effect previously obtained with a smaller dose, i.e. reduced sensitivity. By contrast, the term tachyphylaxis describes the phenomenon of progressive lessening of effect (refractoriness) in response to frequently administered doses (see Receptors, p. 75); it tends to develop more rapidly than tolerance.
Accelerated metabolism by enzyme induction (see p. 93) also leads to tolerance, as experience shows with alcohol, taken regularly as opposed to sporadically. There is commonly cross-tolerance between drugs of similar structure.
Failure of certain individuals to respond to normal doses of a drug, e.g. resistance to warfarin, vitamin D, constitutes a form of natural tolerance (see Pharmacogenetics, p. 101).
Pharmacokinetics
Pharmacokinetics3 is concerned with the rate at which drug molecules cross cell membranes to enter the body, to distribute within it and to leave the body, as well as with the structural changes (metabolism) to which they are subject within it.
The discussion covers the following topics:
• Drug passage across cell membranes.
• Order of reaction or process (first and zero order).
• Time course of drug concentration and effect:
• The individual processes: absorption, distribution, metabolism (biotransformation), elimination.
Drug passage across cell membranes
Passive diffusion
This is the most important means by which a drug enters the tissues and distributes through them. It refers simply to the natural tendency of any substance to move passively from an area of high concentration to one of low concentration. In the context of an individual cell, the drug moves at a rate proportional to the concentration difference across the cell membrane, i.e. it shows first-order kinetics (see p. 81); cellular energy is not required, which means that the process does not become saturated and is not inhibited by other substances.
It is useful to classify drugs in a physicochemical sense into:
• Those that are variably ionised according to environmental pH (electrolytes) (lipid soluble or water soluble).
• Those that are incapable of becoming ionised whatever the environmental pH (un-ionised, non-polar substances) (lipid soluble).
• Those that are permanently ionised whatever the environmental pH (ionised, polar substances) (water soluble).
Drugs that ionise according to environmental pH
• Acidic groups become less ionised in an acidic environment.
• Basic groups become less ionised in a basic (alkaline) environment and vice versa.
This in turn influences diffusibility because:
Permanently ionised drugs
The following are particular examples of the relevance of drug passage across membranes.
Maternal blood bathes the chorionic villi, which consist of a layer of trophoblastic cells that enclose fetal capillaries. Their large surface area and the high placental blood flow (500 mL/min) are essential for gas exchange, uptake of nutrients and elimination of waste products. Thus a lipid barrier separates the fetal and maternal bloodstreams, allowing the passage of lipid-soluble substances but excluding water-soluble compounds, especially those with a molecular weight exceeding 600.4
This exclusion is of particular importance with short-term use, e.g. tubocurarine (mol. wt. 772) (lipid insoluble) or gallamine (mol. wt. 891) used as a muscle relaxant during caesarean section do not affect the infant; with prolonged use, however, all compounds will eventually enter the fetus to some extent (see Index).
Carrier-mediated transport
Some carrier-mediated transport processes operate passively, i.e. do not require cellular energy, and this is facilitated diffusion, e.g. vitamin B12 absorption. Other, energy-requiring processes move substrates into or out of cells against a concentration gradient very effectively, i.e. by active transport; they are subject to saturation, inhibition and induction (see p. 91).
The order of reaction or process
• First-order processes by which a constant fraction of drug is transported/metabolised in unit time.
• Zero-order processes by which a constant amount of drug is transported/metabolised in unit time.
Zero-order processes (saturation kinetics)
As the amount of drug in the body rises, metabolic reactions or processes that have limited capacity become saturated. In other words, the rate of the process reaches a maximum amount at which it stays constant, e.g. due to limited activity of an enzyme, and any further increase in rate is impossible despite an increase in the dose of drug. In these circumstances, the rate of reaction is no longer proportional to dose, and exhibits rate-limited or dose-dependent5 or zero-order or saturation kinetics. In practice, enzyme-mediated metabolic reactions are the most likely to show rate limitation because the amount of enzyme present is finite and can become saturated. Passive diffusion does not become saturated. There are some important consequences of zero-order kinetics.
An illustration. Consider a man of average size who drinks about half (375 mL) a standard bottle of whisky (40% alcohol), i.e. 150 mL alcohol, over a short period, absorbs it and goes drunk to bed at midnight with a blood alcohol concentration of about 250 mg/dL. If alcohol metabolism were subject to first-order kinetics, with a t½ of 1 h throughout the whole range of social consumption, the subject would halve his blood alcohol concentration each hour (see Fig. 8.2). It is easy to calculate that, when he drives his car to work at 08.00 hours the next morning, he has a negligible blood alcohol concentration (less than 1 mg/dL) though, no doubt, a hangover might reduce his driving skill.
Time course of drug concentration and effect
Plasma half-life and steady-state concentration
Decrease in plasma concentration after an intravenous bolus injection
Following an intravenous bolus injection (a single dose injected in a period of seconds as distinct from a continuous infusion), plasma concentration rises quickly as drug enters the blood to reach a peak. There is then a sharp drop as the drug distributes round the body (distribution phase), followed by a steady decline as drug is removed from the blood by the liver or kidneys (elimination phase). If the elimination processes are first order, the time taken for any concentration point in the elimination phase to fall to half its value (the t½) is always the same; see Figure 8.2. Note that the drug is virtually eliminated from the plasma in five t½ periods.
Increase in plasma concentration with constant dosing
With a constant rate infusion, the amount of drug in the body and with it the plasma concentration rise until a state is reached at which the rate of administration to the body is exactly equal to the rate of elimination from it: this is called the steady state. The plasma concentration is then on a plateau, and the drug effect is stable. Figure 8.3 depicts the smooth changes in plasma concentration that result from a constant intravenous infusion. Clearly, giving a drug by regularly spaced oral or intravenous doses will result in plasma concentrations that fluctuate between peaks and troughs, but in time all of the peaks will be of equal height and all of the troughs will be of equal depth; this is also called a steady-state concentration, as the mean concentration is constant.6
Time to reach steady state
of the ultimate steady state.
Change in plasma concentration with change or cessation of dosing
The same principle holds for change from any steady-state plasma concentration to a new steady state brought about by increase or decrease in the rate of drug administration. Provided the kinetics remain first order, increasing or decreasing the rate of drug administration (b and c in Fig. 8.3) gives rise to a new steady-state concentration in a time equal to 5 × t½ periods.
Similarly, starting at any steady-state plasma concentration (100%), discontinuing the dose (d in Fig. 8.3) will cause the plasma concentration to fall to virtually zero in 5 × t½ periods, as described in Figure 8.2.
Some t½ values appear in Table 8.1 to illustrate their range and implications for dosing in clinical practice.
| Drug | t½ |
|---|---|
| Adenosine | < 2 s |
| Dobutamine | 2 min |
| Benzylpenicillin | 30 min |
| Amoxicillin | 1 h |
| Paracetamol | 2 h |
| Midazolam | 3 h |
| Tolbutamide | 6 h |
| Atenolol | 7 h |
| Dosulepin | 25 h |
| Diazepam | 40 h |
| Piroxicam | 45 h |
| Ethosuximide | 54 h |
Therapeutic drug monitoring
Plasma concentration may correlate well with effect
Plasma concentration monitoring has proved useful:
• As a guide to the effectiveness of therapy, e.g. plasma gentamicin and other antimicrobials against sensitive bacteria, plasma theophylline for asthma, plasma ciclosporin to avoid transplant rejection, lithium for mood disorder.
• To reduce the risk of adverse drug effects when therapeutic doses are close to toxic doses (low therapeutic index), e.g. otic damage with aminoglycoside antibiotics; adverse CNS effects of lithium, nephrotoxicity with ciclosporin.
• When the desired effect is suppression of infrequent sporadic events such as epileptic seizures or episodes of cardiac arrhythmia.
• To check patient compliance on a drug regimen, when there is failure of therapeutic effect at a known effective dose, e.g. antiepilepsy drugs.
• To diagnose and manage drug overdose.
• When lack of therapeutic effect and toxicity may be difficult to distinguish. Digoxin is both a treatment for, and sometimes the cause of, cardiac supraventricular tachycardia; a plasma digoxin measurement will help to distinguish whether an arrhythmia is due to too little or too much digoxin.
Interpreting plasma concentration measurements
• The target therapeutic concentration range for a drug is a guide to optimise dosing together with other clinical indicators of progress.
• Take account of the time needed to reach steady-state dosing conditions (see above). Additionally, some drugs alter their own rates of metabolism by enzyme induction, e.g. carbamazepine and phenytoin, and it is best to allow 2–4 weeks between change in dose and meaningful plasma concentration measurement.
• As a general rule, when a drug has a short t½ it is desirable to know both peak (15 min after an intravenous dose) and trough (just before the next dose) concentrations to provide efficacy without toxicity, as with gentamicin (t½ 2.5 h). For a drug with a long t½, it is usually best to sample just before a dose is due; effective immunosuppression with ciclosporin (t½ 27 h) is obtained with trough concentrations of 50–200 micrograms/L when the drug is given by mouth.
Individual pharmacokinetic processes
Drug absorption into, distribution around, metabolism by and elimination from the body are reviewed.
Absorption
• Enteral: by mouth (swallowed) or by sublingual or buccal absorption; by rectum.
• Parenteral: by intravenous injection or infusion, intramuscular injection, subcutaneous injection or infusion, inhalation, topical application for local (skin, eye, lung) or for systemic (transdermal) effect.
[/not-level-membership-for-basic-science-category]
