[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 35 Gastrointestinal and Hepatic Manifestations of Systemic Diseases
RHEUMATOLOGIC AND COLLAGEN VASCULAR DISEASES
Rheumatologic diseases encompass a wide variety of clinical syndromes and are frequently associated with gastrointestinal abnormalities (Table 35-1). In addition, the medications used to treat these diseases often produce gastrointestinal and hepatic toxicity. This section focuses on the more common abnormalities that may be encountered by the gastroenterologist.
Table 35-1 Gastrointestinal Manifestations of Rheumatologic Diseases
| DISEASE | ABNORMALITY/ASSOCIATION | CLINICAL MANIFESTATIONS |
|---|---|---|
| Rheumatoid arthritis | Temporomandibular arthritis | Impaired mastication |
| Esophageal dysmotility | Dysphagia, GERD | |
| Visceral vasculitis | Abdominal pain, cholecystitis, intestinal ulceration and infarction | |
| Amyloidosis | Pseudo-obstruction, malabsorption, protein-losing enteropathy, intestinal ulceration and infarction, gastric outlet obstruction | |
| Portal hypertension (Felty’s syndrome) | Variceal hemorrhage | |
| Gold enterocolitis | Enteritis, diarrhea, fever, eosinophilia, megacolon | |
| Scleroderma | Esophageal dysmotility | Dysphagia, GERD, stricture, Barrett’s esophagus |
| Gastroparesis | Gastric retention, GERD | |
| Intestinal fibrosis and dysmotility | Constipation, pseudo-obstruction, malabsorption, intussusception, volvulus, pneumatosis intestinalis | |
| Pseudodiverticula | Hemorrhage, stasis, bacterial overgrowth | |
| Arteritis (rare) | Mesenteric thrombosis, infarction, pancreatic necrosis | |
| Pancreatitis | Calcific pancreatitis, pancreatic exocrine insufficiency | |
| SLE | Esophageal dysmotility | Dysphagia, reflux |
| Mesenteric vasculitis | GI ulceration, intestinal infarction, intussusception, pancreatitis, pneumatosis intestinalis | |
| Sjögren’s syndrome | Desiccation of membranes | Oral fissures, oropharyngeal dysphagia |
| Esophageal webs | Dysphagia | |
| Gastric lymphoid infiltrates | ||
| Pancreatitis | Abdominal pain, pancreatic exocrine insufficiency | |
| Primary biliary cirrhosis | Jaundice, hepatic failure, variceal hemorrhage | |
| Polymyositis-dermatomyositis | Skeletal muscle dysfunction | Aspiration, impaired glutition |
| Dysmotility | Dysphagia, GERD, gastroparesis, constipation, diverticula | |
| Mesenteric vasculitis (rare) | GI ulceration, perforation, pneumatosis intestinalis | |
| MCTD | Dysmotility | Dysphagia, GERD, stricture, gastroparesis, bezoars, pseudo- obstruction |
| PAN | Mesenteric vasculitis (rare) | Ulceration, perforation, pancreatitis |
| Mesenteric vasculitis | Cholecystitis, appendicitis, intestinal infarction, pancreatitis, perforation, strictures, mucosal hemorrhage, submucosal hematomas | |
| CSS | Mesenteric vasculitis | Hemorrhage, ulceration, intestinal infarction, perforation |
| Eosinophilic gastritis | Gastric masses | |
| Henoch-Schönlein purpura | Mesenteric vasculitis | Intussusception, ulcers, cholecystitis, hemorrhage, intestinal infarction, appendicitis, perforation |
| Kohlmeier-Degos disease | Mesenteric vasculitis | Hemorrhage, ulceration, intestinal infarction, malabsorption |
| Cogan’s syndrome | Mesenteric vasculitis (infrequent) | Hemorrhage, ulceration, intestinal infarction, intussusception |
| Crohn’s disease | Bloody diarrhea, abdominal pain, fissures, fistulas | |
| Wegener’s granulomatosis | Mesenteric vasculitis | Cholecystitis, appendicitis, ileocolitis, intestinal infarction |
| Cryoglobulinemia | Mesenteric vasculitis (rare) | Intestinal infarction, ischemia |
| Behçet’s disease | Mucosal ulcerations | Hemorrhage, perforation, pyloric stenosis |
| Complications as in rheumatoid arthritis | ||
| Reactive arthritis | Ileocolonic inflammation | Usually asymptomatic |
| Familial Mediterranean fever | Serositis/peritonitis, amyloidosis, PAN, Henoch-Schönlein purpura | Abdominal pain, fever, dysmotility |
| Marfan/Ehlers-Danlos syndromes | Defective collagen | Megaesophagus, hypomotility, diverticula, megacolon, malabsorption, perforation, arterial rupture |
CSS, Churg-Strauss syndrome; GERD, gastroesophageal reflux disease; GI, gastrointestinal; MCTD, mixed connective tissue disease; PAN, polyarteritis nodosa; SLE, systemic lupus erythematosus.
RHEUMATOID ARTHRITIS
Approximately 0.8% of adults worldwide are affected with rheumatoid arthritis (RA), which is a chronic, inflammatory autoimmune disease primarily targeting the synovial tissues with systemic manifestations. Oropharyngeal symptoms may occur in patients with RA as a result of xerostomia, temporomandibular joint (TMJ) arthritis, cervical spine abnormalities, and laryngeal involvement.1 Esophageal dysmotility, characterized by low-amplitude peristaltic waves, has been described in the proximal, middle, and distal esophagus with reduced lower esophageal sphincter (LES) pressure.1,2 Rheumatoid vasculitis typically occurs in the setting of severe RA with rare gastrointestinal manifestations such as ischemic cholecystitis or appendicitis, ulceration, pancolitis, infarction, or intra-abdominal hemorrhage due to a ruptured visceral aneurysm.3,4 Other gastrointestinal complications of RA include amyloidosis (discussed later) and malabsorption. Felty’s syndrome–RA, splenomegaly, and leukopenia have been associated with severe infections, portal hypertension, and variceal hemorrhage.5
Hepatic Abnormalities
Abnormal liver function tests, especially elevations of serum alkaline phosphatase of hepatobiliary origin,6–8 are commonly observed in patients with RA. In one large series of patients with RA,6 18% had elevated levels of serum alkaline phosphatase and 11% were found to have hepatomegaly. Fluctuations in serum alkaline phosphatase levels have also been reported to correlate with disease activity.6,8 However, degrees of alkaline phosphatase elevations are usually modest, with the mean level being less than two-fold abnormal.6 Furthermore, other clinical signs of liver disease are usually absent, and liver biopsy and autopsy studies have not revealed any consistent or specific findings, with the most common abnormalities being fatty change, Kupffer cell hyperplasia, and mild mononuclear cell infiltration of the portal tracts or rare parenchymal foci of hepatocyte necrosis.6,9–11 Periportal fibrosis is also present in a small minority of cases.11 Determination of etiology of hepatic dysfunction in patients with active rheumatoid arthritis is complicated by the fact that many of the agents commonly used as therapy for this disease have known potential for liver injury.7,10–12
In a small subset of patients with RA and/or Sjögren’s syndrome, antimitochondrial antibodies are present along with the biochemical and histologic features of primary biliary cirrhosis.13–15 The incidence of primary biliary cirrhosis or autoimmune hepatitis appears to be much higher in patients with Sjögren’s syndrome alone than in those with Sjögren’s RA.16,17
Because chronic hepatitis C and RA are relatively common diseases of adults, it is not surprising that these entities are found concurrently in some patients. However, in addition, it has been noted that 75% of individuals with chronic hepatitis C infection develop rheumatoid factors,18 and a subset of these rheumatoid factor–positive individuals develop essential mixed cryoglobulinemia that may be manifested in part by development of arthralgias.19,20 Liver disease in such individuals is often asymptomatic and biochemical abnormalities modest or even absent.19,20 Thus some individuals with essential mixed cryoglobulinemia associated with chronic hepatitis C infection may instead be labeled as having RA. However, anticyclic citrulinated peptide (CCP) antibodies are rarely found in subjects with chronic hepatitis C and nonspecific rheumatologic manifestations and thus anti-CCP antibodies appear to be reliable markers of RA.21 Most rheumatic disease patients with progressive liver disease have concomitant chronic viral or autoimmune hepatitis.22 In patients with concomitant hepatitis B infection, the intermittent use of tumor necrosis factor (TNF) inhibitors or other immunosuppressive therapy may be associated with severe flares of hepatitis B23–25 and prophylactic use of antiviral therapy should be considered.25 TNF inhibitor therapy in RA also has been associated with flares of severe liver disease, with characteristics of autoimmune hepatitis.26
Perhaps the most distinctive association between RA and hepatic abnormalities is seen in another subset of patients who develop splenomegaly and neutropenia (Felty’s syndrome). Felty’s syndrome is associated with an even higher incidence of hepatomegaly and liver function test abnormalities than seen in uncomplicated RA.27,28 However, there is little correlation between serum hepatic enzyme abnormalities and histopathologic findings.27,28 Nevertheless, more than half of patients with this syndrome have been found to have hepatic histologic abnormalities that range from sinusoidal lymphocytosis and portal fibrosis to the more distinctive picture of nodular regenerative hyperplasia, which has been reported on multiple occasions in patients with Felty’s syndrome27–30 and in one small prospective series was found to be present in 5 of 18 (28%) patients. Hepatic encephalopathy or other manifestations of liver failure have not been reported in patients with Felty’s syndrome, and nodular regenerative hyperplasia but portal hypertension and esophageal variceal hemorrhage may occur.28–30
Gastrointestinal Abnormalities
The most common gastrointestinal problems encountered in patients with RA are due to drug therapy with nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying antirheumatic drugs (DMARDs). NSAIDs are most commonly associated with upper gastrointestinal complications such as perforation, ulcers, and bleeding (see Chapters 52 and 53). Less commonly recognized complications of NSAIDs include pill esophagitis (Chapter 45), small bowel ulceration (Chapter 115), strictures of the small and large intestine, and exacerbations of diverticular disease and inflammatory bowel disease.31 Significant risk factors for the development of serious upper gastrointestinal events in patients with RA include NSAIDs therapy, age older than 65 years, history of peptic ulcer disease, glucocorticoid therapy, and severe RA.32,33 In patients with RA, the use of certain selective cyclooxygenase-2 inhibitors results in a lower incidence of gastrointestinal complications than that seen with nonselective NSAIDs.34,35 Helicobacter pylori and NSAIDs are independent and possibly synergistic risk factors for peptic ulceration. As such, chronic NSAID users who develop ulcers should be assessed for H. pylori infection and undergo eradication therapy when infection is present.32,33 Although hypergastrinemia has been reported in patients with RA, the incidence of peptic ulcers is no greater than that seen in patients with osteoarthritis.36 As reviewed in Chapter 53, NSAID-associated gastric and duodenal ulcers can be prevented with misoprostol, high-dose histamine (H2) blockers, and proton pump inhibitors.37 Once identified, ulcers may be treated successfully using proton pump inhibitors despite continued NSAID therapy. In the subgroup of patients with a history of bleeding ulcers, therapy with cyclooxygenase-2 inhibitors rather than NSAIDs may be cost effective and less expensive than combining an NSAID with a proton pump inhibitor.38,39
Synthetic DMARDs such as gold and penicillamine are rarely used because of toxicity and marginal efficacy.40 Gold, parenteral as well as oral forms, has been associated with diarrhea, enterocolitis, toxic megacolon, and death. The onset of gold colitis usually occurs within several weeks after the start of therapy and is manifested by nausea, vomiting, diarrhea, and fever. Although the colon is most commonly involved, gold-induced gastrointestinal toxicity may affect the esophagus, stomach, and small bowel, with 25% of patients developing a peripheral eosinophilia.41 Treatment includes dose reduction or discontinuation of gold, antidiarrheals, glucocorticoids, cromolyn sodium, or the chelating agent dimercaprol.41,42
Leflunomide, a synthetic DMARD that inhibits pyrimidine synthesis, can cause diarrhea in up to 32% of patients.43 It may also cause severe hepatic toxicity (see Chapter 86). Biologic DMARDs, which inhibit the action of TNF-α (infliximab, etanercept, and adalimumab) or interleukin-1 (IL-1; anakinra), have not shown significant gastrointestinal adverse effects but may cause hepatic toxicity on occasion (see later).
ADULT-ONSET STILL’S DISEASE
Adult-onset Still’s disease, the adult form of juvenile RA, often has gastrointestinal manifestations such as weight loss, sore throat, hepatosplenomegaly, elevated aminotransferases, and abdominal pain, in addition to fever.44 In contrast to the lack of significant hepatic dysfunction in classic rheumatoid arthritis, adults with Still’s disease present with features of mild hepatitis in the majority of cases and life-threatening acute liver failure in exceptional cases.45–49 Variable degrees of aminotransferase and alkaline phosphatase elevations are typically observed in such patients during symptomatic disease flares. Liver biopsies usually reveal moderate portal mononuclear cell infiltration with occasional evidence of focal hepatocyte necrosis.46 Biopsies obtained in patients with jaundice and biochemical evidence of severe hepatitis have been found to have interface and lobular hepatitis with lymphoplasmocytic inflammation reminiscent of autoimmune hepatitis.49 Most cases of severe hepatitis have been observed in patients previously treated with salicylates or other NSAIDs,46,49 but liver enzyme abnormalities are also commonly noted prior to therapy. Some patients with severe hepatitis have been reported to respond to immunosuppressive therapy,49 whereas others required liver transplantation or have died of liver failure.45,47–49 Although severe hepatitis is a rare complication of adult-onset Still’s disease, liver failure appears to be the most common cause of death related to this disease.45
PROGRESSIVE SYSTEMIC SCLEROSIS
Gastrointestinal manifestations occur in up to 90% of patients with PSS.50 Gastrointestinal tract involvement can occur from the mouth to the anus. Atrophy and fibrosis of the perioral skin may limit mandibular motion. The periodontal ligament may become hypertrophic, and the gingivae, tongue papillae, and buccal mucosa may become friable and atrophic, resulting in impaired sensation and taste.
The esophagus is the most frequently involved gastrointestinal organ.50 On pathology, atrophy of smooth muscle layers and intimal proliferation of arterioles is seen in the distal esophagus.51 Dysphagia occurs as a result of impaired esophageal motility, and gastroesophageal reflux disease (GERD) is related to hypotensive LES pressures, impaired esophageal clearance of acid, and reduced acid-neutralizing capacity due to xerostomia with reduced saliva production.52 The incidence of esophagitis approaches 100% in patients with severe cutaneous involvement.53 The extent of hypomotility varies from occasional uncoordinated contractions to complete paralysis.54 The severity of esophageal dysmotility correlates with the development of interstitial lung disease.55,56 Stricture formation from GERD may contribute to dysphagia, affecting approximately 8% of patients.57 Upper gastrointestinal hemorrhage has been reported from esophageal ulcers, rare esophageoatrial fistulas, and esophageal telangiectasia.58,59 An increased risk of infectious esophagitis with Candida (see Chapter 45) has been attributed to esophageal dysmotility and concomitant immunosuppressive therapy.60 Severe esophagitis typically responds to proton pump inhibitors but may require higher doses for maximal effect.61 A neuropathic achalasia-like syndrome has also been reported.62 The prevalence of Barrett’s metaplasia of the esophagus in patients with PSS ranges between 7% and 38%.63,64 A recent cohort study reported an increased incidence of carcinoma of the tongue and esophagus in patients with PSS.65
Gastric involvement most commonly leads to gastroparesis but other manifestations may include dyspepsia, exacerbation of GERD, or gastric hemorrhage from gastric antral vascular ectasia (GAVE, watermelon stomach). Delayed gastric emptying has been shown using radionucleotide scintigraphy or radiopaque pellets, with cutaneous electrogastrography demonstrating bradygastria and decreased amplitude of electrical activity.56,66,67 Prokinetic agents such as metoclopramide and erythromycin may increase LES pressures and improve gastric emptying in some patients with PSS.57
The pathologic changes in the small bowel of PSS patients consist of smooth muscle atrophy and deposition of collagen in submucosal, muscular, and serosal layers. Small bowel hypomotility is present in as many as 88% of cases.68 In the early stages of the disease, hypomotility is caused by neuropathic involvement, which may be more responsive to prokinetic agents. In advanced cases, hypomotility is more likely a result of “myopathic” and “fibrotic” changes.57 The interdigestive migrating motor complex (IMMC) is frequently absent or markedly diminished in amplitude in PSS patients with symptoms of intestinal dysmotility.69 Small bowel radiographic abnormalities are present in about 60% of PSS patients, but they may not correlate with symptoms. The duodenum is often dilated, especially in its second and third portions, often with prolonged retention of barium.70 Typically the jejunum is dilated and foreshortened because of mural fibrosis, but valvulae conniventes of normal thickness give rise to an accordion-like appearance. Pneumatosis cystoides intestinalis, pseudo-obstruction, pseudodiverticula, sacculations, intussusception, acquired intestinal lymphangiectasia, and small bowel volvulus have been noted.71–73
Symptoms of small intestinal PSS include bloating, borborygmi, anorexia, nausea, and vomiting. Rarely, thrombosis of large mesenteric arteries with extensive bowel necrosis may occur.74 Malabsorption with steatorrhea is present in as many as a third of PSS patients68 and is caused by bacterial overgrowth (see Chapters 101 and 102). Although antibiotic therapy can be effective in these patients, d-xylose malabsorption is often incompletely reversed, suggesting that collagen deposition in PSS may also contribute to malabsorption.75 Although often disappointing, the use of prokinetic agents such as metoclopramide may be effective in some cases. Octreotide in low doses and erythromycin also may provide sustained relief from nausea, abdominal pain, and bloating in some patients with pseudo-obstruction.76
Delayed colonic transit and impaired anal sphincter function are frequently found in constipated patients with PSS.77,78 Cisapride (a drug no longer available in the United States) accelerates colonic transit,79 but refractory cases may require surgery.80 Colonic stricture, volvulus, and bleeding from mucosal telangiectasias have been reported.81,82 Wide-necked diverticula can be seen, especially in the antimesenteric border of the transverse and descending colons. Rectal prolapse worsens anal sphincter function, aggravating fecal incontinence in patients with PSS.83 Rectal bleeding can occur from vascular ectasia.84
Pancreatic exocrine secretion is depressed in a third of patients with PSS, and idiopathic calcific pancreatitis has been reported.85 In addition, arteritis resulting in ischemic pancreatic necrosis has been described in patients with PSS.86,87 Gallbladder motility is not altered in PSS.88
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is a multisystem disease characterized by immune system abnormalities and the production of autoantibodies with tissue damage. Gastrointestinal symptoms are common in patients with active SLE. Oral ulcers (one of the criteria used to diagnose SLE) are most commonly seen in the buccal mucosa, hard palate, and vermilion border.89 In SLE, dysphagia (1% to 13% of patients) and GERD (11% to 50% of patients) poorly correlates with esophageal manometric abnormalities such as hypoperistalsis.90 Dysphagia is typically related to GERD or peptic stricture, with one report of esophageal epidermolysis bullosa acquisita.91 Malabsorption of d-xylose, steatorrhea, hyperplastic gastropathy, and protein-losing enteropathy have been described (see Chapter 28); the latter can be steroid responsive.92,93 Lupus peritonitis is a diagnosis that can be made only after other causes have been carefully excluded. Pneumatosis cystoides intestinalis may be an isolated benign condition or may accompany lupus vasculitis or necrotizing enterocolitis.94,95
One of the most devastating complications of lupus is gastrointestinal vasculitis. Affecting only 2% of patients, it has a fatality rate of more than 50%.96 Common sequelae include ulceration, hemorrhage, perforation, and infarction.97–99 Pancreatitis,100,101 gastritis, hemorrhagic ileocolitis resembling inflammatory bowel disease, and intussusception also have been reported. Although occasional case reports have documented polyarteritis-like changes on visceral arteriograms (described later), the typical pathologic changes are seen in the small vessels of the bowel wall rather than the medium-sized vessels of the bowel wall.94 Computed tomography (CT) scan may help establish the diagnosis of ischemic bowel disease in SLE if there are at least three of the following five CT findings: (1) bowel wall thickening, (2) target sign (a thickened bowel wall with peripheral rim enhancement or an enhancing inner and outer rim with hypoattenuation in the center), (3) dilatation of intestinal segments, (4) engorgement of mesenteric vessels, and (5) increased attenuation of mesenteric fat.102 Because visceral angiography is not routinely helpful, the diagnosis is difficult to establish. The role of endoscopy or upper gastrointestinal series in the diagnosis of lupus vasculitis is not well defined. The diagnosis currently rests on clinical judgment, findings on CT scans, and occasionally from surgical specimens when exploratory laparotomy is undertaken to rule out acute surgical emergencies.103 Treatment of abdominal lupus-induced vasculitis with glucocorticoids has been largely unsatisfactory. Although a controlled clinical trial comparing cyclophosphamide with glucocorticoids has not been performed, anecdotal reports of dramatic responses to intravenous cyclophosphamide are promising.94 Some investigators have suggested that cyclophosphamide be considered early in patients who have not shown significant improvement shortly after high-dose glucocorticoids are started.
Patients with SLE have a 25% to 50% incidence of abnormal liver biochemical tests during the course of their disease, but clinically significant liver disease is rare.104 Abnormal liver tests are commonly associated either with medication use or with mild, predominantly lobular hepatitis associated with periods of SLE activity.104,105 Despite the shared association with antinuclear antibodies, the typical histologic and clinical features of autoimmune hepatitis are rarely observed in adult patients with SLE.104,106 Concurrent SLE and autoimmune hepatitis occur more frequently in pediatric patients.106 In addition, SLE patients with anticardiolipin antibodies or lupus anticoagulants may have thrombotic events in the liver leading to Budd-Chiari syndrome or nodular regenerative hyperplasia manifested by complications of portal hypertension.104,107
POLYMYOSITIS AND DERMATOMYOSITIS
Polymyositis is a syndrome characterized by weakness, high serum levels of striated muscle enzymes (creatine kinase, aldolase), and electromyographic (EMG) or biopsy evidence of an inflammatory myopathy. When accompanied by a characteristic violaceous rash on the extensor surfaces of the hands and periorbital regions, the disease is termed dermatomyositis. The primary gastrointestinal symptoms are due to involvement of the cricopharyngeus, resulting in nasal regurgitation, tracheal aspiration, and impaired deglutition.108 Involvement is not limited to skeletal muscle fibers. Disordered esophageal motility, impaired gastric emptying, and poorly coordinated small intestinal peristalsis have been noted.109 Malabsorption, malnutrition, and pseudo-obstruction rarely occur.110 Pathologically, edema of the bowel wall, muscle atrophy, fibrosis, and mucosal ulcerations or perforation due to vasculitis may be seen at any level of the gut. Symptoms include heartburn, bloating, constipation, and gastrointestinal hemorrhage. Pneumoperitoneum, pneumatosis intestinalis, colonic dilation, and pseudodiverticula also may be seen. Perforations of the esophagus and of duodenal diverticula have been described as rare complications.111,112
In middle-aged to older adult patients, dermatomyositis and possibly polmyositis are associated with an increased prevalence of malignancy.113 The possibility that gastrointestinal symptoms may be the resultl of an underlying malignancy should be considered when evaluating these patients (see Chapter 22).
MIXED CONNECTIVE TISSUE DISEASE
Mixed connective tissue disease (MCTD) is a syndrome with overlapping features of PSS, polymyositis, and SLE, often in the presence of high levels of antibody directed against ribonucleoprotein. Upper gastrointestinal symptoms are seen in most patients.114 Abnormalities include diminished esophageal peristalsis (48%), esophageal stricture (6%), abnormal gastric emptying (6%), and gastric bezoar (2%).114 Small intestinal and colonic involvement includes dilation of proximal bowel, slow transit, intestinal pseudo-obstruction, diverticulosis, and, rarely, intestinal vasculitis. Pancreatitis also has been reported.114 Unlike PSS, the esophageal motility disturbances seen in MCTD appear to improve with the administration of glucocorticoids.
SJÖGREN’S SYNDROME
Sjögren’s syndrome (SS), occurring alone (primary SS) or in association with systemic autoimmune rheumatic diseases (secondary SS), is characterized by lymphocytic tissue infiltration of lacrimal and salivary glands with the clinical findings of keratoconjunctivitis sicca and xerostomia. As reviewed in Chapter 22, excessive dryness of the mouth and pharynx leads to oral symptoms of soreness, adherence of food to buccal surfaces, fissuring of the tongue, and periodontal disease.115 Dysphagia, reported by up to three quarters of patients with SS, can result from esophageal dysmotility and a lack of saliva; however, symptoms do not correlate with manometry or salivary secretion.116–118 Mild atrophic antral gastritis can be seen in 25% of patients with primary SS, but 31% were infected with H. pylori.119 Older studies that reported higher rates and greater severity of gastritis did not control for H. pylori infection. GAVE can occur in patients with SS and is responsive to fulguration therapy.120 A triad of sclerosing cholangitis, chronic pancreatitis, and SS has been reported in eight patients.121 Pancreatic exocrine function is frequently impaired.122 In primary SS, 7% of patients have positive antimitochondrial antibodies and among patients with primary biliary cirrhosis, clinical manifestations of SS are common (see Chapter 89).115
POLYARTERITIS NODOSA AND OTHER VASCULITIDES
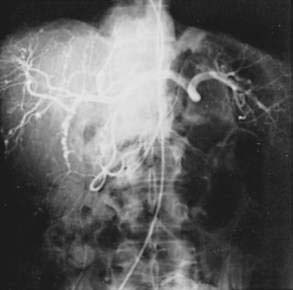
Polyarteritis nodosa (PAN) is a necrotizing vasculitis of small and medium-sized muscular arteries, frequently with visceral involvement (Fig. 35-1). A characteristic feature of this condition is the finding of aneurysmal dilatations up to 1 cm in size seen on visceral angiography (Fig. 35-2). Abdominal complications occur in up to 50% of patients and carries a poor prognosis.123 Other clinical features of PAN include fever, myalgia, arthralgia of the large joints, mononeuritis multiplex, and livedo reticularis. Mesenteric visceral arteriograms are abnormal in up to 80% of patients with gastrointestinal involvement, with the superior mesenteric artery most commonly involved.123 Organ damage resulting from ischemia frequently underlies symptoms. The most common gastrointestinal manifestation is abdominal pain with other common symptoms including nausea, vomiting, and gastrointestinal bleeding.123 Bowel infarction and perforation, aneurysmal rupture, and acute cholecystitis are common causes of acute abdomen in PAN.123 Rarely, PAN can present as acalculous cholecystitis secondary to isolated vasculitis of the gallbladder.124 Pancreatitis,125 appendicitis,126 hemobilia,127 solitary biliary strictures,128 and hepatic infarcts129 also have been reported to complicate PAN. The frequency of hepatitis B infection in PAN has declined from more than 30% to less than 10% because of improved screening of the blood supply and vaccination against hepatitis B.130 Because of the frequent association with hepatitis B infection and potential association with hepatitis C infection (see Chapters 78 and 79), patients with clinical manifestations of PAN should be assessed for evidence of hepatitis B or C infection.
Churg-Strauss syndrome (CSS, allergic granulomatous angiitis) is a small to medium-sized vessel vasculitis characteristically associated with eosinophilia, asthma, sinusitis, and rhinitis. Abdominal pain is the most common gastrointestinal symptom.131 Preceding the vasculitic phase of CSS, patients may present with an eosinophilic gastroenteritis associated with abdominal pain, nausea, vomiting, diarrhea, and bleeding with an absolute eosinophil count of greater than 1500 cells/mm3 (see Chapter 27).132 Additional gastrointestinal manifestations of CSS include pancreatitis, cholecystitis, ascites, small intestinal ulcerations, and perforation.131,133,134 Colonic involvement may present with multiple ulcers or obstruction.134,135
Henoch-Schönlein purpura (HSP) is a systemic vasculitis characterized by nonthrombocytopenic purpura, arthralgias, renal disease, and colicky abdominal pain. Although the disease is frequently seen in children and adolescents, adults of any age may be affected. Colicky abdominal pain and gastrointestinal bleeding are seen in two thirds of cases.136 Colonoscopic and endoscopic findings in bleeding patients include erosive duodenitis, small aphthous ulcerations, and petechial colonic lesions.137 In patients who undergo CT scan, common findings include bowel-wall thickening, dilated intestinal segments, mesenteric vascular engorgement, and regional lymphadenopathy.138 Other reported gastrointestinal complications of HSP include protein-losing enteropathy, esophageal and ileal structures, gastric and small bowel perforations, bowel infarction, pancreatitis, appendicitis, cholecystitis, intramural hematomas, and intussusception.139
Malignant atrophic papulosis (Kohlmeier-Degos disease) is a rare vasculitis that causes nausea, vomiting, bleeding, malabsorption, bowel ischemia, and perforation.140 Scattered on the skin are red papules that become hypopigmented atrophic scars (see Fig. 22-13).
Cogan’s syndrome is characterized by nonsyphilitic interstitial keratitis, audiovestibular symptoms, and large-vessel vasculitis that may involve the gut. Gastrointestinal manifestations include abdominal pain, diarrhea, hepatomegaly, and splenomegaly.141 Crohn’s disease has been reported in association with this rare condition.142
Wegener’s granulomatosis, a systemic vasculitis characterized by pulmonary, sinus, and renal involvement, less commonly affects the gut.143 Inflammatory ileocolitis with hemorrhage, gangrenous cholecystitis, and bowel infarction have been reported.144 Wegener’s granulomatosis may mimic Crohn’s disease with granulomatous gastritis or ileitis.145,146
Mixed immunoglobulin (IgG-IgM) cryoglobulinemia characterized by the triad of purpura, arthralgia, and asthenia may complicate chronic hepatitis C infection (see Chapter 79) and a variety of immune diseases, including inflammatory bowel disease, celiac disease, and postintestinal bypass syndrome. Cryoglobulinemia may cause severe visceral vasculitis with diarrhea, ischemia, and perforation of the small or large intestine.147
BEHÇET’S DISEASE
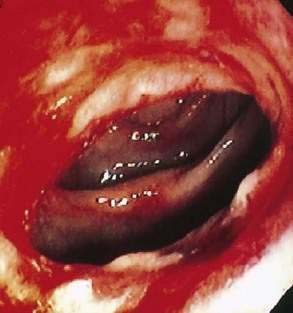
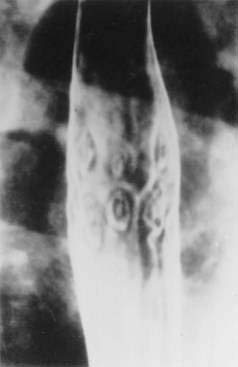
Behçet’s disease is an idiopathic inflammatory disorder characterized by oral aphthous ulcers, genital ulcers, uveitis, and skin lesions, with gastrointestinal involvement occurring in up to 50% of patients.148 As in Crohn’s disease, ulceration may occur throughout the alimentary tract, with the ileocecal region most commonly affected. Differentiating Behçet’s from Crohn’s disease can be difficult because of similarities in gastrointestinal symptoms, endoscopic findings, histology, and extraintestinal manifestations. Involvement of the esophagus includes ulcers (Fig. 35-3), varices, and perforation.149 In the stomach, which is infrequently involved, aphthous ulcers may be seen. The typical intestinal involvement in Behçet’s disease includes “punched-out” ileocecal ulcerations, the most common finding on colonoscopy. Additional manifestations of Behçet’s disease include abdominal pain, diarrhea, bleeding, perforation, and fistulas (perianal, rectovaginal, and enteroenteric).150 Hepatic or portal vein thrombosis may occur in patients with Behçet’s, and this syndrome should be included in the differential diagnosis of patients presenting with Budd-Chiari syndrome.151,152 Medical therapy of the gastrointestinal lesions of Behçet’s disease includes mesalamine, glucocorticoids, immunomodulators such as azathioprine and 6-mercaptopurine, infliximab, and thalidomide.153,154 Surgical intervention is associated with a high rate of recurrence, with nearly 50% requiring repeat surgery.155
SERONEGATIVE SPONDYLOARTHROPATHIES (REACTIVE ARTHRITIDES)
The term seronegative spondyloarthropathy is used to describe an interrelated group of inflammatory disorders that include ankylosing spondylitis, reactive arthritis (formerly called Reiter’s syndrome), and psoriatic arthritis. The term has also been used to describe the enteropathic spondylitis associated with Crohn’s disease and ulcerative colitis.156 These disorders are characterized by the absence of rheumatoid factor, an association with human leukocyte antigen-B27 (HLA-B27), and inflammation at the site of bony insertion of ligaments and tendons (enthesitis). There is a high prevalence of clinically silent inflammatory colon lesions in patients with these seronegative spondyloarthropathies.157 Capsule endoscopy may yield more small bowel abnormalities than ileocolonoscopy.158 Conversely, 22% of patients with inflammatory bowel disease have evidence of a seronegative spondyloarthropathy, with ankylosing spondylitis most commonly seen.159 Although infliximab has been shown to induce remissions in some patients with ankylosing spondylitis as well as in Crohn’s disease, the effect of infliximab on gastrointestinal inflammatory lesions in typical seronegative spondyloarthropathies has not yet been studied.
MARFAN’S AND EHLERS-DANLOS SYNDROMES
Owing to defective collagen synthesis, patients with Marfan’s or Ehlers-Danlos syndrome develop skin fragility, megaesophagus, small intestine hypomotility, giant jejunal diverticula, bacterial overgrowth, and megacolon.160 Mesenteric arterial rupture and intestinal perforation also can occur.161
FAMILIAL MEDITERRANEAN FEVER
Familial Mediterranean fever (FMF) is an autosomal recessive inherited disease characterized by recurrent self-limiting attacks of fever, joint pain, and abdominal pain. Acute attacks typically last three to five days. FMF is most commonly seen in people of Mediterranean origin including Sephardic Jews, Arabs, Turks, Greeks, Italians, and Armenians, although FMF has been described in Cubans, and Belgians. The gene responsible for FMF in Mediterranean patients, designated MEFV, has been mapped to chromosome 16, which encodes a 781-amino acid protein called pyrin or marenostrin.162
Gastrointestinal symptoms, typically manifest as episodic abdominal pain, are seen in 95% of patients, and this may be the presenting symptom in as many as 50% of cases.163 Abdominal pain may be diffuse or localized and may range from mild bloating to acute peritonitis with boardlike rigidity, rebound tenderness, and air-fluid levels on upright radiographs. The acute presentation may be confused with acute appendicitis, cholecystitis, or pelvic inflammatory disease, whereas relapsing and remitting attacks may be confused with other diseases such as porphyrias. Small bowel obstruction from adhesions may occur as a consequence of recurrent sterile peritonitis or as a result of previous exploratory surgery. In patients with obstruction due to adhesions, abdominal attacks without other typical symptoms (arthralgias, fever) should tip off the clinician to consider an obstruction.164 The diagnosis of FMF is based on validated clinical criteria including fever, serositis, location of pain, and response to colchicine.165
In FMF, the long-term prognosis is poor in patients who develop nephrotic syndrome and chronic kidney disease from amyloid A deposition163 (amyloidosis is discussed later in this chapter). Prophylactic colchicine has been shown to reduce the frequency of attacks, prevent amyloidosis, and avoid renal failure.166 Vasculitis in the form of HSP, PAN, protracted febrile myalgia, or Behçet’s is encountered in 3% of FMF patients.163
ONCOLOGIC AND HEMATOLOGIC DISEASES
METASTASES
Metastasis to the gut can occur by direct invasion from adjacent organs, by intraperitoneal seeding, or by hematogenous or lymphatic spread. About 20% of all patients with nongastrointestinal malignancies have metastases to the gastrointestinal tract, the most common of which are breast, lung, and ovarian cancers and melanoma (Fig. 35-4).167 Patterns of metastases are not random but reflect the location and histologic type of the primary tumor. The esophagus is most frequently affected by direct extension from tumors arising from adjacent structures (bronchus and stomach). The stomach is a particularly common site of breast cancer metastases, and the small intestine can be involved by tumor extension from the stomach, pancreas, biliary system, kidney, or retroperitoneum. The pancreas is usually an asymptomatic site of metastasis, with the most common primary tumors being lung, gastrointestinal, and renal.168 The ileum may be affected by cancers arising in the colon or pelvis. Metastases to the gut typically begin in the serosa or submucosa and produce intraluminal lesions that can lead to obstruction, submucosal polypoid masses that can result in intussusception or ulcerated mucosal lesions. The most common presenting clinical condition in patients with metastatic lesions to the gut is small bowel obstruction. Lobular breast cancer, malignant melanoma, and non–small cell lung cancer are the most common neoplasms to cause small bowel obstruction from isolated metastases.169 In addition, pain, fever, ascites, gastrointestinal bleeding, and perforation have been described.
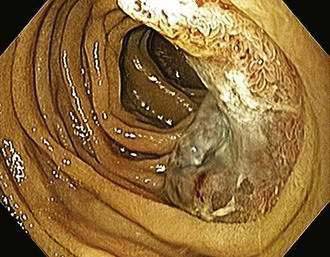
Metastases to the gastrointestinal tract may be difficult to diagnose. Barium contrast studies may reveal extramural masses, mucosal ulcerations, or a rigid stomach with the appearance of linitis plastica. CT may be helpful in determining the primary tumor, in tumor staging, and in detecting large serosal implants. Small bowel metastases, however, are detectable radiographically in only 50% of cases.170
When feasible, surgical resection should be used to treat gastrointestinal metastases that result in obstruction, perforation, or significant hemorrhage. If a solitary bowel metastasis is the only evident site of disseminated malignancy, segmental bowel resection should be performed, offering a small chance for cure. In aggressive resections of melanoma metastases, the mesenteric nodes draining the involved segment of bowel should be resected because they frequently contain tumors.171
PARANEOPLASTIC SYNDROMES
Paraneoplastic syndromes affecting the gut include the hormonal effects of carcinoid tumors, vasoactive intestinal polypeptide-secreting tumors (VIPomas), gastrinomas, and somatostatinomas (see Chapters 31 and 32), as well as the gastrointestinal effects of hypercalcemia (constipation, nausea, and vomiting). A watery diarrhea syndrome with elevated serum immunoreactive VIP has been described accompanying nonpancreatic tumors such as bronchogenic carcinomas, ganglioneuromas, pheochromocytomas, and a rare mastocytoma.172 Elevated serum levels of somatostatin, calcitonin, gastrin, and corticotropin also have been reported in pheochromocytoma.173
Paraneoplastic gastrointestinal dysmotility may occur in some patients with occult or established malignancy and specific serum antibodies. Clinically the patient may present with pseudoachalasia, gastroparesis, intestinal pseudo-obstruction, or constipation. Intestinal pseudo-obstruction (see Chapter 120) is most frequently associated with small cell carcinoma of the lung but has been described with other tumors such as squamous cell lung carcinoma, lymphoma, melanoma, and cancers of the kidney, breast, and prostate.174–176 Patients with paraneoplastic intestinal pseudo-obstruction characteristically suffer from constipation and obstipation and from symptoms of intestinal obstruction. In addition, dysphagia, gastroparesis, early satiety, autonomic insufficiency, and peripheral neuropathy have been described.177 The onset of symptoms may precede the discovery of the primary tumor by several years. The gastrointestinal pathology in this syndrome is confined to the myenteric plexus, in which an inflammatory lymphocytic infiltrate is variably seen accompanying neuronal degeneration.178 Cross-reacting autoantibodies found in the sera of these patients bind to the primary tumor cells and to neural cells in the myenteric plexus, resulting in inflammation and destruction of the myenteric plexus.179 In the setting of pseudo-obstruction, detection in the serum of circulating antineuronal nuclear antibodies (ANNA-1 or anti-Hu), type 1 Purkinje cell antibodies (PCA-1), or N-type calcium channel binding antibodies should suggest a paraneoplastic process and prompt further evaluation for an underlying malignancy.177 ANNA-1 are postulated to induce neuronal apoptosis leading to gut dysmotility.180 Although the symptoms of paraneoplastic pseudo-obstruction may resolve with successful treatment of the primary tumor, persistence of gastrointestinal symptoms despite effective anticancer treatment is more common. Attempts to alleviate the symptoms of pseudo-obstruction with prokinetic agents have been disappointing.
HEMATOLOGIC MALIGNANCIES
Liver involvement during hematologic malignancies is only rarely life threatening or a source of great morbidity. Nevertheless, the liver is a major component of the reticuloendothelial system, and thus it is not surprising that malignant infiltration of the liver commonly occurs in such diseases.181 As detailed in Table 35-2, the frequency of malignant infiltration varies from less than 10% to nearly 100% depending on the nature of the underlying hematologic malignancy. In addition to histologic and biochemical abnormalities related to malignant infiltration, a variety of other hepatic abnormalities are observed in a significant fraction of such patients. Many of these abnormalities are related to toxicity of pharmacologic or radiation therapies or to the secondary opportunistic or transfusion-related infections commonly observed in such patients. In addition, a variety of nonspecific histologic abnormalities of uncertain etiology such as steatosis, fibrosis, hemosiderosis, and nonspecific portal lymphocytic infiltrates are observed commonly in treated as well as untreated patients. Other hepatic manifestations relatively unique to selected malignancies also may occur. Such notable paraneoplastic manifestations include granuloma formation or development of pronounced intrahepatic cholestasis in patients with Hodgkin’s disease and deposition of amyloid in patients with multiple myeloma.
Hodgkin’s Lymphoma (see Chapter 29)
As detailed in Table 35-2, malignant infiltration of the liver is observed in only a minority of patients with untreated Hodgkin’s disease.182,183 However, autopsy series have noted hepatic involvement in as many as 55% of patients,184 suggesting that hepatic involvement increases with disease progression. Although Reed-Sternberg cells have been reported in only 8% of liver biopsies at the time of initial evaluation, fully a third of specimens exhibit nonspecific mononuclear cell infiltrates in portal tracts and approximately 10% to 25% have noncaseating hepatic granulomas not associated with malignant histiocytes or infectious etiologies.182,185 Moderate elevations of serum alkaline phosphatase activity are often observed, especially in febrile patients or patients with advanced stage disease.186 Although such elevations almost invariably appear related to elevations of the hepatic fraction of serum alkaline phosphatase activity,186 not all patients with elevated alkaline phosphatase levels are found to have tumor infiltration of the liver.182,186 All patients with hepatic involvement have been reported to have splenic involvement,182 but again the presence of splenic infiltration does not invariably imply liver involvement.
Although Hodgkin’s disease may involve extrahepatic bile ducts or lymph nodes in the porta hepatis and cause extrahepatic obstruction, multiple reports describe an additional syndrome of idiopathic intrahepatic cholestasis unrelated to hepatic infiltration, extrahepatic obstruction, or other identifiable causes.187–189 The degree of cholestasis is often disproportionate to apparent tumor load.187,189 However, cholestasis has been reported to resolve with response to systemic therapy,187 although in other cases this syndrome has been associated with intractable, fatal liver damage. Recently, progressive loss of small intrahepatic bile ducts has been documented in some of these patients,189 suggesting that this syndrome may be caused by destruction of bile duct epithelial cells either by direct effects of tumor cells invading the intrahepatic bile ducts or by indirect effects of cytokines released from lymphoma cells.
As liver involvement with Hodgkin’s disease defines a patient as having stage IIIE or IV disease, correct interpretation of causes of abnormal liver biochemistries in patients with this disease is often of significant importance in determining prognosis and therapy. Numerous studies have noted the superiority of laparotomy or peritoneoscopy to blind percutaneous liver biopsy in detecting hepatic involvement with Hodgkin’s disease. Presumably this relates to the relatively small volume of tissue obtained at percutaneous liver biopsy and the difficulty in finding diagnostic Reed-Sternberg cells in the liver. Laparoscopy provides a diagnostic yield equal to that obtained at laparotomy, and laparoscopy with or without laparoscopic splenectomy has become the standard approach to diagnostic staging in the majority of patients.183,190
Non-Hodgkin’s Lymphoma (see Chapter 29)
Lymphoma involves the gastrointestinal tract either as the primary site or secondarily from systemic lymphomas. Lymphomas may affect any organ and must be included in the differential diagnosis of any gastrointestinal symptom, especially in patients with advanced acquired immunodeficiency syndrome. As noted in Table 35-2, the frequency of liver involvement at initial clinical staging is significantly higher in patients with non-Hodgkin’s lymphomas than in those with Hodgkin’s disease. When evaluated by percutaneous liver biopsy, 16% to 26% of patients with non-Hodgkin’s lymphomas are found to have liver infiltration191 with significantly higher percentages found to have hepatic involvement when evaluated by laparoscopy.192 In both Hodgkin’s and non-Hodgkin’s lymphomas, the majority of infiltrative lesions are portal in location.193 Although the overall frequency of hepatic involvement appears similar in different histologic types of lymphoma,191 primary hepatic lymphoma is an unusual variant that occurs more often in diffuse large cell lymphomas of B cell origin than in T cell or non–B, non–T cell lymphomas.194,195 In contrast to secondary lymphomatous involvement of the liver that is often only detected by histologic evaluation, patients with primary lymphoma are commonly found to have evidence of mass lesions on CT, magnetic resonance imaging, or other hepatic imaging procedures that may mimic primary or metastatic carcinoma.194–196 Some reports have suggested an association between primary hepatic lymphomas and immunosuppression or chronic viral hepatitis, but such comorbid conditions are noted in only a minority of cases.194,195
Recently, hepatosplenic γδT cell lymphoma has been recognized as a distinct lymphoma entity.197 This extremely rare form of lymphoma occurs most frequently in young males who present with hepatosplenomegaly secondary to diffuse hepatic sinusoidal and splenic sinus infiltration with clonal populations of T cell receptors, gamma delta (γδTCR) expressing cells. Lymphadenopathy is absent, but bone marrow involvement common at the time of presentation and cytogenetic analysis commonly reveals an isochromosome 7q and trisomy 8.19
The most common liver test abnormality reported in patients with non-Hodgkin’s lymphoma is a moderately elevated serum alkaline phosphatase. Overall liver test abnormalities are poorly predictive of the presence or absence of lymphomatous infiltration of the liver.192 This likely relates in part to the presence of a variety of nonspecific histologic abnormalities191,193 including portal lymphocytic infiltrates, hemosiderosis, and steatosis that may be associated with liver test abnormalities in patients without hepatic involvement. Other patients with lymphomatous liver infiltrates may have normal liver tests.
Noncaseating granulomas also have been found in the portal tracts of patients with non-Hodgkin’s lymphoma though at a much lower frequency than observed in Hodgkin’s disease.198 Extrahepatic obstruction secondary to nodal involvement in the porta hepatis also may occur,192 and in some cases bile duct involvement may mimic the features of cholangiocarcinoma.199 Percutaneous liver biopsies have been found to be of value in detecting hepatic involvement with lymphoma, and if such specimens are properly processed, immunotyping can be performed to better characterize the phenotype of the malignant cells.200 However, quantity of tissue obtained appears very important in determining diagnostic sensitivity, with biopsy at laparotomy being superior to either blind percutaneous or laparoscopic biopsies in obtaining a diagnosis of hepatic infiltration by non-Hodgkin’s lymphoma.192
Leukemia
Approximately 10% of patients with leukemia suffer significant gastrointestinal complications, either from the leukemia itself or as the result of chemotherapy (Table 35-3).201 Examination of autopsy specimens reveals gastrointestinal involvement in less than 15% of all patients with leukemia.202 Acute myelogenous leukemia is the type most likely to affect the gut. Lesions result from four major causes: leukemia cell infiltration, immunodeficiency, coagulation disorders, and drug toxicities. Radiologically, leukemic lesions assume many forms. Infiltration of the bowel may produce polypoid masses (chloromas), plaquelike thickenings, ulcers, and diffuse masses. Esophageal filling defects with clot and debris have been described.203 Gastric mucosal folds can assume a “brainlike” deeply convoluted appearance resembling adenocarcinoma. Diffuse intestinal leukemoid polyposis may produce obstruction, hemorrhage, or intussusception.
| Leukemic Invasion of the Bowel and Related Structures |
BMT, bone marrow transplantation; CMV, cytomegalovirus; EBV, Epstein-Barr virus; HSV, herpes simplex virus; SOS, sinusoidal obstruction syndrome.
Immunodeficiency and immunocytopenia may lead to agranulocytic ulcers with bacterial invasion and bleeding. Coagulation defects can produce intramural hematomas and hemorrhagic necrosis of the bowel. Clinical syndromes are myriad. Common oral symptoms (see Chapter 22) are gingival bleeding, hypertrophy, inflammation, and focal ulcerations. Oral mucositis (stomatitis) is a severe inflammatory condition seen in the setting of recent chemotherapy, radiation therapy, or bone marrow transplantation. Treatment consists of appropriate antifungal, antiviral, or antibacterial therapy as well as viscous lidocaine and systemic analgesia. Esophageal lesions, usually caused by candidiasis or herpesviruses, may cause odynophagia, dysphagia, or bleeding (see Chapter 45). Gastric acid hypersecretion with peptic ulcers has been reported in a patient with hyperhistaminemia secondary to basophilic granulocytic leukemia.204 Massive gastrointestinal hemorrhage may result from infectious lesions, agranulocytic ulcers, or primary leukemic lesions of the gastrointestinal tract. The treatment of bleeding gastric and colonic leukemic lesions with radiation therapy has occasionally met with success and has been advocated by some investigators.205
A dire complication, seen in 5% of patients with acute leukemia and 3% of those with chronic leukemia, is the development of an acute abdomen. Acute appendicitis, abdominal abscesses, and perforation are noted with increased frequency. Necrotizing ileocecal enterocolitis and leukemic typhlitis are relatively infrequent but life-threatening problems in neutropenic leukemia patients. Typhlitis (i.e., inflammation of the cecum) complicates 6.5% of cases of acute myeloid leukemia and 4.6% of cases of acute lymphoblastic leukemia.206 Typhlitis typically manifests after induction chemotherapy and is usually preceded by neutropenia.207 Rarely, typhlitis can be the presenting manifestation of acute leukemia.208 Although the cause of this condition is not entirely clear, multiple factors such as chemotherapy, radiotherapy, neutropenia, and altered gastrointestinal flora are implicated in its pathogenesis.207 Cecal superinfection with fungi and with cytomegalovirus also has been associated with typhlitis. Patients usually present with fever, severe right lower quadrant pain, and occasionally an acute abdomen. Bloody diarrhea accompanies typhlitis in 35% of patients.209 The diagnosis can be inferred indirectly by the finding of symmetrical cecal thickening on abdominal ultrasonography or CT.207 Bowel wall thickness greater than 10 mm is associated with a 60% mortality.210 Most patients with leukemic typhlitis can be managed conservatively with the administration of intravenous fluids, packed red blood cells, and, as needed, granulocyte colony-stimulating factor (G-CSF), platelets, and broad-spectrum antibiotics. On rare occasions surgery may be required if dire complications arise.
Pseudomembranous colitis may complicate leukemia even in the absence of antibiotic therapy.211 Other rare complications are listed in Table 35-3. Proctologic problems can include stercoral ulcers, neutropenic ulcers, and perirectal abscesses (see Chapter 125).
At initial presentation, hepatomegaly is present in the majority of patients with acute lymphocytic leukemia (ALL) and in a significant minority of patients with acute myelogenous leukemia (AML). In patients with advanced stages of these acute leukemias, incidence of liver involvement has been reported in more than 95% of cases of ALL and in about three quarters of patients with AML at autopsy.212 The hemorrhagic complications of these acute leukemias rarely permit histologic evaluation of the liver in patients with early, active disease. Therefore, it is difficult to discern the relative contributions of leukemic infiltrates, extramedullary hematopoiesis, other infectious or toxic complications of these diseases, or the therapies employed to the development of hepatomegaly and/or liver test abnormalities.
In patients with leukemias that run more chronic courses, sufficient numbers of patients have been biopsied to indicate that hepatic involvement is far more commonly detected on histologic evaluation than initially indicated by clinical or laboratory assessment.213–217 In an autopsy series, 98% of patients with chronic lymphocytic leukemia (CLL) were found to have leukemic infiltration consisting predominantly of portal infiltrates that usually left the hepatic limiting plates intact.213 However, in some cases, leukemic infiltrates were observed to bridge adjacent portal tracts and were associated with hepatocellular necrosis, bridging necrosis and occasionally pseudolobule formation. In contrast to the predominantly portal pattern of hepatic infiltration during CLL, liver involvement during hairy cell leukemia (HCL), large granular lymphocyte leukemia (LGLL), or the adult T cell leukemia/lymphoma syndrome associated with human T-lymphotropic virus type 1 (HTLV-1) infection is usually characterized by diffuse sinusoidal infiltration or a mixed pattern of portal and sinusoidal involvement.214–217 As in the case of CLL, all or nearly all patients with HCL including some without hepatomegaly or liver test abnormalities demonstrate hepatic infiltration on histologic evaluation.215,216 Although infiltration by HCL occasionally may be missed on conventional histologic evaluation, use of tartrate-resistant acid phosphatase staining215 and immunotyping by staining with monoclonal antibodies against lymphocyte cell surface markers200 has been reported to enhance diagnostic sensitivity and specificity. HCL has also been associated with angiomatous lesions in the liver created by disruption of the sinusoidal wall, creation of wide areas of communication between the sinusoidal lumen and space of Disse, and replacement of the sinusoidal cell lining by tumor cells in direct contact with hepatocytes.216
SYSTEMIC MASTOCYTOSIS
Systemic mastocytosis, a clonal disorder of the mast cell–progenitor associated with activating mutations in the c-kit gene, is characterized by a dense infiltrate of mast cells in extracutaneous tissue (bone marrow, spleen, liver, lymph nodes, and gastrointestinal tract) (Fig. 35-5).218 The classic dermatologic finding of urticaria pigmentosa may be seen with or without systemic involvement (see Chapter 22). The typical symptoms of mastocytosis (pruritus, flushing, tachycardia, asthma, headache) are believed to result from the release of histamine and prostaglandins (e.g., PGD2) from mast cells.219 Heparin is also released from mast cells and may contribute to a bleeding diathesis.220 Eighty percent of patients have gastrointestinal symptoms that include nausea, vomiting, diarrhea, and abdominal pain.221 Hepatomegaly, portal hypertension, splenomegaly, and ascites may occur frequently.222 These symptoms can be precipitated or provoked by heat, alcohol, aspirin, anticholinergics, NSAIDs, and contrast media.219 Hyperhistaminemia produces gastric hypersecretion in more than 40% of cases,223 and secretion may be as marked as in Zollinger-Ellison syndrome.224 Gastric hyperacidity correlates with the degree of histaminemia and with the presence of acid-peptic disease.223,224 Duodenal ulceration or duodenitis has been reported in more than 40% of cases.223 Gastrointestinal hemorrhage from peptic ulcers and from bleeding esophageal varices has been reported.224,225 Diarrhea has been reported in as many as 60% of cases, and minimal fat malabsorption occurs in some cases.220,223 Decreased absorption of d-xylose and vitamin B12 is also found in patients with mastocytosis.223 The cause of diarrhea is unclear. Some diarrheal symptoms (but not malabsorption) respond to H2-receptor antagonists, but there is no clear correlation between stool output and the degree of plasma histaminemia or gastric acidity.223 It is presumed that diarrhea and malabsorption are the result of morphologic changes in the absorptive mucosa. Jejunal biopsy specimens may show large numbers of mast cells in the lamina propria, muscularis mucosa, and submucosa, with normal villi or mild villous atrophy.226 The colon may also be involved (see Fig. 35-5). Endoscopy may reveal urticaria-like mucosal lesions, thickened gastric folds, and edematous mucosa, whereas colonoscopy has shown purple pigmented lesions.227 Small bowel radiographic abnormalities include bull’s-eye lesions resembling metastases, edema, thickened folds, and a nodular mucosal pattern.228 Abdominal ultrasound and CT may show hepatosplenomegaly, adenopathy, thickening of the omentum and the mesentery, and ascites.229 H1-receptor and H2-receptor antagonists, anticholinergics, oral disodium cromoglycate, and glucocorticoids have been used successfully to relieve the diarrhea and abdominal pain of mastocytosis.219 Imatinib mesylate, a tyrosine kinase inhibitor, is not effective in systemic mastocytosis because a conformational change associated with the most common mutation (Asp816Val) interferes with drug binding.218

MYELOPROLIFERATIVE AND MYELOPHTHISIC DISORDERS
Because the liver is a major site of extramedullary hematopoiesis, hepatomegaly or mild liver test abnormalities secondary to extramedullary hematopoiesis may be appreciated in any of a variety of myeloproliferative disorders or marrow infiltrating malignancies. Benign or malignant proliferations of histiocytes (macrophages) or dendritic cells may be complicated by hepatomegaly or jaundice related to diffuse infiltration of hepatic sinusoids by erythrophagocytic histiocytes, development of peliosis hepatis or intrahepatic, or extrahepatic invasion of bile ducts and portal tracts by histiocytes or Langerhans’ (dendritic) cells.230–232 Erythrophagocytosis may be a manifestation of malignant histiocytosis or represent a reactive benign histiocyte proliferation in patients with advanced T cell lymphomas.197 Thus assessment of involved tissues for malignant cells of T cell or more rarely B cell origin should also be included in the diagnostic evaluation of cases of uncertain origin. Liver biopsies are commonly abnormal in untreated patients with Langerhans cell histiocytosis (formerly termed histiocytosis X). The most common abnormality is mild mononuclear cell infiltration of the portal tracts.231 However, portal triaditis associated with periportal fibrosis, cirrhosis, or extrahepatic cholangiographic evidence of sclerosing cholangitis also may be seen and in some patients may lead to severe cholestatic liver disease.231,232
Primary myelofibrosis (PMF) is a myeloproliferative disease characterized by bone marrow fibrosis with progressive anemia and splenomegaly. Portal hypertension, which occurs in 7% of patients with PMF, results from increased portal venous flow and from infiltration of the liver by foci of extramedullary hematopoiesis.233 Massive gastrointestinal hemorrhage complicates 5% of cases and most often is due to bleeding esophageal varices. Extramedullary hematopoiesis can involve the esophagus, stomach, and small bowel leading to abdominal pain and hemorrhage.234 Increased thrombotic complications have been associated with PMF, polycythemia vera, and essential thrombocytosis.235 Splenic infarction can cause left upper quadrant abdominal pain. As many as 50% of patients with hepatic vein thrombosis, or the Budd-Chiari syndrome, have an overt myelodysplastic syndrome.236 One study237 suggests that 80% of patients with hepatic vein thrombosis may have latent myeloproliferative abnormalities without overt disease (see Chapter 83).
DYSPROTEINEMIAS
Multiple myeloma or plasma cell tumors may directly involve the gastrointestinal tract with amyloidosis or with local infiltration by plasmacytomas. Twenty-one percent of patients with amyloidosis have multiple myeloma.238 As with gastrointestinal involvement by amyloidosis from other causes (see later), bowel wall infiltration and dysmotility underlie most clinical symptoms. Primary extramedullary plasmacytoma account for 3% to 5% of all plasma cell dyscrasias with gut involvement noted from the oral cavity to anus with manifestations including dysphagia, hemorrhage, pseudo-obstruction, and polyposis.239 Hepatomegaly and abnormalities of liver biochemistries are commonly observed in patients with multiple myeloma.240 In up to half of patients with hepatic histologic evaluation, either diffuse sinusoidal or portal infiltration or, less commonly, nodule formation in the liver by malignant plasma cells has been observed.193,240,241 The frequency of jaundice has ranged from 0% to 30% in series of patients with hepatic infiltration by multiple myeloma.240,241 Ascites formation or, more rarely, esophageal varices have been reported to complicate the course of 10% to 35% of patients with massive hepatic infiltration.200,240 Portal hypertension secondary to tumor infiltration appears to be the cause in most patients, although other causes including congestive heart failure, dissemination of myeloma cells into the peritoneal cavity or development of tuberculous peritonitis also have been noted. In addition to direct malignant infiltration and development of such nonspecific hepatic abnormalities as hemosiderosis or portal lymphocytic infiltrates, multiple myeloma is complicated in about 10% of patients by deposition of amyloid or non–amyloid-containing IgG light chain deposits in the space of Disse.200,240–242 Extramedullary hematopoiesis also may contribute to hepatomegaly or liver test abnormalities in these patients.240 Clinical staging and follow-up of patients with multiple myeloma are largely based on assessment of marrow, osseous, and serum or urinary abnormalities, and thus histologic evaluation of the liver is only occasionally considered. As discussed in the section dealing with amyloidosis later in this chapter, potential diagnostic benefits of liver biopsy must be weighed against concerns regarding bleeding complications.
Waldenström’s macroglobulinemia, a neoplasm characterized by malignant proliferation of lymphocytes producing IgM, presents with hepatomegaly or splenomegaly in one third of patients.243 Gastrointestinal IgM deposition may occur in an infiltrative pattern characterized by diffuse infiltration of the bowel wall with neoplastic cells similar to the pattern seen in immunoproliferative diseases. More commonly, acellular macroglobulin is deposited predominantly in the tips of the villi, the interstitium, and the lacteals, leading to lymphangiectasia.244 Small intestinal mucosal IgM deposits may stain weakly with periodic acid–Schiff, simulating the microscopic appearance of Whipple’s disease. Gastric involvement may present with epigastric pain or bleeding, whereas small intestinal disease can present with steatorrhea, diarrhea, protein-losing enteropathy, pseudo-obstruction, or occult bleeding.244
The rare plasma cell proliferative disorder, γ heavy-chain disease, has been associated with abdominal pain, weight loss, and gastric infiltration from malignant plasma cells.245 α Heavy-chain disease, an immunoproliferative small intestinal disease (IPSID), is a mucosa-associated lymphoid-tissue lymphoma characterized by infiltration of the bowel wall resulting in malabsorption and protein-losing enteropathy (see Chapter 29). IPSID is mostly seen in the Mediterranean basin, Middle East, Far East, and Africa, with a recent study suggesting that Campylobacter jejuni may be a causative agent.246
COAGULATION DISORDERS
In hemophiliac individuals, acute abdominal pain can be a manifestation of spontaneous intra-abdominal hemorrhage. Gastrointestinal bleeding may occur from varices related to chronic liver disease secondary to hepatitis C acquired from transfused blood products. von Willebrand’s disease, heparin or warfarin therapy, hepatic failure, qualitative or quantitative platelet defects, and other bleeding diatheses may result in gastrointestinal hemorrhage or intramural hematomas (Table 35-4). Radiologically, intramural bleeding can be recognized by thickened mucosal folds, rigidity, luminal narrowing (Fig. 35-6), and intragastric masses. Intestinal obstruction and intussusception may result.
| Platelet Deficiency |
GI, gastrointestinal.
Hemolytic-uremic syndrome (HUS) consists of a triad of acute kidney injury, microangiopathic hemolytic anemia, and thrombocytopenia without the consumption of humoral clotting factors through defibrination. In children, idiopathic, sporadic, and epidemic cases have variously been described. In adults, HUS occurs in conjunction with complications during childbirth or chemotherapy, with mitomycin C being the most common implicated agent.247 More commonly, adult HUS is preceded by a mild diarrheal illness. Enteric pathogens associated with the HUS prodrome (“HUS colitis”) include Shigella, Salmonella, Yersinia, Campylobacter, and the “hemorrhagic” 0157:H7 strain of Escherichia coli (see Chapter 107).248–252 Undercooked hamburger is the most common vector for 0157:H7 infection with apple juice, radish sprouts, and sausages also implicated in the spread of this infection.248 Several studies suggest that antibiotic therapy of E. coli 0157:H7 with antibiotics increases the risk of development of HUS in children and adults; however, this assertion has been challenged in a meta-analysis.253 Empirical therapy of diarrhea with antimicrobial agents may be appropriate for certain subsets of patients, such as those who are quite clearly at high risk of invasive infections.254 Once HUS appears, colonic involvement is common owing to microangiopathic thrombosis of submucosal vessels and intramural hemorrhage.255 Pancreatitis has also been described.256 Radiographic abnormalities include mucosal irregularities, intestinal dilation, filling defects, bowel wall edema, and findings that may resemble those of idiopathic ulcerative colitis, or vasculitis.257 Because HUS is usually self-limited, therapy consists of hemodialysis and supportive gastrointestinal care. Severe complications may include hemoperitoneum, transmural bowel necrosis with perforation, or colonic stricture.258,259
Thrombotic thrombocytopenic purpura (TTP) is an idiopathic disorder consisting of thrombocytopenia, microangiopathic hemolytic anemia (without significant consumption of clotting factors), fever, renal insufficiency, and profound neurologic dysfunction. Compared with HUS, central nervous system (CNS) symptoms predominate in TTP and renal failure is less severe than in HUS—20% of patients have nonspecific abdominal complaints. The bleeding diathesis of TTP can lead to gastrointestinal hemorrhage, but TTP may also cause thrombosis of intestinal vessels that resembles HUS, clinically and pathologically. Acute colitis, cholecystitis, and pancreatitis have been described.260,261 Plasmapheresis allows 90% of patients with TTP to survive an episode without permanent organ damage.255
Venous thromboembolism (VTE), comprising deep venous thrombosis (DVT) and pulmonary embolism, may be the first manifestation of malignancy. Trousseau’s syndrome refers to migratory superficial thrombophlebitis or DVT occurring in patients with occult cancer. In patients with VTE without a predisposing factor, the incidence of previously undiagnosed cancer was 6% at baseline and 10% from baseline to one year.262 In patients with newly diagnosed VTE, an extensive screening strategy for occult malignancy, which included abdominal imaging and colonoscopy, was superior to a limited screening strategy of medical history, physical examination, and basic blood work in the detection of cancer.263
RED BLOOD CELL DYSCRASIAS
Sickle Cell Disease
Sickle cell disease is an autosomal recessive disorder of hemoglobin structure that is characterized by chronic hemolytic anemia and recurrent episodes of vascular occlusion leading to ischemia and distal tissue infarction in multiple organs. Eight percent of African Americans are heterozygous for the hemoglobin S trait and homozygotes comprise 0.2% of African Americans. Patients with sickle cell anemia and other hemoglobinopathies may develop splenic infarction and liver disease (see following), likely from ischemic injury due to intrasinusoidal sickling and impairment of intrahepatic blood flow and delivery of oxygen to hepatocytes.264,265 Chronic anemia due to hemolysis is typically present and predisposes to an indirect-reacting bilirubin elevation and to the formation of pigmented gallstones.266 Patients with other hereditary defects involving red blood cell cytoskeletal proteins, hereditary spherocytosis, and hereditary elliptocytosis, also have diminished red blood cell survival, leading to an increased incidence of pigmented gallstones. Transfusions are frequently used in the therapy for sickle cell anemia, and therefore such patients who were transfused prior to 1992 are at increased risk for hepatitis C.267 Multitransfused teenage and adult patients with sickle cell anemia also have been found to have degrees of excess hepatic iron stores that are comparable to that noted in thalassemia major.268–270
Sickle cell crisis, an acute manifestation of this disease, is characterized by severe skeletal pain and fever. Abdominal pain is also commonly present, and it is important to distinguish vaso-occlusive crises from surgical conditions such as cholecystitis, bowel infarction, appendicitis, and pancreatitis. Abdominal pain from vaso-occlusive crises tends to be more diffuse and associated with remote pain such as limb and chest pain. The pain of vaso-occlusive crises is typically relieved with hydration and oxygen within 48 hours.271 Sickle cell hepatopathy, a syndrome characterized by severe hyperbilirubinemia out of proportion to degree of ongoing hemolysis, is a rare complication of sickle cell anemia that can be associated with coagulaopthy and encephalopathy, leading to death from acute liver failure. Because treatment with exchange transfusions is associated with improved outcome,272 it is important to distinguish this complication of sickle cell anemia from other causes of liver disease that are common in this patient population.273
When histologic evaluation of the liver has been performed in patients with sickle cell anemia at autopsy, cholecystectomy or diagnostic percutaneous liver biopsy, dilated sinusoids, erythrophagocytosis by Kupffer cells (Fig. 35-7), and varying degrees of parenchymal atrophy in the central zones of the liver have been observed frequently.265,274–277 In association with hepatic sinusoids engorged by phagocytosed, sickled red blood cells, adjacent areas of ischemic necrosis have been reported in patients with acute episodes of jaundice, right upper quadrant pain, fever, and leukocytosis thought to be secondary to intrahepatic sickle cell crises or sickle cell hepatopathy.264,265,274,276,278 Accumulation of collagen or thin basement membranes within the space of Disse,274 peri-sinusoidal fibrosis,276 and an apparently high incidence of cirrhosis in patients with sickle cell anemia264 has suggested that recurrent ischemic injury secondary to intrahepatic sickling may also be a cause of chronic liver disease.
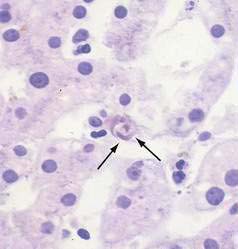
Although early reports suggested that viral hepatitis was an unusual cause of acute or chronic liver disease in such patients,274,275 studies conducted in the 1980s and 1990s suggested that hepatitis B276,277 and hepatitis C267 are common infections in patients with sickle cell anemia and may account for many episodes of acute or chronic liver disease previously attributed to sickle cell hepatopathy. More recent studies of adult sickle cell anemia patients presenting with acute hepatic dysfunction have found that viral or autoimmune hepatitis plays a role in about a third of such episodes.273 Of special note, intrasinusoidal sickling and Kupffer cell erythrophagocytosis (see Fig. 35-7) almost invariably are found in all patients with sickle cell disease irrespective of apparent cause of liver disease or degree of serum alanine aminotransferase (ALT) elevation.277 Intrasinusoidal sickling was also found in two liver biopsies performed after recovery from acute viral hepatitis. To some extent the presence of nonphagocytosed sickled red blood cells in hepatic sinusoids could be attributed to the fact that formalin fixation was noted to induce irreversible sickling of red blood cells in patients with hemoglobin SS or sickle cell disease.277,279 In addition, Omata and colleagues277 have suggested that Kupffer cell erythrophagocytosis may reflect the role of Kupffer cells in clearance of sickled red cells in functionally asplenic patients with sickle cell disease. Thus, assessment of degree of intrasinusoidal sickling or even Kupffer cell erythrophagocytosis is a poor indicator of possible ischemic liver injury in patients with sickle cell disease. In contrast, other features of vascular insufficiency such as acute ischemic necrosis, sinusoidal dilatation and perisinusoidal fibrosis appear to be more specific markers of vascular injury in patients with symptomatic liver dysfunction in the absence of viral hepatitis or other causes for hepatocellular injury.276
Diggs265 reported in 1965 that 10% of patients presenting with acute sickle crises were jaundiced. More recent assessment of prevalence of liver disease in patients with sickle cell anemia have found persistent abnormalities of one or more liver enzyme tests in 24% of patients followed for sickle cell anemia.279 In addition, 48 of 72 (67%) patients without other biochemical evidence of liver disease had total serum bilirubin levels of greater than 2 mg/dL (greater than 34 µmol/L). Thus laboratory abnormalities suggesting possible liver disease are relatively common in patients with sickle cell disease and frequently lead to diagnostic evaluations as detailed in Figure 35-8. Of note, however, a high rate of complications has been reported in association with liver biopsies performed during acute sickling crises280 and thus invasive diagnostic procedures should be used judiciously in this patient population.
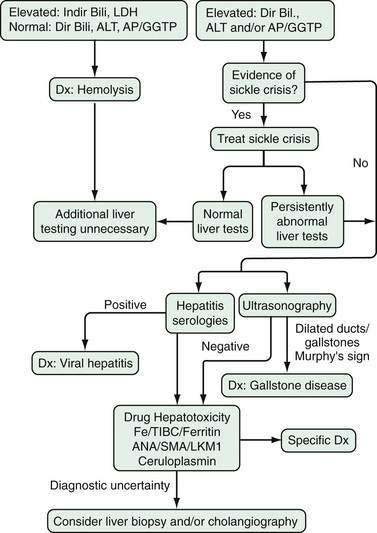
In sickle cell disease patients without liver disease, hyperbilirubinemia is exclusively unconjugated or indirect and only uncommonly exceeds levels of 4.5 mg/dL (77 µmol/L). In more severely jaundiced patients, higher serum lactic dehydrogenase levels are also seen and suggest higher rates of hemolysis.279 However, in the setting of acute viral hepatitis or other causes of liver dysfunction, extreme levels of hyperbilirubinemia consisting of relatively equal amounts of direct and indirect bilirubin are observed.279,281 Although early reports suggested that a total serum bilirubin level of greater than 25 mg/dL (greater than 428 µmol/L) was a grave prognostic sign,265,282 relatively benign courses have been noted in patients with extreme degrees of hyperbilirubinemia during the course of acute viral hepatitis or presumed intrahepatic sickle cell crises.279,281,283 The degree of serum ALT or aspartate aminotransferase (AST) elevation in patients with sickle cell anemia and acute viral hepatitis is similar to that observed in other patients with acute viral hepatitis, with most symptomatic patients having elevations more than 10-fold the upper limit of normal.275,281 However, among patients with jaundice and other symptoms such as fever, leukocytosis and intense right upper quadrant pain attributed to intrahepatic sickling or sickle cell hepatopathy, serum AST and ALT values often have been found to be only modestly elevated272,275,282,283 although in other cases, elevations in excess of 15 times the upper limit of normal have been noted.274 Thus, in patients with jaundice and prominent ALT elevations, both acute viral hepatitis and ischemic injury related to sickle cell disease itself must be considered as possible etiologies. In addition, coincidental causes of liver disease such as autoimmune hepatitis284,285 have been reported in patients with sickle cell anemia and clinically apparent liver disease that was initially incorrectly ascribed to complications of sickle cell disease. Thus when evaluating liver disease in these patients, care must be taken to consider the full spectrum of possible etiologies.
Although the majority of patients with acute hepatocellular dysfunction thought secondary to either viral hepatitis or intrahepatic sickle cell crises recover following supportive care, cases of acute hepatic failure have been reported.264,275,278,286 Most such patients have presented with right upper quadrant pain, jaundice, modest aminotransferase elevations (less than 10-fold elevated) and progressive coagulopathy, and at postmortem examination have had histologic findings suggesting that the initiating cause of liver failure was centrilobular necrosis secondary to vascular complications of sickle cell disease. Recovery from severe cholestasis and coagulopathy has been reported after exchange transfusions.287 It also has been suggested that sickle cell disease may be a predisposing factor to the development of fulminant hepatic failure in children with acute viral hepatitis.286 Thus even in cases of liver failure with an apparent viral etiology, aggressive measures directed at reversal or prevention of intrahepatic sickling may be warranted.
In all sickle cell disease patients with direct hyperbilirubinemia and especially those with right upper quadrant pain, fever, or leukocytosis, acute cholecystitis with or without choledocholithiasis must be investigated as possible primary or contributing causes. A number of reports indicate that bile duct stones are found in a significant fraction of sickle cell disease patients undergoing cholecystectomy for symptomatic biliary tract disease.288 However, other studies have noted no objective evidence of choledocholithiasis or acute or chronic cholecystitis in many sickle cell disease patients undergoing cholecystectomy for presumed symptomatic biliary tract disease. This has led to speculation that events related to intrahepatic ischemia might better explain the signs and symptoms of liver disease in many of these patients.276
A significant rate of operative and anesthetic complications have been reported in patients with sickle cell disease.276,288,289 Thus cholecystectomy is not recommended in asymptomatic patients with cholelithiasis. However, several reports have noted that in sickle cell patients with recurrent bouts of right upper quadrant pain and jaundice, a marked decrease in such symptomatic episodes is noted after cholecystectomy.276,288,289 Thus in recurrently symptomatic patients with gallstones in whom there is difficulty in distinguishing between cholecystitis and intrahepatic crisis, cholecystectomy is recommended.289 However, in such patients special attention should be directed toward minimizing risks of anoxic injury during surgery by preoperative transfusion of red blood cells and expansion of intravascular volume and by intra- and postoperative oxygen therapy.
Chronic liver disease related to chronic viral hepatitis or to post-transfusional iron overload is being recognized with increased frequency in adult patients with sickle cell disease.267–269,290,291 Ferritin levels should be monitored in sickle cell patients who are recipients of multiple red cell transfusions and iron chelation therapy considered for those with evidence of significant iron overload.269 In recipients of chelation therapy and in the general sickle cell disease patient population, zinc deficiency related to excess renal losses is prevalent and via effects on ornithine transcarbamylase may potentiate hyperammonemia. Thus zinc therapy should be considered in sickle cell disease patients with evidence of hepatic encephalopathy.292
Thalassemia
Patients with thalassemia typically develop hepatomegaly from extramedullary hematopoiesis, with CT revealing well-defined hypodense lesions that enhance in the portovenous phase of contrast injection.293 These patients can also develop iron overload due to multiple transfusions, with the resulting end-organ dysfunction in the liver, gonads, and pancreas. The early parenteral use of the iron-chelating agent desferoxamine, in an amount proportional to the iron load, has been shown to be effective in halting the progression of fibrosis in patients with thalassemia major.294 However, deferiprone, an orally acting iron-chelating agent, has not been as effective in reducing the body iron burden and may worsen the fibrosis.295 Further studies are needed to better define the role of orally active iron chelators in thalassemic patients with iron overload.
ENDOCRINE DISEASES (see Table 35-5)
DIABETES MELLITUS
Gastrointestinal symptoms are more prevalent in diabetic patients in comparison to the general population and have a negative effect on the quality of life.296,297 These symptoms are independently associated with poor glycemic control and peripheral neuropathy.298 Autonomic dysfunction in diabetics (diabetic autonomic neuropathy [DAN]) can manifest itself in one or more organ systems. The pathogenesis of DAN is related to hyperglycemia, neurovascular insufficiency, autoimmune damage, and neurohormonal growth factor deficiency.299 In type 1 diabetes, enteric neurotransmission may be modulated by a functional IgG autoantibody that acts as an agonist at the L-type calcium channels of smooth muscle of the colon.300 Constipation, abdominal pain, nausea, vomiting, dysphagia, diarrhea, and fecal incontinence are symptoms of enteric DAN that are more commonly seen in older patients with long-standing type 1 diabetes, poor glucose control, and symptoms of cardiovascular or peripheral neuropathy.301 Although motility disturbances are common in these patients, they do not correlate well with the presence or severity of symptoms. This suggests that other manifestations of DAN may play a role in the development of symptoms.
Table 35-5 Gastrointestinal Manifestations of Endocrine Diseases
| DISEASE | ABNORMALITY/ASSOCIATION | GASTROINTESTINAL MANIFESTATIONS |
|---|---|---|
| Hyperthyroidism | Lymphocytic mucosal infiltrates | Superficial gastritis, steatorrhea |
| Accelerated intestinal transit | Diarrhea | |
| Ulcerative colitis | Bloody diarrhea | |
| Minor histologic changes in liver | Aminotransferase(s) elevation, mild indirect hyperbilirubinemia | |
| Hypothyroidism | Rare chronic hepatitis (with thyroiditis) | Increased ALP, aminotransferase(s) |
| Impaired LES function | Reflux esophagitis | |
| Gastric hypomotility | Bezoars | |
| Decreased intestinal transit | Constipation, fecal impaction, volvulus, pseudo-obstruction, rectal prolapse, diarrhea, steatorrhea (bacterial overgrowth) | |
| Liver biochemical test abnormalities | ||
| Primary biliary cirrhosis | Features of hepatic cirrhosis | |
| Cronkhite-Canada syndrome | Intestinal polyps | |
| Celiac disease | Diarrhea, steatorrhea | |
| Familial polyendocrine failure | Esophageal candidiasis, adrenal insufficiency, hypogonadism, diabetes mellitus, hypothyroidism | |
| MCT | Increased serum calcitonin | Watery diarrhea (increased intestinal secretion due to calcitonin?) |
| MEN-IIA, MEN-IIB | Pheochromocytoma (see below), mucosal neuromas, ileus, megacolon | |
| Adrenal insufficiency | Corticosteroid deficiency | Nausea, vomiting, anorexia, diarrhea, malabsorption |
| Familial polyendocrine failure | Esophageal candidiasis, hypothyroidism, hypogonadism, diabetes mellitus, hypoparathyroidism | |
| Pheochromocytomas | Increased plasma catecholamines | Paralytic ileus, megacolon |
| Cholelithiasis | Biliary pain, cholecystitis | |
| MEN-IIA | MCT (see above) | |
| Hypercortisolism (Cushing’s disease) | Increased pituitary ACTH | Gastric ulceration |
| Acromegaly | Increased pituitary GH | Colorectal polyps |
| Panhypopituitarism | Adrenal insufficiency/hypothyroidism | As for adrenal insufficiency and hypothyroidism |
| Hyperparathyroidism | Increased serum calcium | Constipation, nausea, vomiting |
| Peptic ulceration | Bleeding, abdominal pain, perforation | |
| Pancreatitis | Acute pancreatitis | |
| MEN-I | Gastrinoma, VIPoma, others | |
| Hypoparathyroidism | Familial polyendocrine failure | Esophageal candidiasis, hypothyroidism, hypogonadism, diabetes mellitus, adrenal insufficiency |
| Malabsorption | Diarrhea, steatorrhea | |
| Intestinal lymphangiectasia | Protein-losing enteropathy | |
| Diabetes mellitus | Esophageal dysmotility | Dysphagia, reflux esophagitis |
| Esophageal candidiasis | Odynophagia, dysphagia | |
| Gastroparesis | Nausea, vomiting, gastric outlet obstruction, bezoars | |
| Small intestinal dysmotility | Bacterial overgrowth, malabsorption, diarrhea | |
| Impaired intestinal fluid reabsorption | “Diabetic” diarrhea | |
| Colonic dysmotility | Constipation, megacolon, fecal incontinence | |
| Intestinal ischemia | Ischemic colitis, bowel infarction | |
| Pancreatic disease | Acute pancreatitis, pancreatic carcinoma | |
| Cholelithiasis | Biliary sepsis | |
| Sclerosing cholangitis | Biliary obstruction, sepsis | |
| Hepatic steatonecrosis | Abnormal liver biochemical tests, hepatic fibrosis | |
| Hepatocellular carcinoma | 2.5-fold increased risk | |
| Diabetic radiculopathy | Abdominal pain | |
| Familial polyendocrine failure | Candidiasis, hypothyroidism, hypogonadism, hypoparathyroidism, adrenal insufficiency | |
| Celiac disease | Diarrhea, steatorrhea |
ACTH, adrenocorticotropic hormone; ALP, alkaline phosphatase; GH, growth hormone; LES, lower esophageal sphincter; MCT, medullary carcinoma of the thyroid; MEN, multiple endocrine neoplasia; VIP, vasoactive intestinal polypeptide.
Esophageal Dysfunction (see Chapter 42)
Diabetic patients have been shown to have numerous esophageal motility abnormalities (although these may be clinically silent) such as hypotensive lower esophageal pressure, decreased amplitude of contractions, and simultaneous prolonged aperistaltic contractions in the body of the esophagus (see Chapter 42).301 Esophageal dysmotility in diabetes has been attributed to DAN mediated by vagal nerve dysfunction but recent motor nerve conduction studies suggest a motor neuropathy.302 Esophageal scintigraphy has demonstrated prolonged esophageal transit time.303 GERD is seen more frequently in diabetics and associated with the cardiovascular autonomic dysfunction, increased body mass index, disease duration, and poor glycemic control.304,305 Odynophagia in a diabetic patient should suggest possible Candida infection (see Chapter 45).306
Gastric Dysfunction
Abnormal gastric motility (see also Chapter 48) results in disordered gastric emptying, or gastroparesis diabeticorum (GD), which affects as many as 30% to 60% of diabetic patients.307 In this disorder, the normal physiology of gastric emptying, largely under the control of the vagus nerve, is grossly disturbed. Liquid emptying may be normal, but solid emptying is frequently delayed. There is an increased frequency of postcibal antral dysrhythmias such as antral tachygastria. Phase 3 contractions of the IMMC, which normally stimulate antral contractions, are frequently absent, resulting in poor antral expulsion of indigestible solids, predisposing to bezoars. Furthermore, maintenance of the gastroduodenal pressure gradient, as well as receptive relaxation of the stomach, is abnormal. Prolonged pyloric contractions (pylorospasm) may cause functional resistance to gastric outflow.301 The pathophysiology of these motor disturbances is unclear. Hyperglycemia can cause delayed gastric emptying in diabetic patients as well as in normal volunteers.308 As noted, vagal parasympathetic function, which is involved in gastric emptying, may not be entirely normal. High plasma levels of the gut peptide motilin are reported in patients with GD.309 Because motilin stimulates the initiation of phase 3 activity, the elevation of this peptide in diabetic patients with GD may, in part, be compensatory. This is consistent with the observation that the treatment of GD with prokinetic agents is associated with a fall in plasma motilin levels.309
Epigastric discomfort, nausea, vomiting, pyrosis, early satiety and weight loss are symptoms associated with GD that typically are most severe postprandially. Only abdominal bloating or fullness has been shown to be an independent predictor of delayed gastric emptying.310 Markedly delayed gastric emptying may make the regulation of blood glucose levels difficult. Hyperglycemia further impairs gastric emptying and may accelerate the onset of diabetic ketoacidosis, particularly when it is associated with severe vomiting. Although many diabetics have abnormal gastric emptying, few develop overt clinical symptoms. Furthermore, an occasional patient may have symptoms suggestive of GD but little or no delay in gastric emptying.
The diagnosis of GD should be strongly suspected from the history. Physical examination may reveal gastric dilation with a succussion splash. A saline load test is not a sensitive test in GD because liquid emptying is frequently normal. The usual method for diagnosis is exclusion of structural lesions by esophagogastroduodenoscopy or by standard barium radiographic examination. Food remnants may be noted in stomach. Such studies should be followed by more quantitative measurements of the degree of delay. Radiolabeled scintigraphy is the preferred way to confirm the diagnosis and to quantify the response to therapy.311 When interpreting these studies, it should be noted that anticholinergics, tricyclic antidepressants, benzodiazepines, and ganglionic-blocking agents may contribute to delayed emptying in these patients. In July 2006, the U.S. Food and Drug Administration (FDA) approved the SmartPill Capsule (SmartPill, Buffalo, NY) for evaluation of gastric emptying by measurement of luminal pressure, pH, and temperature data after ingestion of a radiotelemetry capsule.
The management of GD requires a multimodal approach. Most importantly, glycemic control should be optimized. Dietary changes include a low-fat, low-fiber soft diet with frequent small meals. Sometimes patients may require a mostly liquid diet, at least temporarily. Antiemetics and prokinetics are the two primary classes of medical therapy for GD. Antiemetics such as promethazine or prochlorperazine can be given orally or in suppository form but long-term use is limited because of side effects. Other treatments for nausea and vomiting include scopolamine patch, 5-hydroxytryptamine (5-HT3) receptor antagonists such as odansetron, dronabinol, or low-dose tricyclic antidepressants to modify visceral hypersensitivity.312 Prokinetic agents, which increase gastric motor activity, are frequently used to treat GD. Metoclopramide (10 to 20 mg, 30 minutes before each meal and at bedtime) and domperidone (10 to 30 mg four times a day also given 30 minutes before meal and bedtime) are dopamine antagonists that increase antral contractions and decrease receptive relaxation of the proximal stomach.312 Metoclopramide crosses the blood-brain barrier whereas domperidone does not. Domperidone is not approved for use by the FDA—40% of patients cannot tolerate metoclopramide because of CNS side effects, and 5% of patients taking domperidone develop symptoms of hyperprolactinemia such as gynecomastia in men and breast enlargement and lactation in women.312 A third prokinetic drug is cisapride which, although efficacious in GD, has been severely restricted due to life-threatening proarrhythmic cardiac side effects. The macrolide antibiotic erythromycin, a motilin agonist, has been found to be effective in accelerating gastric emptying in GD but data regarding symptomatic relief are limited.313 Erythromycin can be given orally (125 mg two or three times daily) or intravenously (200 mg over 5 to 10 minutes every eight hours) but its use limited by nausea and abdominal cramping as well as loss of effectiveness over time secondary to downregulation of the motilin receptor.312 In a small pilot study, the infusion of ghrelin, an appetite stimulating hormone synthesized within the stomach, was shown to improve gastric emptying.314 Small case series suggested that endoscopic therapy with injection of botulinum toxin into the pyloric sphincter resulted in improvement in both gastric emptying and subjective symptoms; however, a double-blind randomized controlled trial showed no significant benefit.315 In severe or refractory cases, a venting gastrostomy and feeding jejunostomy tube can be placed. Surgical therapy had been limited to partial or complete gastric resection in medically refractory cases, with often disappointing results. Enterra (Medtronic, Minneapolis, Minn), a gastric electrical stimulator (GES) device, was approved in 2000 for treatment of refractory gastroparesis. At laparoscopy or laparotomy, two electrodes are placed into the muscularis propria of the greater curvature 10 cm from the pylorus and attached to a neurostimulator placed subcutaneously in the abdominal wall. The GES device delivers high-frequency (12 cycles per minute [CPM]), low-energy pacing, with 81% and 63% reduction in vomiting frequency at 6 and 12 months, respectively, in patients with GD.316 The most common significant complication was pacemaker hardware infection, which was seen in 5% of patients and required GES device removal. In long-term follow-up of up to five years, GES device placement has been shown to improve glycemic control and nutritional parameters, enhance quality of life, and decrease health care costs.316
Acute erosive gastritis is common in diabetic ketoacidosis and is frequently accompanied by bleeding. A postulated association between diabetes and H. pylori has been called into question.317 The incidence of duodenal ulcer in diabetes is lower than expected. Autoimmune chronic gastritis and gastric atrophy also may be seen with long-standing diabetes. In type 1 diabetes, 15% to 20% of patients have serologic evidence antiparietal cell antibodies, and this subset of patients has an increased prevalence of autoimmune gastritis with pernicious anemia, iron deficiency anemia, hypochlorhydria, and hypergastrinemia.318
Diabetic Diarrhea
Diarrhea is a common symptom of autonomic neuropathy, affecting 3.7% of diabetic patients, predominantly those with type 1 diabetes (see also Chapter 15).319 A common cause of diarrhea in diabetic patients is drug therapy, but diarrhea or increased stool frequency may occur because of coexistent celiac disease, pancreatic insufficiency, bacterial overgrowth, consumption of artificial sweeteners, islet cell tumors or fecal incontinence.320 Metformin, a biguanide derivative with structural similarity to 5-HT3–receptor agonists, is associated with diarrhea, usually with the initiation of treatment but also late in therapy.321–323 Extended release metformin is less likely to cause diarrhea than immediate release metformin and may be an alternative to discontinuing metformin therapy.324 Acarbose and miglitol are α-glucosidase inhibitors that competitively inhibit the breakdown of oligo- and disaccharides to monosaccharides in the small intestinal brush border. Thirty percent of patients treated with acarbose develop abdominal discomfort, flatulence, and diarrhea.325 Acarbose may cause diarrhea by an increase in colonic butyrate production, which increases prostaglandin E production leading to water and electrolyte loss.326
The pathogenesis of diabetic diarrhea is unclear. Marked abnormalities are found in the motor pattern of the small intestine. Phase 3 contractions during the IMMC are shorter, and phase 2 activity of the stomach and upper small intestine is abnormal. No significant differences between diabetic patients and control subjects, however, can be observed in mouth-to-cecum or whole-gut transit times. In patients treated with prokinetic agents, fasting IMMC and fed motor patterns in the small intestine may be normalized, but the symptomatic improvement of diarrhea is no better than with placebo.327 Sympathetic denervation of the gut is common in diabetic patients with autonomic neuropathy. Because adrenergic nerves normally stimulate intestinal absorption of fluids and electrolytes, decreased intestinal absorption, rather than intestinal dysmotility, may underlie the pathogenesis of diabetic diarrhea.
The long-acting somatostatin analog octreotide (50 to 100 µg subcutaneously, twice daily) may be used in the treatment of refractory diabetic diarrhea.328 It may, however, predispose to intestinal bacterial overgrowth owing to decreased small bowel transit time, and it may aggravate steatorrhea by inhibiting pancreatic exocrine function. Symptomatic measures that may be used include the prescription of codeine sulfate (30 mg every six to eight hours), diphenoxylate with atropine (Lomotil), or loperamide. In some patients, psyllium hydrophilic mucilloid may be helpful.
Fecal Incontinence
A troublesome symptom of DAN is fecal incontinence (see Chapter 17). Incontinence often coincides with the onset of diabetic diarrhea, but in most cases the total stool volume is normal. Steatorrhea is present in as many as 30% of cases.329 Autonomic dysfunction is thought to be responsible for the impairment of normal internal anal sphincter resting tone and reflexive internal sphincter relaxation. Primary management is empiric including antidiarrheal therapy and biofeedback training, whereas more severe cases may benefit from surgery or sacral nerve stimulation.330 In some patients incontinence remits spontaneously.
Constipation and Megacolon
The colon is frequently involved in diabetes mellitus. The most common gastrointestinal complaint of diabetics is constipation (see Chapter 18), related in some cases to autonomic neuropathy.331 Occasionally, severe constipation with megacolon may be encountered. Rarely, chronic intestinal pseudo-obstruction may result.332 High-fiber diets have not proved to be of great benefit, and anorectal myectomy has not been adequately evaluated. Complications of severe constipation include stercoral ulcer, perforation, volvulus, and anal overflow diarrhea. Treatment is aimed at symptomatic relief with enemas, laxatives, and cathartics.
Unexplained Abdominal Pain
Diabetic radiculopathy or diabetic plexus neuropathy of thoracic nerve roots may cause otherwise unexplained upper abdominal pain in patients with diabetic neuropathy. Pain may be associated with anorexia and weight loss, which mimics intra-abdominal malignancy. The diagnosis may be strengthened by an abnormal EMG of the anterior abdominal wall muscles when compared with an EMG of thoracic paraspinal muscles.333
Biliary Tree and Liver
Cholelithiasis, cholecystitis, and cholangitis are thought to occur more frequently in diabetic patients. Lithogenic bile composition and stasis of bile in the gallbladder may contribute to stone formation in patients with diabetes, and it is generally thought that diabetic patients have an increased incidence of cholelithiasis. As with infections in general, hepatobiliary sepsis tends to be more severe in diabetic patients. In addition to severe bouts of cholecystitis and ascending cholangitis, unusual infections with gas-producing organisms and rare abscesses due to Yersinia enterocolitica have been reported.334,335 An increased incidence of sclerosing cholangitis also has been reported in diabetic patients.336 Despite the increased severity of cholecystitis and cholangitis in these patients, however, it is not recommended that diabetic patients with asymptomatic gallstones undergo prophylactic cholecystectomy. The most prominent hepatic complication of type 2 diabetes is nonalcoholic fatty liver disease, discussed in Chapter 82.
Pancreatic Disease
The prevalence of acute pancreatic disease (see Chapter 58) and pancreatic insufficiency (see Chapter 59) is increased in patients with diabetes. Acute pancreatitis is twice as frequent in young, type 1 diabetic patients. Acute pancreatitis causing diabetic ketoacidosis (DKA) has a particularly serious prognosis, with a high mortality rate.337 In the setting of DKA, nonspecific elevations (less than three times the upper limits of normal) in serum amylase and lipase occur in 16% to 25% of cases.338 The incidence of clinically apparent chronic pancreatitis, however, is not increased. Diabetes is a risk factor pancreatic cancer (see Chapter 59) and associated with increased mortality rate.339,340 Diabetes of new onset may also be an early sign of pancreatic cancer.
THYROID DISEASE
Hyperthyroidism
Hyperthyroidism may underlie a number of important gastrointestinal symptoms, owing to its own effects on almost all organs of the gastrointestinal system. In addition, these symptoms sometimes occur in the absence of the cardinal features of hyperthyroidism (“apathetic” hyperthyroidism). Apathetic thyrotoxicosis may present with protracted abdominal pain, recurrent vomiting (thyrotoxic vomiting), marked weight loss, and altered bowel habits. Patients affected by thyroid storm may display a constellation of symptoms involving high fever, marked tachycardia, agitation, and delirium along with intestinal manifestations that include acute abdominal pain, vomiting, jaundice and severe diarrhea. Even in the absence of overt congestive heart failure or thyroid storm, jaundice, mild aminotransferase elevations (less than 250 IU/L) and prolonged prothrombin times may be observed.341
Hyperthyroidism clearly affects gastrointestinal motility. Excess thyroid hormone may cause myopathy, resulting in dysfunction of the striated muscles of the pharynx and the cervical esophagus. This is a potential mechanism that may explain dysphagia.342 Dysphagia is a rare manifestation of hyperthyroidism and can be readily reversible with correction of the thyrotoxic state.343
More than 25% of hyperthyroid patients have mild to moderate diarrhea. Intestinal transit time inversely correlates with thyroid hormone levels,344 whereas gastric emptying is not significantly increased with the hyperthyroid state.344,345 Although hypermotility is the most likely explanation for diarrhea, thyroid hormone itself can induce secretory diarrhea by increasing intracellular cyclic adenosine monophosphate, akin to the actions of cholera toxin and VIP. Hyperthyroid-associated diarrhea (and steatorrhea, when present) readily responds to treatment with propylthiouracil. Treatment with propylthiouracil can lead to a euthyroid state with concomitant normalization of orocecal transit times and relief of gastrointestinal symptoms.346 The relationship between transient hyperthyroidism and hyperemesis gravidarum during pregnancy is discussed in Chapter 38.
Infrequently, hyperthyroidism may coexist with ulcerative colitis. Hyperthyroidism may intensify the symptoms of ulcerative colitis, and it may impair the response to therapy.347
Hypothyroidism
Hypothyroidism is most commonly caused by an autoimmune mechanism or as a consequence of therapy for hyperthyroidism. It is occasionally seen in association with other diseases such as ulcerative colitis,347 pernicious anemia,348 and primary biliary cirrhosis.349 Hypothyroidism is seen in approximately 20% of patients with primary biliary cirrhosis.349 Rarely, celiac disease or diabetes mellitus is also associated with autoimmune thyroiditis.
Hypothyroidism is associated with hypomotility of the gastrointestinal tract. Disturbances of esophageal peristalsis and LES function resulting in reflux and esophagitis may be seen with severe hypothyroidism. Replacement therapy can normalize sphincter tone and restore peristalsis. Hypothyroidism also may result in gastric and intestinal hypomotility. In rare instances, phytobezoars may form and result in gastrointestinal obstructions.350 Severely impaired colonic motility may manifest with constipation, obstipation, sigmoid volvulus, rectal prolapse, fecal impaction, and rarely megacolon. Hypothyroidism can be a cause of ileus and should be considered as an etiology of pseudo-obstruction.351 Diarrhea, although rare in hypothyroidism, may be due to bacterial overgrowth from bowel hypomotility. Antibiotic treatment can result in resolution of diarrheal symptoms.352 Myxedema has also been found in association with Cronkhite-Canada syndrome (see Chapter 122).353
Medullary Carcinoma of the Thyroid
Medullary carcinoma of the thyroid (MCT) is a calcitonin-producing tumor of the C cells of the thyroid gland. Diarrhea is seen in one third of patients with MCT. Diarrhea may occur presumably due to the effects of high circulating calcitonin on the gut.354 MCT also may produce VIP and prostaglandins that contribute to diarrhea. Decreased colonic transit time due to as yet unknown humoral agents may also underlie the diarrhea of MCT.355 MCT is also associated with multiple endocrine neoplasia (MEN) syndromes IIA and IIB. These syndromes can be complicated by hyperparathyroidism and pheochromocytomas and in MEN-IIB with mucosal neuromas.
ADRENAL DISEASE
Adrenal insufficiency, or Addison’s disease, is associated with gastrointestinal symptoms or pathology in more than half of cases. A constellation of symptoms including anorexia, weight loss, nausea, vomiting, diarrhea, and abdominal pain may be present. Patients with Addison’s disease may also present with chronically elevated aminotransferase levels.356 Cyclical vomiting in children may rarely be due to adrenal insufficiency.357 Malabsorption and diarrhea seen in some patients with Addison’s disease are apparently due to functional defects in enterocytes that can be readily reversed with the administration of glucocorticoids. Atrophic gastritis, achlorhydria, and pernicious anemia may be present in association with autoimmune Addison’s disease.
Pheochromocytomas are tumors arising from the adrenal medulla and chromaffin tissue that secrete high levels of catecholamines, leading to hypertension. The humoral effects of high circulating levels of catecholamines may result in ileus or pseudo-obstruction.358 Gastrointestinal manifestations of pheochromocytoma also include ischemic colitis, diarrhea, acute abdominal pain, and, rarely, gastrointestinal bleeding.359 For unclear reasons, pheochromocytoma is associated with an increased incidence of cholelithiasis. Some patients also have MEN-IIA or MEN-IIB syndrome (see earlier).
PITUITARY DISEASE
Pituitary disorders infrequently affect the gastrointestinal tract, except in association with MEN-I syndrome (see Chapter 32). Hypercortisolism, caused by the inappropriate secretion of corticotropin in Cushing’s disease, may be associated with an increased incidence of gastric ulceration when concomitant NSAIDs are used.360 Panhypopituitarism may present with addisonian crisis, with hypotension, nausea, vomiting, abdominal pain, and diarrhea. The excessive secretion of pituitary growth hormone with concomitant elevation of insulin-like growth factor I results in acromegaly. The incidence of adenomatous colonic polyps may be increased in patients with acromegaly and the adenomas tend to be larger, multiple, and right-sided.361 The risk of colon cancer is approximately two-fold higher in acromegaly, but screening recommendations have been contentious.362
PARATHYROID DISEASE
Hyperparathyroidism
Gastrointestinal problems are common in patients with hyperparathyroidism.363 Most common complaints are constipation, diffuse abdominal discomfort, or nausea and vomiting. A minority (5% to 15%) have peptic ulcer disease, and a small percentage (1% to 2%) develop pancreatitis. Remission of pancreatitis after parathyroidectomy has been reported.364 Severe pancreatitis may also occur immediately following parathyroidectomy has been reported.365 Gastrointestinal symptoms associated with hypercalcemia include nausea, vomiting, anorexia, and abdominal pain. Some patients with hyperparathyroidism have MEN-I or MEN-II syndrome (see earlier).
Hypoparathyroidism
Hypoparathyroidism with hypocalcemia may be associated with malabsorption and mild to moderate steatorrhea. Constipation, and in rare instances, even pseudo-obstruction may be important gastrointestinal disturbances in this disease. In the familial polyendocrine failure syndrome (candidiasis, endocrinopathy, or polyendocrine autoimmune disease, type 1), patients have hypoparathyroidism, adrenal insufficiency, hypogonadism, and, in many cases, diabetes mellitus. From 4% to 29% also have malabsorption. Varying degrees of gastric atrophy with antiparietal cell antibodies, hypochlorhydria, autoimmune hepatitis, dental enamel hypoplasia, and severe oral and esophageal candidiasis are also seen.366 Intestinal lymphangiectasia with protein-losing enteropathy also has been reported in association with malabsorption and hypoparathyroidism.367 Idiopathic hypoparathyroidism may also coexist with celiac disease likely due to autoimmune reactivity. When this occurs, a gluten-free diet may lead to the disappearance of parathyroid immunoreactivity.368
DISORDERS OF LIPID METABOLISM
HYPERLIPOPROTEINEMIAS AND DYSLIPIDEMIAS
In familial hyperchylomicronemia (type I phenotype), the plasma is lactescent, with marked elevation of chylomicrons and triglycerides due to a deficiency of lipoprotein lipase. Manifestations can appear early in life and include recurrent episodes of abdominal pain, fever, peritonitis, and pancreatitis (see Chapter 58). In most patients the cause of recurrent attacks of pain is not known.369 Patients with familial hyperbetalipoproteinemia (type IV phenotype) suffer from premature atherosclerosis, hyperuricemia, and attacks of pancreatitis that generally occur when plasma triglyceride values are greater than 2000 mg/dL.370 The hyperlipidemia may mask elevated plasma amylase values. Type IV patients also have an increased incidence of cholelithiasis and cholecystitis.371 Patients with familial hyperlipoproteinemia (type V phenotype) are prone to bouts of abdominal pain, with or without pancreatitis. Exacerbation of endogenous hypertriglyceridemia by diabetes, diet, alcohol, or medications can also cause pancreatitis.370
ABETALIPOPROTEINEMIA
Abetalipoproteinemia is an autosomal recessive disorder characterized by acanthotic erythrocytes, serum lipid abnormalities, ataxia, atypical retinitis pigmentosa, and steatorrhea.372 The typical laboratory feature is complete absence in plasma of all lipoproteins containing apolipoprotein B: chylomicrons, low-density lipoprotein (LDL), and very-low-density lipoprotein (VLDL). The histologic appearance of the small intestine after an overnight fast is marked by mucosal epithelial cells loaded with lipid droplets (Fig. 35-9).373 By contrast, the submucosa and lamina propria show practically no lipid, and the lymphatics are empty. The villi are normal in length and configuration. Mild steatorrhea with onset during the first 2 years of life is seen (see Chapter 101). Cholesterol malabsorption with increased endogenous cholesterol synthesis has also been reported.374 The intestinal mucosa may appear yellowish on endoscopy, reflecting the presence of mucosal lipid.375 Therapy consists of substituting medium-chain for long-chain triglycerides.
TANGIER DISEASE
Tangier disease is an autosomal recessive disorder characterized by accumulation of cholesterol esters in macrophages in tonsils, thymus, lymph nodes, marrow, liver, and the gut. Tangier disease is caused by a mutation in the adenosine triphosphate-binding cassette protein, ABCA1, which mediates the efflux of excess cellular sterol to apolipoprotein A-I (apo A-I), a step leading to the formation of beneficial high-density lipoprotein (HDL).376 These patients have very low levels of plasma cholesterol and HDLs, owing to a lack of apo A-I. The gene encoding apo A-I is normal in Tangier disease, but a defect in post-translational processing results in rapid degradation of apo A-I.376 The striking clinical findings include yellow-orange “streaked” tonsils in 80% of cases, hepatosplenomegaly, and peripheral neuropathy. Patients may have diarrhea without steatorrhea. Colonoscopy reveals orange-brown mucosal spots throughout the colon and rectum, and laparoscopy reveals similar yellow patches on the surface of the liver due to cholesterol esters in hepatic reticuloendothelial cells.377
NEUTRAL GLYCOSPHINGOLIPIDOSES
Fabry’s disease is an X-linked disorder of glycolipid metabolism due to the deficiency or absence of the enzyme α-galactosidase A, resulting in globotriaosylceramide deposition in many tissues and subsequent organ dysfunction. Impaired motility is the prominent gastrointestinal abnormality.378 Electron microsopic examination of biopsy specimens from the small intestine and rectum reveals large sphingolipid-filled vacuoles in the ganglion cells of Meissner’s plexus within smooth muscle cells of the muscularis mucosa and within endothelial cells lining the blood vessels. Mucosal enterocytes are normal.378 The prevalence of gastrointestinal symptoms was 52%, with abdominal pain and diarrhea being the most frequent symptoms.379 Delayed gastric emptying, bacterial overgrowth, increased fecal bile acid loss, cholelithiasis, and jejunal diverticulosis with perforation have been documented. Successful treatment of the gastrointestinal component of this disorder with metoclopramide and tetracycline has been reported. Glycolipid deposition in small vessels can induce severe vasculitis and thrombosis, resulting in ischemic bowel lesions. Ileal perforation also has been reported.380 Thirty percent to 60% of obligate carrier women and 60% of men have nonspecific gastrointestinal symptoms that can improve with agalsidase alfa enzyme replacement therapy.381
Gaucher’s disease is a rare, usually autosomal recessive deficiency of the enzyme acid β-glucosidase resulting in the deposition of glucosylceramide in the cells of the reticuloendothelial system, including the liver and spleen. In the adult form of the disease gastrointestinal complications predominate, including hepatosplenomegaly, hepatic cirrhosis, ascites, and esophageal variceal hemorrhage.382,383
Niemann-Pick disease is a rare autosomal-recessive disease with a predilection for Ashkenazi Jews. Types A and B result from defects in sphingomyelinase. In type C Niemann-Pick disease, mutation in Niemann-Pick protein C results in defective transport of cholesterol across the lysosomal membrane.384 Sphingomyelinase deficiency results in the deposition of sphingomyelin and cholesterol in the liver and spleen, the CNS, and the lungs and skin. Gastrointestinal complications include hepatosplenomegaly and liver failure. Rare adult survivors of Niemann-Pick disease have been reported.385
RENAL DISEASES
Upper gastrointestinal tract symptoms are common in patients with chronic kidney disease (CKD) who require peritoneal dialysis (PD) or hemodialysis (HD). Anorexia, singultus (hiccups), nausea, vomiting, epigastric pain, and heartburn are common manifestations of azotemia. Delayed gastric emptying is common in CKD.386 Although the prevalence of peptic ulcer is only 2%, which is not significantly different from that in the general population, gastritis, duodenitis, and mucosal erosions are commonly seen.387 A variety of data suggests that neither hyperacidity, hypergastrinemia, nor H. pylori plays major roles in the pathogenesis of uremic gastropathy, although these data have recently been called into question.388 Impaired mucosal cytoprotection has been postulated but not proved. Also seen on esophagogastroduodenoscopy are esophagitis, Brunner’s gland hyperplasia, gastric fold thickening, and nodular duodenitis (which resolves after renal transplantation) and angiodysplasia.389–391 GERD may be related to the absence of H. pylori infection, amyloidosis, and PD, which increases intra-abdominal pressure.392 In controlled studies, the incidence of gallstones in CKD is similar to healthy controls.393
It is possible that angiodysplastic lesions in the upper and lower gastrointestinal tract are no more common in patients with CKD than in the general population but are discovered more frequently because of their greater tendency to bleed. Angiodysplasia in renal failure is acquired lesions formed by repeated episodes of submucosal venous outflow obstruction resulting in incompetent precapillary sphincters with subsequent arteriovenous communication.394 Angiodysplastic lesions are much more likely to bleed in patients with CKD than in patients with normal renal function,395 perhaps because of uremic platelet dysfunction. In a series of CKD patients with upper gastrointestinal hemorrhage, gastric ulcer (37%) and duodenal ulcer (23%) were the two most common bleeding lesions, but angiodysplasia of the upper gastrointestinal tract was the cause of bleeding in 13%. In contrast, angiodysplasia was only responsible for 1.3% of upper intestinal tract bleeding in control patients. In CKD patients with recurrent hemorrhage, angiodysplasia was the most frequent cause of bleeding. Angiodysplasia as a cause of bleeding was most closely associated with the duration of renal failure and the need for hemodialysis.396 In CKD, peptic lesions may be managed successfully with standard medical treatments in appropriate “renal” doses (see Chapter 53), and angiodysplasia may be treated with laser, electrocoagulation, or surgery (see Chapters 19 and 36). Small intestinal complications of CKD include ileus, ulceration, and nonocclusive ischemic bowel disease.397–399 Diarrhea may occur secondary to bacterial overgrowth related to abnormal small intestinal motility.400 In addition, exocrine pancreatic insufficiency has been documented in a number of hemodialysis patients.401 The cause of the condition is not known, but patients may improve clinically with pancreatic enzyme replacement.
Patients with CKD appear more likely to develop colonic perforation from ruptured diverticula, fecalomas (secondary to the use of aluminum-containing antacids or barium), or cecal ulcers that may bleed profusely.402 Life-threatening hemorrhage from rectal ulcers also has been reported.403 Colonic intussusception and ileus are also encountered in CKD. The diarrhea experienced by some patients with CKD appears to be related to abnormal bile acid metabolism.404 Ischemic colitis in patients receiving HD tends to be more right-sided in anatomic distribution, which is associated with poor outcome.405
See Chapter 34 for a discussion of gastroin-testinal and hepatic complications associated with renal transplantation.
NEUROLOGIC DISEASES
Because of the importance of nerves and neurotransmitters on gastrointestinal function (see Chapter 1), it is not surprising that neurologic diseases are frequently associated with gastrointestinal symptoms. Some of the more common disorders affecting the CNS (brain and spinal cord), cranial nerves, autonomic nervous system, neuromuscular junction, and musculature are presented in Table 35-6 and in the following sections.
Table 35-6 Gastrointestinal Manifestations of Neuromuscular Diseases
| LOCATION/DISEASE | GASTROINTESTINAL MANIFESTATIONS |
|---|---|
| Cerebrum/Cerebellum | |
| Cerebrovascular accident | Oropharyngeal dysphagia, gastroparesis, constipation, peptic ulceration, anorectal dysfunction |
| Multiple sclerosis | Oropharyngeal dysphagia, gastroparesis, constipation, anorectal dysfunction |
| Cerebral palsy | Oropharyngeal dysphagia |
| Migraine headache | Nausea, vomiting, abdominal pain (abdominal migraine), peptic ulceration |
| “Abdominal epilepsy”/viscerosensory auras | Abdominal pain, bloating, diarrhea |
| Pseudotumor cerebri | Nausea, vomiting |
| Brainstem/Cranial Nerve | |
| Cerebrovascular accident, other brainstem disorders | Oropharyngeal dysphagia, dysgeusia |
| Multiple sclerosis | Oropharyngeal dysphagia, dysgeusia |
| Pseudobulbar palsy | Oropharyngeal dysphagia |
| Diphtheria | Oropharyngeal dysphagia |
| Spinal Cord/Peripheral Nerve | |
| Spinal cord injury/transection | Gastroparesis, constipation, incontinence, megacolon, ileus, autonomic dysreflexia |
| Amyotrophic lateral sclerosis | Oropharyngeal dysphagia, ileus |
| Charcot-Marie-Tooth syndrome | Oropharyngeal dysphagia, delayed gastric emptying |
| Tabes dorsalis | Abdominal pain crises, diarrhea |
| Poliomyelitis | Ileus, gastric atony, megacolon |
| Alcoholic and amyloid neuropathy | Esophageal and gastrointestinal dysmotility |
| Extrapyramidal/Autonomic | |
| Parkinson’s disease | Oropharyngeal dysphagia, constipation, fecal incontinence |
| Huntington’s chorea | Oropharyngeal dysphagia, gastroparesis, constipation |
| Familial dysautonomia (Riley-Day syndrome) | Esophageal dysmotility, vomiting crises, gastric atony, diarrhea, megacolon |
| Shy-Drager syndrome | Postprandial orthostatic hypotension, esophageal dysmotility, achlorhydria, constipation |
| Chagas’ disease | Achalasia, megaesophagus, megaduodenum, megacolon |
| Paraneoplastic neuropathy | Achalasia, megacolon, intestinal pseudo-obstruction |
| Ganglioneuromatosis | Constipation, megacolon |
| Diabetic neuropathy | See Table 35-5 |
| Neuromuscular Junction | |
| Myasthenia gravis | Oropharyngeal dysphagia, autoimmune hepatitis, primary biliary cirrhosis |
| Muscle Disease | |
| Stiff-man syndrome | Oropharyngeal dysphagia |
| Oculopharyngeal muscular dystrophy | Oropharyngeal dysphagia |
| Mitochondrial neurogastrointestinal encephalomyopathy | Dysmotility, achalasia, malabsorption, pseudo-obstruction, diarrhea |
| Duchenne’s muscular dystrophy | Oropharyngeal dysphagia, gastric atony, malabsorption, megacolon, pseudo-obstruction |
| Familial visceral myopathy | Dysphagia, pseudo-obstruction |
| Myotonic dystrophy | Oropharyngeal dysphagia, esophageal dysmotility, gastric atony, megacolon, pseudo-obstruction, volvulus, gallbladder dysfunction |
| Polymyositis/dermatomyositis | See Table 35-1 |
NEUROGENIC ABDOMINAL PAIN (see Chapters 10 and 11)
Neurogenic causes of abdominal pain originate in either the CNS or peripheral nervous system. Central neurogenic abdominal pain can result from abdominal migraines. Although classic migraine headaches are often associated with nausea and vomiting, abdominal migraine is a migraine variant characterized by recurrent gastrointestinal symptoms, including vomiting and epigastric pain.406 It is often seen in children and is not always associated with headache. The pathophysiology of the gastrointestinal symptoms is unclear, and some have questioned the validity of this controversial diagnosis. In one Greek study, most pediatric patients with abdominal migraine had evidence of esophagitis, gastritis, or duodenitis at endoscopy.407 A more recent study of 53 children with abdominal migraine and no underlying gastrointestinal pathology demonstrated symptomatic improvement or successful prophylaxis using standard antimigraine therapies such as propranolol or cyproheptadine.408 Adults with migraine do not tend to report abdominal pain,409 although some investigators believe that a subset of adult patients with recurrent nonorganic abdominal pain may suffer from abdominal migraine. Unfortunately, the lack of a precise definition of abdominal migraine has made research in this area difficult.
Abdominal epilepsy is an uncommon cause of central neurogenic abdominal pain. One retrospective study describes the spectrum and clinical course of 10 patients with abdominal epilepsy.410 These patients, each with temporal lobe electroencephalographic (EEG) abnormalities, experienced a variety of paroxysmal gastrointestinal symptoms that included periumbilical and right upper quadrant pain, bloating, and diarrhea. All of the patients also experienced CNS symptoms such as headaches, confusion, dizziness, syncope, or blindness, occurring daily in association with the gastrointestinal complaints. Anticonvulsant therapy resulted in resolution of gastrointestinal and CNS disturbances. Even patients with classic epileptic seizure disorders may experience viscerosensory auras (vague epigastric sensations and nausea). Although they can occur in patients with temporal lobe mass lesions, epigastric auras are more frequently associated with hippocampal sclerosis. These epigastric auras can be unpleasant or even debilitating. Unfortunately, 25% of patients who are rendered seizure free after anteromedial temporal lobe resection continue to have persistent epigastric auras.411 Although epilepsy with gastrointestinal symptoms is uncommon, an EEG should be considered in the diagnostic workup of patients with unexplained paroxysmal gastrointestinal complaints associated with CNS symptoms.
GASTROINTESTINAL COMPLICATIONS OF ACUTE HEAD INJURY AND STROKE
Increased intracranial pressure from any cause may lead to episodic projectile vomiting, which may precede other signs. In addition to direct effects of CNS injury on the control of oropharyngeal muscle movement during swallowing, acute head trauma and stroke (cerebrovascular accidents) are associated with a high incidence of upper gastrointestinal tract pathology.412 In one prospective study, acute gastrointestinal erosive lesions were found at endoscopy in 75% of patients. Erosive gastritis was seen in 69% of cases, gastric ulcer in 23%, esophagitis in 11%, and duodenal inflammation in 8%.413 Most lesions were present within a week of injury. There was no correlation between glucocorticoid administration and development of upper gastrointestinal tract lesions. Although these lesions fall within the spectrum of stress gastropathy (see Chapters 52 and 53), an additional pathogenetic mechanism may play a role in their development. Serum gastrin levels are elevated in patients with head injury,414 presumably through a direct neurogenic reflex, and gastric hypersecretion has been reported.415 Because these injuries are largely preventable, patients with acute neurologic injury due to trauma, severe strokes, neurosurgery, or some other condition should receive prophylactic antiulcer therapy (see Chapter 53).
GASTROINTESTINAL PROBLEMS AFTER SPINAL CORD INJURY
The effect of spinal cord injuries on the gastrointestinal tract depends on the level of the lesion. Delayed gastric emptying is seen in patients with cervical spinal cord injuries.416 In the early postinjury period, severe gastric stasis, gastric dilation, and ileus are often present.417 Nasogastric suction is frequently required. Promotility agents such as metoclopramide can be effective because the enteric nervous system and smooth muscle layers are intact. A frequent problem in the first weeks after injury is peptic ulceration, although the mechanism is unclear. Upper gastrointestinal hemorrhage is more common with cervical cord injuries, with the use of oral anticoagulants, or when there is respiratory distress.418 Ulcer perforation and peritonitis may not be detected initially because of myelopathy involving sensory fibers or because of concomitant glucocorticoid therapy. When ulcer surgery is required, gastric resection or simple closure of the perforation is sufficient; truncal vagotomy is not performed because of the risk of severe gastric retention.419 Pancreatitis, another early complication of spinal injuries, also may be related to the effects of glucocorticoids.417,420 Autonomic dysreflexia, a life-threatening condition, sometimes affects patients whose lesion lies above the fifth thoracic root, the upper level of greater splanchnic flow. The pathophysiology involves an abnormal autonomic reflex that is initiated by fecal impaction or bladder distention and leads to severe hypertension and tachycardia.421 If untreated, seizures, subarachnoid hemorrhage, and stroke may result. Routine bladder catheterization and avoidance of constipation are preventive.
Patients often face a different set of problems in the months to years following permanent spinal cord damage. With chronic loss of function, patients with quadriplegia are more likely to have gut complications than are patients with paraplegia. The incidence of gastroesophageal reflux is increased.417 There may be decreased bioavailability of orally administered drugs owing to impaired gastric emptying.422 Secondary amyloidosis involving the gastrointestinal tract is more common (see later), especially when chronic pyelonephritis and renal failure complicate spinal cord disease.417,423 Other complications include cholelithiasis, the superior mesenteric artery syndrome (see Chapters 14 and 36), hemorrhage due to solitary colonic ulcer, and the precocious appearance of diverticulosis.78,417 Many patients with spinal cord injury have marked impairment of their bowel function. Fecal incontinence and urgency can have a significant effect on quality of life.424,425
Chronic constipation plagues many patients with spinal cord injury. Damage to neurons in the spinal cord eliminates the sensation of rectal fullness and the voluntary control of defecation. There may be decreased splanchnic outflow, impairing the coordination of intestinal and colonic motility. Prolonged transit time may be explained by decreased colonic activity, colonic contractions, and intraluminal pressure. Further, the gastrocolic reflex after feeding may be regionally diminished in the rectosigmoid.426 Fortunately, the lower motor neurons of the second, third, and fourth sacral roots, which provide the sensory and motor fibers for the defecation reflex, are usually intact.
Rehabilitation of bowel function is individualized to each patient’s disability. Physical exercise, adequate fluid intake, and stool softeners are prescribed, as for constipation of any origin. Most patients can learn to distend the rectum digitally on a regular schedule to initiate the defecation reflex. Stimulatory laxatives such as bisacodyl suppositories occasionally are necessary. It is not clear which patients, if any, may benefit from promotility agents. In the setting of major bowel dysfunction, colostomy may be a viable option.427 Patients with spinal cord injuries often have subtle symptoms and signs of colorectal emergencies that may lead to a delayed diagnosis and increased morbidity and mortality.428
DISEASES OF THE AUTONOMIC NERVOUS SYSTEM
Congenital and neurodegenerative diseases of the autonomic nervous system may affect the gastrointestinal tract and are listed in Table 35-6. These include familial dysautonomia, or the Riley-Day syndrome. Patients present at birth with feeding difficulties, poor temperature control, and motor incoordination. Common gastrointestinal symptoms include dysphagia, gastroesophageal reflux, and abnormal swallowing reflexes. A large percentage of patients require feeding gastrostomy or fundoplication. Lower gastrointestinal tract symptoms are less common, but patients may develop diarrhea as a result of decreased motility and bacterial overgrowth.429
Idiopathic autonomic neuropathy is a relatively uncommon acquired cause of autonomic neuropathy.429 It can affect sympathetic as well as parasympathetic function (pandysautonomia), or it may be limited to a parasympathetic deficit (cholinergic dysautonomia). The onset of symptoms can be acute or subacute. Cholinergic dysautonomia usually affects patients in their teens, whereas pandysautonomia manifests in early middle age.430 The etiology of this condition is unknown, but most cases develop after nonspecific viral infections. The syndrome also has been seen in association with mononucleosis,431 Stevens-Johnson syndrome,432 and herpes zoster or herpes simplex infections.433 In nearly 70% of cases, the initial manifestation of the disease is in the gastrointestinal tract. The most common symptoms are due to excessive cholinergic activity, such as diarrhea, hyperhidrosis, and hypersalivation. Although a large proportion of these patients has manometric abnormalities, complications of motility disorders such as bacterial overgrowth are usually not seen.429
A poorly understood autonomic condition is postprandial orthostatic hypotension, which occurs commonly in older adults and in patients with autonomic dysfunction. Direct gastrointestinal symptoms are infrequent, but abdominal pain and nausea may occur after meals. Other clinical manifestations include postprandial presyncope and syncope. The diagnosis is established by demonstrating a 20-mm Hg or more decrease in systolic blood pressure after a standardized test meal.434
Secondary causes of autonomic neuropathy include the porphyrias (variegate, acute intermittent, hereditary coproporphyria), discussed in Chapter 76, diabetes mellitus (discussed earlier), paraneoplastic autonomic neuropathy, amyloidosis (discussed following), and Chagas’ disease (see Chapter 109).
EXTRAPYRAMIDAL DISORDERS
Huntington’s chorea is a hereditary neurodegenerative basal ganglia disease characterized by chorea, dementia, and emotional changes. Dysphagia is a common symptom that may lead to fatal complications from aspiration pneumonia.435 Gastroparesis and constipation have been reported.
Parkinson’s disease is frequently associated with gastrointestinal symptoms. Oropharyngeal dysphagia and drooling can be particularly distressing. Abnormalities are found in the oral, pharyngeal, and esophageal stages of deglutition. Poor voluntary control of the tongue results in lingual hesitancy, poor bolus formation, and delayed transit into the pharynx. This defect in bolus propulsion persists in the poorly contracting pharynx. Food is often retained in the valleculae, and tracheal aspiration frequently occurs even in asymptomatic patients. Incomplete upper esophageal sphincter relaxation can be seen in 21% of patients.436 Manometric studies suggest that between 61% and 73% of patients have abnormal esophageal function, including aperistalsis, simultaneous or ineffective contractions, and decreased LES tone.437 Treatment of swallowing difficulties in patients with Parkinson’s disease should include optimization of therapy directed at the tremor and associated depression and treatment of underlying gastroesophageal reflux if it exists. Levodopa has beneficial effects on the oral and pharyngeal aspects of swallowing and should be taken at mealtimes. Anticholinergics may impair esophageal motility, and excessive dosing of levodopa itself may cause nausea. Botulinum toxin injection of the cricopharyngeal muscle leads to significant short-term improvement in swallowing and weight gain.438 Finally, smaller and more frequent meals of soft food and posterior spoon placement may help in the oral phase of swallowing. Delayed gastric emptying, constipation due to delayed colonic transit, and external sphincter dysfunction also can be seen in Parkinson’s disease.437
MULTIPLE SCLEROSIS
Multiple sclerosis (MS) is associated with fecal incontinence and constipation in up to 70% of patients (see Chapters 17 and 18).439 Impaired external and internal anal sphincter function contributes to fecal incontinence in MS.440 Defecography in MS patients with intractable constipation can demonstrate rectal intussusception, rectal outlet obstruction, and failure of the puborectalis and anal sphincter muscles to relax appropriately.441,442 Biofeedback training may be beneficial in patients with limited disability and a nonprogressive disease course.443 Cranial nerve involvement in MS may lead to oropharyngeal dysphagia.
NEUROMUSCULAR DISORDERS
Degenerative diseases of peripheral motor neurons can present with gastrointestinal symptoms. In amyotrophic lateral sclerosis, bulbar dysfunction leads to oropharyngeal dysphagia with complications of malnutrition and aspiration. Patients often need gastrostomy or jejunostomy placement; a forced vital capacity of less than 50% is associated increased postprocedural mortality.444 Charcot-Marie-Tooth degenerative peripheral neuropathy also has been reported to cause pharyngeal dysphagia and abnormal gastrointestinal motility with delayed gastric emptying and disordered motility of the esophageal body.445
Motor end-plate disorders such as myasthenia gravis, an autoimmune disorder of the neuromuscular junction, are frequently associated with oropharyngeal dysphagia. Autoimmune hepatitis and primary biliary cirrhosis also may be associated with myasthenia gravis.446,447
Inherited muscular dystrophies, including myotonic dystrophies (see later), are generally believed to involve only skeletal muscles. However, several muscular (and neuromuscular) disorders have been associated with motor disturbances of the gastrointestinal system. Duchenne’s muscular dystrophy (DMD) is an X-linked recessive disease that is the most common neuromuscular disease of childhood. Patients with DMD may experience nausea and vomiting, as well as abdominal distention, constipation, pseudo-obstruction, and gastric dilation. Even when gastrointestinal symptoms are absent, dysfunction of smooth muscle in the upper gastrointestinal tract is detectable.448 Patients with DMD as well as those with oculopharyngeal muscular dystrophy may experience cervical dysphagia. Also, gastroparesis and intestinal pseudo-obstruction have been reported in DMD.449
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare autosomal recessive mitochondrial myopathy defined by the constellation of peripheral neuropathy, opthalmoparesis, and gastrointestinal dysmotility; muscle biopsy reveals histologic features of a mitochondrial myopathy.450 Gastrointestinal symptoms include dysmotility, diarrhea, pseudo-obstruction, achalasia, and malabsorption. A possible variant of MNGIE may be the “3-A” syndrome of familial achalasia, alacrima, and ACTH (corticotropin) sensitivity, characterized by postural hypotension, achalasia, decreased sweating and tears, and denervation hypersensitivity of the pupils.451
Stiff-man syndrome manifests as symmetrical stiffness and painful spasm of the axial musculature. Dysphagia, perhaps due to spasm of the cricopharyngeus and upper esophagus, and delayed gastric emptying have been reported.452 Myotonic dystrophy, an autosomal dominant disease of striated and smooth muscle, is considered a rare cause of gastrointestinal dilation and abnormal peristalsis. Gastrointestinal problems can be the presenting feature in 28% of patients with common symptoms including abdominal pain, dysphagia, emesis, diarrhea, and fecal incontinence.449
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 35 Gastrointestinal and Hepatic Manifestations of Systemic Diseases
RHEUMATOLOGIC AND COLLAGEN VASCULAR DISEASES
Rheumatologic diseases encompass a wide variety of clinical syndromes and are frequently associated with gastrointestinal abnormalities (Table 35-1). In addition, the medications used to treat these diseases often produce gastrointestinal and hepatic toxicity. This section focuses on the more common abnormalities that may be encountered by the gastroenterologist.
Table 35-1 Gastrointestinal Manifestations of Rheumatologic Diseases
| DISEASE | ABNORMALITY/ASSOCIATION | CLINICAL MANIFESTATIONS |
|---|---|---|
| Rheumatoid arthritis | Temporomandibular arthritis | Impaired mastication |
| Esophageal dysmotility | Dysphagia, GERD | |
| Visceral vasculitis | Abdominal pain, cholecystitis, intestinal ulceration and infarction | |
| Amyloidosis | Pseudo-obstruction, malabsorption, protein-losing enteropathy, intestinal ulceration and infarction, gastric outlet obstruction | |
| Portal hypertension (Felty’s syndrome) | Variceal hemorrhage | |
| Gold enterocolitis | Enteritis, diarrhea, fever, eosinophilia, megacolon | |
| Scleroderma | Esophageal dysmotility | Dysphagia, GERD, stricture, Barrett’s esophagus |
| Gastroparesis | Gastric retention, GERD | |
| Intestinal fibrosis and dysmotility | Constipation, pseudo-obstruction, malabsorption, intussusception, volvulus, pneumatosis intestinalis | |
| Pseudodiverticula | Hemorrhage, stasis, bacterial overgrowth | |
| Arteritis (rare) | Mesenteric thrombosis, infarction, pancreatic necrosis | |
| Pancreatitis | Calcific pancreatitis, pancreatic exocrine insufficiency | |
| SLE | Esophageal dysmotility | Dysphagia, reflux |
| Mesenteric vasculitis | GI ulceration, intestinal infarction, intussusception, pancreatitis, pneumatosis intestinalis | |
| Sjögren’s syndrome | Desiccation of membranes | Oral fissures, oropharyngeal dysphagia |
| Esophageal webs | Dysphagia | |
| Gastric lymphoid infiltrates | ||
| Pancreatitis | Abdominal pain, pancreatic exocrine insufficiency | |
| Primary biliary cirrhosis | Jaundice, hepatic failure, variceal hemorrhage | |
| Polymyositis-dermatomyositis | Skeletal muscle dysfunction | Aspiration, impaired glutition |
| Dysmotility | Dysphagia, GERD, gastroparesis, constipation, diverticula | |
| Mesenteric vasculitis (rare) | GI ulceration, perforation, pneumatosis intestinalis | |
| MCTD | Dysmotility | Dysphagia, GERD, stricture, gastroparesis, bezoars, pseudo- obstruction |
| PAN | Mesenteric vasculitis (rare) | Ulceration, perforation, pancreatitis |
| Mesenteric vasculitis | Cholecystitis, appendicitis, intestinal infarction, pancreatitis, perforation, strictures, mucosal hemorrhage, submucosal hematomas | |
| CSS | Mesenteric vasculitis | Hemorrhage, ulceration, intestinal infarction, perforation |
| Eosinophilic gastritis | Gastric masses | |
| Henoch-Schönlein purpura | Mesenteric vasculitis | Intussusception, ulcers, cholecystitis, hemorrhage, intestinal infarction, appendicitis, perforation |
| Kohlmeier-Degos disease | Mesenteric vasculitis | Hemorrhage, ulceration, intestinal infarction, malabsorption |
| Cogan’s syndrome | Mesenteric vasculitis (infrequent) | Hemorrhage, ulceration, intestinal infarction, intussusception |
| Crohn’s disease | Bloody diarrhea, abdominal pain, fissures, fistulas | |
| Wegener’s granulomatosis | Mesenteric vasculitis | Cholecystitis, appendicitis, ileocolitis, intestinal infarction |
| Cryoglobulinemia | Mesenteric vasculitis (rare) | Intestinal infarction, ischemia |
| Behçet’s disease | Mucosal ulcerations | Hemorrhage, perforation, pyloric stenosis |
| Complications as in rheumatoid arthritis | ||
| Reactive arthritis | Ileocolonic inflammation | Usually asymptomatic |
| Familial Mediterranean fever | Serositis/peritonitis, amyloidosis, PAN, Henoch-Schönlein purpura | Abdominal pain, fever, dysmotility |
| Marfan/Ehlers-Danlos syndromes | Defective collagen | Megaesophagus, hypomotility, diverticula, megacolon, malabsorption, perforation, arterial rupture |
CSS, Churg-Strauss syndrome; GERD, gastroesophageal reflux disease; GI, gastrointestinal; MCTD, mixed connective tissue disease; PAN, polyarteritis nodosa; SLE, systemic lupus erythematosus.
RHEUMATOID ARTHRITIS
Approximately 0.8% of adults worldwide are affected with rheumatoid arthritis (RA), which is a chronic, inflammatory autoimmune disease primarily targeting the synovial tissues with systemic manifestations. Oropharyngeal symptoms may occur in patients with RA as a result of xerostomia, temporomandibular joint (TMJ) arthritis, cervical spine abnormalities, and laryngeal involvement.1 Esophageal dysmotility, characterized by low-amplitude peristaltic waves, has been described in the proximal, middle, and distal esophagus with reduced lower esophageal sphincter (LES) pressure.1,2 Rheumatoid vasculitis typically occurs in the setting of severe RA with rare gastrointestinal manifestations such as ischemic cholecystitis or appendicitis, ulceration, pancolitis, infarction, or intra-abdominal hemorrhage due to a ruptured visceral aneurysm.3,4 Other gastrointestinal complications of RA include amyloidosis (discussed later) and malabsorption. Felty’s syndrome–RA, splenomegaly, and leukopenia have been associated with severe infections, portal hypertension, and variceal hemorrhage.5
Hepatic Abnormalities
Abnormal liver function tests, especially elevations of serum alkaline phosphatase of hepatobiliary origin,6–8 are commonly observed in patients with RA. In one large series of patients with RA,6 18% had elevated levels of serum alkaline phosphatase and 11% were found to have hepatomegaly. Fluctuations in serum alkaline phosphatase levels have also been reported to correlate with disease activity.6,8 However, degrees of alkaline phosphatase elevations are usually modest, with the mean level being less than two-fold abnormal.6 Furthermore, other clinical signs of liver disease are usually absent, and liver biopsy and autopsy studies have not revealed any consistent or specific findings, with the most common abnormalities being fatty change, Kupffer cell hyperplasia, and mild mononuclear cell infiltration of the portal tracts or rare parenchymal foci of hepatocyte necrosis.6,9–11 Periportal fibrosis is also present in a small minority of cases.11 Determination of etiology of hepatic dysfunction in patients with active rheumatoid arthritis is complicated by the fact that many of the agents commonly used as therapy for this disease have known potential for liver injury.7,10–12
In a small subset of patients with RA and/or Sjögren’s syndrome, antimitochondrial antibodies are present along with the biochemical and histologic features of primary biliary cirrhosis.13–15 The incidence of primary biliary cirrhosis or autoimmune hepatitis appears to be much higher in patients with Sjögren’s syndrome alone than in those with Sjögren’s RA.16,17
Because chronic hepatitis C and RA are relatively common diseases of adults, it is not surprising that these entities are found concurrently in some patients. However, in addition, it has been noted that 75% of individuals with chronic hepatitis C infection develop rheumatoid factors,18 and a subset of these rheumatoid factor–positive individuals develop essential mixed cryoglobulinemia that may be manifested in part by development of arthralgias.19,20 Liver disease in such individuals is often asymptomatic and biochemical abnormalities modest or even absent.19,20 Thus some individuals with essential mixed cryoglobulinemia associated with chronic hepatitis C infection may instead be labeled as having RA. However, anticyclic citrulinated peptide (CCP) antibodies are rarely found in subjects with chronic hepatitis C and nonspecific rheumatologic manifestations and thus anti-CCP antibodies appear to be reliable markers of RA.21 Most rheumatic disease patients with progressive liver disease have concomitant chronic viral or autoimmune hepatitis.22 In patients with concomitant hepatitis B infection, the intermittent use of tumor necrosis factor (TNF) inhibitors or other immunosuppressive therapy may be associated with severe flares of hepatitis B23–25 and prophylactic use of antiviral therapy should be considered.25 TNF inhibitor therapy in RA also has been associated with flares of severe liver disease, with characteristics of autoimmune hepatitis.26
Perhaps the most distinctive association between RA and hepatic abnormalities is seen in another subset of patients who develop splenomegaly and neutropenia (Felty’s syndrome). Felty’s syndrome is associated with an even higher incidence of hepatomegaly and liver function test abnormalities than seen in uncomplicated RA.27,28 However, there is little correlation between serum hepatic enzyme abnormalities and histopathologic findings.27,28 Nevertheless, more than half of patients with this syndrome have been found to have hepatic histologic abnormalities that range from sinusoidal lymphocytosis and portal fibrosis to the more distinctive picture of nodular regenerative hyperplasia, which has been reported on multiple occasions in patients with Felty’s syndrome27–30 and in one small prospective series was found to be present in 5 of 18 (28%) patients. Hepatic encephalopathy or other manifestations of liver failure have not been reported in patients with Felty’s syndrome, and nodular regenerative hyperplasia but portal hypertension and esophageal variceal hemorrhage may occur.28–30
Gastrointestinal Abnormalities
The most common gastrointestinal problems encountered in patients with RA are due to drug therapy with nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying antirheumatic drugs (DMARDs). NSAIDs are most commonly associated with upper gastrointestinal complications such as perforation, ulcers, and bleeding (see Chapters 52 and 53). Less commonly recognized complications of NSAIDs include pill esophagitis (Chapter 45), small bowel ulceration (Chapter 115), strictures of the small and large intestine, and exacerbations of diverticular disease and inflammatory bowel disease.31 Significant risk factors for the development of serious upper gastrointestinal events in patients with RA include NSAIDs therapy, age older than 65 years, history of peptic ulcer disease, glucocorticoid therapy, and severe RA.32,33 In patients with RA, the use of certain selective cyclooxygenase-2 inhibitors results in a lower incidence of gastrointestinal complications than that seen with nonselective NSAIDs.34,35 Helicobacter pylori and NSAIDs are independent and possibly synergistic risk factors for peptic ulceration. As such, chronic NSAID users who develop ulcers should be assessed for H. pylori infection and undergo eradication therapy when infection is present.32,33 Although hypergastrinemia has been reported in patients with RA, the incidence of peptic ulcers is no greater than that seen in patients with osteoarthritis.36 As reviewed in Chapter 53, NSAID-associated gastric and duodenal ulcers can be prevented with misoprostol, high-dose histamine (H2) blockers, and proton pump inhibitors.37 Once identified, ulcers may be treated successfully using proton pump inhibitors despite continued NSAID therapy. In the subgroup of patients with a history of bleeding ulcers, therapy with cyclooxygenase-2 inhibitors rather than NSAIDs may be cost effective and less expensive than combining an NSAID with a proton pump inhibitor.38,39
Synthetic DMARDs such as gold and penicillamine are rarely used because of toxicity and marginal efficacy.40 Gold, parenteral as well as oral forms, has been associated with diarrhea, enterocolitis, toxic megacolon, and death. The onset of gold colitis usually occurs within several weeks after the start of therapy and is manifested by nausea, vomiting, diarrhea, and fever. Although the colon is most commonly involved, gold-induced gastrointestinal toxicity may affect the esophagus, stomach, and small bowel, with 25% of patients developing a peripheral eosinophilia.41 Treatment includes dose reduction or discontinuation of gold, antidiarrheals, glucocorticoids, cromolyn sodium, or the chelating agent dimercaprol.41,42
Leflunomide, a synthetic DMARD that inhibits pyrimidine synthesis, can cause diarrhea in up to 32% of patients.43 It may also cause severe hepatic toxicity (see Chapter 86). Biologic DMARDs, which inhibit the action of TNF-α (infliximab, etanercept, and adalimumab) or interleukin-1 (IL-1; anakinra), have not shown significant gastrointestinal adverse effects but may cause hepatic toxicity on occasion (see later).
ADULT-ONSET STILL’S DISEASE
Adult-onset Still’s disease, the adult form of juvenile RA, often has gastrointestinal manifestations such as weight loss, sore throat, hepatosplenomegaly, elevated aminotransferases, and abdominal pain, in addition to fever.44 In contrast to the lack of significant hepatic dysfunction in classic rheumatoid arthritis, adults with Still’s disease present with features of mild hepatitis in the majority of cases and life-threatening acute liver failure in exceptional cases.45–49 Variable degrees of aminotransferase and alkaline phosphatase elevations are typically observed in such patients during symptomatic disease flares. Liver biopsies usually reveal moderate portal mononuclear cell infiltration with occasional evidence of focal hepatocyte necrosis.46 Biopsies obtained in patients with jaundice and biochemical evidence of severe hepatitis have been found to have interface and lobular hepatitis with lymphoplasmocytic inflammation reminiscent of autoimmune hepatitis.49 Most cases of severe hepatitis have been observed in patients previously treated with salicylates or other NSAIDs,46,49 but liver enzyme abnormalities are also commonly noted prior to therapy. Some patients with severe hepatitis have been reported to respond to immunosuppressive therapy,49 whereas others required liver transplantation or have died of liver failure.45,47–49 Although severe hepatitis is a rare complication of adult-onset Still’s disease, liver failure appears to be the most common cause of death related to this disease.45
PROGRESSIVE SYSTEMIC SCLEROSIS
Gastrointestinal manifestations occur in up to 90% of patients with PSS.50 Gastrointestinal tract involvement can occur from the mouth to the anus. Atrophy and fibrosis of the perioral skin may limit mandibular motion. The periodontal ligament may become hypertrophic, and the gingivae, tongue papillae, and buccal mucosa may become friable and atrophic, resulting in impaired sensation and taste.
The esophagus is the most frequently involved gastrointestinal organ.50 On pathology, atrophy of smooth muscle layers and intimal proliferation of arterioles is seen in the distal esophagus.51 Dysphagia occurs as a result of impaired esophageal motility, and gastroesophageal reflux disease (GERD) is related to hypotensive LES pressures, impaired esophageal clearance of acid, and reduced acid-neutralizing capacity due to xerostomia with reduced saliva production.52 The incidence of esophagitis approaches 100% in patients with severe cutaneous involvement.53 The extent of hypomotility varies from occasional uncoordinated contractions to complete paralysis.54 The severity of esophageal dysmotility correlates with the development of interstitial lung disease.55,56 Stricture formation from GERD may contribute to dysphagia, affecting approximately 8% of patients.57 Upper gastrointestinal hemorrhage has been reported from esophageal ulcers, rare esophageoatrial fistulas, and esophageal telangiectasia.58,59 An increased risk of infectious esophagitis with Candida (see Chapter 45) has been attributed to esophageal dysmotility and concomitant immunosuppressive therapy.60 Severe esophagitis typically responds to proton pump inhibitors but may require higher doses for maximal effect.61 A neuropathic achalasia-like syndrome has also been reported.62 The prevalence of Barrett’s metaplasia of the esophagus in patients with PSS ranges between 7% and 38%.63,64 A recent cohort study reported an increased incidence of carcinoma of the tongue and esophagus in patients with PSS.65
Gastric involvement most commonly leads to gastroparesis but other manifestations may include dyspepsia, exacerbation of GERD, or gastric hemorrhage from gastric antral vascular ectasia (GAVE, watermelon stomach). Delayed gastric emptying has been shown using radionucleotide scintigraphy or radiopaque pellets, with cutaneous electrogastrography demonstrating bradygastria and decreased amplitude of electrical activity.56,66,67 Prokinetic agents such as metoclopramide and erythromycin may increase LES pressures and improve gastric emptying in some patients with PSS.57
The pathologic changes in the small bowel of PSS patients consist of smooth muscle atrophy and deposition of collagen in submucosal, muscular, and serosal layers. Small bowel hypomotility is present in as many as 88% of cases.68 In the early stages of the disease, hypomotility is caused by neuropathic involvement, which may be more responsive to prokinetic agents. In advanced cases, hypomotility is more likely a result of “myopathic” and “fibrotic” changes.57 The interdigestive migrating motor complex (IMMC) is frequently absent or markedly diminished in amplitude in PSS patients with symptoms of intestinal dysmotility.69 Small bowel radiographic abnormalities are present in about 60% of PSS patients, but they may not correlate with symptoms. The duodenum is often dilated, especially in its second and third portions, often with prolonged retention of barium.70 Typically the jejunum is dilated and foreshortened because of mural fibrosis, but valvulae conniventes of normal thickness give rise to an accordion-like appearance. Pneumatosis cystoides intestinalis, pseudo-obstruction, pseudodiverticula, sacculations, intussusception, acquired intestinal lymphangiectasia, and small bowel volvulus have been noted.71–73
Symptoms of small intestinal PSS include bloating, borborygmi, anorexia, nausea, and vomiting. Rarely, thrombosis of large mesenteric arteries with extensive bowel necrosis may occur.74 Malabsorption with steatorrhea is present in as many as a third of PSS patients68 and is caused by bacterial overgrowth (see Chapters 101 and 102). Although antibiotic therapy can be effective in these patients, d-xylose malabsorption is often incompletely reversed, suggesting that collagen deposition in PSS may also contribute to malabsorption.75 Although often disappointing, the use of prokinetic agents such as metoclopramide may be effective in some cases. Octreotide in low doses and erythromycin also may provide sustained relief from nausea, abdominal pain, and bloating in some patients with pseudo-obstruction.76
Delayed colonic transit and impaired anal sphincter function are frequently found in constipated patients with PSS.77,78 Cisapride (a drug no longer available in the United States) accelerates colonic transit,79 but refractory cases may require surgery.80 Colonic stricture, volvulus, and bleeding from mucosal telangiectasias have been reported.81,82 Wide-necked diverticula can be seen, especially in the antimesenteric border of the transverse and descending colons. Rectal prolapse worsens anal sphincter function, aggravating fecal incontinence in patients with PSS.83 Rectal bleeding can occur from vascular ectasia.84
Pancreatic exocrine secretion is depressed in a third of patients with PSS, and idiopathic calcific pancreatitis has been reported.85 In addition, arteritis resulting in ischemic pancreatic necrosis has been described in patients with PSS.86,87 Gallbladder motility is not altered in PSS.88
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is a multisystem disease characterized by immune system abnormalities and the production of autoantibodies with tissue damage. Gastrointestinal symptoms are common in patients with active SLE. Oral ulcers (one of the criteria used to diagnose SLE) are most commonly seen in the buccal mucosa, hard palate, and vermilion border.89 In SLE, dysphagia (1% to 13% of patients) and GERD (11% to 50% of patients) poorly correlates with esophageal manometric abnormalities such as hypoperistalsis.90 Dysphagia is typically related to GERD or peptic stricture, with one report of esophageal epidermolysis bullosa acquisita.91 Malabsorption of d-xylose, steatorrhea, hyperplastic gastropathy, and protein-losing enteropathy have been described (see Chapter 28); the latter can be steroid responsive.92,93 Lupus peritonitis is a diagnosis that can be made only after other causes have been carefully excluded. Pneumatosis cystoides intestinalis may be an isolated benign condition or may accompany lupus vasculitis or necrotizing enterocolitis.94,95
One of the most devastating complications of lupus is gastrointestinal vasculitis. Affecting only 2% of patients, it has a fatality rate of more than 50%.96 Common sequelae include ulceration, hemorrhage, perforation, and infarction.97–99 Pancreatitis,100,101 gastritis, hemorrhagic ileocolitis resembling inflammatory bowel disease, and intussusception also have been reported. Although occasional case reports have documented polyarteritis-like changes on visceral arteriograms (described later), the typical pathologic changes are seen in the small vessels of the bowel wall rather than the medium-sized vessels of the bowel wall.94 Computed tomography (CT) scan may help establish the diagnosis of ischemic bowel disease in SLE if there are at least three of the following five CT findings: (1) bowel wall thickening, (2) target sign (a thickened bowel wall with peripheral rim enhancement or an enhancing inner and outer rim with hypoattenuation in the center), (3) dilatation of intestinal segments, (4) engorgement of mesenteric vessels, and (5) increased attenuation of mesenteric fat.102 Because visceral angiography is not routinely helpful, the diagnosis is difficult to establish. The role of endoscopy or upper gastrointestinal series in the diagnosis of lupus vasculitis is not well defined. The diagnosis currently rests on clinical judgment, findings on CT scans, and occasionally from surgical specimens when exploratory laparotomy is undertaken to rule out acute surgical emergencies.103 Treatment of abdominal lupus-induced vasculitis with glucocorticoids has been largely unsatisfactory. Although a controlled clinical trial comparing cyclophosphamide with glucocorticoids has not been performed, anecdotal reports of dramatic responses to intravenous cyclophosphamide are promising.94 Some investigators have suggested that cyclophosphamide be considered early in patients who have not shown significant improvement shortly after high-dose glucocorticoids are started.
Patients with SLE have a 25% to 50% incidence of abnormal liver biochemical tests during the course of their disease, but clinically significant liver disease is rare.104 Abnormal liver tests are commonly associated either with medication use or with mild, predominantly lobular hepatitis associated with periods of SLE activity.104,105 Despite the shared association with antinuclear antibodies, the typical histologic and clinical features of autoimmune hepatitis are rarely observed in adult patients with SLE.104,106 Concurrent SLE and autoimmune hepatitis occur more frequently in pediatric patients.106 In addition, SLE patients with anticardiolipin antibodies or lupus anticoagulants may have thrombotic events in the liver leading to Budd-Chiari syndrome or nodular regenerative hyperplasia manifested by complications of portal hypertension.104,107
POLYMYOSITIS AND DERMATOMYOSITIS
Polymyositis is a syndrome characterized by weakness, high serum levels of striated muscle enzymes (creatine kinase, aldolase), and electromyographic (EMG) or biopsy evidence of an inflammatory myopathy. When accompanied by a characteristic violaceous rash on the extensor surfaces of the hands and periorbital regions, the disease is termed dermatomyositis. The primary gastrointestinal symptoms are due to involvement of the cricopharyngeus, resulting in nasal regurgitation, tracheal aspiration, and impaired deglutition.108 Involvement is not limited to skeletal muscle fibers. Disordered esophageal motility, impaired gastric emptying, and poorly coordinated small intestinal peristalsis have been noted.109 Malabsorption, malnutrition, and pseudo-obstruction rarely occur.110 Pathologically, edema of the bowel wall, muscle atrophy, fibrosis, and mucosal ulcerations or perforation due to vasculitis may be seen at any level of the gut. Symptoms include heartburn, bloating, constipation, and gastrointestinal hemorrhage. Pneumoperitoneum, pneumatosis intestinalis, colonic dilation, and pseudodiverticula also may be seen. Perforations of the esophagus and of duodenal diverticula have been described as rare complications.111,112
In middle-aged to older adult patients, dermatomyositis and possibly polmyositis are associated with an increased prevalence of malignancy.113 The possibility that gastrointestinal symptoms may be the resultl of an underlying malignancy should be considered when evaluating these patients (see Chapter 22).
MIXED CONNECTIVE TISSUE DISEASE
Mixed connective tissue disease (MCTD) is a syndrome with overlapping features of PSS, polymyositis, and SLE, often in the presence of high levels of antibody directed against ribonucleoprotein. Upper gastrointestinal symptoms are seen in most patients.114 Abnormalities include diminished esophageal peristalsis (48%), esophageal stricture (6%), abnormal gastric emptying (6%), and gastric bezoar (2%).114 Small intestinal and colonic involvement includes dilation of proximal bowel, slow transit, intestinal pseudo-obstruction, diverticulosis, and, rarely, intestinal vasculitis. Pancreatitis also has been reported.114 Unlike PSS, the esophageal motility disturbances seen in MCTD appear to improve with the administration of glucocorticoids.
SJÖGREN’S SYNDROME
Sjögren’s syndrome (SS), occurring alone (primary SS) or in association with systemic autoimmune rheumatic diseases (secondary SS), is characterized by lymphocytic tissue infiltration of lacrimal and salivary glands with the clinical findings of keratoconjunctivitis sicca and xerostomia. As reviewed in Chapter 22, excessive dryness of the mouth and pharynx leads to oral symptoms of soreness, adherence of food to buccal surfaces, fissuring of the tongue, and periodontal disease.115 Dysphagia, reported by up to three quarters of patients with SS, can result from esophageal dysmotility and a lack of saliva; however, symptoms do not correlate with manometry or salivary secretion.116–118 Mild atrophic antral gastritis can be seen in 25% of patients with primary SS, but 31% were infected with H. pylori.119 Older studies that reported higher rates and greater severity of gastritis did not control for H. pylori infection. GAVE can occur in patients with SS and is responsive to fulguration therapy.120 A triad of sclerosing cholangitis, chronic pancreatitis, and SS has been reported in eight patients.121 Pancreatic exocrine function is frequently impaired.122 In primary SS, 7% of patients have positive antimitochondrial antibodies and among patients with primary biliary cirrhosis, clinical manifestations of SS are common (see Chapter 89).115
POLYARTERITIS NODOSA AND OTHER VASCULITIDES
Polyarteritis nodosa (PAN) is a necrotizing vasculitis of small and medium-sized muscular arteries, frequently with visceral involvement (Fig. 35-1). A characteristic feature of this condition is the finding of aneurysmal dilatations up to 1 cm in size seen on visceral angiography (Fig. 35-2). Abdominal complications occur in up to 50% of patients and carries a poor prognosis.123 Other clinical features of PAN include fever, myalgia, arthralgia of the large joints, mononeuritis multiplex, and livedo reticularis. Mesenteric visceral arteriograms are abnormal in up to 80% of patients with gastrointestinal involvement, with the superior mesenteric artery most commonly involved.123 Organ damage resulting from ischemia frequently underlies symptoms. The most common gastrointestinal manifestation is abdominal pain with other common symptoms including nausea, vomiting, and gastrointestinal bleeding.123 Bowel infarction and perforation, aneurysmal rupture, and acute cholecystitis are common causes of acute abdomen in PAN.123 Rarely, PAN can present as acalculous cholecystitis secondary to isolated vasculitis of the gallbladder.124 Pancreatitis,125 appendicitis,126 hemobilia,127 solitary biliary strictures,128 and hepatic infarcts129 also have been reported to complicate PAN. The frequency of hepatitis B infection in PAN has declined from more than 30% to less than 10% because of improved screening of the blood supply and vaccination against hepatitis B.130 Because of the frequent association with hepatitis B infection and potential association with hepatitis C infection (see Chapters 78 and 79), patients with clinical manifestations of PAN should be assessed for evidence of hepatitis B or C infection.
Churg-Strauss syndrome (CSS, allergic granulomatous angiitis) is a small to medium-sized vessel vasculitis characteristically associated with eosinophilia, asthma, sinusitis, and rhinitis. Abdominal pain is the most common gastrointestinal symptom.131 Preceding the vasculitic phase of CSS, patients may present with an eosinophilic gastroenteritis associated with abdominal pain, nausea, vomiting, diarrhea, and bleeding with an absolute eosinophil count of greater than 1500 cells/mm3 (see Chapter 27).132 Additional gastrointestinal manifestations of CSS include pancreatitis, cholecystitis, ascites, small intestinal ulcerations, and perforation.131,133,134 Colonic involvement may present with multiple ulcers or obstruction.134,135
Henoch-Schönlein purpura (HSP) is a systemic vasculitis characterized by nonthrombocytopenic purpura, arthralgias, renal disease, and colicky abdominal pain. Although the disease is frequently seen in children and adolescents, adults of any age may be affected. Colicky abdominal pain and gastrointestinal bleeding are seen in two thirds of cases.136 Colonoscopic and endoscopic findings in bleeding patients include erosive duodenitis, small aphthous ulcerations, and petechial colonic lesions.137 In patients who undergo CT scan, common findings include bowel-wall thickening, dilated intestinal segments, mesenteric vascular engorgement, and regional lymphadenopathy.138 Other reported gastrointestinal complications of HSP include protein-losing enteropathy, esophageal and ileal structures, gastric and small bowel perforations, bowel infarction, pancreatitis, appendicitis, cholecystitis, intramural hematomas, and intussusception.139
Malignant atrophic papulosis (Kohlmeier-Degos disease) is a rare vasculitis that causes nausea, vomiting, bleeding, malabsorption, bowel ischemia, and perforation.140 Scattered on the skin are red papules that become hypopigmented atrophic scars (see Fig. 22-13).
Cogan’s syndrome is characterized by nonsyphilitic interstitial keratitis, audiovestibular symptoms, and large-vessel vasculitis that may involve the gut. Gastrointestinal manifestations include abdominal pain, diarrhea, hepatomegaly, and splenomegaly.141 Crohn’s disease has been reported in association with this rare condition.142
Wegener’s granulomatosis, a systemic vasculitis characterized by pulmonary, sinus, and renal involvement, less commonly affects the gut.143 Inflammatory ileocolitis with hemorrhage, gangrenous cholecystitis, and bowel infarction have been reported.144 Wegener’s granulomatosis may mimic Crohn’s disease with granulomatous gastritis or ileitis.145,146
Mixed immunoglobulin (IgG-IgM) cryoglobulinemia characterized by the triad of purpura, arthralgia, and asthenia may complicate chronic hepatitis C infection (see Chapter 79) and a variety of immune diseases, including inflammatory bowel disease, celiac disease, and postintestinal bypass syndrome. Cryoglobulinemia may cause severe visceral vasculitis with diarrhea, ischemia, and perforation of the small or large intestine.147
BEHÇET’S DISEASE
Behçet’s disease is an idiopathic inflammatory disorder characterized by oral aphthous ulcers, genital ulcers, uveitis, and skin lesions, with gastrointestinal involvement occurring in up to 50% of patients.148 As in Crohn’s disease, ulceration may occur throughout the alimentary tract, with the ileocecal region most commonly affected. Differentiating Behçet’s from Crohn’s disease can be difficult because of similarities in gastrointestinal symptoms, endoscopic findings, histology, and extraintestinal manifestations. Involvement of the esophagus includes ulcers (Fig. 35-3), varices, and perforation.149 In the stomach, which is infrequently involved, aphthous ulcers may be seen. The typical intestinal involvement in Behçet’s disease includes “punched-out” ileocecal ulcerations, the most common finding on colonoscopy. Additional manifestations of Behçet’s disease include abdominal pain, diarrhea, bleeding, perforation, and fistulas (perianal, rectovaginal, and enteroenteric).150 Hepatic or portal vein thrombosis may occur in patients with Behçet’s, and this syndrome should be included in the differential diagnosis of patients presenting with Budd-Chiari syndrome.151,152 Medical therapy of the gastrointestinal lesions of Behçet’s disease includes mesalamine, glucocorticoids, immunomodulators such as azathioprine and 6-mercaptopurine, infliximab, and thalidomide.153,154 Surgical intervention is associated with a high rate of recurrence, with nearly 50% requiring repeat surgery.155
SERONEGATIVE SPONDYLOARTHROPATHIES (REACTIVE ARTHRITIDES)
The term seronegative spondyloarthropathy is used to describe an interrelated group of inflammatory disorders that include ankylosing spondylitis, reactive arthritis (formerly called Reiter’s syndrome), and psoriatic arthritis. The term has also been used to describe the enteropathic spondylitis associated with Crohn’s disease and ulcerative colitis.156 These disorders are characterized by the absence of rheumatoid factor, an association with human leukocyte antigen-B27 (HLA-B27), and inflammation at the site of bony insertion of ligaments and tendons (enthesitis). There is a high prevalence of clinically silent inflammatory colon lesions in patients with these seronegative spondyloarthropathies.157 Capsule endoscopy may yield more small bowel abnormalities than ileocolonoscopy.158 Conversely, 22% of patients with inflammatory bowel disease have evidence of a seronegative spondyloarthropathy, with ankylosing spondylitis most commonly seen.159 Although infliximab has been shown to induce remissions in some patients with ankylosing spondylitis as well as in Crohn’s disease, the effect of infliximab on gastrointestinal inflammatory lesions in typical seronegative spondyloarthropathies has not yet been studied.
MARFAN’S AND EHLERS-DANLOS SYNDROMES
Owing to defective collagen synthesis, patients with Marfan’s or Ehlers-Danlos syndrome develop skin fragility, megaesophagus, small intestine hypomotility, giant jejunal diverticula, bacterial overgrowth, and megacolon.160 Mesenteric arterial rupture and intestinal perforation also can occur.161
FAMILIAL MEDITERRANEAN FEVER
Familial Mediterranean fever (FMF) is an autosomal recessive inherited disease characterized by recurrent self-limiting attacks of fever, joint pain, and abdominal pain. Acute attacks typically last three to five days. FMF is most commonly seen in people of Mediterranean origin including Sephardic Jews, Arabs, Turks, Greeks, Italians, and Armenians, although FMF has been described in Cubans, and Belgians. The gene responsible for FMF in Mediterranean patients, designated MEFV, has been mapped to chromosome 16, which encodes a 781-amino acid protein called pyrin or marenostrin.162
Gastrointestinal symptoms, typically manifest as episodic abdominal pain, are seen in 95% of patients, and this may be the presenting symptom in as many as 50% of cases.163 Abdominal pain may be diffuse or localized and may range from mild bloating to acute peritonitis with boardlike rigidity, rebound tenderness, and air-fluid levels on upright radiographs. The acute presentation may be confused with acute appendicitis, cholecystitis, or pelvic inflammatory disease, whereas relapsing and remitting attacks may be confused with other diseases such as porphyrias. Small bowel obstruction from adhesions may occur as a consequence of recurrent sterile peritonitis or as a result of previous exploratory surgery. In patients with obstruction due to adhesions, abdominal attacks without other typical symptoms (arthralgias, fever) should tip off the clinician to consider an obstruction.164 The diagnosis of FMF is based on validated clinical criteria including fever, serositis, location of pain, and response to colchicine.165
In FMF, the long-term prognosis is poor in patients who develop nephrotic syndrome and chronic kidney disease from amyloid A deposition163 (amyloidosis is discussed later in this chapter). Prophylactic colchicine has been shown to reduce the frequency of attacks, prevent amyloidosis, and avoid renal failure.166 Vasculitis in the form of HSP, PAN, protracted febrile myalgia, or Behçet’s is encountered in 3% of FMF patients.163
ONCOLOGIC AND HEMATOLOGIC DISEASES
METASTASES
Metastasis to the gut can occur by direct invasion from adjacent organs, by intraperitoneal seeding, or by hematogenous or lymphatic spread. About 20% of all patients with nongastrointestinal malignancies have metastases to the gastrointestinal tract, the most common of which are breast, lung, and ovarian cancers and melanoma (Fig. 35-4).167 Patterns of metastases are not random but reflect the location and histologic type of the primary tumor. The esophagus is most frequently affected by direct extension from tumors arising from adjacent structures (bronchus and stomach). The stomach is a particularly common site of breast cancer metastases, and the small intestine can be involved by tumor extension from the stomach, pancreas, biliary system, kidney, or retroperitoneum. The pancreas is usually an asymptomatic site of metastasis, with the most common primary tumors being lung, gastrointestinal, and renal.168 The ileum may be affected by cancers arising in the colon or pelvis. Metastases to the gut typically begin in the serosa or submucosa and produce intraluminal lesions that can lead to obstruction, submucosal polypoid masses that can result in intussusception or ulcerated mucosal lesions. The most common presenting clinical condition in patients with metastatic lesions to the gut is small bowel obstruction. Lobular breast cancer, malignant melanoma, and non–small cell lung cancer are the most common neoplasms to cause small bowel obstruction from isolated metastases.169 In addition, pain, fever, ascites, gastrointestinal bleeding, and perforation have been described.
Metastases to the gastrointestinal tract may be difficult to diagnose. Barium contrast studies may reveal extramural masses, mucosal ulcerations, or a rigid stomach with the appearance of linitis plastica. CT may be helpful in determining the primary tumor, in tumor staging, and in detecting large serosal implants. Small bowel metastases, however, are detectable radiographically in only 50% of cases.170
When feasible, surgical resection should be used to treat gastrointestinal metastases that result in obstruction, perforation, or significant hemorrhage. If a solitary bowel metastasis is the only evident site of disseminated malignancy, segmental bowel resection should be performed, offering a small chance for cure. In aggressive resections of melanoma metastases, the mesenteric nodes draining the involved segment of bowel should be resected because they frequently contain tumors.171
PARANEOPLASTIC SYNDROMES
Paraneoplastic syndromes affecting the gut include the hormonal effects of carcinoid tumors, vasoactive intestinal polypeptide-secreting tumors (VIPomas), gastrinomas, and somatostatinomas (see Chapters 31 and 32), as well as the gastrointestinal effects of hypercalcemia (constipation, nausea, and vomiting). A watery diarrhea syndrome with elevated serum immunoreactive VIP has been described accompanying nonpancreatic tumors such as bronchogenic carcinomas, ganglioneuromas, pheochromocytomas, and a rare mastocytoma.172 Elevated serum levels of somatostatin, calcitonin, gastrin, and corticotropin also have been reported in pheochromocytoma.173
Paraneoplastic gastrointestinal dysmotility may occur in some patients with occult or established malignancy and specific serum antibodies. Clinically the patient may present with pseudoachalasia, gastroparesis, intestinal pseudo-obstruction, or constipation. Intestinal pseudo-obstruction (see Chapter 120) is most frequently associated with small cell carcinoma of the lung but has been described with other tumors such as squamous cell lung carcinoma, lymphoma, melanoma, and cancers of the kidney, breast, and prostate.174–176 Patients with paraneoplastic intestinal pseudo-obstruction characteristically suffer from constipation and obstipation and from symptoms of intestinal obstruction. In addition, dysphagia, gastroparesis, early satiety, autonomic insufficiency, and peripheral neuropathy have been described.177 The onset of symptoms may precede the discovery of the primary tumor by several years. The gastrointestinal pathology in this syndrome is confined to the myenteric plexus, in which an inflammatory lymphocytic infiltrate is variably seen accompanying neuronal degeneration.178 Cross-reacting autoantibodies found in the sera of these patients bind to the primary tumor cells and to neural cells in the myenteric plexus, resulting in inflammation and destruction of the myenteric plexus.179 In the setting of pseudo-obstruction, detection in the serum of circulating antineuronal nuclear antibodies (ANNA-1 or anti-Hu), type 1 Purkinje cell antibodies (PCA-1), or N-type calcium channel binding antibodies should suggest a paraneoplastic process and prompt further evaluation for an underlying malignancy.177 ANNA-1 are postulated to induce neuronal apoptosis leading to gut dysmotility.180 Although the symptoms of paraneoplastic pseudo-obstruction may resolve with successful treatment of the primary tumor, persistence of gastrointestinal symptoms despite effective anticancer treatment is more common. Attempts to alleviate the symptoms of pseudo-obstruction with prokinetic agents have been disappointing.
HEMATOLOGIC MALIGNANCIES
Liver involvement during hematologic malignancies is only rarely life threatening or a source of great morbidity. Nevertheless, the liver is a major component of the reticuloendothelial system, and thus it is not surprising that malignant infiltration of the liver commonly occurs in such diseases.181 As detailed in Table 35-2
[/not-level-membership-for-gastroenterology-and-hepatology-category]
