Chapter 685 General Considerations
The genetically and clinically heterogeneous group of disorders of skeletal development and growth are referred to as skeletal dysplasias, bone dysplasias, and osteochondrodysplasias. Their prevalence is estimated to be about 1/4,000 births. They can be divided into the osteodysplasias typified by osteogenesis imperfecta (Chapter 692) and the chondrodysplasias. The latter result from mutations of genes that are essential for skeletal development and growth. The clinical picture is dominated by skeletal abnormalities. The manifestations may be restricted to the skeleton, but in most cases nonskeletal tissues are also involved. The disorders may be lethal in utero or mild with features that go undetected.
The chondrodysplasias are distinguished from other forms of short stature by a disproportionality of skeletal manifestations. The importance of cartilage in bone formation is noted in Figure 685-1. There are two basic categories: predominantly with short limbs and predominantly with short trunks. Efforts to define the extent of clinical heterogeneity resulted in the delineation of >100 distinct entities. Many of these disorders result from mutations of a relatively small group of genes, the “chondrodysplasia genes.” An International Working Group on Bone Dysplasias has named and classified these disorders into groups based on genetic cause if known or on similarities of clinical and radiographic manifestations, which often imply a common pathogenesis and a common genetic basis, if the cause is unknown (Table 685-1). The better-defined chondrodysplasia groups, such as the achondroplasia and type II collagenopathy groups, contain graded series of disorders that range from very severe to very mild. This may be true for other groups as more mutations are found and the full spectrum of clinical phenotypes associated with mutations of a given gene is defined. These disorders are clinical phenotypes distributed along spectra of phenotypic abnormality associated with mutations of particular genes. For mutations of some genes such as COL2A1, the distribution is fairly continuous, with clinical phenotypes merging into one another across a broad range. There is much less clinical overlap for mutations of some other genes, such as FGFR3, in which the distribution is discontinuous. Because most clinicians and most reference materials refer to the disorders as distinct entities, this vernacular continues to be used.
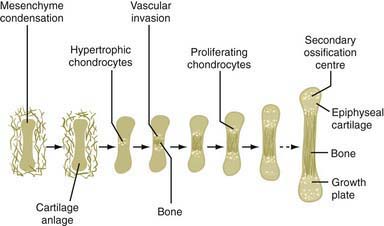
Figure 685-1 The importance of cartilage in bone formation.
(From Horton WA: Skeletal development: Insights from targeting the mouse genome, Lancet 362:560, 2005.)
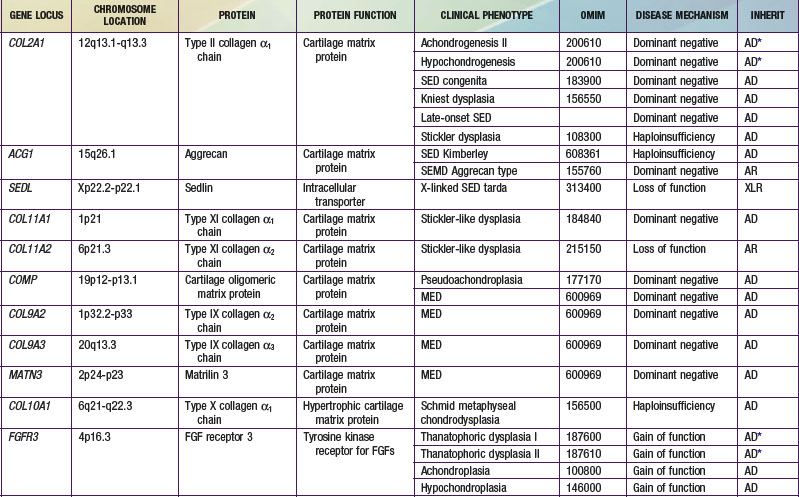
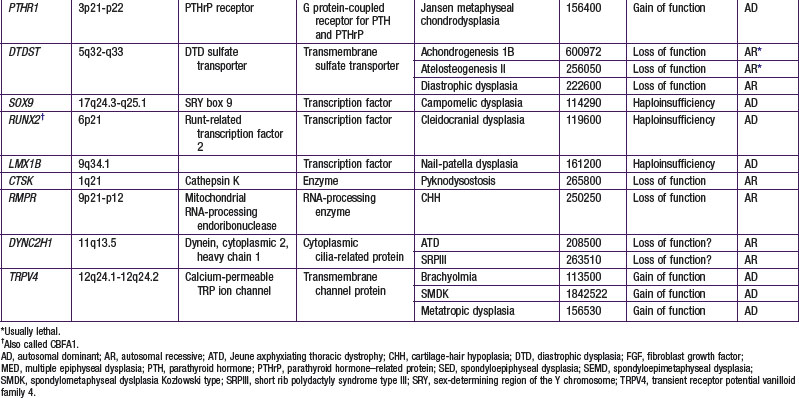
Most chondrodysplasias require the analysis of information from the history, physical examination, skeletal radiographs, family history, and laboratory testing to make a diagnosis. The process involves recognizing complex patterns that are characteristic of the different disorders (Tables 685-2, 685-3, 685-4, and 685-5). Comprehensive descriptions of disorders and references are at the Online Mendelian Inheritance in Man (OMIM) Internet site (see the references).
Table 685-2 MAJOR PROBLEMS ASSOCIATED WITH SKELETAL DYSPLASIAS
| PROBLEM | EXAMPLE |
|---|---|
| Lethality* | Thanatophoric dysplasia |
| Associated anomalies† | Ellis-van Creveld syndrome |
| Short stature | Common to almost all |
| Cervical spine dislocations | Larsen syndrome |
| Severe limb bowing | Metaphyseal dysplasia, Schmid type |
| Spine curvatures | Metatropic dysplasia |
| Clubfeet | Diastrophic dysplasia |
| Fractures | Osteogenesis imperfecta |
| Pneumonias, aspirations | Campomelic dysplasia |
| Spinal cord compression | Achondroplasia |
| Joint problems (hips, knees) | Most skeletal dysplasias |
| Hearing loss | Common (greatest with cleft palate) |
| Myopia/cataracts | Stickler syndrome |
| Immunodeficiency‡ | Cartilage-hair hypoplasia, Schimke immuno-osseous dysplasia |
| Poor body image | Variable, but common to all |
| Sex reversal | Campomelic dysplasia |
* Mostly due to severely reduced size of thorax.
‡ At least four additional disorders, all involving the metaphyses, can have immunodeficiency.
Table 685-3 ASSOCIATED ANOMALIES IN SKELETAL DYSPLASIAS
| ANOMALY | EXAMPLE |
|---|---|
| Heart defects | Ellis-van Creveld syndrome, Jeune syndrome |
| Polydactyly | Short rib polydactyly, Majewski type |
| Cleft palate | Diastrophic dysplasia |
| Ear cysts | Diastrophic dysplasia |
| Spinal cord compression | Achondroplasia |
| Encephalocele | Dyssegmental dysplasia |
| Hemivertebrae | Dyssegmental dysplasia |
| Micrognathia | Campomelic dysplasia |
| Nail dysplasia | Ellis-van Creveld syndrome |
| Conical teeth, oligodontia | Ellis-van Creveld syndrome |
| Multiple oral frenulae | Ellis-van Creveld syndrome |
| Dentinogenesis imperfecta | Osteogenesis imperfecta |
| Pretibial skin dimples | Campomelic dysplasia |
| Cataracts, retinal detachment | Stickler syndrome |
| Intestinal atresia | Saldino-Noonan |
| Renal cysts | Saldino-Noonan |
| Campodactyly | Diastrophic dysplasia |
| Craniosynostosis | Thanatophoric dysplasia |
| Ichthyosis | Chondrodystrophica punctata |
| Hitchhiker thumb | Diastrophic dysplasia |
| Sparse scalp hair | Cartilage-hair hypoplasia |
| Hypertelorism | Robinow syndrome |
| Hypoplastic nasal bridge | Acrodysostosis |
| Clavicular agenesis | Cleidocranial dysplasia |
| Genital hypoplasia | Robinow syndrome |
| Tail | Metatropic dysplasia |
| Omphalocele | Beemer-Langer syndrome |
| Blue sclera | Osteogenesis imperfecta |
Table 685-4 LETHAL NEONATAL DWARFISM
USUALLY FATAL*
OFTEN FATAL
OCCASIONALLY FATAL
* A few prolonged survivors have been reported in most of these disorders.
Table 685-5 USUALLY NONLETHAL DWARFING CONDITIONS RECOGNIZABLE AT BIRTH OR WITHIN FIRST FEW MONTHS OF LIFE
MOST COMMON
LESS COMMON
Clinical Manifestations
Growth Related
With some exceptions, there is a strong correlation between the age at onset and the clinical severity. Many of the lethal neonatal chondrodysplasias are evident during routine fetal ultrasound examinations performed at the end of the 1st trimester of gestation (see Table 685-4). Gestational standards exist for long-bone lengths; discrepancies are often detected between biparietal diameter of the skull and long-bone lengths. Many disorders become apparent around the time of birth; others manifest during the 1st yr of life. A number of disorders manifest in early childhood and a few in late childhood or later.
Non–Growth Related
Manifestations may be unrelated to the skeleton; they reflect expression of mutant genes in nonskeletal tissues. Examples include retinal detachment in spondyloepiphyseal dysplasia congenita, sex reversal in campomelic dysplasia, congenital heart malformations in Ellis-van Creveld syndrome, immunodeficiency in cartilage-hair hypoplasia, and renal dysfunction in asphyxiating thoracic dystrophy. These nonskeletal problems provide valuable clues to specific diagnoses and must be managed clinically (see Table 685-3).
Diagnosis
Laboratory testing has not been useful in diagnosing chondrodysplasias except in osteogenesis imperfecta, in which analysis of collagen synthesis by skin fibroblasts or genes whose products are involved in collagen biosynthesis, has helped establish a diagnosis. Osteogenesis imperfecta is not a chondrodysplasia, but it is often in the differential diagnosis, especially for newborns with severe skeletal deformities (Chapter 692). Reduced plasma levels of COMP (cartilage oligomeric matrix protein) have been detected in patients with pseudoachondroplasia and multiple epiphyseal dysplasias in which COMP mutations have been found.
Molecular Genetics
A number of chondrodysplasia genes have been identified (see Table 685-1). They encode several categories of proteins, including cartilage matrix proteins, transmembrane receptors, ion transporters, and transcription factors. The number of identified gene loci is smaller than anticipated from the number of recognized clinical phenotypes. The majority of patients have disorders that map to <10 loci; mutations at 2 loci (COL2A1 and FGFR3) account for more than half of all cases. There may be a limited number of genes whose function is critical to skeletal development, especially linear bone growth; mutations in these genes give rise to a wide range of chondrodysplasia clinical phenotypes. However, new genes harboring mutations that cause chondrodysplasias continue to be identified with advances in detection technology.
Pathophysiology
Regardless of genetic mechanism, the mutations ultimately disrupt endochondral ossification, the biologic process responsible for the development and linear growth of the skeleton (see Fig. 685-1). Indeed, a wide range of morphologic abnormalities of the skeletal growth plate, the anatomic structure in which endochondral ossification occurs, have been described in the chondrodysplasias.
Treatment
The first step is to establish the correct diagnosis. This allows one to predict a prognosis and to anticipate the medical and surgical problems associated with a particular disorder. Establishing a diagnosis helps to distinguish between lethal disorders and nonlethal disorders in a premature or newborn infant (see Tables 685-4 and 685-5). A poor prognosis for long-term survival might argue against initiating extreme lifesaving measures for thanatophoric dysplasia or achondrogenesis types Ib or II, whereas such measures may be indicated for infants with spondyloepiphyseal dysplasia congenita or diastrophic dysplasia, which have a good prognosis if the infant survives the newborn period.
Because there is no definitive therapy to normalize bone growth in any of the disorders, management is directed at preventing and correcting skeletal deformities, treating nonskeletal complications, providing genetic counseling, and helping patients and families learn to cope. Each disorder has its own unique set of problems, and consequently management must be tailored to each disorder. Medical information for a few disorders can be found at the Medical Information on Dwarfism website (see references).
Apajasalo M, Sintonen H, Rautonen J, Kaitila I. Health-related quality of life of patients with genetic skeletal dysplasias. Eur J Pediatr. 1998;157:114-121.
Gripp K, Slavotinek A, Hall JG, et al. Handbook of normal physical measurements, ed 2. Oxford, UK: Oxford University Press; 2006.
Horton WA. Skeletal development: insights from targeting the mouse genome. Lancet. 2003;362:560-569.
Horton WA, Hecht JT. Chondrodysplasias: general concepts and diagnostic and management considerations. In: Royce PM, Steinmann B, editors. Connective tissue and its heritable disorders: molecular, genetic, and medical aspects. ed 2. New York: Wiley-Liss; 2002:901-908.
Kozlowski K, Masel J, Sillence DO, et al. Gracile bone dysplasias. Pediatr Radiol. 2002;32:629-634.
Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med. 2009;11:127-133.
Lachman RS. Neurologic abnormalities in the skeletal dysplasias: a clinical and radiological perspective. Am J Med Genet. 1997;69:33-43.
Lachman RS. Taybi and Lachman’s radiology of syndromes, metabolic disorders and skeletal dysplasias, ed 2. New York: Elsevier; 2007.
Schramm T, Gloning KP, Minderer S, et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet Gynecol. 2009;34(2):160-170.
Smits P, Bolton AD, Funari V, et al. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med. 2010;362:206-216.
Spranger J, Brill PW, Poznanski A. Bone dysplasias. An atlas of genetic disorders of skeletal development, ed 2. New York: Oxford University Press; 2002.
Spranger J, Maroteaux P. The lethal osteochondrodysplasias. Adv Hum Genet. 1990;19:1-103. 331-332
Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1-18.
Unger S, Lachman RS, Rimoin DL. Chondrodysplasias. In: Rimoin DL, Conner JM, Pyeritz RE, et al, editors. Emery and Rimoin’s principles and practice of medical genetics. New York: Churchill Livingstone; 2007:3709.





