[level-membership-for-pediatrics-category]
Chapter 85 Gastrointestinal Structure and Function
Abnormalities in gastrointestinal (GI) and hepatobiliary function occur frequently in the pediatric intensive care unit (PICU) from primary GI disease, following surgery, or from complications of systemic disease. The pediatric intensivist is called on for expertise in the preoperative and postoperative management of numerous GI tract and hepatobiliary diseases, as well as secondary dysfunction that may complicate diseases involving other organ systems. The GI tract subserves a wide range of functions beyond simple digestion that have an impact on systemic immunological, endocrinological, and microbiological functions.1 Intact gut function beyond digestive function is of vital importance for the maintenance of health. Recent attention to alterations in GI function, interactions between liver and lung and between liver and kidneys have led to the view that the gut plays a role as an “engine” of multiple organ dysfunction.2 Thus a practical working knowledge of the GI system and its integrated function is essential for the management of critically ill patients.
Intestinal Structure, Digestion, and Absorption of Nutrients, Water, and Electrolytes
The functional absorptive unit of the intestines consists of villi and crypts. The cells of the small intestine are separated from one another by specialized junctions that serve as gaskets to prevent back diffusion of material into the intestinal lumen. A mucus layer secreted by goblet cells in crypts separates enterocytes from direct contact with the luminal contents (Table 85-1).
| Structure | Function |
|---|---|
| Enterocyte | Formed in crypts; migrate to villus over 2–3 days; lifespan of 6 days |
| Villi | Absorption |
| Crypts | Secretion |
| Microvilli | Amplify surface area; contain enzymes and transport systems |
Stem cells in crypts produce enterocytes and other specialized epithelial cells that migrate up the villous axis as they become differentiated. This migratory process takes 48 to 72 hours. Mature villous cells live 6 days. They have microvilli making up the brush border that contain digestive enzymes and membrane bound transport systems for nutrient and electrolytes. Small bowel enterocyte microvilli are estimated to increase the luminal surface area of the cell 14- to 40-fold.3 The cells at the villous tip have a predominantly absorptive function, while crypt cells are primarily secretory. Rotavirus infections cause villous loss resulting in small intestinal mucosa composed largely of crypts and immature villi. This causes a net secretory state leading to malabsorption and osmotic diarrhea. Malabsorption of nutrients is another manifestation of villous injury.
Water and Solute Transport Across the Intestinal Epithelium
Surface area and integrity of intercellular junctions are the major determinants of water and solute flux across epithelium. The transport of solute and water across epithelium occurs either by active or passive transport or by facilitated diffusion (Table 85-2). The gut conserves large volumes of endogenously secreted material associated with digestion. The average luminal fluid input of the gut is about 9 L/day and composed of oral intake and endogenous secretions. Approximately 8.8 L is absorbed, about 7 L in the small intestine and 1.8 L in the colon. Less than 0.2 L is excreted as a component of the normal stool output. When rapid changes in dietary intake or endogenous secretions occur, the intestinal mucosa can adapt transport functions in order to compensate for the changes. The loss of mucosal surface area through disease or surgical resection alters net flux of solute and water in GI tract. Furthermore, loss of specialized absorptive function may occur following loss of specific areas of gut. An example of this occurs in the setting of short bowel syndrome from resection of terminal ileum, with loss of ability to absorb bile acids and intrinsic factor.3 Malfunction of absorptive mechanisms may lead to life threatening loss of fluid and electrolytes.
| Active | Passive | Facilitated Diffusion |
|---|---|---|
| Against electrochemical gradient | Ionic specificity | Saturable kinetics |
| Saturable kinetics | May be associated with transport of a nonelectrolyte | Substrate specific |
| Requires ATP | Proceeds down electrochemical gradient | Depends on carrier molecules (glucose, amino acid) |
| Steady state based on concentration differences | ||
| Displays first-order kinetics | ||
| May occur by convection via osmotic or hydrostatic gradient |
In contrast, critical illness alters energy requirements. In general, the prolonged physiological stress during critical illness causes an increased metabolic rate as well as gluconeogenesis in excess of that needed to maintain serum glucose. Proteolysis and peripheral oxidation of amino acids with increased ureagenesis is also observed. This state leads rapidly to malnutrition as a result of so-called autocannibalism, characterized by levels of glucagon higher than insulin levels and by elevated catecholamines and cortisol that drive the relentless breakdown of peripheral tissues. These hormonal changes result in catabolism of endogenous stores of protein, carbohydrates and fat to provide metabolic substrate necessary to support the metabolic stress response.4 Children with burn injury demonstrate extreme hypermetabolism in the initial phases of illness.5 Children on dialysis have a negative nitrogen balance and loss of folate and selenium.6 Use of catecholamines or neuromuscular blocking agents represent risk factors for undernutrition in critically ill children on dialysis.7 During high physiologic stress, nutrition delivered to patients must be changed to allow for decreased reliance on fat and carbohydrate for energy production. Proteins are increasingly relied on and must be replaced with additional amino acids to support ongoing synthesis of protein including immunoglobulins by the liver and immune system. Protein requirements for various age groups are as follows: 0 to 2 years, 2 to 3 g/kg/day; 2 to 13 years, 1.5 to 2 g/kg/day; and 13 to 18 years, 1.5 g/kg/day.8 Depressed levels of specific amino acids have been found in critically ill children.9
Enteral nutrition is the preferred method of feeding for critically ill children. Restriction of fluid intake seems to be the main barrier to the delivery of adequate nutrition, particularly in infants undergoing cardiac surgery.10 Early transpyloric enteral nutrition is well tolerated in critically ill children and is not associated with an increase in incidence of complications.11 Small-bowel feeds allow a greater amount of nutrition to be successfully delivered to critically ill children compared with gastric feeds. However, small-bowel feeds do not prevent aspiration of gastric contents.12 The institution of a feeding protocol has been found to achieve goal feedings quickly and also improve tolerance of enteral feedings in patients admitted to the pediatric intensive care unit.13 Enteral nutrition may also have some anti-inflammatory effects by lowering expression of specific cytokines.14 The use of specialized formulas is not currently recommended in critically ill children. No effect from adequacy of feeding has been seen on hormone levels such as insulinlike growth factor-1 (IGF-1) and thyroid hormones.15
Digestion of Carbohydrates
Dietary carbohydrates may be classified in several ways (Box 85-1). Monosaccharides, such as glucose and fructose, are present in fruits, sweet corn, corn syrup, and honey. Disaccharides are simple sugars, such as sucrose (glucose + fructose), maltose (glucose + glucose), and lactose (glucose + galactose), the principal mammalian sugar. Polysaccharides, such as starch, are polymers of glucose and are abundantly present in wheat, grains, potatoes, peas, beans, and vegetables. Fiber consists of nondigestible complex polysaccharides of plant origin. This includes both water insoluble and water soluble fiber. The average American diet is 3:1 soluble to insoluble fiber. Some studies have shown that a water-soluble fiber, partially hydrolyzed guar gum can be used to decrease stool output in cholera.16 Insoluble fibers affect fecal bulk, whereas soluble fibers have viscous effects in the upper GI tract including delayed gastric emptying, decreased postprandial glycemic response, and a constipating effect.
Carbohydrates are a major source of calories in healthy children. Carbohydrates are generally broken down by means of glycolysis and the Krebs cycle pathway (see Chapter 74). Carbohydrates may be stored as glycogen and lipids when ingested beyond momentary energy needs or converted to structural materials. A person’s requirement for energy is highly dependent on activity level or, in hospitalized patients, the degree of hypermetabolism accompanying illness. The maximal ability to utilize carbohydrates may be limited during periods of high physiological stress as a result of the complex effects of hormonal mediators of the stress response.
Digestion of carbohydrates begins with the process of chewing that decreases the size of food particles, thereby increasing the total surface area for subsequent action by digestive juices. Salivary secretions are necessary for lubrication and also contain salivary amylase, an endoenzyme that cleaves oligosaccharides. Salivary amylase is rapidly inactivated by gastric acid, leaving the majority of starch digestion to occur in the duodenum under the action of pancreatic amylase and the intestinal brush border disaccharidase enzymes. Amylase contained in human milk facilitates starch digestion in breast-fed infants because of their low levels of endogenous salivary and pancreatic amylase.17 Pancreatic amylase is the major enzyme of starch digestion resulting in short oligosaccharides, maltotriose, maltose, and α-limit dextrins. The amylase concentration becomes limiting in cases of pancreatic insufficiency when amylase levels become less than 10% of normal.18
The enterocyte is incapable of absorbing carbohydrates larger than monosaccharides. Therefore, further hydrolysis to monosaccharides is performed by intestinal brush-border disaccharidases of which the clinically important ones are lactase, which breaks down lactose, and sucrase, which breaks down sucrose. These enzymes are synthesized in the enterocyte and are subsequently inserted into the apical brush border membrane. With the exception of lactase and occasionally sucrase, the disaccharidases are rarely rate-limiting for complete carbohydrate digestion. Deficiencies of any of the disaccharidase enzymes, either acquired or hereditary, may result in carbohydrate malabsorption. This is characterized by osmotic diarrhea with elevated fecal reducing sugars, abdominal distension and flatulence secondary to fermentation of undigested oligosaccharides by colonic bacteria. An example of this is congenital sucrase isomaltase deficiency, an autosomal recessive disorder that is associated with absence of sucrase and maltase.19,20
Absorption of carbohydrates occurs in several ways. During periods of high luminal carbohydrate concentration, simple diffusion of monosaccharides may occur. Additionally, two transport mechanisms exist in the brush border for the absorption of monosaccharides.21 First, glucose, galactose and xylitol are transported with sodium by the Na+/glucose cotransporter. A low intracellular sodium concentration is created by the sodium-potassium-adenosine triphosphatase (Na+-K+ adenosine triphosphatase [ATPase]) pump located on the basolateral membrane. The resulting concentration gradient leads to movement of luminal sodium across the apical membrane, bringing with it glucose or galactose in a one-to-one molar ratio. Glucose-galactose malabsorption is a deficiency of this transport mechanism leading to neonatal onset of severe diarrhea.22 The second mechanism is a non‑energy-dependent facilitated transport system for fructose. The intestinal transport mechanisms are summarized in Table 85-2. The capacity to absorb fructose is limited, and excess ingestion has been found to cause symptoms of carbohydrate malabsorption.23
Clinical conditions that cause loss of the epithelium and brush border system may lead to symptoms of carbohydrate malabsorption. These conditions include: rotoviral gastroenteritis, inflammatory bowel disease, celiac disease, sprue, ischemia/hypoxia, bacterial overgrowth of the proximal gut as a result of either stasis or use of antacids, and malnutrition. Severe mucosal damage requires 7 to 10 days for recovery of brush border function. Several infant and enteral formulas rely on starch as a carbohydrate source to minimize reliance on lactase. Recent studies have observed that lactose digestive capability with lactase can be maintained despite small bowel mucosal damage.24 Lactose absorption is primarily related to lactase activity as opposed to mucosal growth.25
Digestion of Proteins
The GI tract has developed efficient mechanisms for processing exogenous peptides and complex proteins (Box 85-2). It is also very efficient at recycling endogenous proteins such as digestive enzymes, mucus, sloughed cells, and plasma proteins that leak into the alimentary tract.26 The recommended dietary protein intakes in healthy children range from 2.5 to 3.5 g/kg/day in early infancy to 1.2 g/kg/day during childhood, and 0.8 to 0.9 g/kg/day in adolescence.8 The enteral processing of proteins may be divided into digestive and transport phases.
In the digestive phase, gastric acid secretion initiates denaturation of complex proteins making them more susceptible to the actions of proteolytic enzymes. The chief cells of the stomach release pepsinogens that are converted to active pepsins under the influence of gastric acid. The pepsins are endopeptidases that release relatively large peptides and are inactivated when the pH rises above 4 as the food enters the duodenum. The completeness of gastric proteolysis depends in part on the rate of gastric emptying, the pH of intragastric contents, and the types of protein ingested.26 It is noteworthy that patients with achlorhydria or those receiving antacids, H2 blockers, or both agents have no evidence of impaired protein digestion ability. In addition to initiating protein digestion in the mature subject, pepsins act as milk clotting factors, which are important in the neonate for curd formation and provide bulk to the infant’s stools.
Luminal digestion proceeds in the small intestine mediated by five pancreatic peptidases that are secreted by the pancreatic acinar cells as proenzymes and activated by enterokinase and trypsin. Each peptidase possesses proteolytic activity at specific internal or external peptide bonds. Proteins are degraded typically into mixtures of one-third free amino acids and two-thirds peptides containing two to six amino acid residues,27 which are suitable substrates for the brush border peptidases. The brush border peptidases convert the oligopeptides into monopeptides, dipeptides, and tripeptides suitable for transport into the enterocyte.
In the transport phase, specific membrane-associated transport mechanisms exist for the uptake of amino acids and dipeptides.28 They involve simple diffusion, facilitated transport, and carrier-mediated active transport (Table 85-3). Na+-coupled active transport is an energy-dependent process associated with the uptake of luminal Na+ and an amino acid (or glucose) and exchange of the sodium and associated molecule for K+ through the basolateral membrane on the serosal side.29 Peptide transport may also occur using an H+/peptide transport protein which moves according to an H+ gradient in the acidic pH microclimate of the intestinal brush border. This microclimate is maintained by Na+-H+ exchange in the brush border and Na+-K+-ATPase in the basolateral membrane.30 An important characteristic of these transporters is that many amino acids are absorbed more rapidly as dipeptides than as free amino acids. This fact has been capitalized on in the development of enteral nutritional formulas because oligopeptide mixtures have a lower osmolarity and are more efficiently absorbed than single amino acid solutions of equal nitrogen content. Because of the efficient gastrointestinal absorption of dipeptides, patients with specific amino acid transport defects (e.g. Hartnup disease [defective tryptophan transport] and lysinuric protein intolerance [defect in dibasic amino acid transport: lysine, arginine]) infrequently have GI symptoms related to dietary protein malabsorption and instead more commonly manifest with non-GI symptoms such as aminoaciduria.
Once inside the enterocyte, peptides are quickly degraded into their constituent amino acids by cytoplasmic peptidases that complement the activity of the brush border peptidases. Only minute quantities of intact peptide and protein gain access to the systemic circulation. The cytoplasmic amino acids derived from digested proteins are a major source of free amino acids used directly by the enterocyte. When absorbed beyond cellular needs, the free amino acids are released to the portal venous circulation for hepatic and systemic use. Only 23% of absorbed amino acid nitrogen passes to the periphery without modification.31 Of the remaining nitrogen, 57% is converted to urea with the carbon skeleton salvaged for synthesizing other substances and 20% of the total ingested amino acids are used directly for hepatic protein synthesis.
During periods of fasting, the enterocyte derives the majority of its nourishment from the mesenteric arterial vascular supply, whereas during digestion, the enterocyte derives a significant part of its nutrient requirements from the luminal contents. Experience with mucosal recovery and adaptation after injury reveals that an enteral route of nutrition permits optimal recovery. In the premature infant and neonate, the small intestine is capable of absorbing intact milk proteins by pinocytosis. These proteins may include secretory immunoglobulins from breast milk as well as food antigens.32 Peptidase inhibitors have been demonstrated in colostrum and breast milk, partially explaining the failure of normal digestive mechanisms to degrade some of these complex dietary proteins. Both antibodies and antigens ingested with maternal milk create an important part of the immune repertory developed during early infancy.33 Although the exact time of “closure” of the intestinal mucosa to the uptake of macromolecules has not been defined in human infants, other mammals demonstrate marked intestinal impermeability to foreign proteins by the time of weaning34 from breastfeeding.
Digestion of Lipids
Dietary fat accounts for approximately 50% to 70% of the nonprotein calories consumed by infants and approximately 30% of nonprotein calories consumed after age 2 years (Box 85-3).35 Dietary fat is ingested principally in the form of triglycerides containing the fatty acids palmitate and oleate (C16:0 and C18:1, respectively). Dietary triglycerides of animal origin predominantly contain long-chain (i.e., longer than C14 chain length) saturated fatty acids. Polyunsaturated fatty acids are mostly of vegetable origin and include linoleic and linolenic acid, also referred to as essential fatty acids because of absent de novo synthesis in humans. Other dietary lipids include fat-soluble vitamins, cholesterol, prostaglandins, waxes, and phospholipids.
In healthy adults, digestion and absorption of fat is complete with only 5% to 7% of ingested fat escaping absorption. Under normal physiological conditions healthy infants up to age 9 to 12 months fail to absorb 15% to 35% of dietary fat. Digestion and absorption of dietary fat is generally completed by the middle third of jejunum; however, the presence of dietary fiber may reduce the rate and extent of absorption. Loss of dietary fat places children at significant risk for calorie and fat-soluble vitamin malnutrition.
Digestion of Fat
Fat digestion begins with formation of emulsions, which increase the surface area for enzyme interaction. Emulsification begins with release of fat by mastication and gastric “milling” of chyme. Bile salts and coating by phospholipid derived from the diet results in a stable emulsion droplet with a hydrophobic center consisting of triglyceride, cholesterol esters and diglyceride in a hydrophilic envelope. Mammary, lingual, and gastric lipases play an important role in direct lipolysis of long- and medium-chain triglycerides that are present in maternal milk.36 Lingual and gastric lipases are active at pH <5 and begin digestion of fat in the stomach; however, overall only play a limited role in the digestion of lipids. Intragastric lipolysis is consistent across all age groups.37
Most of the enzymatic degradation of dietary lipids to fatty acids and monoglyceride is by the action of pancreatic lipase and co-lipase, and requires an alkaline environment (pH 6 to 8). This underscores the importance of secretion of bicarbonate by the pancreas and biliary tree in order to neutralize gastric acid. Co-lipase is an essential cofactor for lipase action. Co-lipase’s role is to displace the bile salt-triglyceride interaction in emulsion droplets and micelles to facilitate lipase hydrolysis of the triglyceride. Triglyceride hydrolysis occurs at the interface between the emulsion droplet and aqueous phase within the lumen. This is a two-step process. The first step is the enzymatic hydrolysis of long-chain triglycerides and liberation of fatty acids from the glycerol backbone. The second step is formation of fatty acid micelles, that is most efficiently accomplished with the aid of bile salts38 to traffic the fatty acids across the unstirred water layer to the mucosa for absorption.
Transit through the unstirred water layer adjacent to the epithelial surface is considered the rate-limiting step in lipid absorption. Intrinsic gut brush border lipase enzymes are involved as well. The milieu of the unstirred water layer is acidic (pH 5 to 6) owing to the activity of the brush border membrane sodium-hydrogen (Na+/H+) exchanger. The acid environment facilitates dissociation of fatty acids from micelles resulting in a high concentration of fatty acids necessary for diffusion across the mucosal membrane.39,40
Once inside the enterocyte, long-chain fatty acids and monoglycerides are resynthesized into triglycerides and packaged as chylomicrons. Lipoproteins (e.g., apo-A, apo-B) and cholesterol are attached to the intestinal chylomicrons and confer important properties for the subsequent systemic uptake and metabolism of the chylomicrons. This process appears to be defective in cystic fibrosis and may account for some of the fat malabsorption seen in this disease.41 Chylomicrons are exported into the intercellular space and transported through the intestinal lacteals to become part of the intestinal lymph. On entering the bloodstream through the thoracic duct, the chylomicrons are associated with other apolipoproteins that allow them to be recognized by specific peripheral tissues.42
Regulation of Electrolyte and Water Movement
Movement of water is closely linked to the movement of solute in the form of electrolytes and nutrients. Water transport is a largely passive process that occurs through paracellular routes in the intestine coupled with solute movement. Expression of transporters involved in intestinal water and electrolyte transport is regionally specific. Electrolytes are taken up by enterocytes at the apical membrane and extruded through the basolateral membrane into the paracellular space. The relatively hypertonic paracellular fluid pulls water into this space, increasing the hydrostatic pressure locally. Because the tight junction between enterocytes is more impermeable to fluid flux than the capillary membranes, fluid and electrolytes are preferentially driven in the direction of the vascular space.1,43,44 Tight junctions are selective and dynamic in function and are regulated by a number of signaling pathways and cellular processes that can determine the size, selectivity, and flow of molecules across this barrier.3
The gut responds to both systemic and local stimuli to regulate motility, transport, and digestive functions. Secretion and motility are mediated through typical agonist membrane receptor mechanisms, by local autocrine and paracrine action, or through remote endocrine and neurocrine actions. Regulation of intestinal motility is crucial for keeping the chyme in contact with the epithelial surface long enough for efficient absorption of nutrients while permitting removal of unusable material and bacteria from the alimentary tract on a regular basis. GI smooth muscle demonstrates phasic and tonic patterns of contraction. Numerous factors affecting the frequency of contractions include changes in autonomic tone, stimulation of the gut by neurohormonal peptides or pharmacologic agents, and noxious stimuli associated with infectious or inflammatory processes. Hypoxia and ischemia decrease motility, frequently leading to paralytic ileus. Neural regulation of the GI tract integrates the processes of intestinal water and electrolyte transport, motility and blood flow. The augmentation of water and electrolyte absorption after a meal in the jejunum is neurally mediated.45 The enteric nervous system is capable of functioning independently but also may be modified by autonomic nervous system.3
Many other factors alter the functions of the gut. The terminal ileum and colon are particularly important in this respect. The presence of an ileostomy increases the risk of excessive sodium losses, dehydration, and electrolyte abnormalities. Terminal ileal resection or other diseases of the terminal ileum such as Crohn disease or radiation enteritis may result in bile acid malabsorption. In patients with bile acid malabsorption, bile acids reach the colon, which stimulates electrolyte and chloride secretion. Patients with mild to moderate malabsorption present with watery diarrhea and may respond to a bile acid binder such as cholestyramine.46 Impairment of water and ion absorption in inflammatory bowel disease may occur because of numerous mechanisms including alteration of epithelial integrity, augmented secretion, and reduced absorption. In addition, intestinal inflammation is associated with defects in epithelial barrier function. It is not known what effect inflammation may have on ionic function.47 Hyperosmolality of the ileal and colonic contents leads to an osmotic diarrhea. This state is seen when unabsorbed nutrients enter the distal alimentary tract and are broken down by enteric bacteria, resulting in increased luminal osmotic activity and osmotic diarrhea.
Electrolyte Transport
Several basic mechanisms exist for the transport of electrolytes by the epithelia. Na+ may be transported by numerous mechanisms. The regulation of Na+ absorption is closely regulated such that anion secretion is closely aligned with Na+ absorption.48 A Na+-H+ exchange-mechanism present throughout the intestine results in a 1:1 exchange of luminal sodium for protons. Coupled Na-Cl absorption, Na-Cl co-transport, Na-K-Cl co-transport, and movement of Na+ down its electrochemical gradient as in Na-Glucose co-transport are other mechanisms by which Na+ moves across the intestinal epithelium. The Na+-H+ exchanger plays a role in regulation of intracellular pH, regulation of cell volume, initiation of cell growth in response to various trophic factors, and metabolic response to insulin. To maintain electrical neutrality, the epithelium simultaneously exchanges Na+ for H+ and Cl− for HCO3. The presence of glucose in the lumen of the small intestine stimulates increased sodium absorption through coupled transport. Uptake of glucose is carrier mediated although the coupled transport of glucose with sodium is electrogenically driven by the Na+ gradient across the cell membrane.49 Absorption of glucose by Na-Glucose transport results in activation of myosin light chain kinase to regulate tight junction-permeability.50 Backflow of sodium into the lumen is a passive process since a major task for the GI tract is sodium conservation. Systemic acidosis increases Na+ and Cl− absorption in the ileum and colon, whereas alkalosis has the opposite effect. As seen in other epithelial tissues, aldosterone increases ileal and colonic absorption of Na+ and can increase absorption of water in colon three- to fourfold.51 Spironolactone blocks this effect. Glutamine has been shown to stimulate water and electrolyte absorption in the jejunum.52 Glucocorticoids increase sodium and water absorption in the distal colon. Opiate receptor stimulation increases active sodium and chloride absorption in the ileum, and opiate antagonists decrease basal absorption of water and electrolytes. The primary antidiarrheal effect of opiates, however, is mediated through a slowing of transit time.1
In the colon, active absorption and secretion of K+ occurs in a manner consistent with K+-H+ exchange, is electroneutral and independent of Na+-Cl− exchange. After the ions have entered the enterocyte at the luminal surface, extrusion occurs through the basolateral membrane into the paracellular spaces. The process of sodium extrusion depends on Na+-K+-ATPase pumping function located at the basolateral membranes. Oxidative stress inhibits water and electrolyte absorption in the jejunum through inhibition of Na-K ATPase on the basolateral membrane.53
Extrusion of Cl− occurs along an electrochemical potential difference.54 The intraluminal secretion of water and other electrolytes appears to follow active secretion of Cl− from the crypt cells of the jejunum, ileum, and colon. This is a common physiological pattern in the liver, pancreas, and kidney. Numerous substances such as muscarinic receptor agonists, serotonin, and substance P likely work through second messengers and signaling cascades to induce active chloride secretion. Vasoactive intestinal peptide mediates increased secretion of electrolytes and water by increased cyclic adenosine monophosphate production that stimulates active chloride secretion and inhibits sodium-chloride absorption. Certain arachidonic acid metabolites, especially prostaglandins (e.g., prostaglandin E1), have been shown to increase active chloride secretion leading to increased loss of electrolytes and fluid. Many laxatives and antacids may affect fluid and electrolyte balance by stimulating active electrolyte and fluid secretion in the terminal ileum. In addition, these agents may increase mucosal permeability and stimulate motility.1
Disruption of normal Na+-K+-ATPase activity results in the net secretion of fluid and electrolytes. This mechanism is the final common pathway in a number of secretory diarrheal states such as cholera, enterotoxigenic Escherichia coli, Salmonella spp., Campylobacter jejuni, and Clostridium perfringens that appear to act via second messenger pathways via their toxins.55 Rotavirus appears to have several mechanisms of causing diarrhea. The first appears to be a mechanism of increasing chloride secretion with a resulting secretory diarrhea mediated by Ca++, a different mechanism than most bacterial mediated diarrheal diseases. The second is an osmotic diarrhea caused by villus destruction and resultant malabsorption.56 In addition, the effects of various paracrine and endocrine mediators alter intestinal adenyl cyclase activity and lead to changes in electrolyte and water balance.54
Zinc
Normal zinc homeostasis is required for a functional immune system, adequate antioxidant capacity, glucose homeostasis, and wound healing. In addition, zinc is a required co-factor for many enzymes, transcription factors, and replication factors. In noncritically ill patients, zinc supplementation is associated with improvement in markers of immune function. Plasma zinc concentrations are low in critically ill children, correlate with measures of inflammation and are associated with the degree of organ failure.57 Large intestinal losses of zinc with and without complexed proteins often occur in association with high intestinal fluid losses through gastrointestinal stomas and fistulae. Although some patients with sepsis have demonstrated significantly faster recovery with zinc supplementation among other anti-oxidants.58 there is currently inadequate evidence to recommend the routine use of high-dose zinc supplementation in the critically ill.59
Hydrogen Ions
Hydrochloric acid secretion by the gastric parietal cell is necessary for pepsinogen activation (pH <5.0) and to reduce bacterial colonization. H+ and bicarbonate are produced from water and carbon dioxide by the action of carbonic anhydrase within the parietal cell. The bicarbonate is secreted into the bloodstream in exchange for chloride at the basolateral membrane. Chloride and potassium are both secreted along with H+ across the apical membrane against a large concentration gradient. This is an active process, mainly because of the action of the proton pump, H+/K+-ATPase, that represents the final step in gastric acid secretion.60
Histamine, gastrin, and acetylcholine are the main stimulants of gastric acid secretion.61 Gastric distension, dietary amino acids, and amines stimulate gastrin hormone secretion by G cells located in the gastric antrum. Gastrin is the most potent endogenous stimulant of gastric acid secretion and stimulates release of histamine by the enterochromaffin-like cells. Histamine then binds to histamine-2 (H2) receptors on parietal cells leading to acid secretion. Prostaglandins and somatostatin have an inhibitory effect on gastric acid secretion via specific receptors located on the parietal cell.62
H2 receptor antagonists (e.g., ranitidine, famotidine, cimetidine) block histamine-mediated gastric acid secretion found in postprandial acid secretion, Zollinger-Ellison syndrome and other disorders associated with hypergastrinemia. Proton pump inhibitors (PPIs) (e.g., omeprazole, lansoprazole, pantoprazole) block gastric acid secretion by inhibiting the parietal cell H+/K+-ATPase. The PPIs bind irreversibly to the enzyme and subsequent secretion of acid can occur only with the synthesis of new proton pump enzyme, a process that takes 12 to 24 hours. For these reasons, PPIs have revolutionized gastric acid suppression therapy.61,63 H2 receptor antagonists and PPIs are similarly effective for preventing bleeding in the upper part of the GI tract in patients receiving mechanical ventilation.64 Recent studies have shown that some oral PPIs suppress acid in intensive care unit (ICU) patients to a greater extent than IV.65 Patients at risk of stress ulcer–related bleeding are most likely to benefit from prophylaxis.66
Pancreas
The pancreas exhibits both endocrine and exocrine functions and acts in concert with the liver to regulate blood glucose levels. Endocrine-secreting cells of the pancreas are aggregated in the islets of Langerhans. There are approximately 1 million islets in the human pancreas. Four distinct cell types in the islets which serve the pancreatic endocrine function include (1) B-cells that secrete insulin (50% to 80%), (2) A-cells that secrete glucagon (5% to 20%), (3) D-cells that secrete somatostatin (5%), and (4) PP cells that secrete pancreatic polypeptide.21,67
Pancreatic Exocrine Secretory Function
The functional unit of the exocrine pancreas consists of an acinus composed of specialized cells containing secretory granules that drain into ductules, which coalesce to form the pancreatic duct. In contrast to the pancreatic endocrine cells that demonstrate specialized function, each acinar cell is capable of secreting all the pancreatic digestive enzymes. The basolateral membrane has receptors for hormones and neurotransmitters that stimulate pancreatic secretion of the digestive enzymes stored in zymogen granules near the apical membrane of each acinar cell.68 Ultimately, the pancreatic duct joins with the common bile duct and drains into the duodenum through the ampulla of Vater. Anatomic variation exists. 74% of people have a common channel, whereas 19% have a separate opening and 7% have an interposed septum.69
Pancreatic juice is an isotonic fluid, containing primarily Na+, K+, Cl, and HCO3−. The total volume of secretion is approximately 2.5 L daily. Cystic fibrosis transmembrane conductance regulator is the main channel for chloride secretion in the pancreas and may be involved in other ion transport.70 Secretion of bicarbonate and water is mediated through the actions of the gut hormones secretin, cholecystokinin, and vasoactive intestinal peptide. Stimulation of the vagus nerves or the administration of acetylcholine induces digestive enzyme secretion. These effects may be blocked with atropine.
There are four phases of pancreatic secretion: Basal secretion represents approximately 2% of the potential maximum HCO3. The cephalic phase is mediated by the vagal nerves in response to the sight and smell of food. The gastric phase consisting of secretion of a protein-rich pancreatic juice of low volume and HCO3 occurs following either distention of the stomach71 or after the ingestion of food. The intestinal phase is characterized by marked output of digestive enzymes, fluid, and HCO3.
Presence of bicarbonate is essential to achieve an optimal pH (>5) for pancreatic digestive enzyme activity and to ensure solubility of bile salts. In addition to bicarbonate, the primary secretory products of the exocrine pancreas are amylase, lipase, and the proteases. The secondary digestive enzymes consist of nucleases, colipase, and lecithinase. The roles of the pancreatic digestive enzymes are discussed in the sections on carbohydrate digestion (amylase), lipid digestion (lipase), and protein digestion (proteases). Cholecystokinin is the major humoral mediator of meal-stimulated enzyme secretion. It is released from the small intestinal mucosa in response to presence of fat, protein and starch. The response is related to total load rather than concentration. Cholecystokinin activates afferent neurons in duodenal mucosa that leads to secretin release via a vasovagal reflex.72 Inhibitors of exocrine pancreatic secretion include somatostatin, pancreatic polypeptide and peptide YY.73 Octreotide, a somatostatin analogue has been used for its antisecretory effect in clinical management of pancreatic-pseudocysts and fistulae, but its use in acute or chronic pancreatitis remains controversial.74 Inflammation of the pancreas both from infectious and non-infectious causes can produce a dramatic systemic inflammatory response resulting in generalized permeability changes and acute lung injury.75
Hepatobiliary System
Examination
Palpation of the liver provides information about the hepatobiliary tract as well as function of the right side of the heart. Normally, the liver is palpable roughly 1 to 3 cm below the right costal margin in the mid-clavicular line; however, assessment of liver span, and not palpation alone, is the only reliable non-radiological method for determining liver size. Liver span is determined by percussion, palpation, and auscultation along the right mid-clavicular line with the patient supine and breathing quietly. Dullness of the upper border is determined by percussion. Either palpation or auscultation is used to establish the lower border. The liver span increases with body weight and age in both sexes, ranging from 4.5 to 5.0 cm at 1 week of age to 7 to 8 cm in boys and 6.0 to 6.5 cm in girls by age 12 years.76
Examination of the liver should note consistency, contour, tenderness, the presence of any masses or bruits, and assessment of spleen size. Documentation of the presence of ascites and stigmata of chronic liver disease is important. Tenderness over the liver suggests inflammation or stretching of the fibrous capsule through rapid enlargement. Conditions associated with downward displacement of a normal liver include hyperinflated lungs, pneumothorax, retroperitoneal masses, and subdiaphragmatic abscess. End-stage liver disease and cirrhosis are associated with a reduced liver span corresponding to decreased hepatic cell mass. The spleen tip may be palpable normally in children, especially during inspiration. Enlargement of the spleen generally represents elevated portal venous pressures or invasive processes such as sequestration, malignancy, extramedullary hematopoiesis, or hyperplasia of the reticuloendothelial system.77
Anatomy: Microanatomy, Structure, and Function
The liver is the largest organ in the body and is composed of 60% hepatocytes, approximately 17% to 20% endothelial cells and Kupffer cells (reticuloendothelial cells), 3% to 5% bile ducts, and 1% hepatic stellate cells and oval cells. The liver has a dual vascular supply derived from the hepatic artery branches of the celiac axis providing about 30% of the blood supply and the portal vein providing approximately 70%. Innervation of the liver is by the parasympathetic branches derived from both vagi and sympathetic branches, which also carry afferent fibers deriving from thoracic segments. Denervation of the liver, such as seen after liver transplantation, does not affect function.78
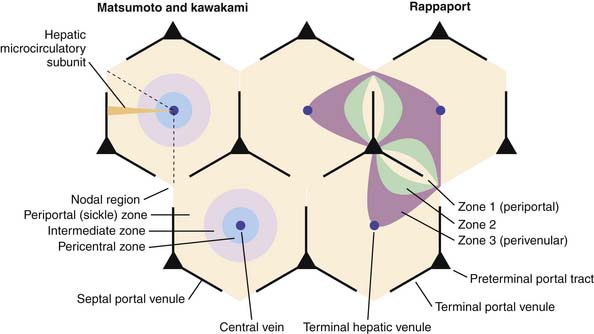
The “liver lobule,” the functional unit of the liver is composed of interconnected hepatocytes (hepatic plates) one to two cells thick and 20 to 25 cells in length separated by a venous sinusoidal space and radiating around the central vein like spokes in wheel (see Figure 85-1). The narrow tissue space between the endothelial cells and hepatic plates is called the space of Disse, which in turn connects with lymphatic vessels in the interlobular septa. Hepatic sinusoidal endothelial cells are flat cells that do not form intracellular junctions and overlap one another. They are fenestrated, allowing plasma to enter into the space of Disse and come into direct contact with the surface of hepatocytes.79 This facilitates bidirectional exchange between the hepatocytes and the sinusoidal space. Macrophage-derived Kupffer cells line the sinusoidal space, have a phagocytic function, and mediate the hepatic inflammatory response.
Hepatic stellate sells, also known as ito cells, lie within the space of Disse, serve as the hepatic storage site of vitamin A, are effectors of fibrogenesis, and play a role in extracellular matrix remodeling after recovery from injury. Chronic activation and proliferation of hepatic stellate cells may lead to noncirrhotic portal hypertension, fibrosis, and cirrhosis.80 Bile canaliculi lie between adjacent hepatocytes and drain into small terminal bile ducts, that successively drain into larger bile ductules, intralobular bile ducts and eventually the extrahepatic bile ducts. Tight junctions between the hepatocytes at the canalicular space permit unidirectional transport of substances from the hepatocytes into the canalicular space. Several different carriers, receptors and transport proteins facilitate movement of compounds across the sinusoidal, hepatocyte and canalicular membranes. ATP-binding cassette proteins are expressed in the canalicular membrane and play an important role in transportation of organic ions. Alkaline phosphatase, leucine aminopeptidase and γ-glutamyl transpeptidase are transaminase enzymes selectively localized in the bile canaliculi.
The microcirculatory “path” within the lobules leads along a declining hydrostatic pressure gradient from the terminal hepatic arterioles and portal venules within the portal triad towards the central vein representing the terminal branch of the hepatic vein, resulting in three hepatocyte functional zones (see Figure 85-1). Zone 1 hepatocytes closest to the portal triad are exposed to sinusoidal blood containing the highest concentration of solutes and oxygen. In contrast, zones 2 and 3 represent hepatocytes more distant from the portal blood supply and are exposed to a declining oxygen and solute gradient. In addition, zone 3 hepatocytes actively participate in drug metabolism and disposition. Ischemic injury and drug hepatotoxicity impacts zone 3 hepatocytes to the greatest degree.81
Portal Circulation
The portal venous system drains the intestines, pancreas, and spleen with numerous collateral anastomoses to other venous beds of the abdomen. There is a mixing of portal and systemic blood circulation within the sinusoids, and all the blood eventually drains from the liver via the hepatic veins to the inferior vena cava. The liver has a high blood flow (~27% of the resting cardiac output) and low vascular resistance. The portal pressure gradient (PPG) is the pressure difference between the inferior vena cava and the portal vein and averages 0 to 5 mm Hg. Portal hypertension is defined as a PPG between 6 and 10 mm Hg. A PPG greater than 10 mm Hg carries risk for development of esophageal varices and a PPG greater than 12 mm Hg predisposes towards ascites formation.82 Obstruction of the portal venous drainage at any level leads to portal hypertension. Portal hypertension may be classified as prehepatic, intrahepatic, or posthepatic, according to the level at which the obstruction to flow occurs. Determination of the location of obstruction is critical for instituting appropriate therapy.
Ascites formation is multifactorial. The central event in ascites formation in cirrhosis is splanchnic arterial vasodilatation secondary to portal hypertension. This creates an increase in capillary pressure because of increased blood inflow leading to leakage of fluids. Additionally, impairment of systemic hemodynamics and renal function leads to sodium and water retention with intravascular volume expansion.83–85 Ascites may form in the absence of portal hypertension as the result of low plasma oncotic pressure associated with malnutrition, with renal or enteral protein losses, or through impaired thoracic duct lymph drainage. Rarely, arterial-portal venous malformations may lead to portal hypertension as a result of excess portal blood flow. One additional factor predisposing to ascites is an elevated central venous pressure that increases formation and impairs resorption of interstitial fluid often associated with generalized anasarca.
Hepatic Function
Albumin has a half-life of approximately 20 days, and is a significant contributor to colloid oncotic pressure. Decreased serum levels may predispose to edema formation and decreased binding of bilirubin, calcium, xenobiotics, and other highly protein-bound molecules. Low serum albumin levels can be secondary to impaired synthesis from protein-calorie malnutrition, chronic liver disease, cachexia, or cytokines. Alternatively, increased losses from proteinuria, protein losing enteropathy, burns, or other iatrogenic losses including paracentesis may cause hypoalbuminemia.86
Prealbumin, also known as transthyretin, is a visceral protein with a short half-life of 1.9 days. Because hepatic synthesis is exquisitely sensitive to both the adequacy and levels of protein and energy intakes it may be used a nutritional marker and for monitoring short-term response to nutritional intervention.87
α1-Antitrypsin is an important antiprotease with regulatory activity for elastase and other proteases. α1-Antitrypsin can be important in regulating elastase-induced tissue injury in certain lung diseases and its absence leads to uncontrolled proteolytic activity in the lung.88 Because α1-Antitrypsin is an endogenous protein that is relatively resistant to hydrolysis by enteric bacteria, elevated levels detected in feces suggest protein losing enteropathy.89
Hepatic synthesis of transferrin facilitates iron transport in the plasma by binding two molecules of iron. Ferritin is the primary storage molecule of iron with each molecule storing up to 4500 atoms of iron. Many coagulation factors are synthesized in the liver. They include plasminogen, fibrinogen, and factors II, V, VII, IX, X, XI, XII, and XIII. Factors II, VII, IX, and X are the so-called vitamin K–dependent factors that require vitamin K for synthesis and secretion in active form.90 In addition, the anticlotting proteins antithrombin III, protein C, and protein S are synthesized largely in the liver and may be vitamin K dependent. Several additional common plasma proteins are synthesized by the liver including haptoglobin, ceruloplasmin, lipoproteins, α-fetoprotein, and the C3 component of complement.
Alterations in plasma proteins frequently occur during acute and chronic liver disease. Although the levels of many of these proteins may rise as part of the systemic inflammatory response (acute phase reactants), plasma levels are generally reduced during liver disease, depending on the duration of hepatic insufficiency and the half-lives of the specific proteins. Thus a decrease in albumin with its half-life of 16 to 21 days generally represents a chronic disease state, whereas a prolonged prothrombin time may be seen within hours of acute hepatic failure because of the short half-life of factor VII (about 6 hours).91
Detoxification and catabolism of ammonia, bilirubin, and xenobiotics is essential to life. Ammonia arises through bacterial degradation of nitrogenous compounds in the intestine, as well as from other physiological sources including the kidneys and peripheral tissues such as skeletal muscle and the brain. Ammonia is transported to the liver via the portal vein in high concentrations and is quickly degraded in the liver to urea. High levels of ammonia are incompatible with life and during hepatic failure, hyperammonemia represents a life threatening aspect of liver disease.92
Enterohepatic Circulation
Bile acids represent a family of steroid molecules derived from cholesterol. They act to eliminate cholesterol from the body and to solubilize dietary fats through a detergent-like action. Enterohepatic circulation acts to conserve bile acids since a minimum concentration of bile acids are required for micelle formation. Bile salts are secreted into the duodenum with 97% reuptake in the terminal ileum, undergoing recycling 4 to 12 times per day. The distal and terminal ileum have specialized transport mechanisms for absorption of bile salts and vitamin B12, that are adversely affected by terminal ileal resection, jejunostomies, inflammatory bowel disease, or other acquired lesions in this anatomical region (e.g., necrotizing enterocolitis). Functional loss of the distal and terminal ileum results in malabsorption of vitamin B12, bile salt deficiency, and impaired digestion and absorption of fat-soluble vitamins and long-chain fats. Furthermore, unresorbed bile acids have a detergent effect on the colonic epithelium resulting in secretory diarrhea.1
Hepatic insufficiency, either from immaturity or as a result of disease affects the elimination of many drugs (see Chapter 87). A large number of commonly used drugs of all classes including aminophylline preparations, narcotics, barbiturates, H2-blockers, vasodilators, antidysrhythmics, and others demonstrate significant hepatic elimination. The hepatic P-450 system plays a central role in many of the mixed-function oxidative reactions responsible for converting lipophilic compounds into more water-soluble ones. In addition, the liver may conjugate drug metabolites to form products that can be eliminated easier in bile or through the kidney. The half-life of many drugs may be prolonged during hepatic insufficiency as a result of a decrease in the total number of functioning hepatocytes. In addition, the half-life of many drugs may be prolonged through competitive inhibition by the presence of other drugs, or may be shortened by induction of elimination reactions. For example, phenobarbital decreases the half-life of xanthines and may increase the toxicity of acetaminophen. Adjustment of medication dosage and schedule must be considered for those drugs with significant hepatic elimination when impaired liver function exists.93
Hepatic regulatory function involves (1) interconversion of amino acids to maintain physiological plasma levels, (2) gluconeogenesis to maintain adequate glucose serum levels for glucose-dependent tissues, and (3) regulation of numerous plasma hormones (Box 85-4).
Box 85–4 Hepatic Endocrine Regulation
From Johnston DG, Alberti KGMM: The liver and the endocrine system. In Wright R, Millward-Sadler GH, Alberti KGMM, et al, editors: Liver and biliary disease, ed 2, Philadelphia, 1985, WB Saunders.
The direct secretion of insulin and glucagon into the portal circulation exposes the liver to much higher concentrations of these hormones than peripheral tissues. This relationship amplifies the hepatic influence over carbohydrate metabolism. Approximately 50% of secreted insulin and a large portion of glucagon are degraded on a first-pass basis by the liver. Both of these hormones are known to have hepatotrophic effects and are thought to be important for differentiation and regeneration of hepatocytes. Intensive insulin therapy in the critical care setting has been shown to have a beneficial effect on liver function.94
The last category of hepatic function involves storage of glycogen, triglycerides, folic acid, vitamin B12, and vitamins A and D. The liver uses glycogenolysis to mobilize hepatic glycogen stores and provide an almost immediate source of glucose to maintain serum levels. The liver glycogen stores contain up to a 2-day supply of glucose.95 Synthesis of vitamin D3 (cholecalciferol) occurs in the skin with subsequent accumulation of D3 in the liver. Hydroxylation at the 25-position that occurs in the liver results in a large pool of circulating 25-(OH)D3, the precursor of the active 1,25-(di OH)D3. Defective storage and absorption of dietary vitamin D and 25-hydroxylation may be present in chronic liver failure.96
Host-Defense Mechanisms of the Gut: Immunology and Microbiology
Both the intestine and the hepatic-based macrophages can serve as a major source of nitric oxide following injury or stimulation.97 The recently recognized role of nitric oxide on immune function further emphasizes the role of the gastrointestinal tract in systemic responses. The frequent association of hepatic dysfunction with the acute respiratory distress syndrome has led to intensive investigation of the lung-liver axis during critical illness.98–100 A major unifying theme in these organ interactions is the regional activation of macrophages and platelets and damage to the endothelium after injury leading to both localized and remote organ function disturbance. The gut has become one of the important focuses in our evolving understanding of the syndrome of multiple organ dysfunction.101
Innate mechanisms exist to prevent infection by bacterial pathogens. In the upper GI tract, action of salivary enzymes, gastric acid, pancreatic enzymes, and the detergent effect of bile acids all act to limit the amount of viable bacteria in the small intestine. The secreted mucous layer preventing attachment and adherence of bacteria combined with intestinal peristalsis continually prevents establishment of infection. In the colon, with its heavy colonization of bacteria, competitive inhibition acts to prevent infection with pathogenic species. Other innate mechanisms include tight junctions between intestinal epithelial cells that act to exclude bacteria, antigens and other macromolecules.102 Clostridium difficile toxin and enteropathogenic E. coli act by disrupting these tight junctions.103,104 Antibiotics profoundly reduce the number of anaerobic and coliform bacteria in the alimentary tract with particular reduction of Bifidobacterium and Lactobacillus species.105 Routine stress ulcer prophylaxis with acid suppressing medications such as H2 blockers and PPIs create a risk of nosocomial pneumonia due to loss of protective gastric acid. However, a recent meta-analysis and systematic review failed to show a significant association between PPI use and respiratory infections.106 Intestinal motility is a critical factor for clearing antigens and bacteria from the gut lumen and thus reducing bacterial overgrowth with its resultant malabsorption of nutrients. Decreased motility leading to bacterial overgrowth may be seen in premature infants and may be associated with many disorders seen in older children, including the use of narcotics and some neuromuscular blockers (e.g., pancuronium bromide).
A physical barrier composed of mucin and the glycocalyx is formed over the intestinal epithelium by secretions from the goblet cells. It forms a gel that can change from a semisolid to a semifluid state under varying intraluminal conditions and thus provides either a relatively impervious barrier protecting the epithelium from osmolar forces or a fluid medium helping to propel bacteria and antigens aborally.1
Mucosal blood flow appears to be an important mechanism for maintaining mucosal integrity in both the stomach and the small intestine. Inadequate microcirculation in the gastric mucosa, that provides cytoprotection, appears to be a major factor contributing to stress ulceration in critically ill patients. As submucosal and mucosal blood flow diminishes, the buffering ability for acid that back diffuses into the tissues is reduced and leads to tissue necrosis. Hypoxia, hypotension, and states of high circulating catecholamines are commonly associated with altered mucosal circulation and stress ulceration.107 Finally, the small intestine, especially the terminal ileum, of newborns and infants is more sensitive to hypoxia and ischemia than that of adults, predisposing them to mucosal injury and necrotizing enterocolitis.108
Bacterial translocation is defined as the migration of bacteria or bacterial products from the intestinal lumen to mesenteric lymph nodes or other extraintestinal organs, representing a disruption of the normal host flora equilibrium that leads to a self-perpetuating inflammatory response and ultimately to infection. This process is increasingly being recognized as potential source of pathogens producing bacteremia and sepsis in a variety of premorbid disease conditions such as cirrhosis and prematurity.109,110 Translocation may occur directly through the M cells that cover the Peyer’s patches or it may occur by ingestion of viable pathogenic material by the mobile phagocytic system with transport into the host bypassing the previously outlined barrier mechanisms. The three main mechanisms predisposing a host to bacterial translocation are (1) disruption of the ecological equilibrium allowing intestinal bacterial overgrowth, (2) deficiencies in the host immune defenses, and (3) damage to the intestinal mucosa vasculature that causes increased permeability.111 Certain aerobic enteric bacteria including E. coli, Proteus mirabilis, and Klebsiella pneumoniae appear to be more commonly associated with translocation from the gastrointestinal tract, presumably because oxygen in the blood may exert an inhibitory effect on anaerobic organisms. Intra-abdominal inflammatory foci are susceptible to invasion by translocating bacteria, suggesting one mechanism for abdominal abscess formation.112 Some studies have demonstrated that administration of glutamine improves the prognosis of critically ill patients by maintaining the physiologic intestinal barrier and by reducing frequency of infection.113
Of central importance to the immune function of the gut are the gut-associated lymphoid tissues consisting of Peyer patches and single lymphoid nodules of the intestinal mucosa and appendix. In addition to mounting protective responses to invading microbial pathogens and detecting and eliminating neoplastic cells, the mucosal immune response has two unique characteristics: secretion of antibodies that complex antigen in the lumen without activating complement and induction of tolerogenic T lymphocytes that maintain controlled local responses to commensal bacteria or dietary constituents with no systemic reactions (oral tolerance).102
Kupffer cells, which account for the largest pool of mononuclear phagocytes with direct access to the blood, play a major role in clearing portal bacteria. In addition, Kupffer cells are key participants in response to tissue injury or organ invasion through the elaboration of cytokine mediators, such as tumor necrosis factor and interleukin-1, and the release of nitric oxide, leading to many of the systemic responses seen in sepsis.97 Through their intimate proximity to the hepatocyte, Kupffer cells interact directly with hepatocytes by means of cell-cell and paracrine interactions. In response to tumor necrosis factor and interleukin-1, well-documented alterations in hepatic function occur including the inhibition of albumin synthesis, gluconeogenesis, and P-450‑mediated detoxification. Acute-phase reactant synthesis is also induced by tumor necrosis factor and interleukin-1.114
Gastrointestinal and Hepatobiliary Testing in the Intensive Care Unit
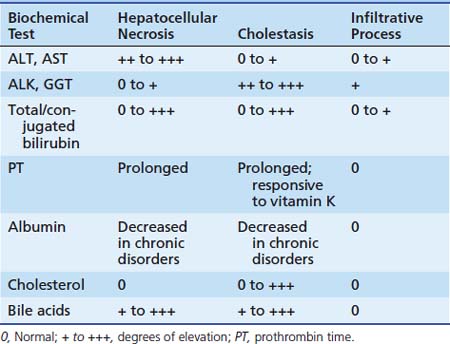
Diagnostic testing in the ICU permits the identification of organ system injury and dysfunction and assists in monitoring the course of a disease as well as response to therapies. Laboratory testing is helpful in detecting and monitoring hepatocellular injury and dysfunction. In the ICU setting, impaired synthetic function is the hallmark of liver failure. This is of more immediate concern than hepatocellular injury alone. Decreased synthesis of the liver dependent clotting factors I (fibrinogen), II (prothrombin), V, VII, IX and X, results in a prolonged prothrombin time. In the absence of vitamin K deficiency or related inhibitors, this represents liver failure. Because factor VII has the shortest half-life shortest half-life (2 to 6 hours) compared to the other factors, it acts as the rate-limiting step for conversion of prothrombin to thrombin. The use of recombinant activated factor VII in acute liver failure remains under debate.91 Other less specific measures of liver dysfunction include increased bilirubin, decreased serum albumin and decreased prealbumin.
Liver failure may also be associated with life-threatening hypoglycemia from a variety of mechanisms including decreased hepatic glycogenolysis, gluconeogenesis and release of glucose, hyperinsulinemia from impaired hepatic degradation, and increased glucose utilization secondary to anaerobic metabolism. A frequent finding in advanced liver failure is elevated serum ammonia reflecting impaired ureagenesis and/or other ammonia clearance mechanisms. Ammonia elevation results from urea cycle defects, portal systemic shunting, and events such as large gastrointestinal bleeds that generate substrate for increased ammonia production by enteric bacteria. In the setting of impaired liver function, this leads to hyperammonemia.115
In children, elevations in ALK may be seen with rickets or during periods of rapid skeletal growth. Isoenzymes may distinguish between liver or bony sources. ALT and AST are hepatic cytosolic enzymes that catalyze the reversible transfer of the α-amino group of the amino acids alanine and glutamate producing pyruvic and oxaloacetic acids respectively. Elevations in serum activities of ALT and AST suggest hepatocellular injury (see Table 85-3). AST is also present in myocardial tissue, skeletal muscle, kidney, pancreas and erythrocytes. Therefore increased serum AST is not specific for hepatocellular injury. Alanine transaminase is present in only relatively low concentrations in tissues other than liver, thus providing greater specificity for hepatocellular injury than AST. Falling levels of AST and ALT in the setting of rising levels of conjugated bilirubin represent worsening destruction of hepatocytes suggesting liver dysfunction rather than recovery. Elevated serum lactate dehydrogenase activity lacks specificity and may be seen in association with hepatocellular injury, hemolysis and myopathy; however, when in association with elevated serum creatinine phosphokinase or aldolase indicates myopathy or a rhabdomyolysis injury.
Of the available biochemical markers of pancreatic disease, serum amylase and lipase determinations are the most widely available. Serum lipase is elevated in about 87% of patients with acute pancreatitis and demonstrates fewer false-positive results than amylase testing. Transient hypocalcemia (<8.0 mg/dL) occurs in about 30% of patients with pancreatitis. Mild to moderate hyperglycemia as a result of islet cell damage is seen in up to 25% of cases, often necessitating the administration of exogenous insulin.
Esophageal pH monitoring is useful for evaluating the efficacy of antisecretory therapy. It may be useful to correlate symptoms (e.g., cough, chest pain) with acid reflux episodes and to select those infants and children with wheezing or respiratory symptoms in whom gastroesophageal reflux is an aggravating factor. The sensitivity, specificity, and clinical utility of pH monitoring for diagnosis and management of possible extraesophageal complications of gastroesophageal reflux are not well established.116
See Table 85-4 for suggested diagnosis tests for selected hepatobiliary disorders.
| Form of Liver Injury | Supportive History/Laboratory Data |
|---|---|
| PREDOMINANTLY HEPATOCELLULAR | |
| Viral hepatitis | Viral serologies: hepatitis A, B, C, E; Epstein-Barr virus; cytomegalovirus, varicella zoster virus, herpes simplex virus |
| Drug-induced hepatitis | History of toxic/excess ingestion ± elevated eosinophil count |
| Ischemia | Shock, status post–cardiac surgery |
| Autoimmune hepatitis | Increased globulin ratio, antinuclear antibody, anti–smooth muscle antibody, anti-liver kidney microsomal antibody |
| Wilson disease | Serum ceruloplasmin, 24-hour urine copper |
| α-Antitrypsin deficiency | α1 antitrypsin phenotyping |
| CHOLESTATIC | |
| Bacterial sepsis | Proteus, E. coli urinary tract infection |
| Galactosemia | Urine-reducing substances, E. coli sepsis, red blood cell galactose-1-phosphate uridyltransferase |
| Tyrosinemia | Urine succinylacetone |
| Biliary atresia | Intraoperative cholangiogram |
| Anatomic anomalies: choledochal cysts, biliary stricture, cholelithiasis, congenital hepatic fibrosis, Caroli disease | Ultrasonography, cholangiogram |
| Alagille syndrome | Butterfly vertebrae, posterior embryotoxon on eye exam, echocardiogram |
| Cystic fibrosis | Sweat test, genetic testing |
| Graft-versus-host disease, venoocclusive disease | History of bone marrow transplant, high-dose busulfan |
| Ischemia | Extracorporeal membrane oxygenation |
| INFILTRATIVE | |
| Hepatocellular carcinoma, hepatoblastoma | α-Feto protein |
| PREDOMINANTLY COAGULOPATHIC | |
| Neonatal hematochromatosis | Serum iron and ferritin |
References are available online at http://www.expertconsult.com.
1. Thomson A.B., Keelan M., Thiesen A., et al. Small bowel review: normal physiology part 1. Dig Dis Sci. 2001;46:2567-2587.
2. Clark J.A., Coopersmith C.M. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. 2007;28:384-393.
3. Harrell L., Chang E. Intestinal water and electrolyte transport. In: FMFLS M.H., editor. Sleisenger and Fordtran’s gastrointestinal and liver disease. ed 8. Philadelphia: Saunders Elsevier; 2006:2128-2146.
4. de Groof F., Joosten K.F., Janssen J.A., et al. Acute stress response in children with meningococcal sepsis: important differences in the growth hormone/insulin-like growth factor I axis between nonsurvivors and survivors. J Clin Endocrinol Metab. 2002;87:3118-3124.
5. Suman O.E., Mlcak R.P., Chinkes D.L., Herndon D.N. Resting energy expenditure in severely burned children: analysis of agreement between indirect calorimetry and prediction equations using the Bland-Altman method. Burns. 2006;32:335-342.
6. Zappitelli M., Juarez M., Castillo L., et al. Continuous renal replacement therapy amino acid, trace metal and folate clearance in critically ill children. Intensive Care Med. 2009;35:698-706.
7. de Neef M., Geukers V.G., Dral A., et al. Nutritional goals, prescription and delivery in a pediatric intensive care unit. Clin Nutr. 2008;27:65-71.
8. Mehta N.M., Compher C. A.S.P.E.N. clinical guidelines: nutrition support of the critically ill child. JPEN J Parenter Enteral Nutr. 2009;33:260-276.
9. van Waardenburg D.A., de Betue C.T., Luiking Y.C., et al. Plasma arginine and citrulline concentrations in critically ill children: strong relation with inflammation. Am J Clin Nutr. 2007;86:1438-1444.
10. Rogers E.J., Gilbertson H.R., Heine R.G., Henning R. Barriers to adequate nutrition in critically ill children. Nutrition. 2003;19:865-868.
11. Sanchez C., Lopez-Herce J., Carrillo A., et al. Early transpyloric enteral nutrition in critically ill children. Nutrition. 2007;23:16-22.
12. Meert K.L., Daphtary K.M., Metheny N.A. Gastric vs small-bowel feeding in critically ill children receiving mechanical ventilation: a randomized controlled trial. Chest. 2004;126:872-878.
13. Petrillo-Albarano T., Pettignano R., Asfaw M., Easley K. Use of a feeding protocol to improve nutritional support through early, aggressive, enteral nutrition in the pediatric intensive care unit. Pediatr Crit Care Med. 2006;7:340-344.
14. Sanderson I.R., Croft N.M. The anti-inflammatory effects of enteral nutrition. JPEN J Parenter Enteral Nutr. 2005;29:S134-138.
15. Hulst J.M., van Goudoever J.B., Visser T.J., et al. Hormone levels in children during the first week of ICU-admission: is there an effect of adequate feeding? Clin Nutr. 2006;25:154-162.
16. Alam N.H., Ashraf H., Sarker S.A., et al. Efficacy of partially hydrolyzed guar gum-added oral rehydration solution in the treatment of severe cholera in adults. Digestion. 2008;78:24-29.
17. Heitlinger L.A., Lee P.C., Dillon W.P., Lebenthal E. Mammary amylase: a possible alternate pathway of carbohydrate digestion in infancy. Pediatr Res. 1983;17:15-18.
18. Layer P., Zinsmeister A.R., DiMagno E.P. Effects of decreasing intraluminal amylase activity on starch digestion and postprandial gastrointestinal function in humans. Gastroenterology. 1986;91:41-48.
19. Jacob R., Zimmer K.P., Schmitz J., Naim H.Y. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J Clin Invest. 2000;106:281-287.
20. Ritz V., Alfalah M., Zimmer K.P., et al. Congenital sucrase-isomaltase deficiency because of an accumulation of the mutant enzyme in the endoplasmic reticulum. Gastroenterology. 2003;125:1678-1685.
21. Stefan Y., Orci L., Malaisse-Lagae F., et al. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes. 1982;31:694-700.
22. Wright E.M., Martin M.G., Turk E. Intestinal absorption in health and disease–sugars. Best Pract Res Clin Gastroenterol. 2003;17:943-956.
23. Montes R.G., Gottal R.F., Bayless T.M., et al. Breath hydrogen testing as a physiology laboratory exercise for medical students. Am J Physiol. 1992;262:S25-28.
24. Koetse H.A., Vonk R.J., Gonera-De Jong G.B., et al. Low lactase activity in a small-bowel biopsy specimen: should dietary lactose intake be restricted in children with small intestinal mucosal damage? Scand J Gastroenterol. 2006;41:37-41.
25. Shulman R.J., Wong W.W., Smith E.O. Influence of changes in lactase activity and small-intestinal mucosal growth on lactose digestion and absorption in preterm infants. Am J Clin Nutr. 2005;81:472-479.
26. Farrell J.Digestion and absorption of nutrients and vitamins Feldman M., editor Sleisenger & Fordtran’s gastrointestinal and liver disease 2006:vol;2147-2199 1 Saunders Elsevier Philadelphia
27. Silk D.B., Grimble G.K., Rees R.G. Protein digestion and amino acid and peptide absorption. Proc Nutr Soc. 1985;44:63-72.
28. Leibach F.H., Ganapathy V. Peptide transporters in the intestine and the kidney. Ann Rev Nutr. 1996;16:99-119.
29. Kekuda R., Torres-Zamorano V., Fei Y.J., et al. Molecular and functional characterization of intestinal Na(+)-dependent neutral amino acid transporter B0. Am J Physiol. 1997;272:G1463-1472.
30. Mackenzie B., Loo D.D., Fei Y., et al. Mechanisms of the human intestinal H+-coupled oligopeptide transporter hPEPT1. J Biol Chem. 1996;271:5430-5437.
31. Elwyn D: The Role of the liver in regulation of amino acid and protein metabolism. In HN M, editor: Mammalian protein metabolism, vol 4, New York, 1970, Academic Press.
32. Jalonen T. Identical intestinal permeability changes in children with different clinical manifestations of cow’s milk allergy. J Allergy Clin Immunol. 1991;88:737-742.
33. Holsapple M.P., West L.J., Landreth K.S. Species comparison of anatomical and functional immune system development. Birth Defects Res B Dev Reprod Toxicol. 2003;68:321-334.
34. Udall J.N., Colony P., Fritze L., et al. Development of gastrointestinal mucosal barrier. II. The effect of natural versus artificial feeding on intestinal permeability to macromolecules. Pediatr Res. 1981;15:245-249.
35. Hardy S.C., Kleinman R.E. Fat and cholesterol in the diet of infants and young children: implications for growth, development, and long-term health. J Pediatr. 1994;125:S69-77.
36. Hamosh M. The milky way: from mammary gland to milk to newborn–Macy-Gyorgy Award presentation (1999). Adv Exp Med Biol. 2002;503:17-25.
37. Roman C., Carriere F., Villeneuve P., et al. Quantitative and qualitative study of gastric lipolysis in premature infants: do MCT-enriched infant formulas improve fat digestion? Pediatr Res. 2007;61:83-88.
38. Woollett L.A., Wang Y., Buckley D.D., et al. Micellar solubilisation of cholesterol is essential for absorption in humans. Gut. 2006;55:197-204.
39. Shiau Y.F. Mechanism of intestinal fatty acid uptake in the rat: the role of an acidic microclimate. J Physiol. 1990;421:463-474.
40. Shiau Y.F., Kelemen R.J., Reed M.A. Acidic mucin layer facilitates micelle dissociation and fatty acid diffusion. Am J Physiol. 1990;259:G671-675.
41. Peretti N., Roy C.C., Drouin E., et al. Abnormal intracellular lipid processing contributes to fat malabsorption in cystic fibrosis patients. Am J Physiol Gastrointest Liver Physiol. 2006;290:G609-615.
42. Black D.D. Development and physiological regulation of intestinal lipid absorption. I. Development of intestinal lipid absorption: cellular events in chylomicron assembly and secretion. Am J Physiol Gastrointest Liver Physiol. 2007;293:G519-524.
43. Madara J.L. Sodium-glucose cotransport and epithelial permeability. Gastroenterology. 1994;107:319-320.
44. Lencer D.J. Transport of water and ions. In WA Walker P.D., Hamilton J.R., et al, editors: Pediatric gastrointestinal disease, ed 2, St Louis: Mosby, 1996.
45. Zarroug A.E., Libsch K.D., Houghton S.G., et al. Postprandial augmentation of absorption of water and electrolytes in jejunum is neurally modulated: implications for segmental small bowel transplantation. J Gastrointest Surg. 2006;10:586-592.
46. Westergaard H. Bile acid malabsorption. Curr Treat Options Gastroenterol. 2007;10:28-33.
47. Martinez-Augustin O., Romero-Calvo I., Suarez M.D., et al. Molecular bases of impaired water and ion movements in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:114-127.
48. Gawenis L.R., Hut H., Bot A.G., et al. Electroneutral sodium absorption and electrogenic anion secretion across murine small intestine are regulated in parallel. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1140-1149.
49. Wright E.M., Loo D.D. Coupling between Na+, sugar, and water transport across the intestine. Ann N Y Acad Sci. 2000;915:54-66.
50. Turner J.R., Rill B.K., Carlson S.L., et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol. 1997;273:C1378-1385.
51. Milla P.J. Advances in understanding colonic function. J Pediatr Gastroenterol Nutr. 2009;48(Suppl 2):S43-45.
52. Coeffier M., Hecketsweiler B., Hecketsweiler P., Dechelotte P. Effect of glutamine on water and sodium absorption in human jejunum at baseline and during PGE1-induced secretion. J Appl Physiol. 2005;98:2163-2168.
53. Orsenigo M.N., Faelli A., Porta C., et al. Oxidative stress reduces transintestinal transports and (Na+, K+) -ATPase activity in rat jejunum. Arch Biochem Biophys. 2007;466:300-307.
54. Holtug K., Hansen M.B., Skadhauge E. Experimental studies of intestinal ion and water transport. Scand J Gastroenterol Suppl. 1996;216:95-110.
55. Viswanathan V.K., Hodges K., Hecht G. Enteric infection meets intestinal function: how bacterial pathogens cause diarrhoea. Nat Rev Microbiol. 2009;7:110-119.
56. Lorrot M., Vasseur M. How do the rotavirus NSP4 and bacterial enterotoxins lead differently to diarrhea? Virol J. 2007;4:31.
57. Cvijanovich N.Z., King J.C., Flori H.R., et al. Zinc homeostasis in pediatric critical illness. Pediatr Crit Care Med. 2009;10:29-34.
58. Beale R.J., Sherry T., Lei K., et al. Early enteral supplementation with key pharmaconutrients improves sequential organ failure assessment score in critically ill patients with sepsis: outcome of a randomized, controlled, double-blind trial. Crit Care Med. 2008;36:131-144.
59. Heyland D.K., Jones N., Cvijanovich N.Z., Wong H. Zinc supplementation in critically ill patients: a key pharmaconutrient? JPEN J Parenter Enteral Nutr. 2008;32:509-519.
60. Feldman M. Gastric secretion. In: Feldman M., editor. Sleisenger and Fordtran’s gastrointestinal and liver disease. ed 8. Philadelphia: Saunders Elsevier; 2006:1029-1048.
61. Wolfe M.M., Soll A.H. The physiology of gastric acid secretion. N Engl J Med. 1988;319:1707-1715.
62. DelValle J. The stomach as an endocrine organ. Digestion. 1997;58(Suppl 1):4-7.
63. Pisegna J.R. Pharmacology of acid suppression in the hospital setting: focus on proton pump inhibition. Crit Care Med. 2002;30:S356-361.
64. Ojiako K., Shingala H., Schorr C., Gerber D.R. Famotidine versus pantoprazole for preventing bleeding in the upper gastrointestinal tract of critically ill patients receiving mechanical ventilation. Am J Crit Care. 2008;17:142-147.
65. Olsen K.M., Devlin J.W. Comparison of the enteral and intravenous lansoprazole pharmacodynamic responses in critically ill patients. Aliment Pharmacol Ther. 2008;28:326-333.
66. Quenot J.P., Thiery N., Barbar S. When should stress ulcer prophylaxis be used in the ICU? Curr Opin Crit Care. 2009;15:139-143.
67. Kemp D.M., Thomas M.K., Habener J.F. Developmental aspects of the endocrine pancreas. Rev Endocr Metab Disord. 2003;4:5-17.
68. Williams J.A. Intracellular signaling mechanisms activated by cholecystokinin-regulating synthesis and secretion of digestive enzymes in pancreatic acinar cells. Annu Rev Physiol. 2001;63:77-97.
69. DiMagno E.P., Shorter R.G., Taylor W.F., Go V.L. Relationships between pancreaticobiliary ductal anatomy and pancreatic ductal and parenchymal histology. Cancer. 1982;49:361-368.
70. Marino C.R., Matovcik L.M., Gorelick F.S., Cohn J.A. Localization of the cystic fibrosis transmembrane conductance regulator in pancreas. J Clin Invest. 1991;88:712-716.
71. Kreiss C., Schwizer W., Erlacher U., et al. Role of antrum in regulation of pancreaticobiliary secretion in humans. Am J Physiol. 1996;270:G844-851.
72. Owyang C., Logsdon C.D. New insights into neurohormonal regulation of pancreatic secretion. Gastroenterology. 2004;127:957-969.
73. Morisset J. Negative control of human pancreatic secretion: physiological mechanisms and factors. Pancreas. 2008;37:1-12.
74. Uhl W., Anghelacopoulos S.E., Friess H., Buchler M.W. The role of octreotide and somatostatin in acute and chronic pancreatitis. Digestion. 1999;60(Suppl 2):23-31.
75. Elfar M., Gaber L.W., Sabek O., et al. The inflammatory cascade in acute pancreatitis: relevance to clinical disease. Surg Clin North Am. 2007;87:1325-1340.
76. Boamah L, Balistreri W: Manifestations of liver disease, ed 18, In Kliegman, editor: Nelson textbook of pediatrics, vol 1, Philadelphia, 2007, Saunders Elsevier.
77. Hilmes M.A., Strouse P.J. The pediatric spleen. Semin Ultrasound CT MR. 2007;28:3-11.
78. Kjaer M., Jurlander J., Keiding S., et al. No reinnervation of hepatic sympathetic nerves after liver transplantation in human subjects. J Hepatol. 1994;20:97-100.
79. Roy-Choudhury N., Roy-Choudhury J. Liver physiology and energy metabolism. In: Feldman M., Friedman L., Sleisenger M., editors. Sleisenger and Fordtran’s gastrointestinal and liver disease. ed 8. Philadelphia: Saunders Elsevier; 2006:1551-1574.
80. Jiao J., Friedman S.L., Aloman C. Hepatic fibrosis. Curr Opin Gastroenterol. 2009;25:223-229.
81. Wanless I: Physioanatomic considerations. In Schiff E, editor: Schiff’s diseases of the liver, vol 1, ed 10, Philadelphia, 2007, Lippincott Williams & Wilkins.
82. Bosch J, D’Amico G, Garcia-Pagan J: Portal hypertension and nonsurgical management. In Schiff E, editor: Schiff’s diseases of the liver, vol 1, ed 10, Philadelphia, 2007, Lippincott Williams & Wilkins.
83. Vorobioff J., Bredfeldt J.E., Groszmann R.J. Increased blood flow through the portal system in cirrhotic rats. Gastroenterology. 1984;87:1120-1126.
84. Benoit J.N., Granger D.N. Splanchnic hemodynamics in chronic portal hypertension. Semin Liver Dis. 1986;6:287-298.
85. Ruiz-del-Arbol L., Monescillo A., Arocena C., et al. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439-447.
86. Ballmer P.E. Causes and mechanisms of hypoalbuminaemia. Clin Nutr. 2001;20:271-273.
87. Devakonda A., George L., Raoof S., et al. Transthyretin as a marker to predict outcome in critically ill patients. Clin Biochem. 2008;41:1126-1130.
88. Fairbanks K.D., Tavill A.S. Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol. 2008;103:2136-2141. quiz 2142
89. Umar S.B., Dibaise J.K. Protein-losing enteropathy: case illustrations and clinical review. Am J Gastroenterol. 2010;105(1):43-49.
90. Van Winckel M., De Bruyne R., Van De Velde S., et al. Vitamin K: an update for the paediatrician. Eur J Pediatr. 2009;168:127-134.
91. De Gasperi A., Corti A., Mazza E., et al. Acute liver failure: managing coagulopathy and the bleeding diathesis. Transplant Proc. 2009;41:1256-1259.
92. Clay A.S., Hainline B.E. Hyperammonemia in the ICU. Chest. 2007;132:1368-1378.
93. Teoh N., Farrell G. Liver disease caused by drugs. In: Feldman M., Friedman L., Sleisenger M., editors. Sleisenger and Fordtran’s gastrointestinal and liver disease. ed 8. Philadelphia: Saunders Elsevier; 2006:1807-1853.
94. Mesotten D., Wauters J., Van den Berghe G., et al. The effect of strict blood glucose control on biliary sludge and cholestasis in critically ill patients. J Clin Endocrinol Metab. 2009;94:2345-2352.
95. Pilkis S.J., Granner D.K. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Ann Rev Physiol. 1992;54:885-909.
96. George J., Ganesh H.K., Acharya S., et al. Bone mineral density and disorders of mineral metabolism in chronic liver disease. World J Gastroenterol. 2009;15:3516-3522.
97. Crispe I.N. The liver as a lymphoid organ. Ann Rev Immunol. 2009;27:147-163.
98. Matuschak G.M. Liver-lung interactions in critical illness. New Horiz. 1994;2:488-504.
99. Matuschak G.M. Lung-liver interactions in sepsis and multiple organ failure syndrome. Clin Chest Med. 1996;17:83-98.
100. Matuschak G.M., Henry K.A., Johanns C.A., Lechner A.J. Liver-lung interactions following Escherichia coli bacteremic sepsis and secondary hepatic ischemia/reperfusion injury. Am J Respir Crit Care Med. 2001;163:1002-1009.
101. Hollenberg S.M., Cinel I. Bench-to-bedside review: nitric oxide in critical illness–update 2008. Crit Care. 2009;13:218.
102. Sartor R.Mucosal immunology and mechanisms of gastrointestinal inflammation Feldman M., editor. Sleisenger & Fordtran’s gastrointestinal and liver disease, vol 1. Philadelphia: Saunders Elsevier, 2006.
103. Simonovic I., Rosenberg J., Koutsouris A., Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol. 2000;2:305-315.
104. Pothoulakis C., Lamont J.T. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280:G178-183.
105. Monreal M.T., Pereira P.C., de Magalhaes Lopes C.A. Intestinal microbiota of patients with bacterial infection of the respiratory tract treated with amoxicillin. Braz J Infect Dis. 2005;9:292-300.
106. Sultan N., Nazareno J., Gregor J. Association between proton pump inhibitors and respiratory infections: a systematic review and meta-analysis of clinical trials. Can J Gastroenterol. 2008;22:761-766.
107. Ali T., Harty R.F. Stress-induced ulcer bleeding in critically ill patients. Gastroenterol Clin North Am. 2009;38:245-265.
108. Hsueh W., Caplan M.S., Qu X.W., et al. Neonatal necrotizing enterocolitis: clinical considerations and pathogenetic concepts. Pediatr Dev Pathol. 2003;6:6-23.
109. Wiest R., Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422-433.
110. Anand R.J., Leaphart C.L., Mollen K.P., Hackam D.J. The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock. 2007;27:124-133.
111. Lichtman S.M. Bacterial translocation in humans. J Pediatr Gastroenterol Nutr. 2001;33:1-10.
112. Balzan S., de Almeida Quadros C., de Cleva R., et al. Bacterial translocation: overview of mechanisms and clinical impact. J Gastroenterol Hepatol. 2007;22:464-471.
113. De-Souza D.A., Greene L.J. Intestinal permeability and systemic infections in critically ill patients: effect of glutamine. Crit Care Med. 2005;33:1125-1135.
114. Vermeire S., Van Assche G., Rutgeerts P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:661-665.
115. Whitington P., Soriano H., Alonso E. Fulminant hepatic failure in children. In: Suchy F., Sokol R., Balistreri W., editors. Liver disease in children. ed 2. Philadelphia: Lippincott Williams & Wilkins; 2001:63-88.
116. Vandenplas Y., Rudolph C.D., Di Lorenzo C., et al. Pediatric gastroesophageal reflux clinical practice guidelines: joint recommendations of the North American Society of Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society of Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2009;49(4):498-547.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 85 Gastrointestinal Structure and Function
Abnormalities in gastrointestinal (GI) and hepatobiliary function occur frequently in the pediatric intensive care unit (PICU) from primary GI disease, following surgery, or from complications of systemic disease. The pediatric intensivist is called on for expertise in the preoperative and postoperative management of numerous GI tract and hepatobiliary diseases, as well as secondary dysfunction that may complicate diseases involving other organ systems. The GI tract subserves a wide range of functions beyond simple digestion that have an impact on systemic immunological, endocrinological, and microbiological functions.1 Intact gut function beyond digestive function is of vital importance for the maintenance of health. Recent attention to alterations in GI function, interactions between liver and lung and between liver and kidneys have led to the view that the gut plays a role as an “engine” of multiple organ dysfunction.2 Thus a practical working knowledge of the GI system and its integrated function is essential for the management of critically ill patients.
Intestinal Structure, Digestion, and Absorption of Nutrients, Water, and Electrolytes
The functional absorptive unit of the intestines consists of villi and crypts. The cells of the small intestine are separated from one another by specialized junctions that serve as gaskets to prevent back diffusion of material into the intestinal lumen. A mucus layer secreted by goblet cells in crypts separates enterocytes from direct contact with the luminal contents (Table 85-1).
| Structure | Function |
|---|---|
| Enterocyte | Formed in crypts; migrate to villus over 2–3 days; lifespan of 6 days |
| Villi | Absorption |
| Crypts | Secretion |
| Microvilli | Amplify surface area; contain enzymes and transport systems |
Stem cells in crypts produce enterocytes and other specialized epithelial cells that migrate up the villous axis as they become differentiated. This migratory process takes 48 to 72 hours. Mature villous cells live 6 days. They have microvilli making up the brush border that contain digestive enzymes and membrane bound transport systems for nutrient and electrolytes. Small bowel enterocyte microvilli are estimated to increase the luminal surface area of the cell 14- to 40-fold.3 The cells at the villous tip have a predominantly absorptive function, while crypt cells are primarily secretory. Rotavirus infections cause villous loss resulting in small intestinal mucosa composed largely of crypts and immature villi. This causes a net secretory state leading to malabsorption and osmotic diarrhea. Malabsorption of nutrients is another manifestation of villous injury.
Water and Solute Transport Across the Intestinal Epithelium
Surface area and integrity of intercellular junctions are the major determinants of water and solute flux across epithelium. The transport of solute and water across epithelium occurs either by active or passive transport or by facilitated diffusion (Table 85-2). The gut conserves large volumes of endogenously secreted material associated with digestion. The average luminal fluid input of the gut is about 9 L/day and composed of oral intake and endogenous secretions. Approximately 8.8 L is absorbed, about 7 L in the small intestine and 1.8 L in the colon. Less than 0.2 L is excreted as a component of the normal stool output. When rapid changes in dietary intake or endogenous secretions occur, the intestinal mucosa can adapt transport functions in order to compensate for the changes. The loss of mucosal surface area through disease or surgical resection alters net flux of solute and water in GI tract. Furthermore, loss of specialized absorptive function may occur following loss of specific areas of gut. An example of this occurs in the setting of short bowel syndrome from resection of terminal ileum, with loss of ability to absorb bile acids and intrinsic factor.3 Malfunction of absorptive mechanisms may lead to life threatening loss of fluid and electrolytes.
| Active | Passive | Facilitated Diffusion |
|---|---|---|
| Against electrochemical gradient | Ionic specificity | Saturable kinetics |
| Saturable kinetics | May be associated with transport of a nonelectrolyte | Substrate specific |
| Requires ATP | Proceeds down electrochemical gradient | Depends on carrier molecules (glucose, amino acid) |
| Steady state based on concentration differences | ||
| Displays first-order kinetics | ||
| May occur by convection via osmotic or hydrostatic gradient |
In contrast, critical illness alters energy requirements. In general, the prolonged physiological stress during critical illness causes an increased metabolic rate as well as gluconeogenesis in excess of that needed to maintain serum glucose. Proteolysis and peripheral oxidation of amino acids with increased ureagenesis is also observed. This state leads rapidly to malnutrition as a result of so-called autocannibalism, characterized by levels of glucagon higher than insulin levels and by elevated catecholamines and cortisol that drive the relentless breakdown of peripheral tissues. These hormonal changes result in catabolism of endogenous stores of protein, carbohydrates and fat to provide metabolic substrate necessary to support the metabolic stress response.4 Children with burn injury demonstrate extreme hypermetabolism in the initial phases of illness.5 Children on dialysis have a negative nitrogen balance and loss of folate and selenium.6 Use of catecholamines or neuromuscular blocking agents represent risk factors for undernutrition in critically ill children on dialysis.7 During high physiologic stress, nutrition delivered to patients must be changed to allow for decreased reliance on fat and carbohydrate for energy production. Proteins are increasingly relied on and must be replaced with additional amino acids to support ongoing synthesis of protein including immunoglobulins by the liver and immune system. Protein requirements for various age groups are as follows: 0 to 2 years, 2 to 3 g/kg/day; 2 to 13 years, 1.5 to 2 g/kg/day; and 13 to 18 years, 1.5 g/kg/day.8 Depressed levels of specific amino acids have been found in critically ill children.9
Enteral nutrition is the preferred method of feeding for critically ill children. Restriction of fluid intake seems to be the main barrier to the delivery of adequate nutrition, particularly in infants undergoing cardiac surgery.10 Early transpyloric enteral nutrition is well tolerated in critically ill children and is not associated with an increase in incidence of complications.11 Small-bowel feeds allow a greater amount of nutrition to be successfully delivered to critically ill children compared with gastric feeds. However, small-bowel feeds do not prevent aspiration of gastric contents.12 The institution of a feeding protocol has been found to achieve goal feedings quickly and also improve tolerance of enteral feedings in patients admitted to the pediatric intensive care unit.13 Enteral nutrition may also have some anti-inflammatory effects by lowering expression of specific cytokines.14 The use of specialized formulas is not currently recommended in critically ill children. No effect from adequacy of feeding has been seen on hormone levels such as insulinlike growth factor-1 (IGF-1) and thyroid hormones.15
Digestion of Carbohydrates
Dietary carbohydrates may be classified in several ways (Box 85-1). Monosaccharides, such as glucose and fructose, are present in fruits, sweet corn, corn syrup, and honey. Disaccharides are simple sugars, such as sucrose (glucose + fructose), maltose (glucose + glucose), and lactose (glucose + galactose), the principal mammalian sugar. Polysaccharides, such as starch, are polymers of glucose and are abundantly present in wheat, grains, potatoes, peas, beans, and vegetables. Fiber consists of nondigestible complex polysaccharides of plant origin. This includes both water insoluble and water soluble fiber. The average American diet is 3:1 soluble to insoluble fiber. Some studies have shown that a water-soluble fiber, partially hydrolyzed guar gum can be used to decrease stool output in cholera.16 Insoluble fibers affect fecal bulk, whereas soluble fibers have viscous effects in the upper GI tract including delayed gastric emptying, decreased postprandial glycemic response, and a constipating effect.
Carbohydrates are a major source of calories in healthy children. Carbohydrates are generally broken down by means of glycolysis and the Krebs cycle pathway (see Chapter 74). Carbohydrates may be stored as glycogen and lipids when ingested beyond momentary energy needs or converted to structural materials. A person’s requirement for energy is highly dependent on activity level or, in hospitalized patients, the degree of hypermetabolism accompanying illness. The maximal ability to utilize carbohydrates may be limited during periods of high physiological stress as a result of the complex effects of hormonal mediators of the stress response.
Digestion of carbohydrates begins with the process of chewing that decreases the size of food particles, thereby increasing the total surface area for subsequent action by digestive juices. Salivary secretions are necessary for lubrication and also contain salivary amylase, an endoenzyme that cleaves oligosaccharides. Salivary amylase is rapidly inactivated by gastric acid, leaving the majority of starch digestion to occur in the duodenum under the action of pancreatic amylase and the intestinal brush border disaccharidase enzymes. Amylase contained in human milk facilitates starch digestion in breast-fed infants because of their low levels of endogenous salivary and pancreatic amylase.17 Pancreatic amylase is the major enzyme of starch digestion resulting in short oligosaccharides, maltotriose, maltose, and α-limit dextrins. The amylase concentration becomes limiting in cases of pancreatic insufficiency when amylase levels become less than 10% of normal.18
The enterocyte is incapable of absorbing carbohydrates larger than monosaccharides. Therefore, further hydrolysis to monosaccharides is performed by intestinal brush-border disaccharidases of which the clinically important ones are lactase, which breaks down lactose, and sucrase, which breaks down sucrose. These enzymes are synthesized in the enterocyte and are subsequently inserted into the apical brush border membrane. With the exception of lactase and occasionally sucrase, the disaccharidases are rarely rate-limiting for complete carbohydrate digestion. Deficiencies of any of the disaccharidase enzymes, either acquired or hereditary, may result in carbohydrate malabsorption. This is characterized by osmotic diarrhea with elevated fecal reducing sugars, abdominal distension and flatulence secondary to fermentation of undigested oligosaccharides by colonic bacteria. An example of this is congenital sucrase isomaltase deficiency, an autosomal recessive disorder that is associated with absence of sucrase and maltase.19,20
Absorption of carbohydrates occurs in several ways. During periods of high luminal carbohydrate concentration, simple diffusion of monosaccharides may occur. Additionally, two transport mechanisms exist in the brush border for the absorption of monosaccharides.21 First, glucose, galactose and xylitol are transported with sodium by the Na+/glucose cotransporter. A low intracellular sodium concentration is created by the sodium-potassium-adenosine triphosphatase (Na+-K+ adenosine triphosphatase [ATPase]) pump located on the basolateral membrane. The resulting concentration gradient leads to movement of luminal sodium across the apical membrane, bringing with it glucose or galactose in a one-to-one molar ratio. Glucose-galactose malabsorption is a deficiency of this transport mechanism leading to neonatal onset of severe diarrhea.22 The second mechanism is a non‑energy-dependent facilitated transport system for fructose. The intestinal transport mechanisms are summarized in Table 85-2. The capacity to absorb fructose is limited, and excess ingestion has been found to cause symptoms of carbohydrate malabsorption.23
Clinical conditions that cause loss of the epithelium and brush border system may lead to symptoms of carbohydrate malabsorption. These conditions include: rotoviral gastroenteritis, inflammatory bowel disease, celiac disease, sprue, ischemia/hypoxia, bacterial overgrowth of the proximal gut as a result of either stasis or use of antacids, and malnutrition. Severe mucosal damage requires 7 to 10 days for recovery of brush border function. Several infant and enteral formulas rely on starch as a carbohydrate source to minimize reliance on lactase. Recent studies have observed that lactose digestive capability with lactase can be maintained despite small bowel mucosal damage.24 Lactose absorption is primarily related to lactase activity as opposed to mucosal growth.25
Digestion of Proteins
The GI tract has developed efficient mechanisms for processing exogenous peptides and complex proteins (Box 85-2). It is also very efficient at recycling endogenous proteins such as digestive enzymes, mucus, sloughed cells, and plasma proteins that leak into the alimentary tract.26 The recommended dietary protein intakes in healthy children range from 2.5 to 3.5 g/kg/day in early infancy to 1.2 g/kg/day during childhood, and 0.8 to 0.9 g/kg/day in adolescence.8 The enteral processing of proteins may be divided into digestive and transport phases.
In the digestive phase, gastric acid secretion initiates denaturation of complex proteins making them more susceptible to the actions of proteolytic enzymes. The chief cells of the stomach release pepsinogens that are converted to active pepsins under the influence of gastric acid. The pepsins are endopeptidases that release relatively large peptides and are inactivated when the pH rises above 4 as the food enters the duodenum. The completeness of gastric proteolysis depends in part on the rate of gastric emptying, the pH of intragastric contents, and the types of protein ingested.26 It is noteworthy that patients with achlorhydria or those receiving antacids, H2 blockers, or both agents have no evidence of impaired protein digestion ability. In addition to initiating protein digestion in the mature subject, pepsins act as milk clotting factors, which are important in the neonate for curd formation and provide bulk to the infant’s stools.
Luminal digestion proceeds in the small intestine mediated by five pancreatic peptidases that are secreted by the pancreatic acinar cells as proenzymes and activated by enterokinase and trypsin. Each peptidase possesses proteolytic activity at specific internal or external peptide bonds. Proteins are degraded typically into mixtures of one-third free amino acids and two-thirds peptides containing two to six amino acid residues,27 which are suitable substrates for the brush border peptidases. The brush border peptidases convert the oligopeptides into monopeptides, dipeptides, and tripeptides suitable for transport into the enterocyte.
In the transport phase, specific membrane-associated transport mechanisms exist for the uptake of amino acids and dipeptides.28 They involve simple diffusion, facilitated transport, and carrier-mediated active transport (Table 85-3). Na+-coupled active transport is an energy-dependent process associated with the uptake of luminal Na+ and an amino acid (or glucose) and exchange of the sodium and associated molecule for K+ through the basolateral membrane on the serosal side.29 Peptide transport may also occur using an H+/peptide transport protein which moves according to an H+ gradient in the acidic pH microclimate of the intestinal brush border. This microclimate is maintained by Na+-H+ exchange in the brush border and Na+-K+-ATPase in the basolateral membrane.30 An important characteristic of these transporters is that many amino acids are absorbed more rapidly as dipeptides than as free amino acids. This fact has been capitalized on in the development of enteral nutritional formulas because oligopeptide mixtures have a lower osmolarity and are more efficiently absorbed than single amino acid solutions of equal nitrogen content. Because of the efficient gastrointestinal absorption of dipeptides, patients with specific amino acid transport defects (e.g. Hartnup disease [defective tryptophan transport] and lysinuric protein intolerance [defect in dibasic amino acid transport: lysine, arginine]) infrequently have GI symptoms related to dietary protein malabsorption and instead more commonly manifest with non-GI symptoms such as aminoaciduria.
Once inside the enterocyte, peptides are quickly degraded into their constituent amino acids by cytoplasmic peptidases that complement the activity of the brush border peptidases. Only minute quantities of intact peptide and protein gain access to the systemic circulation. The cytoplasmic amino acids derived from digested proteins are a major source of free amino acids used directly by the enterocyte. When absorbed beyond cellular needs, the free amino acids are released to the portal venous circulation for hepatic and systemic use. Only 23% of absorbed amino acid nitrogen passes to the periphery without modification.31 Of the remaining nitrogen, 57% is converted to urea with the carbon skeleton salvaged for synthesizing other substances and 20% of the total ingested amino acids are used directly for hepatic protein synthesis.
During periods of fasting, the enterocyte derives the majority of its nourishment from the mesenteric arterial vascular supply, whereas during digestion, the enterocyte derives a significant part of its nutrient requirements from the luminal contents. Experience with mucosal recovery and adaptation after injury reveals that an enteral route of nutrition permits optimal recovery. In the premature infant and neonate, the small intestine is capable of absorbing intact milk proteins by pinocytosis. These proteins may include secretory immunoglobulins from breast milk as well as food antigens.32 Peptidase inhibitors have been demonstrated in colostrum and breast milk, partially explaining the failure of normal digestive mechanisms to degrade some of these complex dietary proteins. Both antibodies and antigens ingested with maternal milk create an important part of the immune repertory developed during early infancy.33 Although the exact time of “closure” of the intestinal mucosa to the uptake of macromolecules has not been defined in human infants, other mammals demonstrate marked intestinal impermeability to foreign proteins by the time of weaning34 from breastfeeding.
Digestion of Lipids
Dietary fat accounts for approximately 50% to 70% of the nonprotein calories consumed by infants and approximately 30% of nonprotein calories consumed after age 2 years (Box 85-3).35 Dietary fat is ingested principally in the form of triglycerides containing the fatty acids palmitate and oleate (C16:0 and C18:1, respectively). Dietary triglycerides of animal origin predominantly contain long-chain (i.e., longer than C14 chain length) saturated fatty acids. Polyunsaturated fatty acids are mostly of vegetable origin and include linoleic and linolenic acid, also referred to as essential fatty acids because of absent de novo synthesis in humans. Other dietary lipids include fat-soluble vitamins, cholesterol, prostaglandins, waxes, and phospholipids.
In healthy adults, digestion and absorption of fat is complete with only 5% to 7% of ingested fat escaping absorption. Under normal physiological conditions healthy infants up to age 9 to 12 months fail to absorb 15% to 35% of dietary fat. Digestion and absorption of dietary fat is generally completed by the middle third of jejunum; however, the presence of dietary fiber may reduce the rate and extent of absorption. Loss of dietary fat places children at significant risk for calorie and fat-soluble vitamin malnutrition.
Digestion of Fat
Fat digestion begins with formation of emulsions, which increase the surface area for enzyme interaction. Emulsification begins with release of fat by mastication and gastric “milling” of chyme. Bile salts and coating by phospholipid derived from the diet results in a stable emulsion droplet with a hydrophobic center consisting of triglyceride, cholesterol esters and diglyceride in a hydrophilic envelope. Mammary, lingual, and gastric lipases play an important role in direct lipolysis of long- and medium-chain triglycerides that are present in maternal milk.36 Lingual and gastric lipases are active at pH <5 and begin digestion of fat in the stomach; however, overall only play a limited role in the digestion of lipids. Intragastric lipolysis is consistent across all age groups.37
Most of the enzymatic degradation of dietary lipids to fatty acids and monoglyceride is by the action of pancreatic lipase and co-lipase, and requires an alkaline environment (pH 6 to 8). This underscores the importance of secretion of bicarbonate by the pancreas and biliary tree in order to neutralize gastric acid. Co-lipase is an essential cofactor for lipase action. Co-lipase’s role is to displace the bile salt-triglyceride interaction in emulsion droplets and micelles to facilitate lipase hydrolysis of the triglyceride. Triglyceride hydrolysis occurs at the interface between the emulsion droplet and aqueous phase within the lumen. This is a two-step process. The first step is the enzymatic hydrolysis of long-chain triglycerides and liberation of fatty acids from the glycerol backbone. The second step is formation of fatty acid micelles, that is most efficiently accomplished with the aid of bile salts38 to traffic the fatty acids across the unstirred water layer to the mucosa for absorption.
Transit through the unstirred water layer adjacent to the epithelial surface is considered the rate-limiting step in lipid absorption. Intrinsic gut brush border lipase enzymes are involved as well. The milieu of the unstirred water layer is acidic (pH 5 to 6) owing to the activity of the brush border membrane sodium-hydrogen (Na+/H+) exchanger. The acid environment facilitates dissociation of fatty acids from micelles resulting in a high concentration of fatty acids necessary for diffusion across the mucosal membrane.39,40
Once inside the enterocyte, long-chain fatty acids and monoglycerides are resynthesized into triglycerides and packaged as chylomicrons. Lipoproteins (e.g., apo-A, apo-B) and cholesterol are attached to the intestinal chylomicrons and confer important properties for the subsequent systemic uptake and metabolism of the chylomicrons. This process appears to be defective in cystic fibrosis and may account for some of the fat malabsorption seen in this disease.41 Chylomicrons are exported into the intercellular space and transported through the intestinal lacteals to become part of the intestinal lymph. On entering the bloodstream through the thoracic duct, the chylomicrons are associated with other apolipoproteins that allow them to be recognized by specific peripheral tissues.42
Regulation of Electrolyte and Water Movement
Movement of water is closely linked to the movement of solute in the form of electrolytes and nutrients. Water transport is a largely passive process that occurs through paracellular routes in the intestine coupled with solute movement. Expression of transporters involved in intestinal water and electrolyte transport is regionally specific. Electrolytes are taken up by enterocytes at the apical membrane and extruded through the basolateral membrane into the paracellular space. The relatively hypertonic paracellular fluid pulls water into this space, increasing the hydrostatic pressure locally. Because the tight junction between enterocytes is more impermeable to fluid flux than the capillary membranes, fluid and electrolytes are preferentially driven in the direction of the vascular space.1,43,44 Tight junctions are selective and dynamic in function and are regulated by a number of signaling pathways and cellular processes that can determine the size, selectivity, and flow of molecules across this barrier.3
The gut responds to both systemic and local stimuli to regulate motility, transport, and digestive functions. Secretion and motility are mediated through typical agonist membrane receptor mechanisms, by local autocrine and paracrine action, or through remote endocrine and neurocrine actions. Regulation of intestinal motility is crucial for keeping the chyme in contact with the epithelial surface long enough for efficient absorption of nutrients while permitting removal of unusable material and bacteria from the alimentary tract on a regular basis. GI smooth muscle demonstrates phasic and tonic patterns of contraction. Numerous factors affecting the frequency of contractions include changes in autonomic tone, stimulation of the gut by neurohormonal peptides or pharmacologic agents, and noxious stimuli associated with infectious or inflammatory processes. Hypoxia and ischemia decrease motility, frequently leading to paralytic ileus. Neural regulation of the GI tract integrates the processes of intestinal water and electrolyte transport, motility and blood flow. The augmentation of water and electrolyte absorption after a meal in the jejunum is neurally mediated.45 The enteric nervous system is capable of functioning independently but also may be modified by autonomic nervous system.3
Many other factors alter the functions of the gut. The terminal ileum and colon are particularly important in this respect. The presence of an ileostomy increases the risk of excessive sodium losses, dehydration, and electrolyte abnormalities. Terminal ileal resection or other diseases of the terminal ileum such as Crohn disease or radiation enteritis may result in bile acid malabsorption. In patients with bile acid malabsorption, bile acids reach the colon, which stimulates electrolyte and chloride secretion. Patients with mild to moderate malabsorption present with watery diarrhea and may respond to a bile acid binder such as cholestyramine.46 Impairment of water and ion absorption in inflammatory bowel disease may occur because of numerous mechanisms including alteration of epithelial integrity, augmented secretion, and reduced absorption. In addition, intestinal inflammation is associated with defects in epithelial barrier function. It is not known what effect inflammation may have on ionic function.47 Hyperosmolality of the ileal and colonic contents leads to an osmotic diarrhea. This state is seen when unabsorbed nutrients enter the distal alimentary tract and are broken down by enteric bacteria, resulting in increased luminal osmotic activity and osmotic diarrhea.
Electrolyte Transport
Several basic mechanisms exist for the transport of electrolytes by the epithelia. Na+ may be transported by numerous mechanisms. The regulation of Na+ absorption is closely regulated such that anion secretion is closely aligned with Na+ absorption.48 A Na+-H+ exchange-mechanism present throughout the intestine results in a 1:1 exchange of luminal sodium for protons. Coupled Na-Cl absorption, Na-Cl co-transport, Na-K-Cl co-transport, and movement of Na+ down its electrochemical gradient as in Na-Glucose co-transport are other mechanisms by which Na+ moves across the intestinal epithelium. The Na+-H+ exchanger plays a role in regulation of intracellular pH, regulation of cell volume, initiation of cell growth in response to various trophic factors, and metabolic response to insulin. To maintain electrical neutrality, the epithelium simultaneously exchanges Na+ for H+ and Cl− for HCO3. The presence of glucose in the lumen of the small intestine stimulates increased sodium absorption through coupled transport. Uptake of glucose is carrier mediated although the coupled transport of glucose with sodium is electrogenically driven by the Na+ gradient across the cell membrane.49 Absorption of glucose by Na-Glucose transport results in activation of myosin light chain kinase to regulate tight junction-permeability.50 Backflow of sodium into the lumen is a passive process since a major task for the GI tract is sodium conservation. Systemic acidosis increases Na+ and Cl− absorption in the ileum and colon, whereas alkalosis has the opposite effect. As seen in other epithelial tissues, aldosterone increases ileal and colonic absorption of Na+ and can increase absorption of water in colon three- to fourfold.51 Spironolactone blocks this effect. Glutamine has been shown to stimulate water and electrolyte absorption in the jejunum.52 Glucocorticoids increase sodium and water absorption in the distal colon. Opiate receptor stimulation increases active sodium and chloride absorption in the ileum, and opiate antagonists decrease basal absorption of water and electrolytes. The primary antidiarrheal effect of opiates, however, is mediated through a slowing of transit time.1
In the colon, active absorption and secretion of K+
[/not-level-membership-for-pediatrics-category]
