[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 31 Gastrointestinal Carcinoid Tumors (Gastrointestinal Neuroendocrine Tumors) and the Carcinoid Syndrome
In 1888, Lubarsch described what is now recognized to be the entity of carcinoid tumors when he reported the autopsy finding of two patients with multiple tumors in the distal ileum.1 The term carcinoid was introduced by Oberndorfer in 1907 in his description of a class of malignant tumors that behaved less aggressively than the more common adenocarcinomas of the gastrointestinal (GI) tract.2 The traditional classification of carcinoids, based on their embryonic origin into foregut, midgut, and hindgut carcinoids, has been gradually abandoned.3 A tumor biology–based classification system introduced by the World Health Organization (WHO) in 2000 has greater applicability, although more recent amplification and revision of this system by the European Neuroendocrine Tumor Society (TNM classification) appears likely to become the standard (Table 31-1).4,5
Table 31-1 TNM Classification for Endocrine Tumors*
| A. Gastric Tumors | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| Tis | In situ tumor, dysplasia (<0.5 mm) |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or subserosa or is >1 cm |
| T3 | Tumor penetrates serosa |
| T4 | Tumor invades adjacent structures |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastases |
| N1 | Regional lymph node metastases |
| MX | Distant metastases cannot be assessed |
| M0 | No distant metastases |
| M1 | Distant metastases |
| B. Tumors of the Duodenum, Ampulla, and Proximal Jejunum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades pancreas or retroperitoneum |
| T4 | Tumor invades peritoneum or other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
| C. Tumors of Distal Jejunum and Ileum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades mucosa or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades subserosa |
| T4 | Tumor invades peritoneum/other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
T, primary tumor (for any T, an “m” is added for multiple tumors; N, regional lymph nodes; M, metastases).
* European Neuroendocrine Tumor Society.
Adapted from Solcia E, Kloppel G, Sobin L. Histological typing of endocrine tumours. In: Verlag S, editor. World Health Organization Histological Classification of Tumours. 2nd ed. New York: Springer; 2000. p 38; and Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2006; 449:395-401.
Carcinoids arise from cells of the diffuse neuroendocrine system and can arise almost anywhere within the gastrointestinal tract.6 Carcinoids, also called gastrointestinal neuroendocrine tumors (GI NETs), are related to medullary carcinoma of the thyroid, pheochromocytoma and pancreatic neuroendocrine tumors. GI NETs synthesize bioactive amines and peptides, including neuron-specific enolase (NSE), 5-hydroxytryptamine (5-HT, or serotonin), and 5-hydroxytryptophan (5-HTP). They also secrete peptides such as chromogranin A, pancreatic polypeptide, calcitonin, tachykinins (neurokinin A and substance P), and various growth factors, such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), endocrine growth factor (EGF), fibroblast growth factor (FGF), and the vascular endothelial growth factor (VEGF) family of growth factors, including their receptors.7–9 Of clinical relevance is the observation that several different genes and genetic divergences related to tumor development are evident. GI NETs can be sporadic (nonfamilial) or part of a familial syndrome, such as von Hippel-Lindau syndrome or neurofibromatosis (NF). GI NETs comprise 0.5% of all malignancies and, as shown in Figure 31-1, their incidence has increased substantially over the last several decades.10,11 In this chapter, we will use the more familiar term carcinoid, although the term GI NET is the most appropriate, but not as well established.
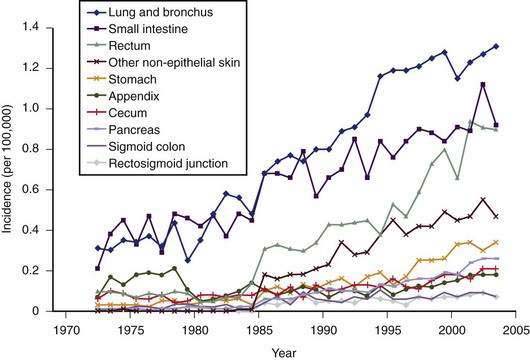
Figure 31-1. Incidence of different subtypes of neuroendocrine tumors, 1970 to 2005, from the Surveillance Epidemiology and End Results (SEER) data base.10,11 Note the significant increase in most subtypes of neuroendocrine tumors since the 1970s.
(From Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9:61-72.)
Carcinoid tumors occur most frequently in the GI tract (67%), with the bronchopulmonary system being the second most common location (25%), followed by considerably less frequent locations, such as the ovaries, testes, and hepatobiliary system.12 The most common location in the GI tract is the ileum (17%). The overall incidence of carcinoid tumors is difficult to determine, because it appears likely that most tumors remain asymptomatic. An autopsy study has estimated the annual incidence to be 8.4/100,000 people.13 The SEER database from 1973 to 2004 indicated an annual incidence of 2.0 to 2.5/100,000/year, with a 3.5% annual increase over this time period.10 Ileal tumors have specifically increased in prevalence—in white males (by 274%), black males (500%), white females (213%), and black females (286%), respectively. A recent evaluation of the SEER database comprising 35,825 cases in the United States has indicated an annual incidence of 5.25/100,000/year for all types of neuroendocrine tumors and a prevalence of 35/100,000.14 The annual incidence of gastrointestinal carcinoids was 2.53/100,000. Overall, the incidence of gastrointestinal carcinoids is higher in African Americans (4.5/100,000) compared with white Americans (2.5/100,000) with striking differences in certain sites, particularly the rectum. As a result of the increasing incidence and prevalence, gastrointestinal neuroendocrine tumors represent a substantial clinical problem. The increasing incidence is probably mostly to the result of the introduction of better diagnostic tools (imaging and immunohistochemical), as well as greater awareness by pathologists and clinicians. There are no known environmental risk factors for carcinoids.
CLINICAL FEATURES
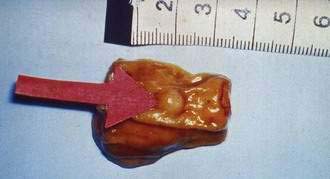
An early and accurate diagnosis is often delayed by four or five years because most small intestinal carcinoids (Fig. 31-2) are small, initially asymptomatic, or misdiagnosed as conditions such as allergy or irritable bowel syndrome. Progressive growth of the tumor may cause vague abdominal discomfort because of intermittent intestinal obstruction. As the tumor gets larger, invading the intestinal wall and occluding the gut lumen, emergency clinical presentations of an acute abdomen (intussusception, obstruction, perforation, bleeding) may arise because of local tumor mass effect or tumor-induced fibrosis.15–18 The frequency of intestinal obstruction that is secondary to a gastrointestinal NET-associated mesenteric fibrosis ranges from 42% to 66%.18,19 Another characteristic feature of these patients is vascular elastosis (thickening of the vessel wall), resulting in ischemic changes of the gut. The cause of this condition is unclear, but a local effect of serotonin or other tumor products, such as TGF-α, have been postulated to have a direct trophic effect on smooth muscle and fibroblasts in the vessel wall. Both mesenteric fibrosis and elastosis may cause abdominal angina.15 A rare cutaneous manifestation of ileal carcinoids is a fibrotic, scleroderma-like condition mostly affecting the lower extremities.20 As the disease advances, gastrointestinal carcinoids frequently metastasize locally to mesenteric lymph nodes and to the liver. A classic carcinoid syndrome is relatively uncommon (10% to 15%), typically consisting of diarrhea, cutaneous flushing, bronchoconstriction and right-sided heart failure.21,22 As many as 15% to 25% of gastrointestinal carcinoids exhibit a synchronous or metachronous association with other tumors, usually adenocarcinomas of the colon.23–25 This may reflect the activity of growth factors produced by the carcinoid tumor. A relatively large percentage of GI NETs are multicentric; for example, up to 33% of carcinoids in the small intestine are multicentric.24–26
PATHOLOGY
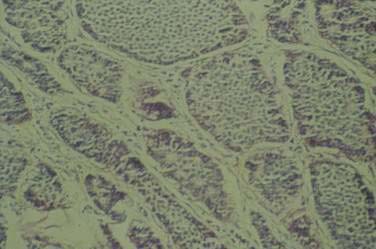
The most common histopathologic type of intestinal carcinoid is the enterochromaffin (EC) cell carcinoid, accounting for more than 98% of cases. The EC cell carcinoid is defined by its argentaffin staining properties, serotonin production, and typical pleomorphic secretory granules. Morphologically, EC cell carcinoids are characterized by medium-sized tumor cells arranged in an organoid pattern and showing only mild to moderate atypia. Tumor necrosis is absent and the mitotic rate is low (<2 mitoses/10 high-powered field [HPF]). Distinct growth patterns have been described in gastrointestinal carcinoids: (1) nodular or insular pattern; (2) trabecular pattern; (3) acinar and tubular pattern; and (4) atypical solid pattern. Mixed patterns may be seen. EC cell carcinoids of the jejunum and ileum predominantly display an insular growth pattern (93%; Fig. 31-3) and, less frequently, mixed insular and glandular (5%) or trabecular (2%) growth patterns. EC cell carcinoids invade the submucosa, muscularis propria, or mesentery. Invasion of lymphatics and veins, or perineural growth, can be seen. EC cell carcinoids may produce multiple hormones. Most of the tumors (>90%) secrete serotonin and tachykinins (substance P, neurokinin A). Production of other hormones such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, or adrenocorticotropic hormone (ACTH) can be demonstrated in less than 5% of cases. Neuroendocrine markers (proteins associated with large dense core vesicles or synaptic-like microvesicles) are abundantly expressed in EC cell carcinoids and can be used to confirm the neuroendocrine phenotype of the these tumors.24,25,27 Chromogranin A, synaptophysin, synaptic vesicle protein 2, neuron-specific enolase, and Leu7 are expressed by 92% to 100% of jejunoileal carcinoid tumors. Expression of the vascular monoamine transporters 1 and 2 (VMAT-1 and -2) has been demonstrated in more than 90% of tumors and is related to the production and storage of serotonin in EC cell carcinoids. Other markers identified in EC cell carcinoids include cytokeratins 8 and 18, carcinoembryonic antigen (CEA), and prostatic acid phosphatase. High expression of the intestinal transcription factor CDX2 has been demonstrated in EC cell carcinoids and may become a useful marker of intestinal origin.6,27–33
Carcinoids (GI NETs) arising in the duodenum and jejunum include gastrin, somatostatin, and EC cell tumors and gangliocytic paraganglioma. Gastrin cell tumors (gastrinomas) are the most common in the duodenum, might be nonfunctioning or functioning (Zollinger-Ellison syndrome [ZES]), and associated with the multiple endocrine neoplasia type I (MEN-I) syndrome (see Chapter 32). Tumors are mainly located in the first and second parts of the duodenum. A small proportion of tumor cells may produce other hormones in addition to gastrin, such as cholecystokinin (CCK), somatostatin, pancreatic polypeptide, neurotensin, and insulin. Morphologically, tumor cells are uniform, with rounded nuclei and abundant cytoplasm. The growth pattern is usually trabecular or cribriform. Necroses are absent and the mitotic rate is low. Most of the tumor cells are positive for neuroendocrine markers, such as chromogranin A, synaptophysin, Leu7, and NSE.34–36
Somatostatin cell tumors (somatostatinomas) represent the second most frequent histopathologic type in the duodenum, accounting for 15% to 27 % of duodenal GI NETs (see Chapter 32). Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin and typical large electron-dense secretory granules. A subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH. Morphologically, tumors are characterized as a mixture of tubular-glandular, insular, and trabecular growth patterns and the presence of psammoma bodies. Tumor cells are uniform and the mitotic rate is low.34,35,37,38
Gangliocytic paraganglioma represent the third most frequent histopathologic type in the duodenum and account for 6% to 9% of duodenal carcinoids. The tumors are preferentially located in the periampullary region and are not associated with familial syndromes. Gangliocytic paragangliomas are identified by their characteristic morphology, which includes a mixture of three different cell types—spindle cells, epithelial cells, and ganglion cells. Spindle cells are arranged in fascicles and stain positive for S-100. Epithelial cells represent the endocrine component and are positive for pancreatic polypeptide, somatostatin, and chromogranin A. Ganglion cells represent the neural component and are positive for synaptophysin and NSE. Gangliocytic paraganglioma are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium.39–42
Endocrine tumors of the stomach are composed of cells with features similar to those of the normal endocrine cell counterparts. The most representative cell types are the histamine-producing enterochromaffin-like (ECL) cell in the corpus and fundus and the gastrin-producing G cell in the antrum, representing about 50% of all endocrine cells at both sites. The other endocrine cell types present throughout the stomach are somatostatin-producing (D), ghrelin-producing (P), and serotonin-producing enterochromaffin (EC) cells. All cell types may be found in gastric tumors, but preneoplastic lesions are known to be comprised mostly of ECL cells. Antral G cell hyperplasia is often observed in chronic atrophic gastritis (see Chapter 51), but it is not considered a preneoplastic lesion. More than 90% of gastric endocrine tumors are well-differentiated tumors–carcinomas (WDET/C), according to the WHO classification. More than 90% of the tumors occur in the corpus-fundus area and are composed of ECL cells, with only rare EC, G, or P cell tumors. Poorly differentiated neuroendocrine carcinomas of the GI tract are highly malignant tumors.43–50 In general, the cytosol markers NSE and PGP 9.5, and the small synaptic-like vesicle marker synaptophysin, are expressed in the poorly differentiated tumors in contrast to well-differentiated neuroendocrine tumors. Chromogranin A and tissue-specific hormones are generally absent or sparse in poorly differentiated neuroendocrine carcinomas. Hormones and chromogranin A are stored in large dense core vesicles, which are rarely observed in poorly differentiated gastric and intestinal tumors.
MOLECULAR GENETICS
Studies have shown that development of various types of GI NETs might involve different genes associated with distinct abnormalities, including point mutations, deletions, methylations, and chromosomal losses and gains (see Chapter 3). The menin gene, a tumor suppressor gene, encodes a protein of 610 amino acids; mutations of this protein cause most cases of MEN-I and a smaller proportion of sporadic and gastric duodenal endocrine tumors.51 Menin is mainly a nuclear protein, but in dividing cells, it interacts in the cytoplasm with several proteins that control transcription, regulation of genome stability, and cell division. Small intestinal carcinoid tumors show deletions on chromosome 18.52,53 Recent studies have also demonstrated overexpression of the neoplasia-related genes NAP1L1 (mitotic regulation), MAGE D2, and MTA1. These genes are thought to regulate the malignant potential of these tumors and their propensity to metastasize.54 Hindgut neuroendocrine tumors express receptors for TGF-α and EGF.51
CLASSIFICATION AND SUBTYPES
Carcinoid tumors were previously classified according to their embryologic region of origin into foregut, midgut, or hindgut tumors. This classification has now largely been abandoned and a WHO classification system is generally accepted. It has been recently updated by the European Neuroendocrine Tumor Society to a TNM and grading system (see Table 31-1).4,5
ESOPHAGUS
Carcinoid tumors of the esophagus are rare.55 However, in a series from Japan, esophageal carcinoids constituted 27% of all GI carcinoids, more frequent than ileal and appendiceal carcinoids.42 The male-to-female ratio was 3.3, indicating a male preponderance for this type of carcinoid tumor. Dysphagia is the most common presenting symptom and is usually localized to the distal portion of the esophagus. Esophageal carcinoids may occur in conjunction with adenocarcinomas arising from Barrett’s esophagus.56
STOMACH
A trend for an increased incidence of gastric neuroendocrine tumors ha been noted in surgical and endoscopic series. In the Surveillance, Epidemiology, and End Results (SEER) database, there was an increase in gastric neuroendocrine tumors, from 2.4% to 8.7% of all GI carcinoids, between 1950 and 1999.12 In mainly endoscopic series, much higher relative incidences were reported, ranging from 11% to 41% of all gastrointestinal neuroendocrine tumors.57 However, the increased relative incidence of gastric neuroendocrine tumors must be put into the context of a wider incremental trend for all types of gastrointestinal well-differentiated neuroendocrine tumors (carcinoids). Gastric neuroendocrine tumors are currently classified as well-differentiated and poorly differentiated lesions on the basis of the differentiation status of the tumor cells. The most common types are tumors derived from histamine-producing ECL cells in the corpus-fundus region, followed by gastrin producing G cell tumors of the antrum.57–59 Rare types of gastric carcinoid are those producing gastrin, ghrelin, and serotonin.43,44
Three clinicopathologic subtypes of ECL cell tumors are recognized (Table 31-2). The type I ECL tumor is associated with diffuse corpus-restricted chronic atrophic gastritis (see Chapter 51). Type II is associated with MEN-I, ZES, and hypertrophic gastropathy. Type III tumors are sporadic are not associated with any distinctive gastric pathology.43,58,59 Types I and II tumors share in common hypergastrinemia, whereas type III tumors are independent of any overt hormonal imbalance. Type I ECL tumors account for the largest fraction of well-differentiated neuroendocrine tumors of the stomach, are especially prevalent in older women, and are associated with antral G cell hyperplasia. Frequently, multiple and multicentric lesions are present and are generally small and limited to the mucosa or submucosa. Metastases are rare and survival is excellent.45 Type II ECL tumors are rare and account for only 6% of gastric carcinoids. Type II tumors arise in adult patients of both genders who have hypergastrinemia, hypertrophic hypersecretory gastropathy, and ECL cell hyperplasia. The ECL tumors are often multiple, multicentric, small in size, and limited to mucosa and submucosa. Despite metastases to local lymph nodes, the survival is excellent and tumor-related death is rare.57 Type III ECL cell tumors are usually single, isolated growths arising in the stomach, without any significant underlying gastric pathology. In one series, they accounted for 14% of all gastric carcinoids. They are more common in men, usually in their sixth decade, and without hypergastrinemia and gastrin-dependent ECL cell hyperplasia. The tumor size may be significantly larger than in types I and II (mean, 3.2 cm), with invasion of the stomach wall and metastases in more than 50% of patients.57 Predictors of malignancy in well-differentiated gastric carcinoids include size, histologic grading, mitotic count and Ki67 index, and p53 overexpression. Poorly differentiated endocrine tumors of the stomach are aggressive, usually large carcinomas and develop with no site predilection in the stomach. They usually occur in patients in the sixth to seventh decade of life. These poorly differentiated tumors are often metastatic at time of diagnosis; their prognosis is invariably poor.

PANCREAS
The differentiation between pancreatic carcinoid and other pancreatic neuroendocrine tumors is primarily a matter of definition (see Chapter 32 for a more detailed discussion). Maurer and colleagues have defined a pancreatic carcinoid as a tumor with typical histologic features of an NET along with evidence of increased serotonin metabolism.60 Using this definition, they found only 29 cases in the literature between 1966 and 1996. In a more recent publication from Japan, Soga found 156 cases of pancreatic carcinoids among 11,343 cases of neuroendocrine tumors (1.4%).61 These tumors were characterized by a high metastatic rate (66%), a large tumor size (averaging almost 7 cm), and a relatively high incidence of the carcinoid syndrome (23%). Serotonin was detected by immunohistochemical methods in 93% of cases. The five-year survival rate was extremely low (29%) when compared with small intestinal carcinoid (82%). Pancreatic carcinoids, like other pancreatic tumors, tend to present later when compared with carcinoids in other locations. Abdominal pain, diarrhea, and weight loss are the most common presenting symptoms.
DUODENUM AND AMPULLA OF VATER
About 4% of all gastrointestinal carcinoids occur in the duodenum and duodenal carcinoids account for 11% of all intestinal carcinoids. The relatively frequency of duodenal carcinoids increased from 3.6% to 16% of all intestinal carcinoids from 1973 to 2002. The annual incidence of duodenal carcinoid is 0.07/100,000. It is higher in males than in females and is higher in blacks than in whites.10–12 Duodenal carcinoids present at a mean age of 48 to 62 years. Most duodenal carcinoids give rise to symptoms related to local growth, such as obstruction, jaundice, abdominal pain with or without pancreatitis, gastrointestinal bleeding, nausea, and vomiting.35,62 A minority of patients with duodenal carcinoids (<10%) present with symptoms and/or signs of hormone overproduction. ZES occurs in approximately 10% of patients with duodenal carcinoids (see Chapter 32). The carcinoid syndrome may occur (4%) and, in rare cases, Cushing’s syndrome and acromegaly may be seen.
Gastrin (G) cell tumors represent the most frequent histopathologic type, accounting for 50% to 60% of all duodenal carcinoids (Table 31-3). G cell tumors can be nonfunctioning or functioning (Zollinger-Ellison syndrome) and may be associated with the MEN-I syndrome.36,62 Somatostatin cell tumors represent the second most frequent histopathologic type (15% to 27%).38 Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin, but a subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH.
| CELL OF ORIGIN/TUMOR TYPE | RELATIVE FREQUENCY (%) |
|---|---|
| Gastrin (G) cell* | 50-60 |
| Somatostatin (D) cell | 15-77 |
| Gangliocytic paraganglioma | 6-9 |
| EC, others | <5 |
EC, enterochromaffin.
* Functional (Zollinger-Ellison syndrome) or nonfunctional.
Gangliocytic paragangliomas represent the third most frequent histopathologic type and account for 6% to 9% of duodenal carcinoids.39,40,62 The tumors are preferentially located in the periampullary region and are not associated with familial syndrome. The tumors are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium. EC cell tumors of the duodenum are rare, and have the same histopathologic features as ECL cell tumors in the lower jejunum and ileum.
The overall five-year survival rate for duodenal carcinoids is reported to be 84%.10–12 However, survival rates vary with histopathologic type, extent of disease, presence of hormonal syndrome, and genetic background. In patients with Zollinger-Ellison syndrome, duodenal localization of the primary tumor carries a much better prognosis than pancreatic localization (see Chapter 32), with 10-year survival rates of 94% and 55%, respectively.10–12,62
SMALL INTESTINE
Forty-four percent of all gastrointestinal carcinoids arise in the small intestine. The ileum is the most frequent location for gastrointestinal carcinoids.10–12,14 The annual incidence of intestinal carcinoids is 0.63/100,000 and 0.04 and 0.31/100,000 for jejunal and ileal carcinoids, respectively. The incidence of intestinal carcinoids has increased four- to fivefold during the past 30 years. Small intestinal carcinoids result from malignant transformation of EC cells. A considerable percentage (17% to 29%) of the patients develop noncarcinoid GI and non-GI tumors, mainly adenocarcinomas of the gastrointestinal tract, suggesting a common pathogenic mechanism for carcinoid and noncarcinoid tumors.23 Familial occurrence of intestinal carcinoid is rare, although a few families have been reported. Jejunal and ileal carcinoids are also rarely associated with MEN-I.27
Patients with carcinoid tumors of the jejunum and ileum have a mean age of 55 to 63 years at the time of diagnosis.15,21 Usually, there is a delay of four to five years for diagnosis of this tumor.17 The most frequent presentations are abdominal pain (16% to 41%) and episodic small bowel obstruction (24% to 41%), which are usually of long duration. Diarrhea, gastrointestinal bleeding, flushing, and weight loss are presenting symptoms in 4% to 30%. The carcinoid syndrome is encountered in 5% to 18% of patients.16,63
Carcinoid tumors occur all along the jejunum and ileum, but increase in frequency distally, with their highest frequency in the terminal ileum.12,13 There is a single primary tumor in 74% of patients, whereas multiple tumors occur in 26% of patients.26 Clonality studies have indicated that multiple tumors are generated by the metastasis of a single primary tumor to different locations in the intestine.64 The size of the primary tumor ranges from 0.3 to 5.5 cm (mean, 2.5 cm). Intestinal carcinoids are usually associated with pronounced fibrosis and tissue scarring, which may cause intestinal obstruction and infarction. The desmoplastic stromal reaction surrounding intestinal carcinoids has been attributed to secretion of hormones and growth factors by tumor cells. Small intestinal carcinoids may produce multiple hormones. Most secrete serotonin and tachykinins (substance P and neurokinin A). Production of other hormones, such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, and ACTH, can be demonstrated in less than 5% of cases.7,63
Data from the SEER registry for period 1973 to 2002, summarized in Table 31-4, reveal an overall five-year survival rate for intestinal carcinoids of 63% and a five-year survival rate of 84%, 72%, and 43% for localized, regional, and distant (metastatic) disease, respectively.10,11,16 However, five-year survival rates of 69% have been reported for patients with intestinal carcinoids metastatic to the liver who were given active interventional treatment.65 Risk factors for mortality include distant metastases, carcinoid syndrome, and female gender.66 Histopathologic markers shown to be associated with poor prognosis are a solid growth pattern and a Ki67 index above 1%.67
APPENDIX
The appendix is one of the most common sites in the GI tract for the development of a carcinoid tumor. The frequency of carcinoid tumors in nonselected series of appendectomy specimens ranges from 0.2% to 0.9%, but more recent data have suggested that the overall trend is toward a decreased prevalence.68,69 It is reported that appendiceal carcinoids occurs twice as often in women as in men. This observation may partly reflect the frequency of incidental appendectomy during gynecologic and gallbladder surgery but the female-to-male ratio is 2 : 1, even after correction for these factors.
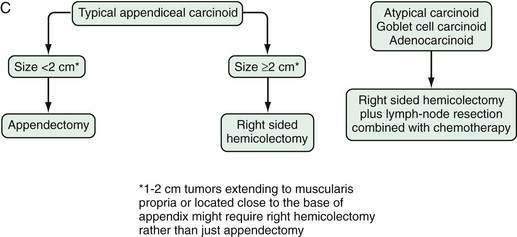
Carcinoid tumors of the appendix have been classified based on their histogenesis, cell types, and histologic pattern, as well as their clinical behavior. All typical appendiceal carcinoid tumors are clinically silent and are discovered incidentally during surgery performed for symptoms of acute appendicitis or during incidental appendectomy. Most typical appendiceal carcinoids occur in the tip of the organ. They are usually small (<1 cm) and rarely exceed 2 cm in diameter. Metastases are rare in typical appendiceal carcinoid.68,69 The overall prognosis for EC cell appendiceal tumors is also favorable, with a 10-year disease specific survival of more than 98%. The incidence of metastases is less than 1% if tumor size is between 1 and 2 cm. Thus, appendectomy alone is likely to be adequate treatment for the most typical appendiceal carcinoid tumors that are smaller than 2 cm.70,71 Conversely, right hemicolectomy should be considered in selected patients with a carcinoid more than 2 cm in diameter, with a tumor that extends into the muscularis propria, and those with a positive resection margin, associated perforation, or evident lymph node metastases. Typical appendiceal carcinoids have the best prognosis of all types of carcinoids, which probably reflects the anatomic site and biologic behavior of the tumor itself; early detection and removal are beneficial.
In contrast to typical carcinoid tumors of the appendix, goblet cell carcinoids of the appendix (Fig. 31-4) have a mixed phenotype, with partial neuroendocrine differentiation and intestinal-type goblet cell morphology.72 The mean age at diagnosis is 50 years, with a range of 29 to 80 years. Goblet cell carcinoids constitute a family of appendiceal carcinoids with various morphologic features and malignant behavior. Some of them are histologically aggressive neoplasms, such as adenocarcinomas of the GI tract. The tumors demonstrate neuroendocrine differentiation, with positive staining for chromogranin and synaptophysin, although the staining is often focal and always much less extensive than in classical carcinoid tumors. The most common and distinctive feature is mucin-containing goblet-shaped epithelial cells arranged in round or oval clusters.73,74 The overall five-year survival rate for different subtypes of goblet cell carcinoids is 42%, with a three-year survival rate of only 17% for the most malignant subtypes.
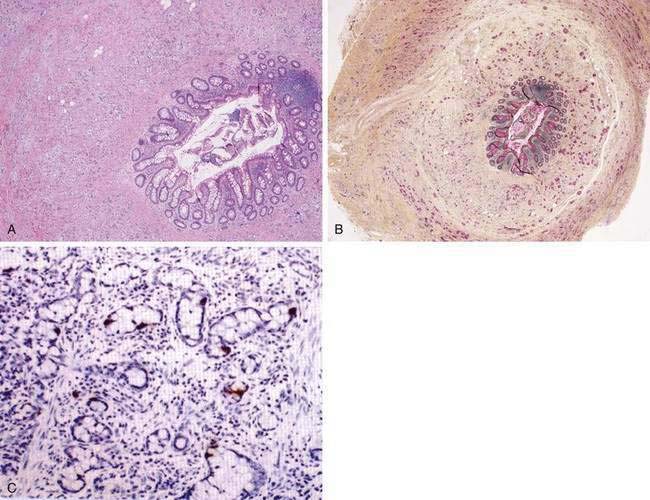
COLON
Colonic carcinoids account for about 5% of all carcinoid tumors.10,11 The average age at diagnosis is 65 years, similar to other colonic cancers. There is a female preponderance. These tumors occur more commonly on the right side of the colon. Colonic carcinoids also tends to present as a larger lesion than most other carcinoids, with an average diameter of approximately 5 cm. Symptoms of colonic carcinoids are usually seen in bulky, advanced lesions. They may cause malaise, anorexia, and weight loss before localizing symptoms are evident. Advanced lesions may cause abdominal pain or colonic obstruction. Approximately one in three patients present with distant metastatic disease.75–77 The overall five-year survival rate is 42%.
RECTUM
The rectum is the second most frequent GI site for NETs (27.4%). Rectal NETs, however, only comprise 1% to 2% of all rectal tumors and exhibit the most benign clinical profile of neuroendocrine tumors, possibly reflecting their early diagnosis by endoscopic examination. The annual increase in incidence of rectal NETs in the United States over the last 30 years is 8.6%. It has increased from 0.12/100 000 in 1973 to 0.93/100 000 in 2004.10,11 The reason for this rapid increase is unknown, but an increased awareness and wider use of endoscopy are believed to play a major role. The average age at diagnosis of rectal carcinoids is 48 to 52 years,75 with an equal gender distribution.
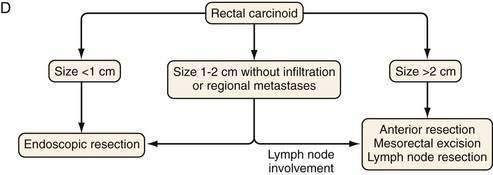
Approximately 50% of patients with rectal NETs are asymptomatic at presentation. Clinical symptoms, when present, include weight loss, constipation, and changes in bowel habits, including diarrhea. Rectal pain may occur as component of late presentation.75,78,79 Rectal NETs often stain for glucagon, pancreatic polypeptide, serotonin, peptide YY, and somatostatin.24 Overall, rectal carcinoids fall into two groups, small solitary tumors, measuring less than 1 cm and larger lesions with the possibility of metastases. Rectal carcinoids present with metastases in only 4% to 18% of cases. Rectal tumors more than 1 cm in diameter metastasize more often than smaller tumors, and those larger than 2 cm have a high rate of metastases (60% to 80%). The five-year survival rate for rectal carcinoids is 87% for localized disease, 41% for regional disease, and 25% for distant (metastatic) disease.12,16
THE CARCINOID SYNDROME
The classic carcinoid syndrome is relatively uncommon in patients with carcinoid tumors, occurring in 10% to 15% of these patients. Symptoms include diarrhea, cutaneous flushing, bronchoconstriction, and right-sided heart failure. Flushing attacks occur in 23% to 65% of patients with carcinoid syndrome at their initial presentation and in 63% to 78% at some time during the disease course. The typical flush is the sudden appearance of a deep red erythema of the upper part of the body, primarily the face and neck. Flushes are often associated with an unpleasant feeling of warmth, occasionally with lacrimation, itching, palpitation, facial or conjunctive edema, and diarrhea (Fig. 31-5). Flushes may be spontaneous or precipitated by stress, alcohol, intake of foods such as cheese and spicy meals, exercise, or by injection of agents such as catecholamines, calcium, or pentagastrin. Flushes may be brief, lasting 2 to 5 minutes, especially initially, or they may be prolonged and last for hours, especially later in the course of the disease.80,81 Flushes are usually seen with carcinoid tumors of the ileum or jejunum, but can also occur in some patients with gastric or duodenal tumors. The flush associated with gastric carcinoids is also reddish, but is patchy in distribution over the neck and face and is sometimes referred to as geographic flushing. It is often provoked by food intake. The flushing is frequently associated with pruritus, often related to release of histamine from the ECL cell gastric carcinoid. The classic flushing seen in ileojejunal carcinoids is related to the release of amines such as serotonin and peptides such as bradykinin and tachykinins (neurokinin A, substance P).80–83
Diarrhea is present in 32% to 73% of patients initially and in 67% to 84% at some time during the disease course. Diarrhea usually occurs with flushing, but it may also occur alone. The diarrhea is described as watery and the number of stools ranges from 2 to 20 daily; 60% of patients have a fecal output of less than 1 L/day. Steatorrhea is present in approximately 60% of cases. Abdominal pain may be present with the diarrhea or be present independently. Diarrhea is usually related to the secretion of serotonin in classic midgut carcinoids; it can also be related to the secretion of calcitonin and prostaglandins.84–86
Carcinoid heart disease occurs in about 50% of patients with the classic carcinoid syndrome.87 In a retrospective study of 200 patients, carcinoid heart disease was diagnosed approximately 1.5 years after the diagnosis of a carcinoid tumor or carcinoid syndrome.88 Carcinoid heart disease is a major cause of morbidity and mortality.80,89 Today, carcinoid heart disease is rare, occurring in about 3% to 4% of patients with carcinoid syndrome. The striking decrease of this manifestation has been suggested to be related to the use of somatostatin analogs and interferon-α, which block the release of serotonin and tachykinins from the tumor.
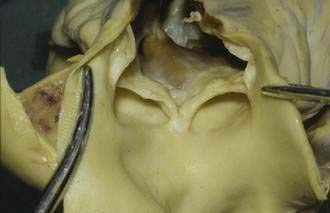
Typically, the carcinoid syndrome occurs when hepatic metastases from a primary gastrointestinal EC cell tumor is evident and the breakdown of hormonally active tumor products by the liver is impaired, allowing these products to reach the systemic circulation. In patients with the carcinoid syndrome, those with the highest levels of tachykinins and serotonin in plasma and of 5-hydroxyindoleacetic acid (5-HIAA) in the urine are more likely to develop cardiac valvulopathy, indicating that one or more of these hormonally active tumor byproducts are involved in the pathogenesis of carcinoid heart disease.90–92 Serotonin has been found to modulate cell proliferation in valvular subendocardial cells and human heart valves have been shown to express mRNA for the 5-HT 1B, 1D, 2A, and 2B receptors.93,94 Fenfluramine, a serotonergic drug once used as an appetite suppressant, was withdrawn from the market in 1997 because it induced a valvular heart disease similar to that seen in the carcinoid syndrome.95 TGF-β, known to affect cell growth and differentiation and to stimulate fibroblasts to produce extracellular matrix protein, is expressed in coronary heart disease lesions and is up-regulated by serotonin.96,97 Another growth factor that may contribute to drive the fibrotic process is connective tissue growth factor (CTGF). CTGF is secreted by the EC cell and acts in concert with TGF-β to drive the overproduction of collagen.98 The cardiac disease in carcinoid syndrome is caused by fibrosis involving the endocardium, primarily on the right side of the heart, although left-sided lesions can also occur. Fibrous plaques are seen on the endocardial surface of the valvular cusps and in the cardiac chambers, as well as on the intima of the great veins and arteries, areas exposed to the highest concentration of tumor products. Proliferation of myofibroblasts with collagen deposition is present in all plaques.87,99 These fibrous deposits tend to cause constriction of the tricuspid and pulmonic valves, resulting in regurgitation (Fig. 31-6).
Transthoracic echocardiography (TTE) is a cornerstone for establishing the diagnosis of carcinoid heart disease, and evidence of cardiac involvement can be identified by TTE in 45% to 77% of patients with carcinoid syndrome.87,90 Left-sided valvular pathology occurs in 10% to 15%, whereas myocardial metastases occur in less than 5%.100 In patients with significant right heart failure, valve replacement is the treatment of choice. The use of bioprosthetic valves has now become more common and widely accepted. The advantages of bioprosthetic valves compared with mechanical ones include a better hemodynamic profile and no need for anticoagulation treatment, which represents a considerable risk for bleeding during hepatic artery embolization or surgical procedures.101 However, when using a bioprosthetic valve, there is a risk for a new fibrotic process, perhaps involving another valve replacement within 10 years.
Other clinical manifestations in patients with carcinoid syndrome are wheezing or asthma-like symptoms, occurring in 3% to 18% of patients, and pellagra-like skin lesions with hyperkeratosis and pigmentation in 2% to 5% of cases.80 Rarely reported are rheumatoid arthritis, changes in mental state, and visual changes during flushing caused by vasospasm. A variety of noncardiac problems caused by increased fibrous tissue have been reported, including retroperitoneal fibrosis leading to a ureteral obstruction, Peyronie’s disease of the penis, intra-abdominal fibrosis, and occlusion of the mesenteric artery or vein. Sexual dysfunction is a common complaint by men with carcinoid syndrome.80,98
PATHOPHYSIOLOGY
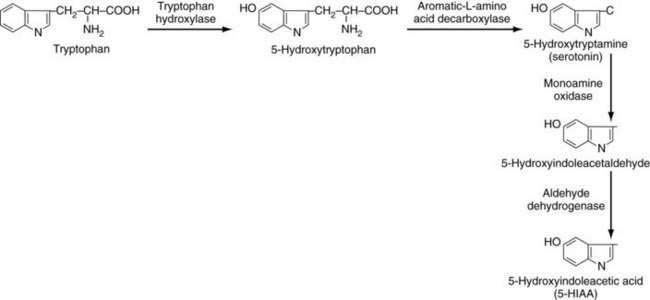
Symptoms of carcinoid syndrome were originally attributed to the secretion of 5-HT (serotonin) by the tumor. In a large review of 748 cases of carcinoid syndrome, 92% had increased circulating serotonin levels.102 Patients may develop a typical or atypical type of carcinoid syndrome. In patients with typical carcinoid syndrome, the conversion of tryptophan to 5-HTP by tryptophan hydroxylase is the rate-limiting step (Fig. 31-7).80,81 Once formed, 5-HTP is rapidly converted to 5-HT in the tumor by aromatic l-amino acid decarboxylase (l-Dopa decarboxylase); it is stored in the neurosecretory tumor granules or released into the vascular compartments, where most is taken up and stored in platelet granules. A small amount of 5-HT remains in the plasma. Most 5-HT in the circulation is converted by monoamine oxidase and aldehyde dehydrogenase to 5-HIAA, which is excreted in large amounts in the urine (see Fig. 31-7).103 The biochemical steps illustrated in Figure 31-7 are the typical pathways for EC cells tumors, which characteristically secrete large amounts of serotonin. Some carcinoid tumors cause an atypical carcinoid syndrome and are thought to be deficient in l– Dopa decarboxylase; thus, they cannot convert 5-HTP to 5-HT and, as a consequence, 5-HTP is secreted into the bloodstream.104 Plasma serotonin levels are normal in these patients, but urinary 5-HT levels are usually elevated because some of the 5-HTP can be decarboxylated in the kidney and excreted as serotonin.
The exact role of serotonin in causing the flushing in carcinoid syndrome remains unclear. Antagonists against serotonin receptor subtypes typically have no effect on the flushing. The precise mediator of the flushing in patients with carcinoid syndrome may differ, depending on the tumor type. In patients with gastric carcinoid tumors, the red, patchy, pruritic flush is thought to be caused by histamine, because this type of flushing can be prevented by the use of histamine H1 and H2 receptor antagonists.105 In addition to serotonin, other candidates for mediators of flushing include the tachykinins (substance P, neuropeptide K, neurokinin A) and bradykinin.106,107 Agents that block the release of tachykinins attacks, such as somatostatin analogs, may prevent flushing attacks.
Patients with carcinoid syndrome often have increased colonic motility, with a shortened colonic transit time and possibly a secretory or absorptive alteration. Serotonin may be predominantly responsible for the diarrhea in some patients through its effect on gut motility and intestinal electrolyte and fluid secretion.85,86 Serotonin receptor antagonists (especially 5-HT3 receptor antagonist) such as ondansetron relieve the diarrhea.
CARCINOID CRISIS
Carcinoid crisis may occur in a number of situations, but most commonly in the setting of surgical or anesthetic stress. The effect of the anesthetic agent or manipulation of the tumor intraoperatively may elicit exacerbation of the carcinoid symptoms, including profound flushing, hyper- or hypotension, tachyarrythmias, hyperglycemia, and refractory bronchospasm.108,109 Patients with known carcinoid syndrome should receive somatostatin analogs before surgery. The use of octreotide before invasive procedures is important to prevent carcinoid crisis. A bolus dose of 250 to 500 µg of octreotide should be given subcutaneously within one to two hours before the surgery. This should be followed by an IV infusion of 50 to 200 µg/hour during the procedure. The postoperative dose should be 50 to 200 µg/hour for 24 hours, followed by resumption of the preoperative treatment schedule. In patients with hypotension, it is important to realize that most pressor substances are ineffective or might aggravate hypotension by increasing serotonin and peptide release from the tumor. A combination of fluid replacement and intravenous octreotide is therefore recommended in this situation. Sometimes, glucocorticoid administration might even further improve the hypotension.9
DIAGNOSIS
BIOCHEMICAL MARKERS
Specific Markers
Carcinoid tumors can synthesize and secrete serotonin, tachykinins, prostaglandins, catecholamines, and histamine. The breakdown product of serotonin, 5-HIAA, is excreted by the kidney and can be measured after a 24-hour urine collection. Restriction of the intake of serotonin-rich food is important for standardization of reference levels (Table 31-5). Serotonin concentrations can also be determined in platelets and platelet-rich plasma. Elevated 24-hour urinary 5-HIAA levels have 23% sensitivity and 100% specificity in predicting the presence of a midgut carcinoid tumor. Measurement of urinary 5-HIAA excretion for diagnosis of carcinoid tumors is the predominant biochemical analytic procedure. In some studies, platelet serotonin levels were more sensitive than urinary 5-HIAA levels. Urinary serotonin levels are not affected by the patient’s diet, as are urinary 5-HIAA levels. Elevation of urinary 5-HIAA levels can occur in intestinal malabsorption and in a number of other conditions (see Table 31-5).110–112 Tachykinins (substance P, neurokinin A and neuropeptide K) can also be measured in the plasma.106 Histamine can be measured in gastric carcinoids and is measured as histamine metabolites in the urine (N-methylhistamine, 1-methylhistamine [MH], and 1-methylimidazole acetic acid [MIAA]).
Table 31-5 Factors That Interfere with Determination of Urinary 5-HIAA
| FOOD | DRUG |
|---|---|
| Factors That May Produce False-Positive Results | |
| Avocado | Acetaminophen |
| Banana | Acetanilid |
| Chocolate | Caffeine |
| Coffee | Fluorouracil |
| Eggplant | Guaifenesin |
| Pecan | l-Dopa |
| Pineapple | Melphalan |
| Plum | Mephenesin |
| Tea | Methamphetamine |
| Walnuts | Methocarbamol |
| Methysergide maleate | |
| Phenmetrazine | |
| Reserpine | |
| Salicylates | |
| Factors That May Produce False-Negative Results | |
| None | Corticotropin (ACTH) |
| p-Chlorophenylalanine | |
| Chlorpromazine | |
| Heparin | |
| Imipramine | |
| Isoniazid | |
| Methenamine mandelate | |
| Methyldopa | |
| Monoamine oxidase inhibitors | |
| Phenothiazines | |
| Promethazine | |
5-HIAA, 5-hydroxyindoleacetic acid; l-Dopa, l-dihydroxyphenylalanine.
Nonspecific Markers
Neuroendocrine cells have vesicles containing peptide hormones, biogenic amines, and neurotransmitters. These vesicles store and release acidic, soluble secretory proteins also known as granins. The granin family consists of the classic granins—chromogranin A (CgA), chromogranin B, and secretogranin II (sometimes called chromogranin C)—as well as secretogranin III (1B1075), secretogranin IV (HISL-19), secretogranin V (7B2), and secretogranin VI (NESP55).113,114 Chromogranin A is an acidic glycoprotein of 439 amino acids, with a molecular mass of 48 kd. CgA is released together with hormones on stimulation from normal endocrine cells and is also released from neuroendocrine tumors. Various assays for measurements of intact CgA and its different cleavage products are currently available. Several CgA-related peptides, or cleavage breakdown products, have been identified in human tissues. Tumors can also release different molecular forms of CgA. CgA levels are increased in most patients with metastatic gastrointestinal carcinoid tumors. A significant correlation between serum CgA levels and tumor mass has been demonstrated. False elevated serum CgA levels have also been documented in patients with impaired renal and liver function because of decreased metabolism and clearance. Chronic atrophic gastritis and inflammatory bowel disease are other conditions with elevated serum CgA levels.113–115
NSE is the neuron-specific isomer of the glycolytic enzyme 2-phospho-d-glycerate hydroxylase or enolase. This isomer is present in neurons and neuroendocrine cells and therefore can serve as a biochemical marker for tumors derived from these cells. Serum NSE levels are frequently elevated in patients with several types of neuroendocrine tumors.116 Like CgA, NSE is a general neuroendocrine marker that cannot differentiate between different subtypes of neuroendocrine tumors. Elevated NSE levels are, however, associated with poor tumor differentiation (high-grade tumors).
Human chorionic gonadotrophin (HCG) is a glycoprotein hormone synthesized during pregnancy by the trophoblastic cells of the placenta. HCG consists of an alpha and beta subunit. The beta subunit is specific for HCG, whereas the alpha subunit is also common to the other hormones of the glycoprotein family (luteinizing hormone [LH], follicle-stimulating hormone [FSH], and thyroid-stimulating hormone [TSH]). Ectopic secretion of alpha subunits is frequently encountered in neuroendocrine tumors, particularly in patients with poorly differentiated carcinoid tumors.117
TUMOR LOCALIZATION
Endoscopy plays a key role in the management of gastric carcinoid tumors, with its ability to identify, biopsy, and even resect the lesions. The advent of endoscopic ultrasound (EUS) has expanded the role of endoscopy in the management of gastric carcinoids through its ability to provide high-resolution ultrasound images of lesions within and adjacent to the GI tract.118 Endoscopy and EUS can be used to identify rectal and colonic carcinoid tumors. Video capsule endoscopy119 should be used with caution in patients with evidence of small bowel obstruction or a history of small bowel resection.
Computed tomography (CT) and magnetic resonance imaging (MRI) have a crucial role to play in the management of patients with neuroendocrine tumors. The introduction of multidetector CT (MDCT) has dramatically altered the performance of CT and its display.120 The most common types of carcinoid tumors usually present as small primary tumors; CT imaging typically demonstrates a secondary feature. Liver metastases are the most frequent findings, followed by tumor-associated desmoplastic fibrosis around the primary tumor and lymph node metastases. MRI has not been widely used for the detection of primary midgut NETs. The best sequence for demonstrating the primary tumor has been the post–gadolinium T1-weighted fat-suppressed image. Hindgut and gastric carcinoids are usually diagnosed at endoscopy, although barium studies may demonstrate a filling defect. CT and MRI can be used to stage lymph node disease and distant (metastatic) spread as part of the preoperative plan. The clinical workup of patients with gastric carcinoid tumors always includes a CT or MRI examination, both for initial workup and for follow-up during different treatment modalities.
Gastrointestinal carcinoid tumors usually possess high-affinity receptors for somatostatin (80% to 100% of cases). Somatostatin receptors are present in the primary tumor and metastases. Five subtypes of somatostatin receptor, SST1 to SST5, have been described. Octreotide, a synthetic somatostatin analog, binds with high affinity to SST2 and SST5 receptors and with lower affinity to the SST3 receptor. Studies have shown that almost all carcinoid tumors (80% to 90%) possess SST2 receptors, and 50% to 60% have SST5 receptors.121 Indium-111 diethylenetriaminepentaacetic acid defenyl alanyl octreotide (111In-DTPA octreotide) is a standard agent for localizing carcinoid tumors using radionuclide scanning. Somatostatin SRS scanning can image the tumor in 73% to 89% of patients with carcinoids (Fig. 31-8).121–123 Numerous studies have demonstrated that SRS has high sensitivity for the localization of gastrointestinal carcinoid tumors, especially the extent of metastatic spread (see Chapter 32). In general, SRS has excellent specificity, but somatostatin receptors may be expressed in other conditions, which can lead to false-positive results. Examples included granulomas in sarcoidosis and tuberculosis, lymphoma cells, and thyroid diseases. One way to assess the importance of SRS in the management of GI carcinoid tumors is to determine its ability to alter the clinical management of these patients. The results of eight different studies have documented the ability of SRS to alter clinical management in 21% to 53% of patients with NETs.124–126
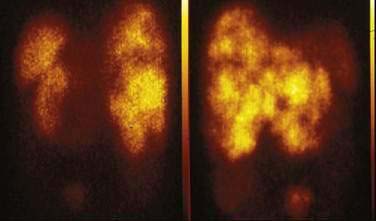
PET using the 18F-labeled glucose analog deoxyglucose (FDG) can be combined with CT (PET-CT) and MRI (PET-MRI) to image tumors.127 FDG-PET has not been shown to be useful for most neuroendocrine gastrointestinal tumors, except for the subset of tumors with a high proliferation rate and poor cell differentiation.128 Based on the amine and precursor uptake and decarboxylation (APUD) diethylenetriaminepentaacetic acid concept, 11C-labeled and 18F-labeled l-Dopa have been used to visualize NETs by PET. In a comparative study in 17 patients with gastrointestinal carcinoids, 18F–l-Dopa PET showed a higher sensitivity (65%) than FDG-PET (29%) and SRS (57%), but morphologic imaging by CT and MRI was found to be most sensitive (73%).129 PET with 11C–5-HTP showed significantly higher sensitivity than that of 11C–l-Dopa. In 38 consecutive patients with various NETs, 11C–5-HTP PET visualized tumors in 95% of patients, SRS in 84%, and CT in 79%.130 More tumors were detected by 11C–5-HTP-PET than SRS and CT. The size of the surgically removed PET-positive tumors ranged from 5 mm to 3 cm in diameter (Fig. 31-9).
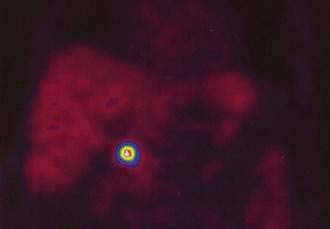
PET with 68Ga-labeled octreotide, with its 68-minute half-life, is available from a generator that produces the radiopharmaceutical independently of on an onsite cyclotron. 68Ga-labeled octreotide has been used in a few published reports for PET imaging of NETs of the GI tract. 68Ga–DOTA (1,4,7,10-tetraaxacyclododecane–1,4,7,10-tetraacetic acid)-TOC PET (Fig. 31-10) has demonstrated a higher sensitivity than SRS in GI carcinoid tumors.131
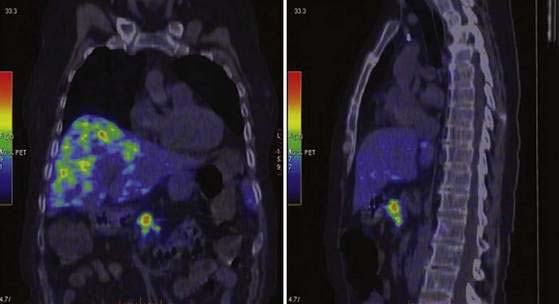
Technetium-labeled somatostatin analogs have also been evaluated. 68Ga-labeled octreotide has advantages, with fast pharmacokinetics and rapid tumor accumulation, allowing PET to be performed approximately 1 hour after injection. Several preparations have been synthesized, such as 68Ga–DOTA-TOC, 68Ga–DOTA-NOC, and 68Ga–DOTA-TATE. Another potential improvement in somatostatin receptor imaging with PET is the better quantification of tumors and normal tissue uptake, thereby allowing the possibility for adequate dosimetry before peptide receptor radionuclide treatment (see later).127
TREATMENT
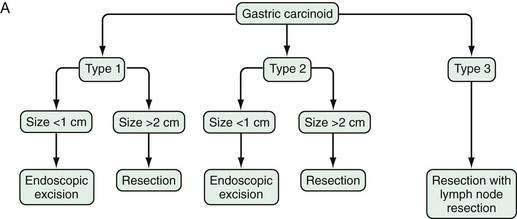
Figure 31-11 presents a treatment algorithm for patients with the more common carcinoid tumors—gastric, small intestinal, appendiceal, and rectal.
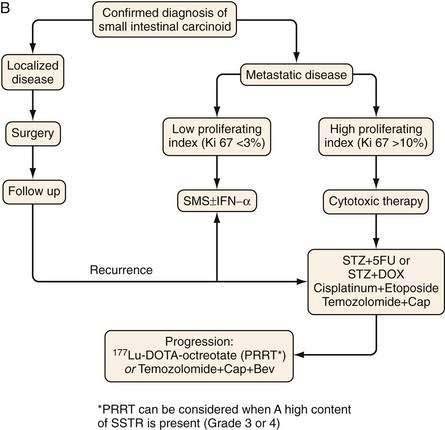
SURGERY
Surgery is the only form of curative therapy for carcinoid tumors. Unfortunately, most symptomatic patients are not candidates for curative treatment. In these individuals, the focus of therapy is palliation of symptoms and facilitating medical treatment.132 In the case of appendiceal tumors smaller than 1 cm and without metastases (the majority), a simple appendectomy is sufficient.133,134 In tumors of the appendix 2 cm or larger, a right hemicolectomy is the operation of choice.
With rectal carcinoid tumors smaller than 1 cm, local resection is usually adequate and results in cure. The depth of invasion is also an important prognostic factor and should also be assessed in all tumors. If no invasion of the muscularis propria is present for rectal carcinoid tumors smaller than 2 cm, local resection is adequate.135 For a rectal tumor larger than 2 cm, an abdominoperineal resection or a low anterior resection with primary anastomosis is recommended by some surgeons.135
In patients with midgut carcinoid tumors, malignancy is independent of size.136 Therefore, a wide en bloc resection of the adjacent lymph node–bearing mesentery is recommended for all small intestinal carcinoid tumors. If the midgut carcinoid is 2 cm or larger, a full-scale cancer operation should be carried out.
In patients with type 1 or 2 gastric carcinoids, lesions smaller than 1 cm can be removed endoscopically.137 For type 1 or 2 lesions between 1 and 2 cm in size, there is no general agreement on treatment, with some recommending endoscopic and others recommending surgical treatment. Type 3 gastric carcinoids tend to be larger and more aggressive so excision and regional lymph node resection are recommended.7
Resection of hepatic metastases may be beneficial (or sometimes curative) in selected patients. For patients with symptomatic tumors treatable by hepatectomy, resection is the treatment of choice if tumors are completely resectable. The role of cytoreductive or debulking surgery in patients from whom all tumors cannot be removed is unclear. No prospective randomized trials have addressed this question, but there are a number of retrospective analyses suggesting that such an approach should be considered in selected cases. In a study of 314 patients with midgut carcinoid tumors who underwent surgery mainly to remove the primary tumor and debulk mesenteric metastases, it was concluded that surgery provided considerable symptomatic relief and improved survival.136 Resection of liver metastases may relieve clinical symptoms, an effect that may last several months. It is recommended that if more than 90% of imaged tumors can be safely removed, resection should be considered.137 Local ablative therapies can also be used in the liver to control tumor growth. In several series, radiofrequency ablation resulted in symptom reduction and improved overall survival.132,138–140
The precise role of liver transplantation has yet to be determined. In a review of 103 patients with malignant NETs who underwent liver transplantation, the two- and five-year survival rates were 60% and 47%, respectively. However, recurrence-free survival was less than 24%. Multivariate analyses have identified age older than 50 years and transplantation combined with Whipple’s resection as adverse prognostic factors.141,142
HEPATIC ARTERY EMBOLIZATION AND CHEMOEMBOLIZATION
Transcatheter arterial chemoembolization (TACE) is an excellent method of treatment of nonresectable metastases from endocrine tumors of digestive origin and yields acceptable results in terms of symptom control and tumor response. Chemoembolization and other ablative therapies should be largely reserved for patients with advanced neuroendocrine tumors not amenable to curative surgery. TACE can be combined with systemic chemotherapy (see later) in selected patients—namely, those with hepatic metastases or other extrahepatic metastatic disease. In multiple series, chemoembolization with doxorubicin (Adriamycin) and streptozotocin, or both in combination, gave symptomatic responses in 67% to 100% of patients with midgut carcinoid tumors.143,144 Urinary 5-HIAA levels decreased more than 50% in 50% to 90% of patients. Inclusion criteria for chemoembolization vary but, in general, include patients with unresectable disease occupying less than 50% of the hepatic volume, a patent portal vein, near-normal liver function, a total serum bilirubin less than 2 mg/dL, and no contraindication to angiography. An uncommon model is to a mix a cytotoxic drug (e.g., doxorubicin) with iodized oil (Lipiodol), which is injected into the branches of the hepatic artery, distal to the origin of the gastroduodenal artery. This is followed by embolization with a gelatin sponge (2- to 3-mm particles or microspheres [embospheres]), which are placed distally in the distribution of the hepatic artery until a marked decrease in blood flow is observed. Usually, one lobe of the liver is embolized at a time and the interval between sessions varies from one to three months. The need for a repeated session should be guided by the individual patient’s response.145 More than three or four embolizations produces a diminishing effect, depending on revascularization from the diaphragm. The most common side effects of the TACE procedure include nausea, vomiting, pain, and increased serum liver aminotransferase levels. Procedure-related complications are rare, but may be life-threatening and include liver failure, cholecystitis, renal failure, and carcinoid crisis.
CHEMOTHERAPY
Chemotherapy for metastatic small intestinal carcinoid tumor has in general been disappointing. Single-agent therapy with doxorubicin, 5-fluorouracil (5-FU), dacarbazine, actinomycin D, cisplatin, etoposide, streptozotocin, or carboplatin has low tumor response rates of 0% to 30%.146 Furthermore, the duration of responses (when they occur) are short, usually less than one year. Combinations of chemotherapeutic agents for metastatic carcinoid tumors has not been shown to have any clear advantaged compared with single-agent chemotherapy. For example, a combination of streptozotocin with 5-FU or doxorubicin produced response rates of 0% to 40%.147,148 Newer cytotoxic regimens with temozolomide have not improved response rates in patients who had small intestinal carcinoids, with low proliferation indices (Ki67 index of 1% to 2%).149 However, in patients with high proliferation indices (Ki67 index > 15%), cytotoxic treatment (e.g., with etoposide and cisplatin) may sometimes produce a significant antitumor response, but such combination therapies should be reserved for advanced tumors with evidence of progression late in the disease course.150,151
SOMATOSTATIN ANALOGS
Somatostatin and its analogs are effective in controlling carcinoid-related symptoms, particularly the carcinoid syndrome, in up to 60% to 80% of patients. They inhibit synthesis and release of tumor-produced amines and peptides and also block their effect on target tissues.152 The two clinically available somatostatin analogs, octreotide and lanreotide, bind to somatostatin receptor subtypes 2 and 5. They are able to control flushing and diarrhea in 50% to 80% of patients.153 Octreotide has immediate-release activity and is used at subcutaneous doses of 100 to 1000 µg, three times daily. However, long-acting prolonged-release formulations are now commonly used, such as Sandostatin LAR or Lanreotide Autogel. The recommended doses are 10 to 30 mg of Sandostatin LAR and 60 to 120 mg of Lanreotide Autogel once a month.154–156 Some patients develop side effects with somatostatin analog treatment (see later). Therefore, it is recommended to begin therapy with the immediate-release form of octreotide at a dose of 100 µg, two or three times daily, for four to five days before switching to a long-acting formulation.
In general, these somatostatin analogs have a weak tumoricidal effect, and a decrease in tumor size is seen in only 3% to 8% of patients.154,157 However, somatostatin analogs appear to have a tumoristatic effect, stabilizing the growth of metastatic disease. In various studies, 30% to 60% of patients with metastatic disease have demonstrated tumor stabilization during treatment with somatostatin analogs. Several trials comparing octreotide and lanreotide have suggested a similar efficacy between the two analogs in the control of diarrhea and flushing.155,158 Treatment of nonfunctioning tumors (i.e., those without any hormone-related symptoms) with somatostatin analogs is controversial.
Side effects of somatostatin analog treatment are mild, with steatorrhea, borborygmus, flatulence, and abdominal pain. A few cases of hypocalcemia and bradycardia, impaired glucose tolerance, and cholelithiasis have been reported.154
INTERFERON-α
Interferon-α was introduced in 1981 for the treatment of small intestinal carcinoid tumors. In multiple studies in more 600 patients worldwide, the subjective and biochemical response rates were 40% to 60%, with a significant tumor reduction in 10% to 15%.153,159,160 Recombinant interferon-α (Intron A, Roferon A) can be given in a dose of 3 to 5 MU subcutaneously three to five times per week, or as pegylated interferon (Pegintron A, Pegasys) in a dose of 80 to 120 µg once a week. The interferon dose should be individually titrated and can be monitored by monitoring the white blood cell count, which should be around 3000/mm3. Treatment with interferon-α is associated with significant side effects, including flu-like symptoms in 80% of patients; fatigue in 60% to 70%; anemia, leukopenia, and thrombocytopenia in 15% to 30%; and autoimmune thyroid disease in approximately 15%.160,161
Because of their separate tumoristatic effects and ability to control symptoms, combinations of somatostatin analogs and interferon-α have been evaluated in a number of patients with malignant carcinoid syndrome, either alone or in combination with other agents. The objective response rates include mostly stabilization of tumor disease in up to 60% of patients, with reduced risk of tumor progression in patients treated with a combination.162–165
NEW BIOLOGIC AGENTS
Molecular targeted treatment has recently been applied in the treatment of gastrointestinal carcinoid tumors. These agents include VEGF inhibitors (bevacizumab) and mTOR inhibitors (RAD001). The number of patients treated is still small, but significant antitumor responses have been obtained in 10% to 20% of patients.166 The precise role of the new agents has yet to be determined with randomized controlled trials. It is also likely that they should not be used as single agents, but in combination with cytotoxic and other agents.
PEPTIDE RECEPTOR RADIONUCLIDE THERAPY
Peptide receptor radionuclide therapy (PRRT) is a new treatment modality for patients with inoperable or metastasized NETs.167 Most NETs overexpress receptors for somatostatin, mainly receptor subtype 2. Somatostatin receptor scintigraphy was introduced in the late 1980s and the radiopharmaceutical agent 111In-DTPA octreotide became the gold standard for NET staging. Radiolabeled somatostatin analogs were first tried in 1992 using high doses of 111In-octreotide. Thereafter, somatostatin peptides with higher receptor affinity were developed and conjugated with DOTA, a chelator that allowed stable labeling with the pure, high-energy beta emitter yttrium-90 and the medium-energy beta emitter lutetium-177.168,169 The overall tumor response rates with 90Y-DOTA octreotate are a partial remission in 6% to 29% and disease stabilization in 50% to 88%. For 177Lu-DOTA octreotate, the partial remission response is 25% to 30% and the rate of disease stabilization is 30% to 40%. Time to disease progression has been very favorable using these agents, with a median overall survival from the date of first diagnosis of 10.5 years for 177Lu-DOTA octreotate.170 The treatment seems to be safe, with less than 5% severe adverse events, including myelodysplasia and renal failure, mainly occurring in patients previously treated with cytotoxic agents. The precise role of PRRT in the treatment algorithm will hopefully be clarified by forthcoming studies.
Akerstrom G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:87-109. (Ref 132.)
de Herder WW. Biochemistry of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:33-41. (Ref 9.)
Eriksson B, Oberg K. Summing up 15 years of somatostatin analog therapy in neuroendocrine tumors: Future outlook. Ann Oncol. 1999;10(Suppl 2):S31-8. (Ref 154.)
Kloppel G, Anlauf M. Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2005;19:507-17. (Ref 27.)
Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog (177 Lu-DOTA 0,Tyr3)octreotate: Toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30. (Ref 170.)
Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-59. (Ref 12.)
Modlin IM, Lye KD, Kidd M. A 50-year analysis of 562 gastric carcinoids: Small tumor or larger problem? Am J Gastroenterol. 2004;99:23-32. (Ref 48.)
Oberg K. Chemotherapy and biotherapy in the treatment of neuroendocrine tumours. Ann Oncol. 2001;12(Suppl 2):S111-14. (Ref 160.)
Oberg K, Eriksson B. Nuclear medicine in the detection, staging and treatment of gastrointestinal carcinoid tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:265-76. (Ref 122.)
Oberg K, Stridsberg M. Chromogranins as diagnostic and prognostic markers in neuroendocrine tumours. Adv Exp Med Biol. 2000;482:329-37. (Ref 115.)
Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395-401. (Ref 5.)
Rockall AG, Reznek RH. Imaging of neuroendocrine tumours (CT/MR/US). Best Pract Res Clin Endocrinol Metab. 2007;21:43-68. (Ref 120.)
Ruszniewski P, Malka D. Hepatic arterial chemoembolization in the management of advanced digestive endocrine tumors. Digestion. 2000;62(Suppl 1):79-83. (Ref 144.)
Solcia E, Kloppel G, Sobin L. Histological typing of endocrine tumours. In: Verlag S, editor. World Health Organization histological classification of tumours. 2nd ed. New York: Springer; 2000:38. (Ref 4.)
Sundin A, Garske U, Orlefors H. Nuclear imaging of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:69-85. (Ref 127.)
Tang L, Shia J, Soslow RA, et al. Pathologic classification and clinical behavior of the spectrum of goblet cell carcinoid tumors of the appendix. Am J Surg Pathol. 2008;32:1429-43. (Ref 74.)
Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72. (Ref 14.)
1. Lubarsch O. Ueber den primaren Krebs des Ileum, nebst Bemerkungen über das gleichzeitige Vorkommen von Krebs und Tuberkuolose. Virchow Archiv Pathol Anatom Physiol Klin Med. 1867;111:280-317.
2. Oberndorfer S. Karzenoide Tumoren des Dünndarms. Frankf Zschr Path. 1907;1:426-30.
3. Williams ED, Sandler M. The classification of carcinoid tumours. Lancet. 1963;1:238-9.
4. Solcia E, Kloppel G, Sobin L. Histological typing of endocrine tumours. In: Verlag S, editor. World Health Organization histological classification of tumours. 2nd ed. New York: Springer; 2000:38.
5. Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch. 2006;449:395-401.
6. Kloppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann N Y Acad Sci. 2004;1014:13-27.
7. Eriksson B, Oberg K, Stridsberg M. Tumor markers in neuroendocrine tumors. Digestion. 2000;62(Suppl 1):33-8.
8. Chaudhry A, Funa K, Oberg K. Expression of growth factor peptides and their receptors in neuroendocrine tumors of the digestive system. Acta Oncol. 1993;32:107-14.
9. de Herder WW. Biochemistry of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:33-41.
10. Surveillance, Epidemiology, and End Results (SEER) Program. SEER*Stat Database: SEER 17 Regs Nov 2006 sub (1973-2004). Bethesda, Md: National Cancer Institute; 2007.
11. US National Cancer Institute. SEER Database. http://seer.cancer.gov/. (accessed Nov 15, 2007).
12. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97:934-59.
13. Berge T, Linell F. Carcinoid tumours. Frequency in a defined population during a 12-year period. Acta Pathol Microbiol Scand A. 1976;84:322-30.
14. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72.
15. Gustafsson B, Kidd M, Modlin I. Small intestinal neuroendocrine tumors. In: Modlin I, Öberg K, editors. A century of advances in neuroendocrine tumor biology and treatment. Hanover, Germany: Felsenstein; 2008:100.
16. Modlin IM, Champaneria MC, Chan AK, Kidd M. A three-decade analysis of 3,911 small intestinal neuroendocrine tumors: The rapid pace of no progress. Am J Gastroenterol. 2007;102:1464-73.
17. Maglinte DD, O’Connor K, Bessette J, et al. The role of the physician in the late diagnosis of primary malignant tumors of the small intestine. Am J Gastroenterol. 1991;86:304-8.
18. Cai YC, Barnard G, Hiestand L, et al. Florid angiogenesis in mucosa surrounding an ileal carcinoid tumor expressing transforming growth factor-alpha. Am J Surg Pathol. 1997;21:1373-7.
19. deVries H, Wijffels RT, Willemse PH, et al. Abdominal angina in patients with a midgut carcinoid, a sign of severe pathology. World J Surg. 2005;29:1139-42.
20. Durward G, Blackford S, Roberts D, Jones MK. Cutaneous scleroderma in association with carcinoid syndrome. Postgrad Med J. 1995;71:299-300.
21. Caplin ME, Buscombe JR, Hilson AJ, et al. Carcinoid tumour. Lancet. 1998;352:799-805.
22. Moyssakis IE, Rallidis LS, Guida GF, Nihoyannopoulos PI. Incidence and evolution of carcinoid syndrome in the heart. J Heart Valve Dis. 1997;6:625-30.
23. Cioffi U, De Simone M, Ferrero S, et al. Synchronous adenocarcinoma and carcinoid tumor of the terminal ileum in a Crohn’s disease patient. BMC Cancer. 2005;5:157.
24. Lundqvist M, Wilander E. A study of the histopathogenesis of carcinoid tumors of the small intestine and appendix. Cancer. 1987;60:201-6.
25. Chejfec G, Falkmer S, Askensten U, et al. Neuroendocrine tumors of the gastrointestinal tract. Pathol Res Pract. 1988;183:143-54.
26. Yantiss RK, Odze RD, Farraye FA, Rosenberg AE. Solitary versus multiple carcinoid tumors of the ileum: A clinical and pathologic review of 68 cases. Am J Surg Pathol. 2003;27:811-17.
27. Kloppel G, Anlauf M. Epidemiology, tumour biology and histopathological classification of neuroendocrine tumours of the gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2005;19:507-17.
28. Burke AP, Thomas RM, Elsayed AM, Sobin LH. Carcinoids of the jejunum and ileum: An immunohistochemical and clinicopathologic study of 167 cases. Cancer. 1997;79:1086-93.
29. Lloyd RV. Practical markers used in the diagnosis of neuroendocrine tumors. Endocrinol Pathol. 2003;14:293-301.
30. Bishop AE, Hamid QA, Adams C, et al. Expression of tachykinins by ileal and lung carcinoid tumors assessed by combined in situ hybridization, immunocytochemistry, and radioimmunoassay. Cancer. 1989;63:1129-37.
31. Jakobsen AM, Ahlman H, Wangberg B, et al. Expression of synaptic vesicle protein 2 (SV2) in neuroendocrine tumours of the gastrointestinal tract and pancreas. J Pathol. 2002;196:44-50.
32. Jakobsen AM, Andersson P, Saglik G, et al. Differential expression of vesicular monoamine transporter (VMAT) 1 and 2 in gastrointestinal endocrine tumours. J Pathol. 2001;195:463-72.
33. La Rosa S, Rigoli E, Uccella S, et al. CDX2 as a marker of intestinal EC-cells and related well-differentiated endocrine tumors. Virchows Arch. 2004;445:248-54.
34. Klein A, Clemens J, Cameron J. Periampullary neoplasms in von Recklinghausen’s disease. Surgery. 1989;106:815-19.
35. Burke A, Federspiel B, Sobin L, et al. Carcinoids of the duodenum: A histologic and immunohistochemical study of 65 tumors. Am J Surg Pathol. 1989;13:828-37.
36. Berna MJ, Hoffmann KM, Long SH, et al. Serum gastrin in Zollinger-Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. Evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore). 2006;85:331-64.
37. Hartel M, Wente MN, Sido B, et al. Carcinoid of the ampulla of Vater. J Gastroenterol Hepatol. 2005;20:676-81.
38. Burke AP, Sobin LH, Shekitka KM, et al. Somatostatin-producing duodenal carcinoids in patients with von Recklinghausen’s neurofibromatosis. A predilection for black patients. Cancer. 1990;65:1591-5.
39. Burke AP, Helwig EB. Gangliocytic paraganglioma. Am J Clin Pathol. 1989;92:1-9.
40. Hamilton S, Aaltonen L. World Health Organization classification of tumours. Pathology and genetics of tumours of the digestive system. Washington, DC: IARC Press; 2000.
41. Perrone T, Sibley RK, Rosai J. Duodenal gangliocytic paraganglioma. An immunohistochemical and ultrastructural study and a hypothesis concerning its origin. Am J Surg Pathol. 1985;9:31-41.
42. Soga J. Early-stage carcinoids of the gastrointestinal tract: An analysis of 1914 reported cases. Cancer. 2005;103:1587-95.
43. Solcia E, Rindi G, Buffa R, et al. Gastric endocrine cells: Types, function and growth. Regul Pept. 2000;93:31-5.
44. Rindi G, Necchi V, Savio A, et al. Characterisation of gastric ghrelin cells in man and other mammals: Studies in adult and fetal tissues. Histochem Cell Biol. 2002;117:511-19.
45. Borch K, Renvall H, Liedberg G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology. 1985;88:638-48.
46. Bordi C, Yu JY, Baggi MT, et al. Gastric carcinoids and their precursor lesions. A histologic and immunohistochemical study of 23 cases. Cancer. 1991;67:663-72.
47. Tsolakis AV, Portela-Gomes GM, Stridsberg M, et al. Malignant gastric ghrelinoma with hyperghrelinemia. J Clin Endocrinol Metab. 2004;89:3739-44.
48. Modlin IM, Lye KD, Kidd M. A 50-year analysis of 562 gastric carcinoids: Small tumor or larger problem? Am J Gastroenterol. 2004;99:23-32.
49. Bordi C, Falchetti A, Azzoni C, et al. Aggressive forms of gastric neuroendocrine tumors in multiple endocrine neoplasia type I. Am J Surg Pathol. 1997;21:1075-82.
50. Gould VE, Jao W, Chejfec G, et al. Neuroendocrine carcinomas of the gastrointestinal tract. Semin Diagn Pathol. 1984;1:13-18.
51. Leotlela PD, Jauch A, Holtgreve-Grez H, Thakker RV. Genetics of neuroendocrine and carcinoid tumours. Endocr Relat Cancer. 2003;10:437-50.
52. Tonnies H, Toliat MR, Ramel C, et al. Analysis of sporadic neuroendocrine tumours of the enteropancreatic system by comparative genomic hybridisation. Gut. 2001;48:536-41.
53. Lollgen RM, Hessman O, Szabo E, et al. Chromosome 18 deletions are common events in classical midgut carcinoid tumors. Int J Cancer. 2001;92:812-15.
54. Kidd M, Modlin IM, Mane SM, et al. The role of genetic markers—NAP1L1, MAGE-D2, and MTA1—in defining small-intestinal carcinoid neoplasia. Ann Surg Oncol. 2006;13:253-62.
55. Hoang MP, Hobbs CM, Sobin LH, Albores-Saavedra J. Carcinoid tumor of the esophagus: A clinicopathologic study of four cases. Am J Surg Pathol. 2002;26:517-22.
56. Lindberg GM, Molberg KH, Vuitch MF, Albores-Saavedra J. Atypical carcinoid of the esophagus: A case report and review of the literature. Cancer. 1997;79:1476-81.
57. Modlin IM, Lye KD, Kidd M. Carcinoid tumors of the stomach. Surg Oncol. 2003;12:153-72.
58. Burkitt MD, Pritchard DM. Review article: Pathogenesis and management of gastric carcinoid tumours. Aliment Pharmacol Ther. 2006;24:1305-20.
59. Borch K, Ahren B, Ahlman H, et al. Gastric carcinoids: Biologic behavior and prognosis after differentiated treatment in relation to type. Ann Surg. 2005;242:64-73.
60. Maurer CA, Baer HU, Dyong TH, et al. Carcinoid of the pancreas: Clinical characteristics and morphological features. Eur J Cancer. 1996;32A:1109-16.
61. Soga J. Carcinoids of the pancreas: An analysis of 156 cases. Cancer. 2005;104:1180-7.
62. Burke AP, Sobin LH, Federspiel BH, et al. Carcinoid tumors of the duodenum. A clinicopathologic study of 99 cases. Arch Pathol Lab Med. 1990;114:700-4.
63. Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128:1717-51.
64. Katona TM, Jones TD, Wang M, et al. Molecular evidence for independent origin of multifocal neuroendocrine tumors of the enteropancreatic axis. Cancer Res. 2006;66:4936-42.
65. Wangberg B, Westberg G, Tylen U, et al. Survival of patients with disseminated midgut carcinoid tumors after aggressive tumor reduction. World J Surg. 1996;20:892-9.
66. Tomassetti P, Campana D, Piscitelli L, et al. Endocrine tumors of the ileum: Factors correlated with survival. Neuroendocrinology. 2006;83:380-6.
67. Cunningham JL, Grimelius L, Sundin A, et al. Malignant ileocaecal serotonin-producing carcinoid tumours: The presence of a solid growth pattern and/or Ki67 index above 1% identifies patients with a poorer prognosis. Acta Oncol. 2007;46:747-56.
68. Sandor A, Modlin IM. A retrospective analysis of 1570 appendiceal carcinoids. Am J Gastroenterol. 1998;93:422-8.
69. Carr NJ, Sobin LH. Neuroendocrine tumors of the appendix. Semin Diagn Pathol. 2004;21:108-9.
70. Koca AM, Druart ML, Mehdi A, Limbosch JM. Surgical attitude towards carcinoid tumour of the appendix. Review of the literature. Acta Chir Belg. 2002;102:338-40.
71. O’Donnell ME, Carson J, Garstin WI. Surgical treatment of malignant carcinoid tumours of the appendix. Int J Clin Pract. 2007;61:431-7.
72. Abt AB, Carter SL. Goblet cell carcinoid of the appendix. An ultrastructural and histochemical study. Arch Pathol Lab Med. 1976;100:301-6.
73. Kabbani W, Houlihan PS, Luthra R, et al. Mucinous and nonmucinous appendiceal adenocarcinomas: Different clinicopathological features but similar genetic alterations. Mod Pathol. 2002;15:599-605.
74. Tang L, Shia J, Soslow RA, et al. Pathologic classification and clinical behavior of the spectrum of goblet cell carcinoid tumors of the appendix. Am J Surg Pathol. 2008;32:1429-43.
75. Federspiel BH, Burke AP, Sobin LH, Shekitka KM. Rectal and colonic carcinoids. A clinicopathologic study of 84 cases. Cancer. 1990;65:135-40.
76. Spread C, Berkel H, Jewell L, et al. Colon carcinoid tumors. A population-based study. Dis Colon Rectum. 1994;37:482-91.
77. Rosenberg JM, Welch JP. Carcinoid tumors of the colon. A study of 72 patients. Am J Surg. 1985;149:775-9.
78. Koura AN, Giacco GG, Curley SA, et al. Carcinoid tumors of the rectum: Effect of size, histopathology, and surgical treatment on metastasis-free survival. Cancer. 1997;79:1294-8.
79. Naunheim KS, Zeitels J, Kaplan EL, et al. Rectal carcinoid tumors—treatment and prognosis. Surgery. 1983;94:670-6.
80. Feldman JM. Carcinoid tumors and syndrome. Semin Oncol. 1987;14:237-46.
81. Tiensuu Janson EM, Oberg KE. Carcinoid tumours. Baillieres Clin Gastroenterol. 1996;10:589-601.
82. Emson PC, Gilbert RF, Martensson H, Nobin A. Elevated concentrations of substance P and 5-HT in plasma in patients with carcinoid tumors. Cancer. 1984;54:715-18.
83. Schaffalitzky De Muckadell OB, Aggestrup S, Stentoft P. Flushing and plasma substance P concentration during infusion of synthetic substance P in normal man. Scand J Gastroenterol. 1986;21:498-502.
84. Jensen RT. Overview of chronic diarrhea caused by functional neuroendocrine neoplasms. Semin Gastrointest Dis. 1999;10:156-72.
85. Donowitz M, Binder HJ. Jejunal fluid and electrolyte secretion in carcinoid syndrome. Am J Dig Dis. 1975;20:1115-22.
86. von der Ohe MR, Camilleri M, Kvols LK, Thomforde GM. Motor dysfunction of the small bowel and colon in patients with the carcinoid syndrome and diarrhea. N Engl J Med. 1993;329:1073-8.
87. Lundin L, Norheim I, Landelius J, et al. Carcinoid heart disease: Relationship of circulating vasoactive substances to ultrasound-detectable cardiac abnormalities. Circulation. 1988;77:264-9.
88. Moller JE, Pellikka PA, Bernheim AM, et al. Prognosis of carcinoid heart disease: Analysis of 200 cases over two decades. Circulation. 2005;112:3320-7.
89. Moller JE, Connolly HM, Rubin J, et al. Factors associated with progression of carcinoid heart disease. N Engl J Med. 2003;348:1005-15.
90. Lundin L, Landelius J, Andren B, Oberg K. Transoesophageal echocardiography improves the diagnostic value of cardiac ultrasound in patients with carcinoid heart disease. Br Heart J. 1990;64:190-4.
91. Robiolio PA, Rigolin VH, Wilson JS, et al. Carcinoid heart disease. Correlation of high serotonin levels with valvular abnormalities detected by cardiac catheterization and echocardiography. Circulation. 1995;92:790-5.
92. Hua X, Lundberg JM, Theodorsson-Norheim E, Brodin E. Comparison of cardiovascular and bronchoconstrictor effects of substance P, substance K and other tachykinins. Naunyn Schmiedebergs Arch Pharmacol. 1984;328:196-201.
93. Roy A, Brand NJ, Yacoub MH. Expression of 5-hydroxytryptamine receptor subtype messenger RNA in interstitial cells from human heart valves. J Heart Valve Dis. 2000;9:256-60.
94. Fitzgerald LW, Burn TC, Brown BS, et al. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Mol Pharmacol. 2000;57:75-81.
95. Connolly HM, Crary JL, McGoon MD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337:581-8.
96. Waltenberger J, Lundin L, Oberg K, et al. Involvement of transforming growth factor-beta in the formation of fibrotic lesions in carcinoid heart disease. Am J Pathol. 1993;142:71-8.
97. Jian B, Xu J, Connolly J, et al. Serotonin mechanisms in heart valve disease I: Serotonin-induced up-regulation of transforming growth factor-beta1 via G-protein signal transduction in aortic valve interstitial cells. Am J Pathol. 2002;161:2111-21.
98. Modlin IM, Shapiro MD, Kidd M. Carcinoid tumors and fibrosis: An association with no explanation. Am J Gastroenterol. 2004;99:2466-78.
99. Walker GA, Masters KS, Shah DN, et al. Valvular myofibroblast activation by transforming growth factor-beta: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253-60.
100. Simula DV, Edwards WD, Tazelaar HD, et al. Surgical pathology of carcinoid heart disease: A study of 139 valves from 75 patients spanning 20 years. Mayo Clin Proc. 2002;77:139-47.
101. Connolly HM, Nishimura RA, Smith HC, et al. Outcome of cardiac surgery for carcinoid heart disease. J Am Coll Cardiol. 1995;25:410-16.
102. Soga J, Yakuwa Y, Osaka M. Carcinoid syndrome: A statistical evaluation of 748 reported cases. J Exp Clin Cancer Res. 1999;18:133-41.
103. Kema IP, de Vries EG, Slooff MJ, et al. Serotonin, catecholamines, histamine, and their metabolites in urine, platelets, and tumor tissue of patients with carcinoid tumors. Clin Chem. 1994;40:86-95.
104. Oates JA, Sjoerdsma A. A unique syndrome associated with secretion of 5-hydroxytryptophan by metastatic gastric carcinoids. Am J Med. 1962;32:333-42.
105. Roberts LJ2nd, Bloomgarden ZT, Marney SRJr, et al. Histamine release from a gastric carcinoid: Provocation by pentagastrin and inhibition by somatostatin. Gastroenterology. 1983;84:272-5.
106. Norheim I, Theodorsson-Norheim E, Brodin E, Oberg K. Tachykinins in carcinoid tumors: Their use as a tumor marker and possible role in the carcinoid flush. J Clin Endocrinol Metab. 1986;63:605-2.
107. Oates JA, Melmon K, Sjoerdsma A, et al. Release of a kinin peptide in the carcinoid syndrome. Lancet. 1964;1:514-17.
108. Rubin J, Ajani J, Schirmer W, et al. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J Clin Oncol. 1999;17:600-6.
109. Vaughan DJ, Brunner MD. Anesthesia for patients with carcinoid syndrome. Int Anesthesiol Clin. 1997;35:129-42.
110. Feldman JM. Urinary serotonin in the diagnosis of carcinoid tumors. Clin Chem. 1986;32:840-4.
111. Mailman RB, Kilts CD. Analytical considerations for quantitative determination of serotonin and its metabolically related products in biological matrices. Clin Chem. 1985;31:1849-54.
112. De Vries EG, Kema IP, Slooff MJ, et al. Recent developments in diagnosis and treatment of metastatic carcinoid tumours. Scand J Gastroenterol Suppl. 1993;200:87-93.
113. Stridsberg M, Oberg K, Li Q, et al. Measurements of chromogranin A, chromogranin B (secretogranin I), chromogranin C (secretogranin II) and pancreastatin in plasma and urine from patients with carcinoid tumours and endocrine pancreatic tumours. J Endocrinol. 1995;144:49-59.
114. Nobels FR, Kwekkeboom DJ, Coopmans W, et al. Chromogranin A as serum marker for neuroendocrine neoplasia: Comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J Clin Endocrinol Metab. 1997;82:2622-8.
115. Oberg K, Stridsberg M. Chromogranins as diagnostic and prognostic markers in neuroendocrine tumours. Adv Exp Med Biol. 2000;482:329-37.
116. Baudin E, Gigliotti A, Ducreux M, et al. Neuron-specific enolase and chromogranin A as markers of neuroendocrine tumours. Br J Cancer. 1998;78:1102-7.
117. Grossmann M, Trautmann ME, Poertl S, et al. Alpha-subunit and human chorionic gonadotropin-beta immunoreactivity in patients with malignant endocrine gastroenteropancreatic tumours. Eur J Clin Invest. 1994;24:131-6.
118. Varas Lorenzo MJ, Miquel Collell JM, Maluenda Colomer MD, et al. Preoperative detection of gastrointestinal neuroendocrine tumors using endoscopic ultrasonography. Rev Esp Enferm Dig. 2006;98:828-36.
119. van Tuyl SA, van Noorden JT, Timmer R, et al. Detection of small-bowel neuroendocrine tumors by video capsule endoscopy. Gastrointest Endosc. 2006;64:66-72.
120. Rockall AG, Reznek RH. Imaging of neuroendocrine tumours (CT/MR/US). Best Pract Res Clin Endocrinol Metab. 2007;21:43-68.
121. Reubi JC, Kvols LK, Waser B, et al. Detection of somatostatin receptors in surgical and percutaneous needle biopsy samples of carcinoids and islet cell carcinomas. Cancer Res. 1990;50:5969-77.
122. Oberg K, Eriksson B. Nuclear medicine in the detection, staging and treatment of gastrointestinal carcinoid tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:265-76.
123. Krenning EP, Kwekkeboom DJ, Bakker WH, et al. Somatostatin receptor scintigraphy with (111In-DTPA-D-Phe1)- and (123I-Tyr3)-octreotide: The Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716-31.
124. Lebtahi R, Cadiot G, Sarda L, et al. Clinical impact of somatostatin receptor scintigraphy in the management of patients with neuroendocrine gastroenteropancreatic tumors. J Nucl Med. 1997;38:853-8.
125. Gotthardt M, Dirkmorfeld LM, Wied MU, et al. Influence of somatostatin receptor scintigraphy and CT/MRI on the clinical management of patients with gastrointestinal neuroendocrine tumors: An analysis in 188 patients. Digestion. 2003;68:80-5.
126. Termanini B, Gibril F, Reynolds JC, et al. Value of somatostatin receptor scintigraphy: a prospective study in gastrinoma of its effect on clinical management. Gastroenterology. 1997;112:335-47.
127. Sundin A, Garske U, Orlefors H. Nuclear imaging of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:69-85.
128. Adams S, Baum R, Rink T, et al. Limited value of fluorine-18 fluorodeoxyglucose positron emission tomography for the imaging of neuroendocrine tumours. Eur J Nucl Med. 1998;25:79-83.
129. Hoegerle S, Altehoefer C, Ghanem N, et al. Whole-body 18F-Dopa PET for detection of gastrointestinal carcinoid tumors. Radiology. 2001;220:373-80.
130. Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392-400.
131. Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: Comparison with somatostatin receptor scintigraphy and CT. J Nucl Med. 2007;48:508-18.
132. Akerstrom G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:87-109.
133. Goede AC, Caplin ME, Winslet MC. Carcinoid tumour of the appendix. Br J Surg. 2003;90:1317-22.
134. Stinner B, Rothmund M. Neuroendocrine tumours (carcinoids) of the appendix. Best Pract Res Clin Gastroenterol. 2005;19:729-38.
135. Vogelsang H, Siewert JR. Endocrine tumours of the hindgut. Best Pract Res Clin Gastroenterol. 2005;19:739-51.
136. Hellman P, Lundstrom T, Ohrvall U, et al. Effect of surgery on the outcome of midgut carcinoid disease with lymph node and liver metastases. World J Surg. 2002;26:991-7.
137. Norton JA, Melcher ML, Gibril F, Jensen RT. Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger-Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgical treatment. Surgery. 2004;136:1267-74.
138. Hodul P, Malafa M, Choi J, Kvols L. The role of cytoreductive hepatic surgery as an adjunct to the management of metastatic neuroendocrine carcinomas. Cancer Control. 2006;13:61-71.
139. Decadt B, Siriwardena AK. Radiofrequency ablation of liver tumours: Systematic review. Lancet Oncol. 2004;5:550-60.
140. Hellman P, Ladjevardi S, Skogseid B, et al. Radiofrequency tissue ablation using cooled tip for liver metastases of endocrine tumors. World J Surg. 2002;26:1052-6.
141. Lehnert T. Liver transplantation for metastatic neuroendocrine carcinoma: An analysis of 103 patients. Transplantation. 1998;66:1307-12.
142. Olausson M, Friman S, Herlenius G, et al. Orthotopic liver or multivisceral transplantation as treatment of metastatic neuroendocrine tumors. Liver Transpl. 2007;13:327-33.
143. Roche A, Girish BV, de Baere T, et al. Trans-catheter arterial chemoembolization as first-line treatment for hepatic metastases from endocrine tumors. Eur Radiol. 2003;13:136-40.
144. Ruszniewski P, Malka D. Hepatic arterial chemoembolization in the management of advanced digestive endocrine tumors. Digestion. 2000;62(Suppl 1):79-83.
145. Therasse E, Breittmayer F, Roche A, et al. Transcatheter chemoembolization of progressive carcinoid liver metastasis. Radiology. 1993;189:541-7.
146. Oberg K. The use of chemotherapy in the management of neuroendocrine tumors. Endocrinol Metab Clin North Am. 1993;22:941-52.
147. Moertel CG, Lavin PT, Hahn RG. Phase II trial of doxorubicin therapy for advanced islet cell carcinoma. Cancer Treat Rep. 1982;66:1567-9.
148. Sun W, Lipsitz S, Catalano P, et al. Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J Clin Oncol. 2005;23:4897-904.
149. Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986-91.
150. Mitry E, Baudin E, Ducreux M, et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br J Cancer. 1999;81:1351-5.
151. Fjallskog ML, Granberg DP, Welin SL, et al. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer. 2001;92:1101-7.
152. Lamberts SW, Krenning EP, Reubi JC. The role of somatostatin and its analogs in the diagnosis and treatment of tumors. Endocr Rev. 1991;12:450-82.
153. Plockinger U, Wiedenmann B. Neuroendocrine tumors. Biotherapy. Best Pract Res Clin Endocrinol Metab. 2007;21:145-62.
154. Eriksson B, Oberg K. Summing up 15 years of somatostatin analog therapy in neuroendocrine tumors: Future outlook. Ann Oncol. 1999;10(Suppl 2):S31-8.
155. O’Toole D, Ducreux M, Bommelaer G, et al. Treatment of carcinoid syndrome: A prospective crossover evaluation of lanreotide versus octreotide in terms of efficacy, patient acceptability, and tolerance. Cancer. 2000;88:770-6.
156. Wymenga AN, Eriksson B, Salmela PI, et al. Efficacy and safety of prolonged-release lanreotide in patients with gastrointestinal neuroendocrine tumors and hormone-related symptoms. J Clin Oncol. 1999;17:1111.
157. Arnold R, Trautmann ME, Creutzfeldt W, et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430-8.
158. Ruszniewski P, Ducreux M, Chayvialle JA, et al. Treatment of the carcinoid syndrome with the longacting somatostatin analogue lanreotide: A prospective study in 39 patients. Gut. 1996;39:279-83.
159. Oberg K, Eriksson B. The role of interferons in the management of carcinoid tumours. Br J Haematol. 1991;79(Suppl 1):74-7.
160. Oberg K. Chemotherapy and biotherapy in the treatment of neuroendocrine tumours. Ann Oncol. 2001;12(Suppl 2):S111-14.
161. Ronnblom LE, Alm GV, Oberg K. Autoimmune phenomena in patients with malignant carcinoid tumors during interferon-alpha treatment. Acta Oncol. 1991;30:537-40.
162. Tiensuu Janson EM, Ahlstrom H, Andersson T, Oberg KE. Octreotide and interferon alfa: A new combination for the treatment of malignant carcinoid tumours. Eur J Cancer. 1992;28A:1647-50.
163. Frank M, Klose KJ, Wied M, et al. Combination therapy with octreotide and alpha-interferon: Effect on tumor growth in metastatic endocrine gastroenteropancreatic tumors. Am J Gastroenterol. 1999;94:1381-7.
164. Faiss S, Pape UF, Bohmig M, et al. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors—the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. 2003;21:2689-96.
165. Fazio N, de Braud F, Delle Fave G, Oberg K. Interferon-alpha and somatostatin analog in patients with gastroenteropancreatic neuroendocrine carcinoma: Single agent or combination? Ann Oncol. 2007;18:13-19.
166. Yao JC. Neuroendocrine tumors. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab. 2007;21:163-72.
167. Forrer F, Valkema R, Kwekkeboom DJ, et al. Neuroendocrine tumors. Peptide receptor radionuclide therapy. Best Pract Res Clin Endocrinol Metab. 2007;21:111-29.
168. Otte A, Herrmann R, Heppeler A, et al. Yttrium-90 DOTATOC: First clinical results. Eur J Nucl Med. 1999;26:1439-47.
169. Kwekkeboom DJ, Teunissen JJ, Bakker WH, et al. Radiolabeled somatostatin analog (177Lu-DOTA0,Tyr3) octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol. 2005;23:2754-62.
170. Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog (177 Lu-DOTA 0,Tyr3) octreotate: Toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124-30.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 31 Gastrointestinal Carcinoid Tumors (Gastrointestinal Neuroendocrine Tumors) and the Carcinoid Syndrome
In 1888, Lubarsch described what is now recognized to be the entity of carcinoid tumors when he reported the autopsy finding of two patients with multiple tumors in the distal ileum.1 The term carcinoid was introduced by Oberndorfer in 1907 in his description of a class of malignant tumors that behaved less aggressively than the more common adenocarcinomas of the gastrointestinal (GI) tract.2 The traditional classification of carcinoids, based on their embryonic origin into foregut, midgut, and hindgut carcinoids, has been gradually abandoned.3 A tumor biology–based classification system introduced by the World Health Organization (WHO) in 2000 has greater applicability, although more recent amplification and revision of this system by the European Neuroendocrine Tumor Society (TNM classification) appears likely to become the standard (Table 31-1).4,5
Table 31-1 TNM Classification for Endocrine Tumors*
| A. Gastric Tumors | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| Tis | In situ tumor, dysplasia (<0.5 mm) |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or subserosa or is >1 cm |
| T3 | Tumor penetrates serosa |
| T4 | Tumor invades adjacent structures |
| NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastases |
| N1 | Regional lymph node metastases |
| MX | Distant metastases cannot be assessed |
| M0 | No distant metastases |
| M1 | Distant metastases |
| B. Tumors of the Duodenum, Ampulla, and Proximal Jejunum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades lamina propria or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades pancreas or retroperitoneum |
| T4 | Tumor invades peritoneum or other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
| C. Tumors of Distal Jejunum and Ileum | |
| TX | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor invades mucosa or submucosa and is ≤1 cm |
| T2 | Tumor invades muscularis propria or is >1 cm |
| T3 | Tumor invades subserosa |
| T4 | Tumor invades peritoneum/other organs |
| NX | Same as for gastric |
| N0 | Same as for gastric |
| N1 | Same as for gastric |
| MX | Same as for gastric |
| M0 | Same as for gastric |
| M1 | Same as for gastric |
T, primary tumor (for any T, an “m” is added for multiple tumors; N, regional lymph nodes; M, metastases).
* European Neuroendocrine Tumor Society.
Adapted from Solcia E, Kloppel G, Sobin L. Histological typing of endocrine tumours. In: Verlag S, editor. World Health Organization Histological Classification of Tumours. 2nd ed. New York: Springer; 2000. p 38; and Rindi G, Kloppel G, Alhman H, et al. TNM staging of foregut (neuro)endocrine tumors: A consensus proposal including a grading system. Virchows Arch 2006; 449:395-401.
Carcinoids arise from cells of the diffuse neuroendocrine system and can arise almost anywhere within the gastrointestinal tract.6 Carcinoids, also called gastrointestinal neuroendocrine tumors (GI NETs), are related to medullary carcinoma of the thyroid, pheochromocytoma and pancreatic neuroendocrine tumors. GI NETs synthesize bioactive amines and peptides, including neuron-specific enolase (NSE), 5-hydroxytryptamine (5-HT, or serotonin), and 5-hydroxytryptophan (5-HTP). They also secrete peptides such as chromogranin A, pancreatic polypeptide, calcitonin, tachykinins (neurokinin A and substance P), and various growth factors, such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), endocrine growth factor (EGF), fibroblast growth factor (FGF), and the vascular endothelial growth factor (VEGF) family of growth factors, including their receptors.7–9 Of clinical relevance is the observation that several different genes and genetic divergences related to tumor development are evident. GI NETs can be sporadic (nonfamilial) or part of a familial syndrome, such as von Hippel-Lindau syndrome or neurofibromatosis (NF). GI NETs comprise 0.5% of all malignancies and, as shown in Figure 31-1, their incidence has increased substantially over the last several decades.10,11 In this chapter, we will use the more familiar term carcinoid, although the term GI NET is the most appropriate, but not as well established.
Figure 31-1. Incidence of different subtypes of neuroendocrine tumors, 1970 to 2005, from the Surveillance Epidemiology and End Results (SEER) data base.10,11 Note the significant increase in most subtypes of neuroendocrine tumors since the 1970s.
(From Modlin IM, Oberg K, Chung DC, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008; 9:61-72.)
Carcinoid tumors occur most frequently in the GI tract (67%), with the bronchopulmonary system being the second most common location (25%), followed by considerably less frequent locations, such as the ovaries, testes, and hepatobiliary system.12 The most common location in the GI tract is the ileum (17%). The overall incidence of carcinoid tumors is difficult to determine, because it appears likely that most tumors remain asymptomatic. An autopsy study has estimated the annual incidence to be 8.4/100,000 people.13 The SEER database from 1973 to 2004 indicated an annual incidence of 2.0 to 2.5/100,000/year, with a 3.5% annual increase over this time period.10 Ileal tumors have specifically increased in prevalence—in white males (by 274%), black males (500%), white females (213%), and black females (286%), respectively. A recent evaluation of the SEER database comprising 35,825 cases in the United States has indicated an annual incidence of 5.25/100,000/year for all types of neuroendocrine tumors and a prevalence of 35/100,000.14 The annual incidence of gastrointestinal carcinoids was 2.53/100,000. Overall, the incidence of gastrointestinal carcinoids is higher in African Americans (4.5/100,000) compared with white Americans (2.5/100,000) with striking differences in certain sites, particularly the rectum. As a result of the increasing incidence and prevalence, gastrointestinal neuroendocrine tumors represent a substantial clinical problem. The increasing incidence is probably mostly to the result of the introduction of better diagnostic tools (imaging and immunohistochemical), as well as greater awareness by pathologists and clinicians. There are no known environmental risk factors for carcinoids.
CLINICAL FEATURES
An early and accurate diagnosis is often delayed by four or five years because most small intestinal carcinoids (Fig. 31-2) are small, initially asymptomatic, or misdiagnosed as conditions such as allergy or irritable bowel syndrome. Progressive growth of the tumor may cause vague abdominal discomfort because of intermittent intestinal obstruction. As the tumor gets larger, invading the intestinal wall and occluding the gut lumen, emergency clinical presentations of an acute abdomen (intussusception, obstruction, perforation, bleeding) may arise because of local tumor mass effect or tumor-induced fibrosis.15–18 The frequency of intestinal obstruction that is secondary to a gastrointestinal NET-associated mesenteric fibrosis ranges from 42% to 66%.18,19 Another characteristic feature of these patients is vascular elastosis (thickening of the vessel wall), resulting in ischemic changes of the gut. The cause of this condition is unclear, but a local effect of serotonin or other tumor products, such as TGF-α, have been postulated to have a direct trophic effect on smooth muscle and fibroblasts in the vessel wall. Both mesenteric fibrosis and elastosis may cause abdominal angina.15 A rare cutaneous manifestation of ileal carcinoids is a fibrotic, scleroderma-like condition mostly affecting the lower extremities.20 As the disease advances, gastrointestinal carcinoids frequently metastasize locally to mesenteric lymph nodes and to the liver. A classic carcinoid syndrome is relatively uncommon (10% to 15%), typically consisting of diarrhea, cutaneous flushing, bronchoconstriction and right-sided heart failure.21,22 As many as 15% to 25% of gastrointestinal carcinoids exhibit a synchronous or metachronous association with other tumors, usually adenocarcinomas of the colon.23–25 This may reflect the activity of growth factors produced by the carcinoid tumor. A relatively large percentage of GI NETs are multicentric; for example, up to 33% of carcinoids in the small intestine are multicentric.24–26
PATHOLOGY
The most common histopathologic type of intestinal carcinoid is the enterochromaffin (EC) cell carcinoid, accounting for more than 98% of cases. The EC cell carcinoid is defined by its argentaffin staining properties, serotonin production, and typical pleomorphic secretory granules. Morphologically, EC cell carcinoids are characterized by medium-sized tumor cells arranged in an organoid pattern and showing only mild to moderate atypia. Tumor necrosis is absent and the mitotic rate is low (<2 mitoses/10 high-powered field [HPF]). Distinct growth patterns have been described in gastrointestinal carcinoids: (1) nodular or insular pattern; (2) trabecular pattern; (3) acinar and tubular pattern; and (4) atypical solid pattern. Mixed patterns may be seen. EC cell carcinoids of the jejunum and ileum predominantly display an insular growth pattern (93%; Fig. 31-3) and, less frequently, mixed insular and glandular (5%) or trabecular (2%) growth patterns. EC cell carcinoids invade the submucosa, muscularis propria, or mesentery. Invasion of lymphatics and veins, or perineural growth, can be seen. EC cell carcinoids may produce multiple hormones. Most of the tumors (>90%) secrete serotonin and tachykinins (substance P, neurokinin A). Production of other hormones such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, or adrenocorticotropic hormone (ACTH) can be demonstrated in less than 5% of cases. Neuroendocrine markers (proteins associated with large dense core vesicles or synaptic-like microvesicles) are abundantly expressed in EC cell carcinoids and can be used to confirm the neuroendocrine phenotype of the these tumors.24,25,27 Chromogranin A, synaptophysin, synaptic vesicle protein 2, neuron-specific enolase, and Leu7 are expressed by 92% to 100% of jejunoileal carcinoid tumors. Expression of the vascular monoamine transporters 1 and 2 (VMAT-1 and -2) has been demonstrated in more than 90% of tumors and is related to the production and storage of serotonin in EC cell carcinoids. Other markers identified in EC cell carcinoids include cytokeratins 8 and 18, carcinoembryonic antigen (CEA), and prostatic acid phosphatase. High expression of the intestinal transcription factor CDX2 has been demonstrated in EC cell carcinoids and may become a useful marker of intestinal origin.6,27–33
Carcinoids (GI NETs) arising in the duodenum and jejunum include gastrin, somatostatin, and EC cell tumors and gangliocytic paraganglioma. Gastrin cell tumors (gastrinomas) are the most common in the duodenum, might be nonfunctioning or functioning (Zollinger-Ellison syndrome [ZES]), and associated with the multiple endocrine neoplasia type I (MEN-I) syndrome (see Chapter 32). Tumors are mainly located in the first and second parts of the duodenum. A small proportion of tumor cells may produce other hormones in addition to gastrin, such as cholecystokinin (CCK), somatostatin, pancreatic polypeptide, neurotensin, and insulin. Morphologically, tumor cells are uniform, with rounded nuclei and abundant cytoplasm. The growth pattern is usually trabecular or cribriform. Necroses are absent and the mitotic rate is low. Most of the tumor cells are positive for neuroendocrine markers, such as chromogranin A, synaptophysin, Leu7, and NSE.34–36
Somatostatin cell tumors (somatostatinomas) represent the second most frequent histopathologic type in the duodenum, accounting for 15% to 27 % of duodenal GI NETs (see Chapter 32). Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin and typical large electron-dense secretory granules. A subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH. Morphologically, tumors are characterized as a mixture of tubular-glandular, insular, and trabecular growth patterns and the presence of psammoma bodies. Tumor cells are uniform and the mitotic rate is low.34,35,37,38
Gangliocytic paraganglioma represent the third most frequent histopathologic type in the duodenum and account for 6% to 9% of duodenal carcinoids. The tumors are preferentially located in the periampullary region and are not associated with familial syndromes. Gangliocytic paragangliomas are identified by their characteristic morphology, which includes a mixture of three different cell types—spindle cells, epithelial cells, and ganglion cells. Spindle cells are arranged in fascicles and stain positive for S-100. Epithelial cells represent the endocrine component and are positive for pancreatic polypeptide, somatostatin, and chromogranin A. Ganglion cells represent the neural component and are positive for synaptophysin and NSE. Gangliocytic paraganglioma are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium.39–42
Endocrine tumors of the stomach are composed of cells with features similar to those of the normal endocrine cell counterparts. The most representative cell types are the histamine-producing enterochromaffin-like (ECL) cell in the corpus and fundus and the gastrin-producing G cell in the antrum, representing about 50% of all endocrine cells at both sites. The other endocrine cell types present throughout the stomach are somatostatin-producing (D), ghrelin-producing (P), and serotonin-producing enterochromaffin (EC) cells. All cell types may be found in gastric tumors, but preneoplastic lesions are known to be comprised mostly of ECL cells. Antral G cell hyperplasia is often observed in chronic atrophic gastritis (see Chapter 51), but it is not considered a preneoplastic lesion. More than 90% of gastric endocrine tumors are well-differentiated tumors–carcinomas (WDET/C), according to the WHO classification. More than 90% of the tumors occur in the corpus-fundus area and are composed of ECL cells, with only rare EC, G, or P cell tumors. Poorly differentiated neuroendocrine carcinomas of the GI tract are highly malignant tumors.43–50 In general, the cytosol markers NSE and PGP 9.5, and the small synaptic-like vesicle marker synaptophysin, are expressed in the poorly differentiated tumors in contrast to well-differentiated neuroendocrine tumors. Chromogranin A and tissue-specific hormones are generally absent or sparse in poorly differentiated neuroendocrine carcinomas. Hormones and chromogranin A are stored in large dense core vesicles, which are rarely observed in poorly differentiated gastric and intestinal tumors.
MOLECULAR GENETICS
Studies have shown that development of various types of GI NETs might involve different genes associated with distinct abnormalities, including point mutations, deletions, methylations, and chromosomal losses and gains (see Chapter 3). The menin gene, a tumor suppressor gene, encodes a protein of 610 amino acids; mutations of this protein cause most cases of MEN-I and a smaller proportion of sporadic and gastric duodenal endocrine tumors.51 Menin is mainly a nuclear protein, but in dividing cells, it interacts in the cytoplasm with several proteins that control transcription, regulation of genome stability, and cell division. Small intestinal carcinoid tumors show deletions on chromosome 18.52,53 Recent studies have also demonstrated overexpression of the neoplasia-related genes NAP1L1 (mitotic regulation), MAGE D2, and MTA1. These genes are thought to regulate the malignant potential of these tumors and their propensity to metastasize.54 Hindgut neuroendocrine tumors express receptors for TGF-α and EGF.51
CLASSIFICATION AND SUBTYPES
Carcinoid tumors were previously classified according to their embryologic region of origin into foregut, midgut, or hindgut tumors. This classification has now largely been abandoned and a WHO classification system is generally accepted. It has been recently updated by the European Neuroendocrine Tumor Society to a TNM and grading system (see Table 31-1).4,5
ESOPHAGUS
Carcinoid tumors of the esophagus are rare.55 However, in a series from Japan, esophageal carcinoids constituted 27% of all GI carcinoids, more frequent than ileal and appendiceal carcinoids.42 The male-to-female ratio was 3.3, indicating a male preponderance for this type of carcinoid tumor. Dysphagia is the most common presenting symptom and is usually localized to the distal portion of the esophagus. Esophageal carcinoids may occur in conjunction with adenocarcinomas arising from Barrett’s esophagus.56
STOMACH
A trend for an increased incidence of gastric neuroendocrine tumors ha been noted in surgical and endoscopic series. In the Surveillance, Epidemiology, and End Results (SEER) database, there was an increase in gastric neuroendocrine tumors, from 2.4% to 8.7% of all GI carcinoids, between 1950 and 1999.12 In mainly endoscopic series, much higher relative incidences were reported, ranging from 11% to 41% of all gastrointestinal neuroendocrine tumors.57 However, the increased relative incidence of gastric neuroendocrine tumors must be put into the context of a wider incremental trend for all types of gastrointestinal well-differentiated neuroendocrine tumors (carcinoids). Gastric neuroendocrine tumors are currently classified as well-differentiated and poorly differentiated lesions on the basis of the differentiation status of the tumor cells. The most common types are tumors derived from histamine-producing ECL cells in the corpus-fundus region, followed by gastrin producing G cell tumors of the antrum.57–59 Rare types of gastric carcinoid are those producing gastrin, ghrelin, and serotonin.43,44
Three clinicopathologic subtypes of ECL cell tumors are recognized (Table 31-2). The type I ECL tumor is associated with diffuse corpus-restricted chronic atrophic gastritis (see Chapter 51). Type II is associated with MEN-I, ZES, and hypertrophic gastropathy. Type III tumors are sporadic are not associated with any distinctive gastric pathology.43,58,59 Types I and II tumors share in common hypergastrinemia, whereas type III tumors are independent of any overt hormonal imbalance. Type I ECL tumors account for the largest fraction of well-differentiated neuroendocrine tumors of the stomach, are especially prevalent in older women, and are associated with antral G cell hyperplasia. Frequently, multiple and multicentric lesions are present and are generally small and limited to the mucosa or submucosa. Metastases are rare and survival is excellent.45 Type II ECL tumors are rare and account for only 6% of gastric carcinoids. Type II tumors arise in adult patients of both genders who have hypergastrinemia, hypertrophic hypersecretory gastropathy, and ECL cell hyperplasia. The ECL tumors are often multiple, multicentric, small in size, and limited to mucosa and submucosa. Despite metastases to local lymph nodes, the survival is excellent and tumor-related death is rare.57 Type III ECL cell tumors are usually single, isolated growths arising in the stomach, without any significant underlying gastric pathology. In one series, they accounted for 14% of all gastric carcinoids. They are more common in men, usually in their sixth decade, and without hypergastrinemia and gastrin-dependent ECL cell hyperplasia. The tumor size may be significantly larger than in types I and II (mean, 3.2 cm), with invasion of the stomach wall and metastases in more than 50% of patients.57 Predictors of malignancy in well-differentiated gastric carcinoids include size, histologic grading, mitotic count and Ki67 index, and p53 overexpression. Poorly differentiated endocrine tumors of the stomach are aggressive, usually large carcinomas and develop with no site predilection in the stomach. They usually occur in patients in the sixth to seventh decade of life. These poorly differentiated tumors are often metastatic at time of diagnosis; their prognosis is invariably poor.
PANCREAS
The differentiation between pancreatic carcinoid and other pancreatic neuroendocrine tumors is primarily a matter of definition (see Chapter 32 for a more detailed discussion). Maurer and colleagues have defined a pancreatic carcinoid as a tumor with typical histologic features of an NET along with evidence of increased serotonin metabolism.60 Using this definition, they found only 29 cases in the literature between 1966 and 1996. In a more recent publication from Japan, Soga found 156 cases of pancreatic carcinoids among 11,343 cases of neuroendocrine tumors (1.4%).61 These tumors were characterized by a high metastatic rate (66%), a large tumor size (averaging almost 7 cm), and a relatively high incidence of the carcinoid syndrome (23%). Serotonin was detected by immunohistochemical methods in 93% of cases. The five-year survival rate was extremely low (29%) when compared with small intestinal carcinoid (82%). Pancreatic carcinoids, like other pancreatic tumors, tend to present later when compared with carcinoids in other locations. Abdominal pain, diarrhea, and weight loss are the most common presenting symptoms.
DUODENUM AND AMPULLA OF VATER
About 4% of all gastrointestinal carcinoids occur in the duodenum and duodenal carcinoids account for 11% of all intestinal carcinoids. The relatively frequency of duodenal carcinoids increased from 3.6% to 16% of all intestinal carcinoids from 1973 to 2002. The annual incidence of duodenal carcinoid is 0.07/100,000. It is higher in males than in females and is higher in blacks than in whites.10–12 Duodenal carcinoids present at a mean age of 48 to 62 years. Most duodenal carcinoids give rise to symptoms related to local growth, such as obstruction, jaundice, abdominal pain with or without pancreatitis, gastrointestinal bleeding, nausea, and vomiting.35,62 A minority of patients with duodenal carcinoids (<10%) present with symptoms and/or signs of hormone overproduction. ZES occurs in approximately 10% of patients with duodenal carcinoids (see Chapter 32). The carcinoid syndrome may occur (4%) and, in rare cases, Cushing’s syndrome and acromegaly may be seen.
Gastrin (G) cell tumors represent the most frequent histopathologic type, accounting for 50% to 60% of all duodenal carcinoids (Table 31-3). G cell tumors can be nonfunctioning or functioning (Zollinger-Ellison syndrome) and may be associated with the MEN-I syndrome.36,62 Somatostatin cell tumors represent the second most frequent histopathologic type (15% to 27%).38 Somatostatin cell tumors are preferentially localized to the periampullary region. They are identified by their content of somatostatin, but a subset of tumor cells may also contain calcitonin, pancreatic polypeptide, and ACTH.
| CELL OF ORIGIN/TUMOR TYPE | RELATIVE FREQUENCY (%) |
|---|---|
| Gastrin (G) cell* | 50-60 |
| Somatostatin (D) cell | 15-77 |
| Gangliocytic paraganglioma | 6-9 |
| EC, others | <5 |
EC, enterochromaffin.
* Functional (Zollinger-Ellison syndrome) or nonfunctional.
Gangliocytic paragangliomas represent the third most frequent histopathologic type and account for 6% to 9% of duodenal carcinoids.39,40,62 The tumors are preferentially located in the periampullary region and are not associated with familial syndrome. The tumors are regarded as benign hamartomatous tumors, derived from the ventral pancreatic primordium. EC cell tumors of the duodenum are rare, and have the same histopathologic features as ECL cell tumors in the lower jejunum and ileum.
The overall five-year survival rate for duodenal carcinoids is reported to be 84%.10–12 However, survival rates vary with histopathologic type, extent of disease, presence of hormonal syndrome, and genetic background. In patients with Zollinger-Ellison syndrome, duodenal localization of the primary tumor carries a much better prognosis than pancreatic localization (see Chapter 32), with 10-year survival rates of 94% and 55%, respectively.10–12,62
SMALL INTESTINE
Forty-four percent of all gastrointestinal carcinoids arise in the small intestine. The ileum is the most frequent location for gastrointestinal carcinoids.10–12,14 The annual incidence of intestinal carcinoids is 0.63/100,000 and 0.04 and 0.31/100,000 for jejunal and ileal carcinoids, respectively. The incidence of intestinal carcinoids has increased four- to fivefold during the past 30 years. Small intestinal carcinoids result from malignant transformation of EC cells. A considerable percentage (17% to 29%) of the patients develop noncarcinoid GI and non-GI tumors, mainly adenocarcinomas of the gastrointestinal tract, suggesting a common pathogenic mechanism for carcinoid and noncarcinoid tumors.23 Familial occurrence of intestinal carcinoid is rare, although a few families have been reported. Jejunal and ileal carcinoids are also rarely associated with MEN-I.27
Patients with carcinoid tumors of the jejunum and ileum have a mean age of 55 to 63 years at the time of diagnosis.15,21 Usually, there is a delay of four to five years for diagnosis of this tumor.17 The most frequent presentations are abdominal pain (16% to 41%) and episodic small bowel obstruction (24% to 41%), which are usually of long duration. Diarrhea, gastrointestinal bleeding, flushing, and weight loss are presenting symptoms in 4% to 30%. The carcinoid syndrome is encountered in 5% to 18% of patients.16,63
Carcinoid tumors occur all along the jejunum and ileum, but increase in frequency distally, with their highest frequency in the terminal ileum.12,13 There is a single primary tumor in 74% of patients, whereas multiple tumors occur in 26% of patients.26 Clonality studies have indicated that multiple tumors are generated by the metastasis of a single primary tumor to different locations in the intestine.64 The size of the primary tumor ranges from 0.3 to 5.5 cm (mean, 2.5 cm). Intestinal carcinoids are usually associated with pronounced fibrosis and tissue scarring, which may cause intestinal obstruction and infarction. The desmoplastic stromal reaction surrounding intestinal carcinoids has been attributed to secretion of hormones and growth factors by tumor cells. Small intestinal carcinoids may produce multiple hormones. Most secrete serotonin and tachykinins (substance P and neurokinin A). Production of other hormones, such as gastrin, glucagon, cholecystokinin, calcitonin, somatostatin, and ACTH, can be demonstrated in less than 5% of cases.7,63
Data from the SEER registry for period 1973 to 2002, summarized in Table 31-4, reveal an overall five-year survival rate for intestinal carcinoids of 63% and a five-year survival rate of 84%, 72%, and 43% for localized, regional, and distant (metastatic) disease, respectively.10,11,16 However, five-year survival rates of 69% have been reported for patients with intestinal carcinoids metastatic to the liver who were given active interventional treatment.65 Risk factors for mortality include distant metastases, carcinoid syndrome, and female gender.66 Histopathologic markers shown to be associated with poor prognosis are a solid growth pattern and a Ki67 index above 1%.67
APPENDIX
The appendix is one of the most common sites in the GI tract for the development of a carcinoid tumor. The frequency of carcinoid tumors in nonselected series of appendectomy specimens ranges from 0.2% to 0.9%, but more recent data have suggested that the overall trend is toward a decreased prevalence.68,69 It is reported that appendiceal carcinoids occurs twice as often in women as in men. This observation may partly reflect the frequency of incidental appendectomy during gynecologic and gallbladder surgery but the female-to-male ratio is 2 : 1, even after correction for these factors.
Carcinoid tumors of the appendix have been classified based on their histogenesis, cell types, and histologic pattern, as well as their clinical behavior. All typical appendiceal carcinoid tumors are clinically silent and are discovered incidentally during surgery performed for symptoms of acute appendicitis or during incidental appendectomy. Most typical appendiceal carcinoids occur in the tip of the organ. They are usually small (<1 cm) and rarely exceed 2 cm in diameter. Metastases are rare in typical appendiceal carcinoid.68,69 The overall prognosis for EC cell appendiceal tumors is also favorable, with a 10-year disease specific survival of more than 98%. The incidence of metastases is less than 1% if tumor size is between 1 and 2 cm. Thus, appendectomy alone is likely to be adequate treatment for the most typical appendiceal carcinoid tumors that are smaller than 2 cm.70,71 Conversely, right hemicolectomy should be considered in selected patients with a carcinoid more than 2 cm in diameter, with a tumor that extends into the muscularis propria, and those with a positive resection margin, associated perforation, or evident lymph node metastases. Typical appendiceal carcinoids have the best prognosis of all types of carcinoids, which probably reflects the anatomic site and biologic behavior of the tumor itself; early detection and removal are beneficial.
In contrast to typical carcinoid tumors of the appendix, goblet cell carcinoids of the appendix (Fig. 31-4) have a mixed phenotype, with partial neuroendocrine differentiation and intestinal-type goblet cell morphology.72 The mean age at diagnosis is 50 years, with a range of 29 to 80 years. Goblet cell carcinoids constitute a family of appendiceal carcinoids with various morphologic features and malignant behavior. Some of them are histologically aggressive neoplasms, such as adenocarcinomas of the GI tract. The tumors demonstrate neuroendocrine differentiation, with positive staining for chromogranin and synaptophysin, although the staining is often focal and always much less extensive than in classical carcinoid tumors. The most common and distinctive feature is mucin-containing goblet-shaped epithelial cells arranged in round or oval clusters.73,74 The overall five-year survival rate for different subtypes of goblet cell carcinoids is 42%, with a three-year survival rate of only 17% for the most malignant subtypes.
