General chest ultrasound in neurocritical care
Overview
Neurocritical care patients often require hemodynamic monitoring to optimize cerebral blood flow and brain tissue oxygen delivery. Hemodynamic monitoring also aids in the management of usual coexisting disorders, such as acute lung injury and acute respiratory distress syndrome (ARDS), as well as shock states.1 General chest ultrasound (see Chapter 1), which consists of cardiovascular and lung ultrasound, is a noninvasive bedside tool that facilitates hemodynamic monitoring in modern neurocritical care. The manipulation of the cardiac output (CO), mean arterial pressure (MAP), systemic filling pressures and volumes, as well as the assessment of dynamic markers of fluid responsiveness and evaluation for pulmonary edema are all essential in optimizing cardiorespiratory function in the acutely brain- or spinal cord–injured patient. This chapter illustrates the role of general chest ultrasound in neurocritical care.
Cardiovascular evaluation in neurocritical care
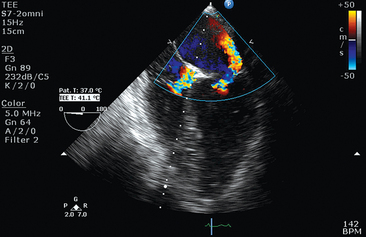
Recommended transthoracic echocardiography (TTE) should include two-dimensional sector scanning, color Doppler flow mapping, as well as pulsed and continuous wave Doppler interrogation (see Chapter 26). The standard parasternal long-axis, short-axis, and apical two- and four-chamber views are used for analysis of left ventricular (LV) dysfunction. LV ejection fraction (EF) can be visually rated as normal (LVEF, 50%-55%) and mildly (LVEF, 40%-54%), moderately (LVEF, 30%-39%), or severely (LVEF < 30%) depressed.
An intriguing syndrome that overlaps substantially with NSM is takotsubo cardiomyopathy (TCM), also known as apical ballooning syndrome or “broken heart” syndrome.2 Originally described in the Japanese population, this syndrome is characterized by transient LV dysfunction that produces a distinctive configuration during systole on the ventriculogram that resembles a Japanese octopus catcher pot. The diagnostic features of takotsubo cardiomyopathy include reversible LV regional wall motion abnormalities beyond a single coronary artery distribution (typically involving the LV apex and midventricle, with relative sparing of the basal segment), ECG abnormalities, minor elevation in cardiac markers, and absence of significant coronary artery disease. LV ballooning depicted by TTE in TCM is usually apical, although midventricular and basal ballooning may be rarely observed. The terminology is still ambivalent because it was lately suggested that TCM should not be referred to as apical ballooning syndrome. Moreover, the overlapping with NSM is obvious. Outside of the neurocritical care setting, both syndromes may occur in postmenopausal women after a stressful event and are accompanied by a massive catecholamine surge.
Of note, the Mayo Clinic criteria for classifying a stress-induced cardiomyopathy consist of transient wall motion abnormalities of the LV midsegments (with the regional wall motion abnormalities extending beyond a single epicardial artery distribution in the absence of obstructive coronary artery disease, pheochromocytoma, or myocarditis), with or without apical involvement, and new ST-segment elevation and/or T-wave inversion or modest elevation in cardiac troponins. NSM fits these criteria with the exception that it allows global LV dysfunction without regional wall motion abnormalities as observed in TCM. Hence this remains largely an issue of classification of various echocardiographic findings. A summary of NSM features is presented in Table 7-1.
TABLE 7-1
| Clinical setting | Acute brain insult High intracranial pressure Sudden emotional stress |
| Clinical presentation | Biventricular failure Cardiogenic shock |
| Echocardiography | Apical hypokinesis/ballooning Hypercontractile base Dynamic left ventricular outflow obstruction Mitral regurgitation |
| Coronary angiography | Absence of occlusive coronary artery disease |
| Serum cardiac markers | Troponin elevation of a lower degree than from coronary artery occlusion for similar cardiac function |
| Lung ultrasound and radiography | Pulmonary edema (cardiogenic features) |
| Hemodynamic monitoring | Depressed biventricular output Elevated filling pressures Dynamic preload variables suggesting plateaued ventricular function (Frank-Starling curve) |
| Clinical management | Vasopressor and inotropic support guided by cardiac ultrasound and invasive hemodynamic monitoring Intraaortic balloon pump to be considered in refractory cases |
| Prognosis | Favorable Reversible |
Despite the aforementioned terminology issues, in neurocritical care, both syndromes have a favorable prognosis because, with supportive measures, LV function spontaneously ameliorates within days, whereas in-hospital mortality remains less than 1%.3–5 However, cardiogenic shock requiring mechanical circulatory support and rare complications, including LV apical thrombus or fatal LV rupture, have been occasionally reported.6,7
Although uncommon, dynamic obstruction within the LV outflow tract should be evaluated because it has been described with apical ballooning syndrome and is believed to be caused by compensatory hyperkinesis of the basal segments. This obstruction can be exacerbated by low filling pressures or decreased afterload. Mitral valve regurgitation (MR) can be encountered, resulting from acceleration of blood through the dynamic obstruction, leading to a Venturi effect, with the anterior mitral leaflet being sucked into the LV outflow tract. As a result, significant hypotension could be expected secondary to both poor LV systolic function, as well as dynamic obstruction of the LV outflow tract. In this setting, avoiding the administration of systemic vasodilators and inotropic stimulation, which can worsen the outflow tract gradient, is a prudent therapeutic strategy. In addition, fluids should be cautiously used because patients often have elevated left-sided filling pressures and evidence of pulmonary edema.8
Preload and responsiveness
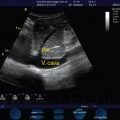
Preload assessment and fluid responsiveness evaluation are essential in the hemodynamic management in the intensive care unit (ICU). The frequently used static preload filling-pressure markers, such as central venous pressure (CVP) and pulmonary artery occlusion pressure (PAOP), have been shown to correlate poorly with ventricular filling volumes and fluid responsiveness in healthy volunteers and critical care patients, leading investigators to discourage their use for the purpose of fluid management.9–11 The goal of volume expansion by the augmentation of stroke volume entails biventricular preload dependence. Stated on Frank-Starling terms, it requires both ventricles to be operating on the ascending part of their performance curves. The finding of systolic obliteration of the LV cross-sectional area (”kissing” papillary muscles sign) by TTE (via the parasternal short-axis view) may indicate severe hypovolemia.12 Also, Feissel et al13 and Barbier et al14 suggested that the quantification of respiratory variation in the diameter of the inferior vena cava (dIVC) by the use of bedside ultrasound is a reliable marker of fluid responsiveness in mechanically ventilated patients. In both studies, with only slightly different cutoffs for dIVC (Feissel, 12% and Barbier, 18%) fluid responders and nonresponders could be discriminated with sensitivity and specificity greater than or equal to 90%. Recently, Moretti and Pizzi15 found a cutoff dIVC greater than16% to be 70% sensitive and 100% specific in predicting fluid responsiveness in 29 patients receiving mechanical ventilation and advanced hemodynamic monitoring for the management of SAH. Traditionally, patients with acute brain injury, and especially patients with SAH, have been managed with a fluid liberal strategy based on daily and cumulative fluid balances, leading to hypervolemic states and significantly positive fluid balances. Based on recent literature, this strategy not only seems unjustified but in fact harmful, leading to respiratory complications, longer ICU and hospital stays, and worse neurologic outcomes.16 Hopefully, the application of dynamic preload variables will lead to rationalization of fluid administration and better outcomes.
Pulmonary evaluation: Neurogenic pulmonary edema
The implementation of lung ultrasound in routine practice offers new opportunities in the respiratory and hemodynamic management of neurocritical patients. Lung ultrasound is analyzed elsewhere in this book. In brief, the most commonly appraised artifacts of lung ultrasound are the lung rockets or B-lines (see Chapter 19). The latter are created mainly by repetitive reflections of the ultrasound wave within the lung parenchyma because of a higher concentration of physiologic or pathologic fluid. B-lines are mainly encountered in the alveolar interstitial syndrome. B-lines separated by less than 3 mm characterize predominantly alveolar abnormalities as opposed to interstitial lung disorders.17 Alveolar abnormalities usually accompany cardiogenic and/or noncardiogenic pulmonary edema.
Although known for over a century, acute neurogenic pulmonary edema (NPE) is a potentially underdiagnosed clinical entity that can be encountered after virtually any form of acute brain injury, such as in patients with SAH, intracerebral/intraventricular hemorrhage, seizures, stroke, and traumatic brain injury.18 The main features of NPE are summarized in Table 7-2. NPE is characterized by an increase in extravascular lung water in patients who have sustained a sudden change in neurologic condition. Its pathophysiologic mechanism remains obscure, and two divergent theories have been proposed to explain its development: increased lung capillary permeability and increased pulmonary vascular hydrostatic pressures. Increased permeability as a mechanism of NPE is supported by animal studies that have shown high interstitial (lung lymphatic) or alveolar protein concentrations and time-dependent ultrastructural changes in pneumocyte type II cells after brain injury. Increased permeability may be caused by damage of the capillary endothelium or by direct neural influences on capillary permeability. Nevertheless, hydrostatically induced pulmonary edema can occur without endothelial damage. One possible sequence leading to NPE is an acute increase in sympathetic tone that abruptly increases LV afterload and causes intense vasoconstriction, thereby elevating LV filling pressures and inducing elevated pulmonary artery capillary pressures, leading to hydrostatic pulmonary edema. This was confirmed by experimental and clinical data.19,20 However, pulmonary edema can occur with normal PAOP, suggesting a neurally mediated increase of capillary permeability. In addition to the above-mentioned hypotheses, pulmonary edema of cardiogenic origin should be considered. In fact, the early hemodynamic changes that occur in the setting of NPE may lead to the conclusion that the pulmonary edema is of cardiac origin. Smith and Matthay21 reported, as have others, that early analysis of NPE fluid reveals a low fluid-serum protein ratio consistent with hydrostatic edema. TTE becomes invaluable in this setting, to investigate both LV and right ventricular function and to rule out a NSM or right ventricular failure. Mutoh et al22 recently studied three patients with SAH-related pulmonary edema by using transpulmonary thermodilution and suggested that its origin can be either due to cardiogenic/hydrostatic or noncardiogenic/high pulmonary vascular permeability mechanisms.
TABLE 7-2
| Clinical setting | Acute brain insult High intracranial pressure |
| Clinical presentation | Hypoxemic respiratory failure |
| Lung ultrasound and radiography | Bilateral infiltrates Pulmonary edema (can be either hydrostatic, permeability, or combined) Preserved ventricular contractility |
| Hemodynamic monitoring | High extravascular lung water Normal cardiac function High pulmonary vascular permeability index (for nonhydrostatic) |
| Clinical management | Mechanical ventilation Diuresis guided by cerebral and systemic hemodynamics (not as effective in the setting of high permeability) Novel use of α-blockers (phentolamine) |
| Prognosis | Favorable |
Lung ultrasound can be used to elucidate the underlying mechanism in NPE. Copetti et al23 reported that demonstration of a heterogeneous alveolar-interstitial syndrome with spared areas, pleural line modifications, and lung consolidations is strongly predictive in an early phase of noncardiogenic pulmonary edema. Targeting fluid and vasoactive medication management to individualized patient pathophysiology enhances the ability to satisfy the often competing clinical goals of cardiorespiratory optimization and cerebral perfusion. A specific diagnosis becomes even more relevant in light of recent work suggesting a potentially novel treatment for NPE with the use of α-receptor blockade and direct antagonism of the “sympathetic storm” that is thought to be the triggering factor behind the syndrome. In 2012, Davison et al24 reported the successful use of phentolamine in a patient who developed refractory hypoxemic respiratory failure after intracerebral hemorrhage and documented the co-linear behavior between serum catecholamines, hypoxemia, pulmonary edema, and its resolution.
Pearls and highlights
• General chest ultrasound comprising of echocardiography and lung ultrasound is an essential tool in the hemodynamic and respiratory monitoring of neurocritical care patients.
• NSM and TCM are overlapping clinical entities with different echocardiographic appearances, which may be seen in patients with SAH and other neurologic disorders.
• Dynamic preload variables, such as the inferior vena cava variation, need to be used in neurocritical care to minimize injudicious fluid administration that has been linked with worse systemic and neurologic outcomes.
• NPE can lead to severe hypoxemic respiratory failure. It should be considered in the patient with severe acute brain injury and noncardiogenic pulmonary edema. Lung ultrasound can diagnose, quantify, classify, and monitor the resolution of NPE. Diagnosis of NPE by means of general chest ultrasound allows the implementation of targeted therapies, such as the recent use of α-blockers.
References
1. Lazaridis, C, Advanced hemodynamic monitoring: principles and practice in neurocritical care (review). Neurocrit Care. 2012;16(1):163–169.
2. Wittstein, IS, Thiemann, DR, Lima, JA, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005; 352(6):539–548.
3. Abe, Y, Kondo, M, Matsuoka, R, et al. Assessment of clinical features in transient left ventricular apical ballooning. J Am Coll Cardiol. 2003; 41(5):737–742.
4. Girod, JP, Messerli, AW, Zidar, F, et al. Takotsubo-like transient left ventricular dysfunction. Circulation. 2003; 107(18):e120–e121.
5. Tsuchihashi, K, Ueshima, K, Uchida, T, et al, Transient left ventricular apical ballooning without coronary artery stenosis: a novel heart syndrome mimicking acute myocardial infarction. Angina pectoris-myocardial infarction investigations in Japan. J Am Coll Cardiol. 2001;38(1):11–18.
6. Lazaridis, C, Pradilla, G, Nyquist, PA, et al. Intra-aortic balloon pump counterpulsation in the setting of subarachnoid hemorrhage, cerebral vasospasm, and neurogenic stress cardiomyopathy. Case report and review of the literature (review). Neurocrit Care. 2010; 13(1):101–108.
7. Akashi, YJ, Tejima, T, Sakurada, H, et al. Left ventricular rupture associated with Takotsubo cardiomyopathy. Mayo Clin Proc. 2004; 79(6):821–824.
8. Tsai, TT, Nallamothu, BK, Prasad, A, et al. Clinical problem-solving. A change of heart. N Engl J Med. 2009; 361(10):1010–1016.
9. Kumar, A, Anel, R, Bunnell, E, et al. Pulmonary artery occlusion pressure and central venous pressure fail to predict ventricular filling volume, cardiac performance, or the response to volume infusion in normal subjects. Crit Care Med. 2004; 32(3):691–699.
10. Osman, D, Ridel, C, Ray, P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med. 2007; 35(1):64–68.
11. Marik, PE, Baram, M, Vahid, B. Does central venous pressure predict fluid responsiveness? A systematic review of the literature and the tale of seven mares (review). Chest. 2008; 134(1):172–178.
12. Beaulieu, Y, Marik, PE, Bedside ultrasonography in the ICU: part 1 (review). Chest. 2005;128(2):881–895.
13. Feissel, M, Michard, F, Faller, JP, et al. The respiratory variation in inferior vena cava diameter as a guide to fluid therapy. Intensive Care Med. 2004; 30(9):1834–1837.
14. Barbier, C, Loubières, Y, Schmit, C, et al. Respiratory changes in inferior vena cava diameter are helpful in predicting fluid responsiveness in ventilated septic patients. Intensive Care Med. 2004; 30(9):1740–1746.
15. Moretti, R, Pizzi, B. Inferior vena cava distensibility as a predictor of fluid responsiveness in patients with subarachnoid hemorrhage. Neurocrit Care. 2010; 13(1):3–9.
16. Contant, CF, Valadka, AB, Gopinath, SP, et al, Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95(4):560–568.
17. Lichtenstein, D, Mezière, G, A lung ultrasound sign allowing bedside distinction between pulmonary edema and COPD: the comet-tail artifact. Intensive Care Med. 1998;24(12):1331–1334.
18. Demling, R, Riessen, R, Pulmonary dysfunction after cerebral injury. Crit Care Med. 1990;18(7):768–774.
19. Simmons, R, Martin, A, Heisterkamp, C, et al. Respiratory insufficiency in combat casualties (pulmonary edema following head injury). Ann Surg. 1969; 170(1):39–44.
20. Faist, E, Baue, AE, Dittmer, H, et al. Multiple organ failure in polytrauma patients. J Trauma. 1983; 23(9):775–787.
21. Smith, WS, Matthay, MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest. 1997; 111(5):1326–1333.
22. Mutoh, T, Kazumata, K, Kobayashi, S, et al, Serial measurement of extravascular lung water and blood volume during the course of neurogenic pulmonary edema after subarachnoid hemorrhage: initial experience with 3 cases. J Neurosurg Anesthesiol. 2012;24(3):203–208.
23. Copetti, R, Soldati, G, Copetti, P, Chest sonography: a useful tool to differentiate acute cardiogenic pulmonary edema from acute respiratory distress syndrome. Cardiovasc Ultrasoun. 2008; 6:16.
24. Davison, DL, Chawla, LS, Selassie, L, et al, Neurogenic pulmonary edema: successful treatment with IV phentolamine. Chest. 2012;141(3):793–795.