CHAPTER 107 Gene- and Viral-Based Therapies for Gliomas
Advances in understanding gene structure/function and in the capacity to manipulate their expression have set the stage to alter genetic material for fighting or preventing disease. As gene products and their role within the cellular environment become increasingly understood, the potential for treating disease by using DNA as a drug is being realized. All proteins are coded for by DNA, and most neoplastic diseases ultimately result from the expression or lack thereof of one or more proteins (e.g., oncogenes or tumor suppressor genes, respectively). In theory, therefore, diseases could be treated by expression of the appropriate protein in the affected cells. Gene therapy is an experimental treatment that involves introducing genetic material (DNA or RNA) into cells, and it has made important advances over the past decade. Within this short time span it has moved from the conceptual laboratory research stage to clinical translational trials for a variety of diseases. Among the diseases being studied are primary genetic disorders1 and acquired diseases (e.g., cancer, neurodegenerative diseases), with brain tumors being among the first human malignancies to be targeted by gene therapy. The most efficient approaches for gene delivery are based on viral vectors, which have proved relatively safe in the central nervous system (CNS) despite occasional morbidity and death in non-neurosurgical trials. Most recently, results from the first clinical trial to test gene therapy for a type of inherited blindness showed that the experimental treatment is safe and can improve sight.2,3 These new developments are a landmark for gene therapy technology and paint a promising future. Although applying gene therapy to inherited CNS disorders remains an extremely difficult challenge, far more promise has been shown in the area of acquired CNS disorders such as brain tumors, spinal cord injury, stroke, and degenerative diseases (Parkinson’s and Alzheimer’s).4 This chapter reviews the basic principles and applications of gene therapy for brain tumors.
What is Gene Therapy?
Two fundamental considerations in gene therapy relate to (1) what gene should be delivered/expressed and (2) how to deliver it. In its simplest form, gene therapy is the process by which nucleic acids are transferred into cells to generate a therapeutic effect. This can be achieved by either replacing defective or missing genes or introducing new functions to the host’s cells. For this purpose, the genetic material is coupled to additional regulatory sequences (promoters, enhancers, and regulatory elements) and packaged inside a gene delivery vehicle to enable transfer and expression of the intended gene product inside the cell (Fig. 107-1).
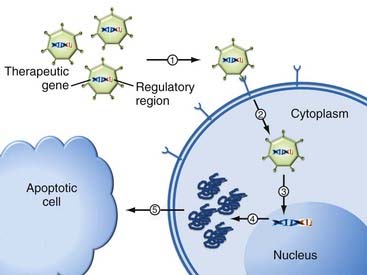
How is Gene Therapy Carried Out?
Although conceptually straightforward, efficient expression of foreign genes is the most critical aspect for the success of in vivo gene therapy. The first step in gene therapy involves gene delivery to facilitate expression of the therapeutic gene in the interior of a cell. The simplest method is direct introduction of therapeutic DNA into target cells by physical (i.e., electroporation) or chemical (i.e., lipofection) techniques.5 This approach still remains limited in application because it is relatively inefficient, can be used only with certain tissues, and requires large amounts of DNA. Furthermore, among the several barriers to successful gene delivery, foreign genes or the vectors used to deliver them, or both, can trigger a range of immune responses. However, sometimes these immune responses have been harnessed behind the concept of using gene therapy as a vaccine (i.e., DNA vaccines).6 The rationale behind this approach is that the immune response against the vector or introduced DNA, or both, could be exploited to provide a vaccine effect. This type of approach is thus used in some gene therapies against cancer or infectious disease.
The next difficulty for the foreign genetic material is that once within the cell, it must escape intracellular degradation and enter the nucleus so that it can be expressed (Fig. 107-1).7,8 Therefore, gene delivery systems (vectors) were designed to protect the genetic material. An ideal vector needs to meet three criteria: (1) it should protect the transgene against degradation by nucleases in the extracellular matrix, (2) it should bring the transgene across the plasma membrane into the nucleus of target cells, and (3) it should have no detrimental effects. Currently, such vectors for gene transfer can be classified into two categories: viral and nonviral.9,10
Nonviral Vectors
There are several methods for nonviral gene transfer. Physical approaches, including needle injection,11 electroporation,12,13 gene gun,14,15 ultrasound,16 and hydrodynamic delivery,17,18 use a physical force that permeates the cell membrane to facilitate intracellular gene transfer. However, these methods have been relatively inefficient in their capacity to transfer genes to a sufficiently elevated number of cells. Other nonviral carriers use cationic lipids, polymers, ceramic-based nanomaterial, carbon nanotubes, metal nanorods, and silica-based nanoparticles19,20 to deliver genes. Chemical approaches use synthetic or naturally occurring compounds as carriers to deliver the genetic material into cells.21 This approach involves the creation of an artificial lipid sphere with an aqueous core.22–24 This liposome, which carries the therapeutic DNA, is capable of passing the DNA through the target cell’s membrane.25,26 Synthetic and naturally occurring polymers represent another category of DNA carriers that have been used widely for gene delivery.27 In addition, lipid-polymer hybrid systems can be used.28,29 There is also new research on its way in which a 47th artificial human chromosome is introduced into target cells. This chromosome would exist along with the standard 46 without affecting their actions or causing any mutations, and it would be a large vector carrying a substantial amount of genes. A problem with this potential method is the difficulty delivering such a large molecule to the nucleus of a target cell.30,31 Although significant progress has been made in the application of various nonviral gene delivery systems, the majority of nonviral approaches remain much less efficient than viral vectors, especially for in vivo gene delivery.
Viral Vectors
Thus far, the most effective means of transferring DNA into somatic cells remains the use of a viral-based vector. Vectors for brain tumor therapy can be divided into two general categories: (1) replication-defective vectors (which we will designate vector from here on) and (2) replication-competent (replication-conditional, oncolytic viruses), which we will designate oncolytic viruses (OVs) from here on. In the first instance, the vector is derived from a virus from which all or most of the viral genes have been removed to minimize virus-mediated toxicity. In the second instance, selected viral genes are deleted or mutated so that viral targeting or replication, or both, can occur selectively in tumor versus endogenous neural cells. Up to now, the replication-defective vectors used in gene therapy trials for brain tumors have been based on retrovirus (RV) and adenovirus (AV). In terms of replication-competent (oncolytic) viruses, the ones used in clinical trials of brain tumors have been herpes simplex virus (HSV), AV, reovirus, and Newcastle disease virus (NDV) (Table 107-1). However, experimentally, almost any type of virus has been used either in a replication-defective or replication-competent fashion.
TABLE 107-1 Comparison of Viral Vectors and Oncolytic Viruses Used for Clinical Trials
| VECTOR OR OV | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Retrovirus (vector) |
FDA, Food and Drug Administration; MEK, mitogen-activated protein kinase/extracellular signal–regulated kinase; OV, oncolytic virus.
Retroviruses
RVs are a class of enveloped viruses containing a single-stranded RNA molecule as the genome. After infection, the viral RNA genome is reverse-transcribed into double-stranded DNA, which can be integrated into the chromosomes of host cells and expressed as proteins.32,33 They have been used primarily as vectors, although there are preclinical studies and a probable future attempt at a clinical trial involving the use of an RV-based OV.34 The advantages of RV vectors are that they are relatively easy to manipulate for gene therapy purposes and have been used widely.35 The available long-term experience and low toxicity to normal brain tissue make this vector a safe candidate for CNS gene therapy.36,37 One of the problems of RV vectors is that the viral genome can be inserted randomly in the genome of the host. If the insertion happens to be in the middle of one of the host genes, this gene will be disrupted (insertional mutagenesis). If the disrupted host gene is involved in regulating cell division, uncontrolled cell division (i.e., cancer) can occur.38,39 Other disadvantages of RV vectors are low titers, instability of the viral particles, and low transgene capacity (the maximal amount of DNA that can be packaged into an RV is just 7.5 kilobases [kb] of foreign DNA). Another drawback of RV vectors is the requirement that the target cell be dividing for integration and expression of viral genes. This restricts gene therapy solely to proliferating cells.40,41 In fact, although RV vectors were used in the initial gene therapy trials, their use for more recent gene therapy trials for cancer has been greatly reduced.
Adenoviral Vectors
AVs are nonenveloped viruses that contain a linear double-stranded DNA genome and cause respiratory and eye infections in humans. For CNS gene therapy, the commonly used AV vectors are derived from a subgroup that can be manipulated to produce replication-deficient vectors and have an insertion capacity of 10 kb of foreign DNA. AV vectors have become the mainstay of gene therapy and, in fact, have become a common tool in the kit of the molecular biology laboratory whenever a gene needs to be expressed in a mammalian cell. The vector can be extensively modified to target it away from its usual receptor, present in only some cells,42 to other receptors present in a desired target cell.43 Once bound to its receptor, the virus enters the cell in endosomal vesicles that fuse to lysosomes.44 Herein, the virus is able to liberate itself and escape to the cell nucleus. Viral DNA does not generally integrate and survives as an extrachromosomal element, from which gene expression derives. Extensive animal studies have revealed that gene expression is usually transient, unless the immune response can be minimized. In fact, long-term gene expression in animal neurons has been achieved by eliminating exposure of gene products/proteins to non-CNS areas45 and using AV vectors completely devoid of all viral genes (gutless vectors).46 In terms of gene delivery to brain tumors, the vector has been used to deliver almost any type of anticancer gene. For glioma clinical trials, however, AV vector–mediated delivery of the genes for HSV thymidine kinase (HSVtk),47 p53,48 and human interferon-β have been published thus far.
In summary, AVs have been used widely as vectors in gene therapy trials, and their advantages are their relative ease of manipulation and ability to be produced at high titer. Furthermore, AV vectors are very efficient at transducing a wide variety of cells, both dividing and nondividing, in vitro and in vivo. AVs do not usually integrate their genetic material into the host genome; rather, they replicate as episomal elements in the nucleus of the host cell and induce transient gene expression, and consequently there is no risk for insertional mutagenesis.49 The limitations of AV vectors are their short-term gene expression and the fact that they can produce toxic acute-phase responses. As clinical trials have begun to progress, it has become apparent that AV vectors may cause a significant innate immune response.50
Oncolytic Adenovirus
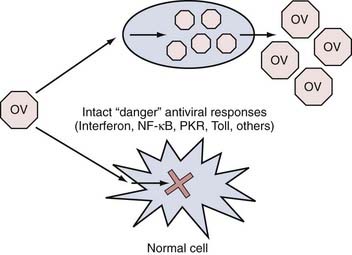
In this paradigm, engineered or naturally occurring strains of virus are created or discovered that appear to replicate better in tumor than in normal cells. This selectivity can occur at three general levels. First, every time a virus infects a cell, a robust antiviral response takes place inside the cell that consists of numerous “stress” or “danger” signals (Fig. 107-2). The overall effect of these signals is to limit the ability of the infecting virus to replicate to high levels and thus limit the number of viral progeny generated that could eventually infect neighboring cells. These “stress” signals consist of genes involved in the interferon, nuclear factor (NF)-κB, Toll-like receptor, PKR (double-stranded RNA-dependent protein kinase), and other pathways.51 In some cases, tumor cells have disabled some of these responses because they tend to be proapoptotic and antiproliferative. Therefore, tumor cells that have such disabled responses provide better targets for viral replication than do normal cells with intact antiviral responses. Second, in tumor cells, several genes involved in cell cycle regulation and apoptosis signaling are disrupted. These disruptions in tumor cell factors can be exploited to rationally engineer viral mutants that cannot replicate well in normal cells with intact cell cycle/apoptosis controls but will replicate in tumor cells that harbor these disruptions. Finally, viral mutants can be re-engineered so that they will target cell surface receptors present on tumor as opposed to normal cells. Thus far, clinical trials of brain tumors with OVs have involved mutants that use primarily the first two mechanisms of selective tumor targeting/lysis.
For instance, an engineered oncolytic AV (named ONYX-015) lacks a viral gene (E1B) that encodes for a protein that inactivates the cellular tumor suppressor protein p53. Initially, this mutant AV was thought to replicate in and lyse p53-deficient human tumor cells, but not cells with functional p53.52,53 Based on gene mutations, this vector could be of use in up to 50% of human glioblastomas carrying a p53 mutation.54 It may possess anticancer effects against the remaining tumors because p53 function is modulated by p14ARF protein, whose gene (CDKN2A) is frequently deleted in glioblastoma multiforme (GBM).55,56 However, subsequent experiments with this virus have revealed that the mechanism of tumor selectivity is not based on the lack of p53 tumor suppressor function but rather on a more complicated mechanism related to the nuclear export of viral mRNA into tumor cells.57 Other oncolytic AVs include those that target defects in the p16INK4a tumor suppressor pathway.58 The p16INK4a protein shares the CDKN2A gene region with p14ARF, thus making it one of the most commonly mutated sites in malignant brain tumors, as discussed in detail in another chapter.
Herpes Simplex Virus Type 1
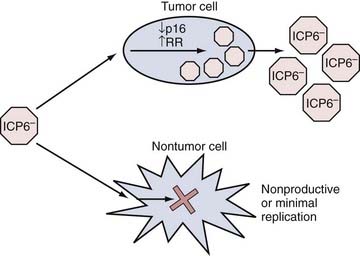
HSV-1 is a human neurotropic virus, which led to initial interest focused on using HSV-1 as a vector for gene transfer to the nervous system but has not yet resulted in clinical trials. HSV-1 is an enveloped DNA virus whose genome spans 152 kb and encodes for more than 80 genes. Wild-type HSV-1 is able to proceed into a lytic life cycle after infection or persist as an intranuclear episome in a latent state. Latently infected neurons function normally and are not rejected by the immune system. Although the latent virus is almost silent transcriptionally, it does possess neuron-specific promoters that are capable of functioning during latency.59 Interest in the use of HSV-1 as a cancer-killing agent intensified in the 1990s after reports showed that a genetically engineered form was oncolytic in brain tumor models.60 Since then, numerous reports have detailed various types of genetically engineered mutants for glioma therapy. The two most commonly used types have revolved around deletions of the viral gene (UL39 or UL40) that encodes for the viral protein ICP6 (Fig. 107-3). This protein possesses the function of a ribonucleotide reductase, and recent reports have linked this defect to an ability to replicate more selectively in cells with a defect in the p16 tumor suppressor gene.61 The other type has focused on mutation of the viral gene that encodes for the viral protein ICP34.5. This viral gene disables/counteracts a host cell response (based on the enzyme PKR) that promotes cellular apoptosis during infection.62 Several studies have investigated the mechanisms of tumor selectivity of this mutant virus. In one case, the authors linked this viral defect to enhanced replication in tumor cells with elevated levels of Ras activity.63 However, others have disputed this claim and have linked mutant OV replication to activity of the MEK (mitogen-activated protein kinase/extracellular signal–regulated kinase) pathway, which is present in tumor more than normal cells.64,65 In addition, it is also possible that tumor cells may have lost some PKR function, thus enabling this mutant virus to replicate.66
Reovirus
This small RNA virus is endemic to the human population, and most individuals become exposed to it in the newborn period. It produces generally self-limited episodes of respiratory and intestinal illness. A naturally occurring strain of the virus was found to replicate in cells with an overactive Ras pathway, and this property has been exploited by attempting to define the safety and efficacy profile of this virus in clinical trials of humans with GBM, as well as other cancers.67 Like most RNA viruses, reovirus can replicate to very high levels in infected cells, and thus in theory it could provide an increased therapeutic “punch” because the large number of viral progeny could infect a large number of neighboring tumor cells. Some of the issues, however, that limit use of reovirus relate to its small size and virologic properties, characteristics that render genetic manipulation difficult. RNA viruses are also notoriously highly mutable, which can also be problematic for production and for use. Finally, clinical experience is currently limited to one biotechnology venture (Oncolytics Biotech, Inc.).
Newcastle Disease Virus
NDV is an avian RNA virus that also possesses multiple strains. Several have been adapted for growth in tumor cells, and recently a phase I clinical trial of intravenous infusion of one such mutant was reported from Israel.68 The advantages and disadvantages of NDV are similar to those of reovirus. However, differences exist: immunity to this avian virus does not exist in humans, and thus initial administration of NDV would not be limited by the adaptive arm of the host’s immune reaction. This could also be a disadvantage in that the zoonotic nature of the virus and its administration to humans could generate mutant species that become adapted to human infection and cause rapid pandemics, such as those that have occurred in the past with coronaviruses and human immunodeficiency virus. The mechanism of selectivity of NDV for tumor cells is also not well understood. It may be due to the impaired interferon or other host cell antiviral responses in tumor versus normal cells (see Fig. 107-2).
Comparison of Vectors
Objective studies evaluating the different delivery systems are rarely carried out, thus making comparisons difficult.69,70 The ideal vector would combine features of all the delivery systems described earlier. Preparation of this ideal vector should be relatively simple and result in high vector concentrations (>1010 particles/mL). Furthermore, there would be no significant immune response to the vector to avoid undesired host reactions and allow later readministration as needed. The lack of an immune response may also allow transgene expression to be prolonged from episomal systems so that readministration is not necessary. Alternatively, integration into a specific site in the host genome would permit the transgene not to be lost during the cell’s life span. However delivered, transcriptional control by the host cell or by the physician would allow greater tissue specificity. As of the present, no single vector meets all these requirements, but improvements are being made and the genomes of viral vectors are progressively being reduced to contain the transgene with a minimum number of maintenance genes. At the same time, liposomes are becoming more like viruses. It is likely that each delivery system will find a niche. Many research groups have invested substantial effort into developing a given system, and they will most likely continue with that vector until a therapeutic application has been found.
Genes Delivered Into Brain Tumors
Prodrug Activating
HSV-1 Thymidine Kinase/Ganciclovir
HSVtk is an enzyme that metabolizes nontoxic nucleoside analogues, such as ganciclovir, acyclovir, or valacyclovir, into a cytotoxic molecule. The ganciclovir metabolite is incorporated into DNA, which leads to termination of the incorporation of deoxynucleosides into DNA and subsequently to cell death.71 Because ganciclovir’s effects are limited to DNA, it targets primarily replicating cells, much like the S phase–specific chemotherapeutic agents, but it also affects the ability of DNA to be repaired by DNA polymerases, and thus it could theoretically also affect quiescent cells with damaged DNA. In vivo efficacy has been demonstrated in multiple animal studies.72–74 Furthermore, preclinical experiments have demonstrated marked tumor elimination despite gene transfer into only a small fraction of the tumor cells.75 This cytotoxic effect of transduced cells on adjacent nontransduced cells is termed the bystander effect.76 The bystander effect is mediated mainly by the transfer of toxic phosphorylated forms of ganciclovir to nontransduced cells, presumably via gap junctions.77 Another presumed mechanism contributing to the bystander effect is the targeting of mitotically active endothelial cells in tumor vessels by the RV vector and the formation of tumor infarction zones after the administration of ganciclovir.78 An immune-associated response against a nonhuman protein, such as HSVtk, leading to diffuse cell death that affects neighboring nontransduced cells has also been suggested.79 In addition to the bystander effect, tumor cells transduced to express HSVtk and treated with the antiviral agent acyclovir display enhanced sensitivity to radiation in culture and in vivo.80 Possible explanations for the radiation enhancement could be that the DNA in which acyclovir has been incorporated may be susceptible to radiation-induced strand breakage and acyclovir might sensitize cells by inhibiting the polymerase activity required for repair of radiation-induced DNA damage.81 The HSVtk gene therapy approach was tested in human glioma trials (phase III) with defective RV vectors and more recently with AV vectors. In fact, a phase III trial in Europe and Israel was recently completed under the sponsorship of a British biotechnology venture (Ark Therapeutics) in which an AVtk vector and ganciclovir were provided to patients with newly diagnosed GBM before the application of standard therapy (radiation or temozolomide, or both). Interim results from this trial are available on the company’s website (www.arktherapeutics.com/main/products.php?content=products_cerepro). To take advantage of the possible synergy between radiation-induced damage of DNA and the inability of DNA with incorporated ganciclovir or valacyclovir metabolites to repair itself, we have recently completed a phase I clinical trial involving patients with newly diagnosed malignant glioma in which AVtk and valacyclovir are provided in conjunction with radiation therapy.
Cytosine Deaminase/5-Fluorocytosine
5-Fluorocytosine (5-FC) is an agent used to treat fungal infections (such as Candida albicans and Cryptococcus neoformans). 5-FC is a prodrug that is converted into the active agent 5-fluorouracil (5-FU) by cytosine deaminase (CD), which is uniquely expressed in certain fungi and bacteria. Although 5-FC is nontoxic to human cells because of the lack of CD, 5-FU is used to treat cancers such as colon, pancreatic, and breast cancer. The toxic effects of 5-FU are mediated by its intracellular metabolites, which cause DNA strand breakage leading to cell death.82 Two preclinical studies of treatment of GBM with an AV vector carrying the CD gene demonstrated promising results.83 However, no trial involving brain tumors has used this gene transfer strategy thus far.
Cytochrome P-450 2B1/Cyclophosphamide
Cyclophosphamide (CPA) is a prodrug that is activated by liver-specific enzymes of the cytochrome P-450 family. The active form of CPA, phosphoramide mustard, is an alkylating agent that generates DNA cross-links and consecutive DNA strand breaks. The efficacy of CPA in treating brain tumors has been limited by the fact that although CPA crosses the blood-brain barrier, its active metabolites are poorly transported across it.84
The rat cytochrome P-450 2B1 (CYP2B1) activates CPA with high efficiency,85 and gene therapy using CYP2B1 to activate CPA was designed primarily for use in brain tumors because other malignancies have ready access to CPA’s active metabolites. Implantation of CYP2B1-expressing RV vectors was shown to induce regression of intracerebral rat glioma cells after intratumoral or intrathecal administration of CPA.86,87
Replacement of the large subunit of the HSV-1 genome with the CYP2B1 gene has led to the design of an HSV-1 vector (rRp450) that can kill tumor cells through three modes: (1) using viral oncolysis (see later) and rendering the infected cell sensitive to (2) CPA and (3) ganciclovir. Subcutaneous tumors established from glioma cell lines in immunodeficient mice regress only when treated with rRp450, CPA, and ganciclovir.82 Currently, no P-450/CPA clinical trials for brain tumors have been conducted.
Tumor Suppressor Genes and Cell Cycle Modulators
Advances in understanding the underlying molecular and genetic mechanisms of neurological diseases have provided a rational framework for the development of new treatments such as gene therapy.88 The p53 tumor suppressor protein regulates cell cycle progression and apoptosis in response to many external insults (e.g., DNA damage and oncogenic mutations).89 Mutation of the p53 gene (TP53) resulting in loss of its function is common in astrocytomas and is associated with progression of tumor from low-grade astrocytoma to GBM.90 Accordingly, the TP53 gene became an attractive candidate for gene transfer in an attempt to restore cell cycle regulation in TP53-mutated cells and induce apoptosis, even in tumors with intact functional genes, by causing enhanced expression of the gene product.91,92
Another key regulatory pathway involved in cell cycle control is the retinoblastoma protein (RB)/cyclin-dependent kinase (CDK)/cyclin-dependent kinase inhibitor (CDKN) circuit.93 This pathway is frequently affected in gliomas.94,95 Preliminary studies to restore the genomic region of CDKN2A/CDKN2B in glioblastoma cell lines have demonstrated tumor growth arrest or apoptosis.96 Another candidate for a gene therapy approach is the epidermal growth factor receptor gene, which shows frequent amplification in primary glioblastomas.97 Currently, a clinical trial using an AV vector to deliver the p53 gene in humans with recurrent GBM has been reported.
Genetic Immune Modulation
Genetic immune modulation enhances the immune response against tumors by expressing cytokines and lymphokines. The cytokines used frequently to achieve genetic immune modulation of tumors are interleukin-2,98,99 interleukin-4,100 interleukin-12,101 interferon-β,102 interferon-γ,103 and granulocyte-macrophage colony-stimulating factor.104
Several studies have been performed in which tumor cells are infected ex vivo with cytokine genes, cell growth is arrested by irradiation, and then the cells are reimplanted to sustain paracrine secretion of cytokines within the tumor.98,105 Another model introduces RV producer cells carrying immune-modulating genes into the tumor so that infection occurs in situ.100
Several phase I clinical trials using this strategy are currently under way. However, for interleukin-2 and interferon-γ, severe CNS toxicity has been reported when these cytokines were secreted by tumor cells intracranially.106 One clinical trial in humans with recurrent malignant glioma has been published in which an AV vector was used to deliver the gene for human interferon-β. Gene therapy can be also used to generate tumor vaccines by inducing tumor antigen presentation by antigen-presenting cells. Antigen-presenting cells (i.e., dendritic cells) can be harvested from peripheral blood or brain tumor biopsy specimens; transduced with the DNA, mRNA, or proteins (or any combination thereof) coding for the tumor antigen or antigens; and then expanded in vitro before administration to the patient. There have been no clinical trials for human GBM involving the use of a gene-based vaccine approach as of yet.
Antiangiogenic Gene Therapy
Neovascularization is an important feature of malignant gliomas and is dependent on several potent angiogenic factors secreted by tumor cells. Vascular endothelial growth factor is an important angiogenic factor overexpressed in gliomas. It has been shown that in vivo transfer of a recombinant AV vector carrying the gene for vascular endothelial growth factor in an antisense orientation into gliomas inhibits tumor growth.107 Antisense oligonucleotides are single strands of DNA or RNA that can bind to a complementary RNA sequence. If binding takes place, this DNA/RNA hybrid can be degraded, which prevents subsequent protein synthesis. Another study showed significant inhibition of glioma by using an RV vector to deliver a signaling-defective vascular endothelial growth factor receptor that forms inactive dimers with wild-type vascular endothelial growth factor receptor.108 In addition to being one of the gene replacement strategies described earlier, p53 gene therapy might also have an antiangiogenic effect based on the discovery that inducing wild-type p53 expression in astrocytomas causes release of an angiogenesis inhibitory factor.109 The intense neovascularization in malignant gliomas may enable a new intravascular modality of gene therapy for this disease. Although genetic vectors administered intravascularly are unlikely to penetrate the blood-brain barrier, intravascular delivery of vectors may target endothelial cells in the brain. Thus, various therapeutic genes for the destruction of tumor vasculature or blockers of angiogenesis could be used to alter tumor blood flow. To date, however, there have been no trials using this approach for GBM.
Oncolytic Viruses
OVs have recently been exploited considerably in clinical trials of brain tumors. A genetically engineered HSV-1 (termed G207) was designed so that it is replication competent in glioblastoma cells, has attenuated neurovirulence, is temperature sensitive, and can be eliminated with ganciclovir.110 It features deletion of the viral genes that encode both ICP6 and ICP34.5, and thus it may selectively target cells with defects in p16 and with overexpression of MEK. Intraneoplastic inoculation with G207 decreased tumor growth and prolonged survival in animal models, with no significant toxicity in normal brain tissue. Based on these results, a phase I study using G207 in patients with anaplastic astrocytoma and glioblastoma was completed and demonstrated promising results.111
Another virus capable of direct cytopathic effect in tumors is an AV mutant deficient in a protein that was thought to inactivate the cellular tumor suppressor protein p53 but has more recently been shown to target cells with altered nuclear export of viral mRNA. A phase I clinical trial of this virus was reported to show safety.112 A version of this virus has been approved for clinical use in China for cancers. A mutant strain of reovirus has also recently been reported in a clinical trial in humans with recurrent glioma.67 Reovirus is a small, relatively nonpathogenic RNA virus that has been shown to replicate preferentially in cells with elevated activity of Ras, an oncogene involved in progression of glioma. The trial also showed this approach to be safe, and a new trial using convection-enhanced delivery of the viral mutant is now in progress.
Clinical Trials of Brain Tumor Gene Therapy
Four viruses (RV, AV, HSV-1, NDV) have been studied in clinical brain tumor trials. RV and AV have been genetically modified to express HSVtk with concurrent ganciclovir administration, and HSV has been used as an oncolytic agent. A selection of clinical trials is summarized in Table 107-2. The first study of brain tumor gene therapy in humans used stereotactic intratumoral inoculation of an RV vector carrying HSVtk in 15 patients with recurrent malignant brain tumors.113 Eight patients showed a 25% reduction in gadolinium enhancement on magnetic resonance imaging (MRI) immediately after administration of ganciclovir, and 4 patients had up to an 18-month reduction after treatment. Although this study showed some promising results in terms of antitumor efficacy, it was not a controlled or randomized protocol. Two subsequent phase I/II studies in patients with recurrent glioblastoma were performed by using an RV/HSVtk vector followed by ganciclovir administration. The virus was delivered via vector-producing cells injected during surgical tumor resection. The first study involved 12 patients, and no treatment-related adverse effects were noted. There was an overall median survival of 6.8 months, with 3 patients surviving longer than 12 months and 1 being recurrence free 2.8 years after treatment.115 A comparable international, multicenter uncontrolled study included 48 patients with recurrent gliomas. The median survival time was 8.6 months, with a 12-month survival rate of 27%. Tumor recurrence was absent on MRI in 7 patients for at least 6 months and in 2 patients for 12 months, and 1 patient remained recurrence free at 24 months.116 A similar phase I study was performed in 12 children 2 to 15 years of age with recurrent malignant supratentorial tumors. This study also used an RV/HSVtk/ganciclovir approach. Disease progression occurred at a median of 3 months after treatment, and the longest time until progression was 24 months, with no adverse effects noted.126 A large controlled phase III trial seemed necessary for ultimate confirmation of the efficacy of the RV/HSVtk/ganciclovir approach. This study used an adjuvant gene therapy protocol in addition to the standard therapy of maximal surgical resection and irradiation for newly diagnosed GBM. After 4 years of follow-up of 248 patients who were divided into a gene therapy and a control arm, survival analysis showed no advantage of gene therapy in terms of tumor progression and overall survival.117
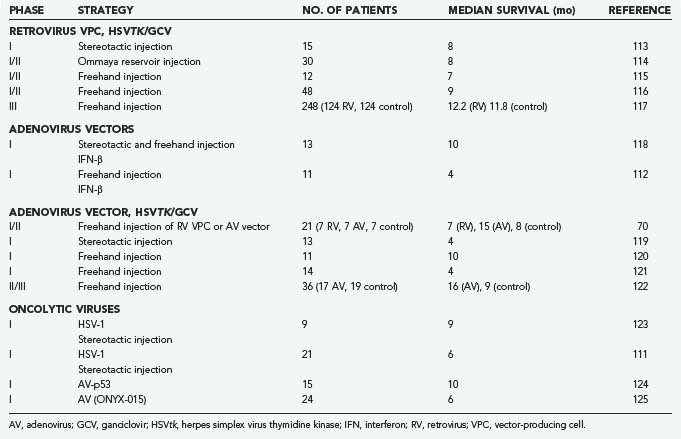
There have been several clinical trials using AV vectors for malignant gliomas. One study, published in 1998,127 compared humans with recurrent gliomas treated by direct stereotactic injection of either an RV or an AV vector that expressed the E. coli LacZ gene. They showed that the latter exhibited more widespread distribution of gene expression and thus concluded that AV vectors may be superior in their ability to distribute the transferred gene into brain tumors. Additional representative AV trials are discussed here briefly.
One recent trial evaluated 11 patients with recurrent high-grade glioma treated with a human interferon-β AV vector introduced by stereotactic injection, followed later by surgical resection and additional injection of the vector into the tumor bed. The vector was well tolerated in all but 1 patient, who experienced confusion after the postoperative injection that was caused by local brain toxicity. Dose-related inflammation and necrosis were identified in tumor specimens resected after treatment,112 findings suggestive of a biologic effect from the transferred gene. A phase I/II trial evaluated an AV vector expressing the HSVtk gene in patients with primary or recurrent high-grade gliomas. This study was performed in a controlled randomized fashion on 36 patients, 17 in the treatment arm and 19 in the control group. All patients underwent surgical resection and AV injection followed by intravenous ganciclovir on postoperative day 5 for 14 days. The control group underwent resection without gene therapy and ganciclovir injections. Median survival in the gene therapy group was significantly longer than in the control group (62 versus 37 weeks).122 As discussed earlier, a large phase III trial has been conducted in Europe and Israel with this agent (Cerepro), and a recent statement from the company (Ark Therapeutics, Inc.) suggested evidence of efficacy, although published data would allow independent evaluation of this claim. A phase I trial of AVtk delivered at the time of surgical resection of newly diagnosed malignant glioma, followed by oral valacyclovir combined with standard therapy (external field irradiation to 6000 cGy and temozolomide), has also recently been concluded and showed relative safety, although the data are still maturing regarding any evidence of efficacy (Chiocca and colleagues, unpublished data). An AV vector delivering a wild-type copy of the p53 transgene was also evaluated in humans with recurrent glioma. Here, the vector was initially introduced via a catheter implanted in the middle of the tumor bed, followed by resection of the tumor to allow studies related to p53 gene delivery and distribution. Again, although the treatment was well tolerated, the distribution of p53 gene into tumor was relatively low.128
The third virus to undergo clinical trials in brain tumors was HSV-1. A phase I trial evaluated the safety of an oncolytic HSV-1 in 9 patients with recurrent glioblastoma. Direct intratumoral injection was performed with no induction of encephalitis, adverse clinical symptoms, or reactivation of latent HSV occurring, thus appearing as though tumor progression was controlled with some efficacy.123 Further investigations of the oncolytic HSV-1 mutant demonstrated this vector to be nontoxic when delivered into the tumor or into adjacent brain. It was also shown that the HSV-1 vector can persist in human glioma cells and has the potential to kill tumor cells over a prolonged period.129 A large phase III trial in Europe is being conducted and sponsored by Crusade Laboratories (www.crusadelabs.com). A clinical trial using a different HSV-1 mutant included 21 patients with recurrent glioma with similar results.111
Conclusion
The rapid evolution of recombinant DNA technology has enabled us to develop new therapeutic modalities, including gene therapy. The completed clinical gene therapy trials for brain tumor and other CNS diseases have offered some promising results. However, the field of gene therapy is in its infancy. Screening of new approaches is based on animal models, which are far from representative of the analogous clinical scenarios, as shown by the discrepancy between experimental animal studies and clinical trials. As for brain tumors, the size, consistency, and extent of tumor models do not reflect the large, necrotic, infiltrative nature of GBMs. Experimental approaches that aim at immune enhancement use animal models that are practically nonsyngeneic for the implanted tumor, and the human response to various viral vectors cannot be predicted in any reliable manner from animal studies. Accordingly, many ingenious gene therapy strategies that are effective in preclinical studies will not fulfill their expectations in the clinic. The problem of delivery of genetic vectors into solid brain tumors and efficient in situ gene transfer remains one of the most significant hurdles in gene therapy. The efficiency of transduction could be improved by modifying vector-producing cells (VPC) that have the ability to track even single tumor cells invading the surrounding brain tissue, such as migratory VPCs.130 The currently used manual injection of vectors, which is limited in the volume that can be injected at any given time and the efflux that occurs along needle tracts, might be improved by the use of three-dimensional neuronavigation techniques and automated slow-speed injection devices. At present, gene therapy is being studied in clinical trials for brain tumors and is not yet available outside a clinical trial.
Aghi M, Kramm CM, Chou TC, et al. Synergistic anticancer effects of ganciclovir/thymidine kinase and 5-fluorocytosine/cytosine deaminase gene therapies. J Natl Cancer Inst. 1998;90:370-380.
Aghi M, Visted T, Depinho RA, et al. Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene. 2008;27:4249-4254.
Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231-2239.
Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373-376.
Boviatsis EJ, Chase M, Wei MX, et al. Gene transfer into experimental brain tumors mediated by adenovirus, herpes simplex virus, and retrovirus vectors. Hum Gene Ther. 1994;5:183-191.
Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008;10:38-45.
Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958-966.
Chiocca EA, Smith KM, McKinney B, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16:618-626.
Culver KW, Ram Z, Wallbridge S, et al. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550-1552.
Fischer A, Cavazzana-Calvo M. Gene therapy of inherited diseases. Lancet. 2008;371:2044-2047.
Freeman SM, Abboud CN, Whartenby KA, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274-5283.
Heise C, Hermiston T, Johnson L, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134-1139.
Heise C, Sampson-Johannes A, McCormick F, et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639-645.
Kim SH, Kim S, Robbins PD. Retroviral vectors. Adv Virus Res. 2000;55:545-563.
Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867-874.
Martuza RL, Malick A, Markert JM, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854-856.
Mineta T, Rabkin SD, Yazaki T, et al. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938-943.
Niidome T, Huang L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 2002;9:1647-1652.
Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389-2401.
Rainov NG, Kramm CM. Recombinant retrovirus vectors for treatment of malignant brain tumors. Int Rev Neurobiol. 2003;55:185-203.
Ram Z, Culver KW, Oshiro EM, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354-1361.
Sandmair AM, Vapalahti M, Yla-Herttuala S. Adenovirus-mediated herpes simplex thymidine kinase gene therapy for brain tumors. Adv Exp Med Biol. 2000;465:163-170.
Vecil GG, Lang FF. Clinical trials of adenoviruses in brain tumors: a review of Ad-p53 and oncolytic adenoviruses. J Neurooncol. 2003;65:237-246.
Verma IM, Somia N. Gene therapy—promises, problems and prospects. Nature. 1997;389:239-242.
Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573-587.
1 Fischer A, Cavazzana-Calvo M. Gene therapy of inherited diseases. Lancet. 2008;371:2044-2047.
2 Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231-2239.
3 Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240-2248.
4 Kaplitt MG, Feigin A, Tang C, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369:2097-2105.
5 Anwer K. Formulations for DNA delivery via electroporation in vivo. Methods Mol Biol. 2008;423:77-89.
6 Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008;8:108-120.
7 Lechardeur D, Lukacs GL. Nucleocytoplasmic transport of plasmid DNA: a perilous journey from the cytoplasm to the nucleus. Hum Gene Ther. 2006;17:882-889.
8 Vaughan EE, DeGiulio JV, Dean DA. Intracellular trafficking of plasmids for gene therapy: mechanisms of cytoplasmic movement and nuclear import. Curr Gene Ther. 2006;6:671-681.
9 Bergen JM, Park IK, Horner PJ, et al. Nonviral approaches for neuronal delivery of nucleic acids. Pharm Res. 2008;25:983-998.
10 Niidome T, Huang L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 2002;9:1647-1652.
11 Wolff JA, Malone RW, Williams P, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465-1468.
12 Heller LC, Ugen K, Heller R. Electroporation for targeted gene transfer. Expert Opin Drug Deliv. 2005;2:255-268.
13 Neumann E, Schaefer-Ridder M, Wang Y, et al. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982;1:841-845.
14 Yang NS, Burkholder J, Roberts B, et al. In vivo and in vitro gene transfer to mammalian somatic cells by particle bombardment. Proc Natl Acad Sci U S A. 1990;87:9568-9572.
15 Yang NS, Sun WH. Gene gun and other non-viral approaches for cancer gene therapy. Nat Med. 1995;1:481-483.
16 Lawrie A, Brisken AF, Francis SE, et al. Microbubble-enhanced ultrasound for vascular gene delivery. Gene Ther. 2000;7:2023-2027.
17 Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258-1266.
18 Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735-1737.
19 Bharali DJ, Klejbor I, Stachowiak EK, et al. Organically modified silica nanoparticles: a nonviral vector for in vivo gene delivery and expression in the brain. Proc Natl Acad Sci U S A. 2005;102:11539-11544.
20 Roy I, Stachowiak MK, Bergey EJ. Nonviral gene transfection nanoparticles: function and applications in the brain. Nanomedicine. 2008;4:89-97.
21 Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7:992-1009.
22 Felgner PL, Gadek TR, Holm M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987;84:7413-7417.
23 Koltover I, Salditt T, Radler JO, et al. An inverted hexagonal phase of cationic liposome–DNA complexes related to DNA release and delivery. Science. 1998;281:78-81.
24 Templeton NS, Lasic DD, Frederik PM, et al. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997;15:647-652.
25 Liu D, Ren T, Gao X. Cationic transfection lipids. Curr Med Chem. 2003;10:1307-1315.
26 Smyth Templeton N. Liposomal delivery of nucleic acids in vivo. DNA Cell Biol. 2002;21:857-867.
27 Hughes JA, Rao GA. Targeted polymers for gene delivery. Expert Opin Drug Deliv. 2005;2:145-157.
28 Gao X, Huang L. Potentiation of cationic liposome–mediated gene delivery by polycations. Biochemistry. 1996;35:1027-1036.
29 Sorgi FL, Bhattacharya S, Huang L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997;4:961-968.
30 Harrington JJ, Van Bokkelen G, Mays RW, et al. Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat Genet. 1997;15:345-355.
31 Grimes BR, Monaco ZL. Artificial and engineered chromosomes: developments and prospects for gene therapy. Chromosoma. 2005;114:230-241.
32 Kim SH, Kim S, Robbins PD. Retroviral vectors. Adv Virus Res. 2000;55:545-563.
33 Young LS, Searle PF, Onion D, et al. Viral gene therapy strategies: from basic science to clinical application. J Pathol. 2006;208:299-318.
34 Tai CK, Kasahara N. Replication-competent retrovirus vectors for cancer gene therapy. Front Biosci. 2008;13:3083-3095.
35 Rainov NG, Kramm CM. Recombinant retrovirus vectors for treatment of malignant brain tumors. Int Rev Neurobiol. 2003;55:185-203.
36 Ram Z, Culver KW, Walbridge S, et al. Toxicity studies of retroviral-mediated gene transfer for the treatment of brain tumors. J Neurosurg. 1993;79:400-407.
37 Long Z, Li LP, Grooms T, et al. Biosafety monitoring of patients receiving intracerebral injections of murine retroviral vector producer cells. Hum Gene Ther. 1998;9:1165-1172.
38 Uren AG, Kool J, Berns A, et al. Retroviral insertional mutagenesis: past, present and future. Oncogene. 2005;24:7656-7672.
39 Fehse B, Roeder I. Insertional mutagenesis and clonal dominance: biological and statistical considerations. Gene Ther. 2008;15:143-153.
40 Roth JA, Nguyen D, Lawrence DD, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2:985-991.
41 Tait DL, Obermiller PS, Redlin-Frazier S, et al. A phase I trial of retroviral BRCA1sv gene therapy in ovarian cancer. Clin Cancer Res. 1997;3:1959-1968.
42 Freimuth P, Philipson L, Carson SD. The coxsackievirus and adenovirus receptor. Curr Top Microbiol Immunol. 2008;323:67-87.
43 Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573-587.
44 Douglas JT. Adenoviral vectors for gene therapy. Mol Biotechnol. 2007;36:71-80.
45 Barcia C, Jimenez-Dalmaroni M, Kroeger KM, et al. One-year expression from high-capacity adenoviral vectors in the brains of animals with pre-existing anti-adenoviral immunity: clinical implications. Mol Ther. 2007;15:2154-2163.
46 Candolfi M, Curtin JF, Ziong WD, et al. Effective high-capacity gutless adenoviral vectors mediate transgene expression in human glioma cells. Mol Ther. 2006;14:371-381.
47 Sandmair AM, Vapalahti M, Yla-Herttuala S. Adenovirus-mediated herpes simplex thymidine kinase gene therapy for brain tumors. Adv Exp Med Biol. 2000;465:163-170.
48 Vecil GG, Lang FF. Clinical trials of adenoviruses in brain tumors: a review of Ad-p53 and oncolytic adenoviruses. J Neurooncol. 2003;65:237-246.
49 Verma IM, Somia N. Gene therapy—promises, problems and prospects. Nature. 1997;389:239-242.
50 Hartman ZC, Appledorn DM, Amalfitano A. Adenovirus vector induced innate immune responses: impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 2008;132:1-14.
51 Chiocca EA. The host response to cancer virotherapy. Curr Opin Mol Ther. 2008;10:38-45.
52 Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373-376.
53 Heise C, Sampson-Johannes A, McCormick F, et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639-645.
54 Hollstein M, Rice K, Greenblatt MS, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551-3555.
55 Rutka JT, Akiyama Y, Lee SP, et al. Alterations of the p53 and pRB pathways in human astrocytoma. Brain Tumor Pathol. 2000;17:65-70.
56 Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731-737.
57 O’Shea CC, Johnson L, Bagus B, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611-623.
58 Heise C, Hermiston T, Johnson L, et al. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134-1139.
59 Taylor TJ, Brockman MA, McNamee EE, et al. Herpes simplex virus. Front Biosci. 2002;7:d752-764.
60 Martuza RL, Malick A, Markert JM, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854-856.
61 Aghi M, Visted T, Depinho RA, et al. Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene. 2008;27:4249-4254.
62 Cassady KA, Gross M, Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J Virol. 1998;72:7005-7011.
63 Farassati F, Yang AD, Lee PW. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol. 2001;3:745-750.
64 Smith KD, Mezhir JJ, Bickenbach K, et al. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1. J Virol. 2006;80:1110-1120.
65 Veerapong J, Bickenbach KA, Shao MY, et al. Systemic delivery of (gamma1)34.5-deleted herpes simplex virus-1 selectively targets and treats distant human xenograft tumors that express high MEK activity. Cancer Res. 2007;67:8301-8306.
66 Balachandran S, Barber GN. PKR in innate immunity, cancer, and viral oncolysis. Methods Mol Biol. 2007;383:277-301.
67 Forsyth P, Roldán G, George D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16:627-632.
68 Freeman AI, Zakay-Rones Z, Gomari JM, et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221-228.
69 Boviatsis EJ, Chase M, Wei MX, et al. Gene transfer into experimental brain tumors mediated by adenovirus, herpes simplex virus, and retrovirus vectors. Hum Gene Ther. 1994;5:183-191.
70 Sandmair AM, Loimas S, Puranen P, et al. Thymidine kinase gene therapy for human malignant glioma, using replication-deficient retroviruses or adenoviruses. Hum Gene Ther. 2000;11:2197-2205.
71 Faulds D, Heel RC, Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs. 1990;39:597-638.
72 Culver KW, Ram Z, Wallbridge S, et al. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550-1552.
73 Ezzeddine ZD, Martuza RL, Platika D, et al. Selective killing of glioma cells in culture and in vivo by retrovirus transfer of the herpes simplex virus thymidine kinase gene. New Biol. 1991;3:608-614.
74 Chen SH, Shine HD, Goodman JC, et al. Gene therapy for brain tumors: regression of experimental gliomas by adenovirus-mediated gene transfer in vivo. Proc Natl Acad Sci U S A. 1994;91:3054-3057.
75 Ram Z, Culver KW, Walbridge S, et al. In situ retroviral-mediated gene transfer for the treatment of brain tumors in rats. Cancer Res. 1993;53:83-88.
76 Freeman SM, Abboud CN, Whartenby KA, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274-5283.
77 Fick J, Barker FG2nd, Dazin P, et al. The extent of heterocellular communication mediated by gap junctions is predictive of bystander tumor cytotoxicity in vitro. Proc Natl Acad Sci U S A. 1995;92:11071-11075.
78 Ram Z, Wallbridge S, Shawker T, et al. The effect of thymidine kinase transduction and ganciclovir therapy on tumor vasculature and growth of 9L gliomas in rats. J Neurosurg. 1994;81:256-260.
79 Barba D, Hardin J, Sadelain M, et al. Development of anti-tumor immunity following thymidine kinase–mediated killing of experimental brain tumors. Proc Natl Acad Sci U S A. 1994;91:4348-4352.
80 Kim JH, Kim Sh, Kolozsvary S, et al. Selective enhancement of radiation response of herpes simplex virus thymidine kinase transduced 9L gliosarcoma cells in vitro and in vivo by antiviral agents. Int J Radiat Oncol Biol Phys. 1995;33:861-868.
81 Nestler U, Wakimoto H, Siller-Lopez F, et al. The combination of adenoviral HSV TK gene therapy and radiation is effective in athymic mouse glioblastoma xenografts without increasing toxic side effects. J Neurooncol. 2004;67:177-188.
82 Aghi M, Kramm CM, Chou TC, et al. Synergistic anticancer effects of ganciclovir/thymidine kinase and 5-fluorocytosine/cytosine deaminase gene therapies. J Natl Cancer Inst. 1998;90:370-380.
83 Miller CR, Williams CR, Buchsbaum DJ, et al. Intratumoral 5-fluorouracil produced by cytosine deaminase/5-fluorocytosine gene therapy is effective for experimental human glioblastomas. Cancer Res. 2002;62:773-780.
84 Wei MX, Tamiva T, Chase M, et al. Experimental tumor therapy in mice using the cyclophosphamide-activating cytochrome P450 2B1 gene. Hum Gene Ther. 1994;5:969-978.
85 Clarke L, Waxman DJ. Oxidative metabolism of cyclophosphamide: identification of the hepatic monooxygenase catalysts of drug activation. Cancer Res. 1989;49:2344-2350.
86 Manome Y, Wen PY, Chen L, et al. Gene therapy for malignant gliomas using replication incompetent retroviral and adenoviral vectors encoding the cytochrome P450 2B1 gene together with cyclophosphamide. Gene Ther. 1996;3:513-520.
87 Wei MX, Tamiya T, Rhee RJ, et al. Diffusible cytotoxic metabolites contribute to the in vitro bystander effect associated with the cyclophosphamide/cytochrome P450 2B1 cancer gene therapy paradigm. Clin Cancer Res. 1995;1:1171-1177.
88 Sathornsumetee S, Reardon PA, Desjardins A, et al. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110:13-24.
89 Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275-283.
90 Louis DN, Holland EC, Cairncross JG. Glioma classification: a molecular reappraisal. Am J Pathol. 2001;159:779-786.
91 Li H, Alonso-Vanegas M, Colicos MA, et al. Intracerebral adenovirus-mediated p53 tumor suppressor gene therapy for experimental human glioma. Clin Cancer Res. 1999;5:637-642.
92 Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2006;6:55-61.
93 Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222-231.
94 von Deimling A, Louis DN, Wiestler OD. Molecular pathways in the formation of gliomas. Glia. 1995;15:328-338.
95 Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97-117.
96 Inoue R, Moghaddam KA, Ranasinghe M, et al. Infectious delivery of the 132 kb CDKN2A/CDKN2B genomic DNA region results in correctly spliced gene expression and growth suppression in glioma cells. Gene Ther. 2004;11:1195-1204.
97 Huang PH, Cavenee WK, Furnari FB, et al. Uncovering therapeutic targets for glioblastoma: a systems biology approach. Cell Cycle. 2007;6:2750-2754.
98 Gansbacher B, Zier K, Daniels B, et al. Interleukin 2 gene transfer into tumor cells abrogates tumorigenicity and induces protective immunity. J Exp Med. 1990;172:1217-1224.
99 Sobol RE, Fakhrai H, Shawler D, et al. Interleukin-2 gene therapy in a patient with glioblastoma. Gene Ther. 1995;2:164-167.
100 Yu JS, Wei MX, Chiocca EA, et al. Treatment of glioma by engineered interleukin 4-secreting cells. Cancer Res. 1993;53:3125-3128.
101 Parker JN, Gillespie GY, Love CE, et al. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci U S A. 2000;97:2208-2213.
102 Qin XQ, Tao N, Dergay A, et al. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci U S A. 1998;95:14411-14416.
103 Gansbacher B, Bannerji R, Daniels B, et al. Retroviral vector–mediated gamma-interferon gene transfer into tumor cells generates potent and long lasting antitumor immunity. Cancer Res. 1990;50:7820-7825.
104 Herrlinger U, Kramm CM, Johnston KM, et al. Vaccination for experimental gliomas using GM-CSF–transduced glioma cells. Cancer Gene Ther. 1997;4:345-352.
105 Fearon ER, Pardoll DM, Itaya T, et al. Interleukin-2 production by tumor cells bypasses T helper function in the generation of an antitumor response. Cell. 1990;60:397-403.
106 Tjuvajev J, Gansbacher B, Desai R, et al. RG-2 glioma growth attenuation and severe brain edema caused by local production of interleukin-2 and interferon-gamma. Cancer Res. 1995;55:1902-1910.
107 Im SA, Gomez-Manzano C, Fuiyo J, et al. Antiangiogenesis treatment for gliomas: transfer of antisense-vascular endothelial growth factor inhibits tumor growth in vivo. Cancer Res. 1999;59:895-900.
108 Millauer B, Shawver LK, Plate KH, et al. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576-579.
109 Van Meir EG, Poherini PJ, Chavin YR, et al. Release of an inhibitor of angiogenesis upon induction of wild type p53 expression in glioblastoma cells. Nat Genet. 1994;8:171-176.
110 Mineta T, Rabkin SD, Yazaki T, et al. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938-943.
111 Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867-874.
112 Chiocca EA, Smith KM, McKinney B, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16:618-626.
113 Ram Z, Culver KW, Oshiro EM, et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med. 1997;3:1354-1361.
114 Prados MD, Valéry CA, Bensimon G, et al. Treatment of progressive or recurrent glioblastoma multiforme in adults with herpes simplex virus thymidine kinase gene vector-producer cells followed by intravenous ganciclovir administration: a phase I/II multi-institutional trial. J Neurooncol. 2003;65:269-278.
115 Klatzmann D, Weber F, Mariani L, et al. A phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma. Study Group on Gene Therapy for Glioblastoma. Hum Gene Ther. 1998;9:2595-2604.
116 Shand N, Raffel C, Villablanca JG, et al. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. GLI328 European-Canadian Study Group. Hum Gene Ther. 1999;10:2325-2335.
117 Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389-2401.
118 Eck SL, Vapalahti M, Agrawal S, et al. Treatment of recurrent or progressive malignant glioma with a recombinant adenovirus expressing human interferon-beta (H5.010CMVhIFN-beta): a phase I trial. Hum Gene Ther. 2001;12:97-113.
119 Trask TW, Trask RP, Aquilar-Cordova E, et al. Phase I study of adenoviral delivery of the HSV-tk gene and ganciclovir administration in patients with current malignant brain tumors. Mol Ther. 2000;1:195-203.
120 Germano IM, Fable J, Gultekin SH, et al. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncol. 2003;65:279-289.
121 Smitt PS, Driesse M, Wolbers J, et al. Treatment of relapsed malignant glioma with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by ganciclovir. Mol Ther. 2003;7:851-858.
122 Immonen A, Vapalahti M, Tyynelä K, et al. AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study. Mol Ther. 2004;10:967-972.
123 Rampling R, Cruickshank G, Papanastassiou V, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7:859-866.
124 Lang FF, Bruner JM, Fuller GN, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508-2518.
125 Chiocca EA, Abbed KM, Tatter S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958-966.
126 Packer RJ, Raffel C, Villablanca JG, et al. Treatment of progressive or recurrent pediatric malignant supratentorial brain tumors with herpes simplex virus thymidine kinase gene vector-producer cells followed by intravenous ganciclovir administration. J Neurosurg. 2000;92:249-254.
127 Puumalainen AM, Vapalahti M, Agrawal S, et al. Beta-galactosidase gene transfer to human malignant glioma in vivo using replication-deficient retroviruses and adenoviruses. Hum Gene Ther. 1998;9:1769-1774.
128 Lang FF, Brunner JM, Fuller GN, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508-2518.
129 Harrow S, Papanastassiou V, Harland J, et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004;11:1648-1658.
130 Herrlinger U, Woiciechowski C, Sena-Esteves M, et al. Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol Ther. 2000;1:347-357.






