[level-membership-for-neurosurgery-category]
CHAPTER 194 General Approaches and Considerations for Pediatric Brain Tumors
Each year in the United States alone, cancer is diagnosed in more than 12,000 children. Of these cancers, pediatric brain tumors are the most common solid tumors and result in the highest overall mortality.1–5 Although survival rates for children with brain tumors continue to increase, studies have shown that these children experience more severe disease and treatment sequelae than do children with other types of cancer and that diagnosis and treatment strongly influence the psychopathology of their families’ functioning.6 More specifically, research has shown that the time of diagnosis is a particular point of vulnerability for families of children in whom brain tumors have been diagnosed and that the most accurate, honest information delivered with compassion is vital during this time.6
Epidemiology
Malignant brain tumors are the most common solid tumors in childhood; they account for 20% to 30% of all childhood cancers and are the leading cause of cancer-related death in this age group.1,5 In this chapter, we provide a general overview of the important clinical, diagnostic, and treatment-related considerations in pediatric brain tumors as a reference for practicing clinicians faced with caring for these patients. The annual incidence has been reported to be 2.4 to 6.3 per 100,000, which has actually risen over the past several decades because of better diagnostic modalities and reporting practices.3,5,7–10 Incidence rates for the pediatric population show that brain tumors are more common in white children than in black ones.3 In both, however, there is an overall slight male preponderance, with studies consistently showing a male-to-female ratio of about 1.5.11,12 More favorable histologic subgroups have repeatedly been found to occur in adolescents, whereas tumors with higher grade, more unfavorable locations, and poorer prognosis have generally occurred in younger children, particularly those younger than 3 years.11
The central nervous system (CNS) tumors that occur most frequently in childhood are vastly different from those in adulthood with regard to both histology and tumor location.12 In contrast to adults, in whom most CNS tumors are located supratentorially, approximately 50% to 55% of all childhood CNS tumors are infratentorial.8 In the first 6 months of age, supratentorial tumors are actually more common; however, by 2 years of age this location reverses, with up to 60% being infratentorial.8 This is in contrast to the third decade of life and beyond, where only 25% to 35% of intracranial tumors are infratentorial.8
The overall 5-year survival rate of children with brain tumors has improved considerably over the past several years. Because of earlier diagnosis and better therapies, survival rates are now between 35% and 65%, depending on several factors, including tumor histology and location.9 Age is also an important prognostic indicator for children with CNS tumors in general. Data consistently show that 10- to 15-year-olds have the longest survival whereas those younger than 2 years have the shortest.11
Classification
Classification schemes are important for all types of cancers because they allow physicians to make accurate predictions regarding the natural history of the disease. The response to certain therapies and prognosis can also be derived from a classification scheme.13 Classification schemes for CNS tumors are categorized according to the cell type from which the abnormal tissue originated.1,12 Subclassifications of tumors derived from a specific cell type exist as well, and such tumors can differ in their histologic, cytologic, and behavioral characteristics.12,13 CNS tumors can also be more broadly categorized into those that occur in certain age groups or locations.12
The cells of the nervous system can roughly be categorized as nerve cells and a variety of supporting cells called neuroglia. Glial cells greatly outnumber nerve cells by approximately 3 : 1 and include astrocytes, oligodendrocytes, ependymal cells, and microglia.1 As a whole, gliomas and, more specifically, astrocytomas are the most common CNS tumors of childhood and account for up to 50% in some series.1,4,11 Astrocytomas vary greatly in their histology, cytology, and behavioral characteristics and occur in several locations in the pediatric population. Pilocytic astrocytomas are the most common and represent up to 25% of pediatric CNS tumors. These tumor are considered benign and typically occur in the posterior fossa but can also be found elsewhere.11 However, other common locations for pediatric gliomas include the hypothalamic and optic pathway regions. The histology of these lesions is typically low grade, but they are difficult to treat surgically as a result of their location.11 Brainstem gliomas are not resectable because of their diffuse infiltrative nature, and their natural history and poor prognosis are similar to glioblastoma multiforme, which is the most common primary supratentorial tumor in adults. Focal lesions of the brainstem may have a better prognosis and can be monitored by serial surveillance with magnetic resonance imaging (MRI). Other focal lesions of the brainstem may have a dorsally exophytic portion that is amenable to surgical debulking.
The second most common CNS tumor in the pediatric population is medulloblastoma, which arises from neural stem cell precursors in the fourth ventricle and accounts for approximately 15% to 20% of tumors in this age group.10,14 Other common CNS tumors in this population are of neuroepithelial origin and include ependymomas, which account for 10% of pediatric CNS tumors; primitive neuroectodermal tumors (PNETs), 1.9%; ganglioglioma, 2.5%; dysembryoplastic neuroepithelial tumors, 0.6%; desmoplastic infantile astrocytomas, 0.6%; and mixed tumors.7,11
Non-neuroepithelial neoplasms consist of germ cell tumors (2.5%) of the pineal or other regions and include germinomas, teratomas, choriocarcinomas, and yolk sac tumors, as well as atypical teratoid/rhabdoid tumors (AT/RTs) (1.3%), choroid plexus tumors (0.9%), and craniopharyngiomas (5.6%).7,11
Overall, the most common CNS tumors seen in childhood are astrocytomas, medulloblastomas, ependymomas, craniopharyngiomas, and germ cell tumors.12 The incidence of these tumors, however, varies greatly within age groups among the pediatric population, and there is a tendency for certain tumors to occur at particular times during a child’s life (Table 194-1).3,11 In infancy (0 to 2 years old), choroid plexus papillomas, desmoplastic infantile astrocytomas, teratomas, PNETs, and AT/RTs predominate. By childhood (3 to 11 years of age), these tumors are rare and astrocytomas and craniopharyngiomas are much more frequently seen. By adolescence (≥12 years), germ cell tumors are commonly encountered, whereas craniopharyngiomas are less commonly seen.11
TABLE 194-1 Statistical Report: Primary Brain Tumors in the United States, CBTRUS, 2000-2004
| AGE (yr) | MOST COMMON HISTOLOGY | SECOND MOST COMMON HISTOLOGY |
|---|---|---|
| 0-4 | Embryonal/primitive/medulloblastoma | Pilocytic astrocytoma |
| 5-9 | Pilocytic astrocytoma | Embryonal/primitive/medulloblastoma |
| 10-14 | Pilocytic astrocytoma | Malignant glioma (NOS) |
| 15-19 | Pilocytic astrocytoma | Pituitary |
| 20-34 | Pituitary | Meningioma |
| 35-44 | Meningioma | Pituitary |
| 45-54 | Meningioma | Glioblastoma |
| 55-64 | Meningioma | Glioblastoma |
| 65-74 | Meningioma | Glioblastoma |
| 75-84 | Meningioma | Glioblastoma |
| 85+ | Meningioma | Glioblastoma |
CBTRUS, Central Brain Tumor Registry of the United States; NOS, not otherwise specified.
Published by the Central Brain Tumor Registry of the United States, 2008.
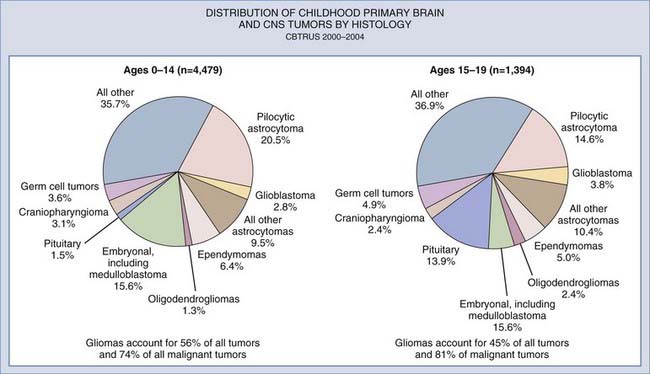
The distribution of astrocytomas also varies by age. As mentioned previously, pilocytic astrocytomas predominate overall and particularly in the childhood and early adolescent age groups. However, the incidence of pilocytic astrocytoma falls, and World Health Organization grades III and IV astrocytomas become more common in children older than 15 years (Fig. 194-1).3,11
Oncogenic Factors
Although population studies have failed to confirm any significant environmental factors associated with the development of CNS malignancies, several syndromes and genetic markers have been shown to be associated with their development.1 Such syndromes include neurofibromatosis types 1 and 2 (NF1 and NF2), tuberous sclerosis, and von Hippel-Lindau (VHL) syndrome, as well as many others. These disorders are associated with specific chromosomal abnormalities and lead to alterations in oncogenes and tumor suppressor genes, which typically regulate normal cell growth but, when altered, lead to tumor genesis. NF1 is associated with genetic alterations on chromosome 17 and predisposes individuals with this disorder to optic gliomas and malignant nerve sheath tumors.1,7,12 The genetic alteration in NF2 is located on chromosome 22 and is associated with the development of meningiomas and vestibular schwannomas.1,7 Tuberous sclerosis is caused by an abnormality on chromosome 9 that predisposes these patients to the development of astrocytomas.7 VHL syndrome is associated with multiple systemic malignancies secondary to an alteration on chromosome 3, including cerebellar hemangioblastomas.1,7
Although not associated with specific syndromes, several common CNS malignancies are linked to certain genetic tumor markers that are prognostic and continue to direct research for therapy. Examples include a strong association between overexpression of the oncogene p53 in childhood gliomas. p53 mutations are found in approximately 40% of malignant gliomas in children older than 3 years, which is much higher than the frequency seen in adult gliomas.1,12,15 In addition, the presence of the p53 mutation is associated with a poor prognosis independent of both the clinical and histologic prognostic features of the tumor at initial evaluation.15 Studies have also shown a relationship between AT/RTs and the INI1 gene on chromosome 22.12 This has become an important diagnostic tool because AT/RTs cannot be distinguished from medulloblastoma by clinical features or neuroimaging.16 Microscopically, these tumors can also be difficult to distinguish as a result of several similar features. Antibody staining for the INI1 gene for immunohistochemical analysis has now been used to differentiate these two entities because inactivating mutations of the INI1 gene located on chromosome 22 are a crucial step in the molecular pathogenesis of AT/RTs.16 Interestingly, choroid plexus tumors, called choroid plexus papillomas and carcinomas, which are intraventricular epithelial tumors arising from the choroid plexus, may also stain for the INI1 gene.
In addition, multiple genetic markers have been linked to medulloblastoma, including Trk-C, whose presence correlates with a good prognosis, whereas overexpression of epidermal growth factor receptor 2 and c-MYC have high predictive power for a poor prognosis.14,15 OTX2 overamplification has also been appreciated in medulloblastomas.17 OTX2 is a transcription factor that plays a role in normal cerebellar development.17 Expression of OTX2 has correlated with higher grade tumors, a poorer prognosis, and medulloblastomas that more frequently localize to the vermis of the cerebellum.17,18 OTX2 may become important in the treatment of this disease as well, inasmuch as a recent study demonstrated that OTX2-expressing medulloblastomas were responsive to all-trans-retinoic acid in vitro.17
Clinical Features
Because CNS malignancies account for approximately a quarter of childhood cancers, it is imperative that health care professionals be able to recognize the signs and symptoms at initial encounter.5 The signs and symptoms at diagnosis are typically a result of tumor location but are most often secondary to the raised intracranial pressure (ICP) that occurs with 40% of all intracranial tumors in childhood.5 Hydrocephalus is principally seen with infratentorial tumors and is due to obstruction of the ventricular system from mass effect by the tumor. The most common symptoms of elevated ICP are headache, nausea, and vomiting, which occur especially in the morning, and lethargy.8,10,12 Less commonly, macrocephaly and a bulging fontanelle can be seen in younger patients whose sutures have not yet closed.10 Infratentorial tumors causing compression on the brainstem can also be manifested as cranial nerve palsies.12 A common example is Parinaud’s syndrome, which is seen with compression of the midbrain tectum from a mass effect or increasing hydrocephalus. Features of Parinaud’s syndrome include paralysis of upgaze, paresis of accommodation, and convergence nystagmus. Additionally, a local mass effect from cerebellar tumors can cause ataxia and subsequent gait disturbances.5
In contrast, supratentorial tumors tend to be manifested as focal neurological deficits because of the local mass effect of the tumor; the deficits vary according to location and include visual disturbances from optic nerve gliomas, hemiplegia, and seizures, but they can also be accompanied by symptoms of increased ICP.12 Seizures may also be the single initial sign, with one study showing that 14% of children had seizures before their first hospitalization for a brain tumor.19 At all ages, the incidence of seizures was higher in children with a supratentorial tumor and increased with age.19 Other less common features can include weight loss, growth retardation, precocious puberty, and excessive thirst and urination, which are more commonly associated with tumors located near the neuro-endocrine structures of the brain, including the pituitary gland and hypothalamus.
It is important to remember that especially in infants, the signs and symptoms may be insidious and can even lead to lengthy investigations of the gastrointestinal tract in a search for causes of failure to thrive and persistent nausea and vomiting. Because early diagnosis is imperative for treatment, a CNS tumor should be part of the differential diagnosis in a child with the aforementioned symptoms.20
Diagnostic Evaluation
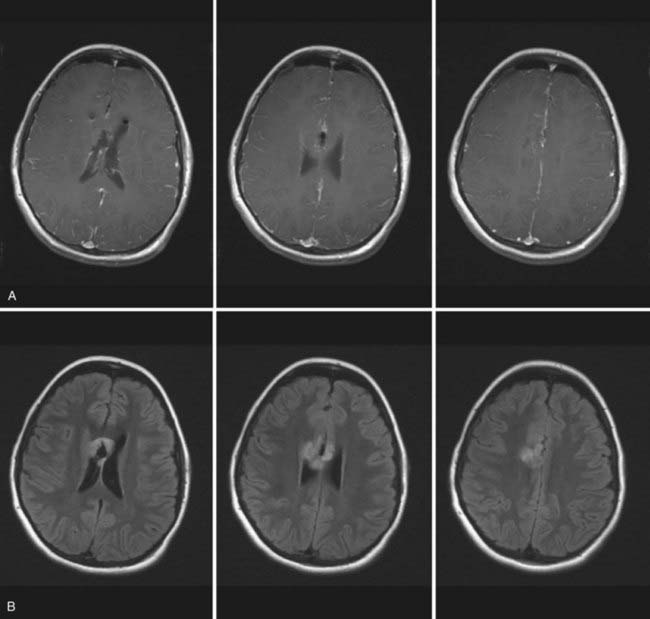
These imaging modalities are also imperative for tumor surveillance in the postoperative period to identify residual or recurrent tumor, secondary malignancies, or secondary effects of therapy such as radiation-induced necrosis. Additional developments in CT and MRI have led to new techniques that can further distinguish abnormal tissue. Fluid-attenuated inversion recovery (FLAIR) MRI sequences blunt the bright signal produced by cerebrospinal fluid (CSF) on T2-weighted images to better differentiate the tumor from adjacent normal brain (Fig. 194-2).21 In addition, magnetic resonance spectroscopy (MRS) has been developed and provides information about the metabolic function of cells by analyzing their biochemical composition.12,21,22 More specifically, MRS provides information about levels of the neuronal marker N-acetylaspartate, choline, which is a marker of membrane turnover, as well as lactate and creatinine, which are markers of cerebral metabolism.12,15,22 The levels of these peaks and their ratios can help characterize brain tumors, as well as differentiate between neoplasia and necrosis.15,22 However, caution must be exercised because similar patterns of these markers can also be found in abscesses and within different tumor types, thus making the use of MRS for diagnostic purposes difficult and more appropriate for long-term follow-up imaging.
Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are additional imaging modalities that can be used to detect CNS abnormalities. PET assesses uptake of the radioactive isotope 18F-fluorodeoxyglucose (FDG), which provides an objective measurement of tissue metabolism. FDG uptake is increased in tissues with high glucose metabolism, such as malignant tumors. SPECT is capable of measuring the distribution of blood flow by detecting the radioactive tracer technetium 99m (99mTc).21,23 Increased levels of 99mTc uptake occur in metabolically active areas as well. Both 99mTc-SPECT and PET can therefore help delineate areas of higher grade in heterogeneous lesions and differentiate postoperative changes from residual or recurrent tumor.4,21,24
The diagnostic evaluation of CNS tumors, however, does not end with standard imaging. Certain CNS lesions have a propensity for dissemination within the CNS, including medulloblastomas, germ cell tumors, ependymomas, and PNETs.10,12 If these lesions are on the differential diagnosis based on tumor location or clinical findings, imaging must be extended to the entire neuraxis to look for drop metastases. If feasible, it is recommended that spinal MRI be performed before surgical intervention to decrease the number of false-positive results that could occur after surgical resection from blood products gravitating down the spinal column. If spinal imaging must be performed after surgery, it is prudent that it be done at a minimum of 2 weeks after surgery to wait until the blood products clear. In addition, assignment into treatment categories for most disseminating tumors mandates that CSF samples be obtained via lumbar puncture for cytologic examination to look for evidence of CSF spread that is unable to be detected by any imaging modalities.12 Furthermore, pineal and suprasellar tumors should also be evaluated with an extensive metabolic work-up. Germ cell tumors can produce several proteins and can be evaluated by determining levels of these markers in CSF and serum. Such markers include β-human chorionic gonadotropin, α-fetoprotein, and placental alkaline phosphatase, whose levels can help establish the diagnosis.12 These markers can also be assessed by immunohistochemistry of tumor tissue after biopsy or resection. The metabolic evaluation, particularly for hypothalamic and sellar lesions, should also include a basic metabolic panel, thyroid panel, and levels of growth hormone, insulin-like growth factor-I, fasting cortisol and adrenocorticotropic hormone, prolactin, and gonadotropins to evaluate for evidence of endocrine dysfunction as a result of compression or a secreting lesion.
Treatment
Over the past century, treatment of children with brain tumors has considerably changed because of the development of better imaging techniques, better intraoperative aids, including the operative microscope, and better adjuvant therapy. The majority of therapy revolves around surgical resection as a mainstay, followed by adjuvant therapy if indicated.8,12 The prognosis for the majority of CNS tumors is related to the extent of surgical resection, which is indicated only if it can safely be performed without significant neurological impairment.5,12 Such is the case for most supratentorial and infratentorial tumors, including medulloblastomas, ependymomas, and the majority of gliomas. There are, however, exceptions to this rule. Germ cell tumors of the pineal region are a special situation. For instance, germinomas have been shown to be incredibly sensitive to radiation, with a long-term survival rate of greater than 90% with craniospinal irradiation alone.25 Hence, there has been a departure from the classic approach of resection followed by adjuvant therapy. If preoperative serum or CSF markers and biopsy findings are consistent with germinoma, removal of the tumor is not necessary or indicated and the patient should proceed with neoadjuvant therapy.10 However, mixed germ cell tumors or those that are marker negative, such as pineoblastoma, are still treated by surgical resection followed by adjuvant chemotherapy and radiation therapy, although the benefit from tumor resection is not widely known. Tumors of this region also defy the recommendations of delaying CSF diversion. If hydrocephalus is present along with a tumor in the pineal region, it must be managed first.10 Endoscopic third ventriculostomy (ETV) is the treatment of choice, particularly to potentially avoid shunt dependence and to reduce the chance of dissemination of tumor by shunting.10 Frequently, an external ventricular drain (EVD) is left in place after ETV to assess ICP, which if still elevated, warrants permanent CSF drainage with a shunt.25 Tumor biopsy can occasionally be achieved at the time of ETV, an important goal because these tumors often have mixed histology, which will dictate the type of adjuvant therapy.10
Additionally, tumors located in particularly eloquent areas, including optic tumors, tumors of the hypothalamic region, and brainstem lesions, are not often amenable to surgical resection and therefore undergo biopsy only in the rare cases in which the diagnosis is questionable on imaging; they are managed by adjuvant treatment and CSF diversion if needed.10 Exceptions to this guideline include brainstem gliomas with a large exophytic component, which can be amenable to tumor debulking. Optic nerve gliomas that are progressive despite chemotherapy may also benefit from tumor debulking.4,10,26
Radiation therapy is frequently avoided in patients younger than 3 years because of its extremely deleterious effects on the developing nervous system during this time, and such children are frequently managed by resection and chemotherapy alone until they are older.12 After the age of 3, radiation therapy is used to treat malignant tumors of the CNS and can be delivered as localized radiotherapy, whole-brain radiation, or irradiation of the entire neuraxis. The choice of radiotherapy is guided by the location and the propensity for the tumor to seed the subarachnoid space.8 For instance, stereotactic radiotherapy, which is designed to deliver high-dose radiation to a specific target without compromising surrounding normal tissue, can be used for small, well-circumscribed tumors in brain tissue.12,15 Conversely, with all posterior fossa medulloblastomas and some ependymomas that have metastasized to the CNS, radiation therapy is provided to the complete neuraxis because of the high tendency of these tumors to metastasize throughout the CNS.8
Although specific chemotherapeutic regimens are beyond the scope of this chapter and many are investigational in nature, several studies have reliably shown that the use of chemotherapy for pediatric brain tumors is better tolerated than in adults, is more effective, and can significantly increase the long-term survival of children with both supratentorial and infratentorial tumors. Various drug regimens are used in combination, depending on the histologic tumor type, to both increase efficacy and decrease drug resistance.9
Despite the success of some of the treatment regimens currently available, optimal therapeutic efficacy from chemotherapy continues to be a dilemma because of the difficulty of formulating a chemotherapeutic agent that can penetrate the blood-brain barrier, a layer of specialized capillary endothelial cells that form a barrier between the brain and blood.9 Therefore, other more investigational therapies are also currently being developed in the hope of improving drug delivery, including the use of immunotherapy in the form of tumor vaccines and monoclonal antibodies to target differences between brain tumor cells and normal tissue, as well as therapies to target the specific genetic abnormalities of some tumors mentioned previously.9,12,15,27
Preoperative Considerations
Vasogenic edema secondary to breakdown of the blood-brain barrier in CNS tumors can result in symptoms of elevated ICP, as well as focal neurological deficits caused by local mass effect on neuroanatomic structures. Additionally, CNS tumors may obstruct the ventricular system, and the associated edema can lead to hydrocephalus and profound symptoms of elevated ICP. Early management of these patients often depends on their neurological status at diagnosis. Those who have focal neurological deficits but no signs of elevated ICP frequently respond well to dexamethasone, which decreases capillary permeability and subsequently decreases the vasogenic edema.28 Typical doses of dexamethasone, a synthetic glucocorticoid, are 0.5 mg/kg per day divided into doses every 6 hours; however, these doses can be increased if needed, depending on the degree of cerebral edema.28 For patients with severe signs of elevated ICP caused by obstruction of CSF flow, such as severe nausea and vomiting and severe declines in mental status that are medically refractory, emergency CSF diversion to relieve the increased ICP is required and is most often done by placement of a temporary EVD.10 Before the current era of advanced neuroimaging, placement of ventriculoperitoneal (VP) shunts was often performed initially for CSF diversion. However, a shunt is now rarely used as an initial management technique for several reasons, including the potential to facilitate metastatic spread of tumor into the peritoneal space and the fact that the child may not require long-term CSF diversion after surgical resection.28
Intraoperative Considerations
For most lesions, surgical resection is the key to long-term survival and should be pursued if the lesion is amenable with low morbidity. However, for tumors not amenable to gross total resection or those that are better managed with adjuvant therapy, biopsy may be the more appropriate option for tissue diagnosis. Biopsy, depending on the location of the lesion, can be done stereotactically under intraoperative image guidance or by open means. Frameless stereotaxy and ultrasound can also be useful for gross total resection and should be considered for intraoperative planning. With certain CNS malignancies, especially tumors of the posterior fossa, an EVD is usually left in place for postoperative ICP monitoring because many children will ultimately require a shunt. As mentioned previously, the exception to this generalization is tumors of the pineal or hypothalamic region that are causing compression on the third ventricle and aqueduct and subsequent obstructive hydrocephalus. Because these patients will frequently need long-term CSF diversion, ETV is often thought to be the optimal choice to avoid a VP shunt.10
Once a surgical plan has been chosen, there are several intraoperative considerations. At the time of surgery a combination of therapies should be considered to facilitate intraoperative relaxation of the brain. Such techniques include hyperventilation, intravenous mannitol and dexamethasone, and CSF diversion if the patient has an EVD in place.28 If the tumor has a cystic component, the surgeon will often decompress the cyst at the beginning of the procedure to further facilitate brain relaxation.28
The use of proper neuroanesthetics that do not increase cerebral blood flow and ICP is critical.28 Furthermore, anesthesiologists are responsible for setting up proper monitoring of the patient, including vital signs, placement of a central venous line for maintenance of proper fluid balance and treatment of air embolism, which can be a complication of neurosurgical procedures, and monitoring of important laboratory values. Of particular importance in pediatric patients is aggressive monitoring of the hematocrit intraoperatively. Pediatric patients have a small circulating blood volume, with neonates averaging approximately 90 mL/kg; infants, 80 mL/kg; children, 70 mL/kg; and adolescents, 60 to 65 mL/kg. Therefore, even small blood losses can drop these values precipitously and require transfusion. In addition, particular attention should be paid to the patient’s temperature because the ratio of body surface area to volume in pediatric patients is higher, which can lead to faster and greater drops in body temperature.
Postoperative Considerations
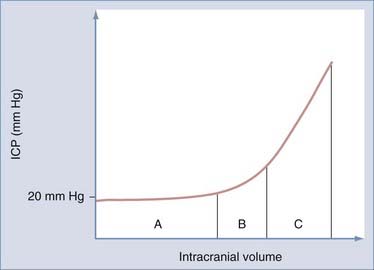
The Monro-Kellie doctrine is important for understanding the potential complications from CNS tumors (Fig. 194-3). This principle recognizes that the brain is enclosed by an inelastic membranous layer, the dura mater, as well as the rigid skull, the intracranial compartments of which are composed of the brain itself, circulating blood, and CSF. An increase in the volume of any of these components, either from increasing tissue mass as a result of a CNS tumor or from increasing CSF space secondary to hydrocephalus, requires displacement of another component.29 The cranial vault can compensate for increases in intracranial volume by displacing other components to maintain normal ICP; however, once a certain threshold is reached, the compensatory mechanisms have been maximized and further increases in intracranial volume will lead to dangerously elevated ICP and secondary loss of cerebral perfusion.29 There is often a misunderstanding that infants are better able to compensate for elevations in ICP because of their open fontanelles, but this is a false impression because this mechanism is limited by the dura, which is still inelastic in infants.29 Increases in ICP are managed by several methods, including elevation of the head of the bed, decreasing PaCO2 with hyperventilation, administration of mannitol or steroids, CSF drainage, or sedation. The incidence of postoperative hydrocephalus is highest in patients with posterior fossa lesions, and it can be seen in up to 30% to 40% of such patients.10,30 In patients in whom postoperative hydrocephalus is a concern, an EVD is often left in place postoperatively to monitor ICP. ICP is monitored over the following few days to determine whether permanent CSF diversion will be required. Certain risk factors such as younger age at diagnosis, extensive preoperative ventricular dilation, and large tumors increase the likelihood of requiring a permanent VP shunt.10
MRI with and without contrast enhancement is imperative in the postoperative period to evaluate for residual tumor. As mentioned previously, it is suggested that postoperative imaging be performed within 24 to 48 hours after surgery to evaluate residual tumor before the effects of surgically induced contrast enhancement cannot be distinguished.12,15
Endocrine abnormalities are also common after neurosurgical procedures. The most common electrolyte disturbance, both before and after neurosurgery, is an imbalance in the sodium concentration.29 This is secondary to the integral role that the CNS plays in the regulation of sodium. Sodium disturbances postoperatively can be manifested as both hyponatremia and hypernatremia, each of which can cause severe consequences, including alterations in mental status, seizures, coma, and cerebral edema.31 Hyponatremia is most commonly caused by the syndrome of inappropriate antidiuretic hormone secretion (SIADH) as a result of excessive release of antidiuretic hormone (ADH) by the hypothalamus and leads to excessive water retention and sodium secretion in urine.31 Treatment of SIADH consists of fluid restriction and, in some cases, replacement of lost sodium with normal saline or hypertonic saline and oral sodium supplements.31 Cerebral salt wasting (CSW) can also be responsible for hyponatremia postoperatively. Although the underlying mechanisms are poorly understood, this entity differs from SIADH in that there is excessive sodium and urine excretion with subsequent hypovolemia.31 CSW is treated with hypertonic saline, oral sodium replacement, and fludrocortisone to help increase renal sodium and water retention.31 Close monitoring of sodium is required, and attention must be paid to the speed at which the sodium levels are corrected to prevent the consequences of correcting sodium imbalances too rapidly. Postoperative hypernatremia can also be seen in this patient population and is usually secondary to diabetes insipidus, which is caused by deficiencies in ADH secretion and leads to increased urine output without the ability to concentrate it.29 It is most often seen with tumors that disrupt the hypothalamic-pituitary axis, such as craniopharyngiomas and hypothalamic gliomas, and it is managed by orally replacing ADH and correcting the patient’s free water deficit.29
Long-Term Effects
The intensive chemotherapeutic regimens and radiotherapies, as well as the overall experience of being treated for a CNS malignancy, including multiple hospitalizations and procedures and lost school and social time, are not without untoward long-term effects. The toxic effects of both chemotherapy and radiation therapy predispose these children to physical, cognitive, social, and emotional underdevelopment.7,32 Studies have repeatedly demonstrated that survivors of childhood brain tumors have intellectual and academic deficits; deficits in memory, processing speed, visual perceptive ability, and attention; and impairments in social functioning and several other areas that begin as soon as 6 months after commencement of therapy but can continue to worsen even after cessation of therapy.2,33,34 Younger children are especially sensitive to these therapies and are affected the most because their nervous systems are still forming synaptic connections and undergoing myelination.2,12 Particular attention to these potential complications and the special needs of these children by health care professionals and educators must be emphasized. There is significant evidence suggesting that the burden of caring for a child with a brain tumor is ongoing and continues well into the off-treatment period. Parents of children surviving a brain tumor have also been reported to be at greater risk for both posttraumatic stress and general distress. In addition, children exposed to these therapies are at high risk for the development of secondary malignancies, permanent endocrinopathies, or CNS vasculopathies that may lead to early strokes,9,12 all of which need to be considered if new neurological complaints develop.
Corbally MT. Supportive care of the pediatric cancer patient. Semin Surg Oncol. 1993;9:461-466.
Kalifa C, Grill J. The therapy of infantile malignant brain tumors: current status? J Neurooncol. 2005;75:279-285.
Puget S, Crimmins DW, Garnett MR, et al. Thalamic tumors in children: a reappraisal. J Neurosurg. 2007;106(5 suppl):354-362.
1 Baldwin RT, Preston-Martin S. Epidemiology of brain tumors in childhood—a review. Toxicol Appl Pharmacol. 2004;199:118-131.
2 Brière ME, Scott JG, McNall-Knapp RY, et al. Cognitive outcome in pediatric brain tumor survivors: delayed attention deficit at long term follow-up. Pediatr Blood Cancer. 2008;50:337-340.
3 Hinsdale IL. Statistical Report (2000-2004 Data) [database online]: Central Brain Tumor Registry of the United States. http://www.cbtrus.org/index.html., 2008. Available at Accessed June 30, 2008
4 Rutka JT, Kuo JS. Pediatric surgical neuro-oncology: current best care practices and strategies. J Neurooncol. 2004;69:139-150.
5 Wilne S, Collier J, Kennedy C, et al. Presentation of childhood CNS tumours: a systemic review and meta-analysis. Lancet Oncol. 2007;8:685-695.
6 Jackson AC, Stewart H, O’Toole M, et al. Pediatric brain tumor patients: their parent’s perceptions of the hospital experience. J Pediatr Oncol Nurs. 2007;24:95-105.
7 Bestak M. Epidemiology of brain tumors. In: Keating RF, Goodrich JT, Packer RJ, editors. Tumors of the Pediatric Central Nervous System. New York: Thieme; 2001:14-21.
8 Duffner PK, Cohen ME, Freeman AI. Pediatric brain tumors: an overview. Cancer J Clinicians. 1985;35:287-301.
9 Khatua S, Jalali R. Recent advances in the treatment of childhood brain tumors. Pediatr Hematol Oncol. 2005;22:361-371.
10 Maher CO, Raffel C. Neurosurgical treatment of brain tumors in children. Pediatr Clin North Am. 2004;51:327-357.
11 Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv Syst. 2001;17:503-511.
12 Ullrich NJ, Pomeroy SL. Pediatric brain tumors. Neurol Clin North Am. 2003;21:897-913.
13 Lim M, Harsh GR. Neuro-oncology: An overview. In: Rengachary SS, Ellenbogen RG, editors. Principles of Neurosurgery. Edinburgh: CV Mosby; 2005:429-449.
14 Rossi A, Caracciolo V, Russo G, et al. Medulloblastoma: from molecular pathology to therapy. Clin Cancer Res. 2008;14:971-976.
15 Robertson RL. Advances in treatment of pediatric brain tumors. J Am Soc Exp Neurotherapeutics. 2006;3:276-291.
16 Takei H, Battacharjee MB, Rivera A, et al. New immunohistochemical markers in the evaluation of central nervous system tumors. Arch Pathol Lab Med. 2007;131:234-241.
17 Di C, Liao S, Adamson DC, et al. Identification of OTX2 as a medulloblastoma oncogene whose product can be targeted by all-trans retinoic acid. Cancer Res. 2005;65:919-924.
18 de Hass T, Oussoren E, Grajkowska W, et al. OTX1 and OTX2 expression correlates with the clinicopathologic classification of medulloblastomas. J Neuropathol Exp Neurol. 2006;65:176-186.
19 Gilles FH, Sobel E, Leviton A, et al. Epidemiology of seizures in children with brain tumors. J Neurooncology. 1992;12:53-68.
20 Reulecke BC, Erker CG, Fiedler BJ, et al. Brain tumors in children: initial symptoms and their influence on the time span between symptom onset and diagnosis. J Child Neurol. 2008;23:178-183.
21 Vezina G, Booth TN. Neuroradiology. In: Keating RF, Goodrich JT, Packer RJ, editors. Tumors of the Pediatric Central Nervous System. New York: Thieme; 2001:27-43.
22 Warren KE. NMR Spectroscopy and pediatric brain tumors. Oncologist. 2004;9:312-318.
23 Spencer SS, Theodore WH, Berkovic SF. Clinical applications: MRI, SPECT, and PET. Magn Reson Imaging. 1995;13:1119-1124.
24 Pirotte B, Acerbi F, Lubansu A, et al. PET imaging in the surgical management of pediatric brain tumors. Childs Nervous System. 2007;23:739-751.
25 Shono T, Natori Y, Morioka T, et al. Results of a long term follow-up after neuroendoscopic biopsy procedure and third ventriculostomy in patients with intracranial germinomas. J Neurosurg. 2007;107:193-198.
26 Recinos PF, Sciubba DM, Jallo GI. Brainstem tumors: where are we today? Pediatr Neurosurg. 2007;43:192-201.
27 Tamber JS, Bansal K, Liang ML, et al. Current concepts in the molecular genetics of pediatric brain tumors: implications for emerging therapies. Childs Nervous System. 2006;22:1379-1394.
28 Duncan JA, Hoffman HJ. Management of brain tumors in the pediatric population. In: Kaye A, Lawes EJr, editors. Brain Tumors. New York: Churchill Livingstone; 1995:405-412.
29 Conway EE. Critical care considerations. In: Keating RF, Goodrich JT, Packer RJ, editors. Tumors of the Pediatric Central Nervous System. New York: Thieme; 2001:111-134.
30 Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: new biological advances. Lancet Neurol. 2007;6:1073-1085.
31 Fraser JF, Stieg PE. Hyponatremia in the neurosurgical patient: epidemiology, pathophysiology, diagnosis and management. Neurosurgery. 2006;59:222-229.
32 Bonner MJ, Hardy KK, Williard VW, et al. Social functioning and facial expression recognition in survivors of pediatric brain tumors. J Pediatr Psychol. 2008;33:1-11.
33 Butler RW, Haser JK. Neurocognitive effects of treatment for childhood cancer. Ment Retard Dev Disabil. 2006;12:184-191.
34 Radcliffe J, Bennet D, Kazak A, et al. Adjustment in childhood brain tumor survival: child, mother, and teacher report. J Pediatr Psychol. 1996;21:529-539.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 194 General Approaches and Considerations for Pediatric Brain Tumors
Each year in the United States alone, cancer is diagnosed in more than 12,000 children. Of these cancers, pediatric brain tumors are the most common solid tumors and result in the highest overall mortality.1–5 Although survival rates for children with brain tumors continue to increase, studies have shown that these children experience more severe disease and treatment sequelae than do children with other types of cancer and that diagnosis and treatment strongly influence the psychopathology of their families’ functioning.6 More specifically, research has shown that the time of diagnosis is a particular point of vulnerability for families of children in whom brain tumors have been diagnosed and that the most accurate, honest information delivered with compassion is vital during this time.6
Epidemiology
Malignant brain tumors are the most common solid tumors in childhood; they account for 20% to 30% of all childhood cancers and are the leading cause of cancer-related death in this age group.1,5 In this chapter, we provide a general overview of the important clinical, diagnostic, and treatment-related considerations in pediatric brain tumors as a reference for practicing clinicians faced with caring for these patients. The annual incidence has been reported to be 2.4 to 6.3 per 100,000, which has actually risen over the past several decades because of better diagnostic modalities and reporting practices.3,5,7–10 Incidence rates for the pediatric population show that brain tumors are more common in white children than in black ones.3 In both, however, there is an overall slight male preponderance, with studies consistently showing a male-to-female ratio of about 1.5.11,12 More favorable histologic subgroups have repeatedly been found to occur in adolescents, whereas tumors with higher grade, more unfavorable locations, and poorer prognosis have generally occurred in younger children, particularly those younger than 3 years.11
The central nervous system (CNS) tumors that occur most frequently in childhood are vastly different from those in adulthood with regard to both histology and tumor location.12 In contrast to adults, in whom most CNS tumors are located supratentorially, approximately 50% to 55% of all childhood CNS tumors are infratentorial.8 In the first 6 months of age, supratentorial tumors are actually more common; however, by 2 years of age this location reverses, with up to 60% being infratentorial.8 This is in contrast to the third decade of life and beyond, where only 25% to 35% of intracranial tumors are infratentorial.8
The overall 5-year survival rate of children with brain tumors has improved considerably over the past several years. Because of earlier diagnosis and better therapies, survival rates are now between 35% and 65%, depending on several factors, including tumor histology and location.9 Age is also an important prognostic indicator for children with CNS tumors in general. Data consistently show that 10- to 15-year-olds have the longest survival whereas those younger than 2 years have the shortest.11
Classification
Classification schemes are important for all types of cancers because they allow physicians to make accurate predictions regarding the natural history of the disease. The response to certain therapies and prognosis can also be derived from a classification scheme.13 Classification schemes for CNS tumors are categorized according to the cell type from which the abnormal tissue originated.1,12 Subclassifications of tumors derived from a specific cell type exist as well, and such tumors can differ in their histologic, cytologic, and behavioral characteristics.12,13 CNS tumors can also be more broadly categorized into those that occur in certain age groups or locations.12
The cells of the nervous system can roughly be categorized as nerve cells and a variety of supporting cells called neuroglia. Glial cells greatly outnumber nerve cells by approximately 3 : 1 and include astrocytes, oligodendrocytes, ependymal cells, and microglia.1 As a whole, gliomas and, more specifically, astrocytomas are the most common CNS tumors of childhood and account for up to 50% in some series.1,4,11 Astrocytomas vary greatly in their histology, cytology, and behavioral characteristics and occur in several locations in the pediatric population. Pilocytic astrocytomas are the most common and represent up to 25% of pediatric CNS tumors. These tumor are considered benign and typically occur in the posterior fossa but can also be found elsewhere.11 However, other common locations for pediatric gliomas include the hypothalamic and optic pathway regions. The histology of these lesions is typically low grade, but they are difficult to treat surgically as a result of their location.11 Brainstem gliomas are not resectable because of their diffuse infiltrative nature, and their natural history and poor prognosis are similar to glioblastoma multiforme, which is the most common primary supratentorial tumor in adults. Focal lesions of the brainstem may have a better prognosis and can be monitored by serial surveillance with magnetic resonance imaging (MRI). Other focal lesions of the brainstem may have a dorsally exophytic portion that is amenable to surgical debulking.
The second most common CNS tumor in the pediatric population is medulloblastoma, which arises from neural stem cell precursors in the fourth ventricle and accounts for approximately 15% to 20% of tumors in this age group.10,14 Other common CNS tumors in this population are of neuroepithelial origin and include ependymomas, which account for 10% of pediatric CNS tumors; primitive neuroectodermal tumors (PNETs), 1.9%; ganglioglioma, 2.5%; dysembryoplastic neuroepithelial tumors, 0.6%; desmoplastic infantile astrocytomas, 0.6%; and mixed tumors.7,11
Non-neuroepithelial neoplasms consist of germ cell tumors (2.5%) of the pineal or other regions and include germinomas, teratomas, choriocarcinomas, and yolk sac tumors, as well as atypical teratoid/rhabdoid tumors (AT/RTs) (1.3%), choroid plexus tumors (0.9%), and craniopharyngiomas (5.6%).7,11
Overall, the most common CNS tumors seen in childhood are astrocytomas, medulloblastomas, ependymomas, craniopharyngiomas, and germ cell tumors.12 The incidence of these tumors, however, varies greatly within age groups among the pediatric population, and there is a tendency for certain tumors to occur at particular times during a child’s life (Table 194-1).3,11 In infancy (0 to 2 years old), choroid plexus papillomas, desmoplastic infantile astrocytomas, teratomas, PNETs, and AT/RTs predominate. By childhood (3 to 11 years of age), these tumors are rare and astrocytomas and craniopharyngiomas are much more frequently seen. By adolescence (≥12 years), germ cell tumors are commonly encountered, whereas craniopharyngiomas are less commonly seen.11
TABLE 194-1 Statistical Report: Primary Brain Tumors in the United States, CBTRUS, 2000-2004
| AGE (yr) | MOST COMMON HISTOLOGY | SECOND MOST COMMON HISTOLOGY |
|---|---|---|
| 0-4 | Embryonal/primitive/medulloblastoma | Pilocytic astrocytoma |
| 5-9 | Pilocytic astrocytoma | Embryonal/primitive/medulloblastoma |
| 10-14 | Pilocytic astrocytoma | Malignant glioma (NOS) |
| 15-19 | Pilocytic astrocytoma | Pituitary |
| 20-34 | Pituitary | Meningioma |
| 35-44 | Meningioma | Pituitary |
| 45-54 | Meningioma | Glioblastoma |
| 55-64 | Meningioma | Glioblastoma |
| 65-74 | Meningioma | Glioblastoma |
| 75-84 | Meningioma | Glioblastoma |
| 85+ | Meningioma | Glioblastoma |
CBTRUS, Central Brain Tumor Registry of the United States; NOS, not otherwise specified.
Published by the Central Brain Tumor Registry of the United States, 2008.
The distribution of astrocytomas also varies by age. As mentioned previously, pilocytic astrocytomas predominate overall and particularly in the childhood and early adolescent age groups. However, the incidence of pilocytic astrocytoma falls, and World Health Organization grades III and IV astrocytomas become more common in children older than 15 years (Fig. 194-1).3,11
Oncogenic Factors
Although population studies have failed to confirm any significant environmental factors associated with the development of CNS malignancies, several syndromes and genetic markers have been shown to be associated with their development.1 Such syndromes include neurofibromatosis types 1 and 2 (NF1 and NF2), tuberous sclerosis, and von Hippel-Lindau (VHL) syndrome, as well as many others. These disorders are associated with specific chromosomal abnormalities and lead to alterations in oncogenes and tumor suppressor genes, which typically regulate normal cell growth but, when altered, lead to tumor genesis. NF1 is associated with genetic alterations on chromosome 17 and predisposes individuals with this disorder to optic gliomas and malignant nerve sheath tumors.1,7,12 The genetic alteration in NF2 is located on chromosome 22 and is associated with the development of meningiomas and vestibular schwannomas.1,7 Tuberous sclerosis is caused by an abnormality on chromosome 9 that predisposes these patients to the development of astrocytomas.7 VHL syndrome is associated with multiple systemic malignancies secondary to an alteration on chromosome 3, including cerebellar hemangioblastomas.1,7
Although not associated with specific syndromes, several common CNS malignancies are linked to certain genetic tumor markers that are prognostic and continue to direct research for therapy. Examples include a strong association between overexpression of the oncogene p53 in childhood gliomas. p53 mutations are found in approximately 40% of malignant gliomas in children older than 3 years, which is much higher than the frequency seen in adult gliomas.1,12,15 In addition, the presence of the p53 mutation is associated with a poor prognosis independent of both the clinical and histologic prognostic features of the tumor at initial evaluation.15 Studies have also shown a relationship between AT/RTs and the INI1 gene on chromosome 22.12 This has become an important diagnostic tool because AT/RTs cannot be distinguished from medulloblastoma by clinical features or neuroimaging.16 Microscopically, these tumors can also be difficult to distinguish as a result of several similar features. Antibody staining for the INI1 gene for immunohistochemical analysis has now been used to differentiate these two entities because inactivating mutations of the INI1 gene located on chromosome 22 are a crucial step in the molecular pathogenesis of AT/RTs.16 Interestingly, choroid plexus tumors, called choroid plexus papillomas and carcinomas, which are intraventricular epithelial tumors arising from the choroid plexus, may also stain for the INI1 gene.
In addition, multiple genetic markers have been linked to medulloblastoma, including Trk-C, whose presence correlates with a good prognosis, whereas overexpression of epidermal growth factor receptor 2 and c-MYC have high predictive power for a poor prognosis.14,15 OTX2 overamplification has also been appreciated in medulloblastomas.17 OTX2 is a transcription factor that plays a role in normal cerebellar development.17 Expression of OTX2 has correlated with higher grade tumors, a poorer prognosis, and medulloblastomas that more frequently localize to the vermis of the cerebellum.17,18 OTX2 may become important in the treatment of this disease as well, inasmuch as a recent study demonstrated that OTX2-expressing medulloblastomas were responsive to all-trans-retinoic acid in vitro.17
Clinical Features
Because CNS malignancies account for approximately a quarter of childhood cancers, it is imperative that health care professionals be able to recognize the signs and symptoms at initial encounter.5 The signs and symptoms at diagnosis are typically a result of tumor location but are most often secondary to the raised intracranial pressure (ICP) that occurs with 40% of all intracranial tumors in childhood.5 Hydrocephalus is principally seen with infratentorial tumors and is due to obstruction of the ventricular system from mass effect by the tumor. The most common symptoms of elevated ICP are headache, nausea, and vomiting, which occur especially in the morning, and lethargy.8,10,12 Less commonly, macrocephaly and a bulging fontanelle can be seen in younger patients whose sutures have not yet closed.10 Infratentorial tumors causing compression on the brainstem can also be manifested as cranial nerve palsies.12 A common example is Parinaud’s syndrome, which is seen with compression of the midbrain tectum from a mass effect or increasing hydrocephalus. Features of Parinaud’s syndrome include paralysis of upgaze, paresis of accommodation, and convergence nystagmus. Additionally, a local mass effect from cerebellar tumors can cause ataxia and subsequent gait disturbances.5
[/not-level-membership-for-neurosurgery-category]
