[level-membership-for-pediatrics-category]
Chapter 8 Gastroenterology
Long Cases
Inflammatory bowel disease
In the last few years, there has been an increase in the understanding of genetic susceptibility to IBD, suggesting that Crohn’s disease (CD) and ulcerative colitis (UC) may represent a continuum of disease. Recently, there has been a shift in the IBD management paradigm. Mucosal healing is now used as an end point rather than a clinical indicator of remission, as it is realised that endoscopic lesions and symptoms may not correlate. Exclusive enteral nutrition (EEN) has emerged as the ideal induction therapy in CD. There is a risk stratification evolving, with those at higher risk of severe CD—predictors of more aggressive disease including younger age of onset, extensive small bowel disease, deep colonic ulcers, perianal disease and early need for corticosteroids—receiving more aggressive therapy, with earlier use of immunosuppressive and biological agents; this is termed ‘top-down’ therapy (as opposed to the traditional approach of escalating, or ‘step up’ therapy). In the case of UC, the best evidence for prevention of dysplasia is for 5-aminosalicylates, not infliximab, so there is no reason for top-down therapy in UC.
History
Extraintestinal symptoms
Interval/current: rashes (e.g. erythema nodosum [more in CD], pyoderma gangrenosum [more in ulcerative colitis, UC]), mouth involvement (e.g. aphthous ulcers), liver involvement (e.g. chronic active hepatitis), joint disease (e.g. arthralgias, spondylo-arthropathies), visual problems (e.g. uveitis), vascular complications (e.g. vasculitis), renal tract involvement (e.g. nephrolithiasis), hypertension, myocarditis, pericarditis, fever, pubertal delay, neurological problems (e.g. peripheral neuropathy), haematological disease (e.g. anaemia), musculoskeletal weakness (e.g. vasculitic myositis, steroid-induced myopathy), pancreatitis, extraintestinal neoplasia (e.g. lymphoma), amyloidosis, thromboembolic disease (e.g. cerebral, retinal).
Investigations
Stool
Mainly to differentiate other causes of symptoms:
1. Microscopy (fresh specimen) for cysts, ova, parasites.
2. Culture for bacteria (e.g. Campylobacter, enteropathogenic E. coli, Shigella).
3. Clostridium difficile toxin.
4. Alpha-1-antitrypsin level (screen for protein loss from bowel).
5. Neutrophil-derived proteins: (a) calprotectin—95% sensitivity, 91% specificity for diagnosis of IBD; (b) lactoferrin—80% sensitivity, 82% specificity for diagnosis of IBD.
Blood
1. Full blood count (leucocytosis found in two thirds of CD; anaemia plus high ESR has 96% specificity in diagnosing IBD in a referred patient population with suspected IBD).
2. CRP (elevated in 70–100% of CD and 50–60% of UC; also useful to prognosticate disease course)/ESR (elevated in 80–90% IBD patients)
3. Liver function tests (liver dysfunction as part of IBD; low albumin, total protein from protein-losing enteropathy).
4. Vitamins: folate (serum and red cell), vitamin B12 (ileal disease), vitamins A, D, E and prothrombin time for vitamin K (fat malabsorption).
5. Minerals: calcium, magnesium, phosphate, iron, ferritin, zinc, copper, selenium.
6. Electrolytes, urea and creatinine (associated renal disease).
7. Serology for Yersinia species (enteropathica and pseudotuberculosis) to exclude this as a diagnosis. Yersinia colitis can present with erythema nodosum and arthralgia, as can CD.
8. Perinuclear antineutrophil cytoplasmic antibodies (pANCAs) to neutrophil proteins, with an atypical staining pattern, may be elevated in UC (in 40–80%) or CD (in 5–25%); for diagnosing UC, sensitivity 55%, specificity 89%. Not tested routinely.
9. Anti-Saccharomyces cerevisiae antibodies (ASCAs) are present in 50–60% of patients with CD, 20% of healthy first-degree relatives of those with CD, 10–15% of patients with UC and 0–5% of controls. Not tested routinely.
10. Antibodies to E. coli (outer membrane porin C protein antibodies), anti-ompC antibodies. These are present in 55% of patients with CD, 5–11% those with UC and 5% of controls. Not tested routinely.
11. Antibodies to I2, a bacterial sequence derived from Pseudomonas fluorescens, are present in 30–50% of CD patients (with a sensitivity of only 42%, and a specificity of 76%), 10% of UC patients, 19% of other inflammatory conditions and 5% of controls. Not tested routinely.
12. Antibodies to CBir1 flagellin, an antigen of enteric microbial flora, are present in 50% of CD patients, but only 6% of UC patients and 8% of controls. Not tested routinely.
13. Three different carbohydrate (glycan) antibodies are highly specific, but not sensitive, for CD. These comprise ALCA, ACCA and AMCA, which stand for: antiaminaribioside carbohydrate IgG antibodies (ALCA); antichitobioside carbohydrate IgA antibodies (ACCA); and antimannobioside antibodies (AMCA). These are present in 44–50% of ASCA-negative CD patients, and have been shown to be associated with the NOD2/CARD15 genotype in CD. Not tested routinely.
14. Two other anticarbohydrate antibodies, anti-chitin IgA (anti-C) and anti-laminarin (anti-L), are associated with complicated CD, and improve differentiation between CD and UC.
Imaging
1. Plain abdominal X-ray, erect and supine (may see evidence of incomplete bowel obstruction with distended bowel loops and air/fluid levels in CD).
2. Fluoroscopic barium studies: small bowel follow through (SBFT), and small bowel enteroclysis (SBE) can detect mucosal ulceration, or irregularities, narrowing or distension of the gut lumen, and the presence of fistulous communications, with a sensitivity of 85–95% and a specificity of 89–94% for SBFT in patients with terminal ileal disease in CD.
3. Double-contrast barium enema (may identify rectal and colonic disease in CD and UC).
4. Chest X-ray (to help detect tuberculosis, a well-known cause of chronic bowel inflammation).
5. Ultrasound of abdomen and pelvis. Ultrasound identifies inflammation by increased bowel wall diameter and altered pattern of mural stratification, and Doppler studies can detect increased blood flow. Ultrasound may be useful in delineating intra-abdominal masses (e.g. solid lesions or bowel loops), including abdominal or pelvic abscesses.
Ultrasound for the initial diagnosis of IBD has a sensitivity of 75–94%, with a specificity of 67–100%, can identify bowel wall thickening and can tell fibrosis from oedema.
6. Contrast-enhanced ultrasound (using the new microbubble contrast agent that lasts a few minutes in the bloodstream) can evaluate the bowel wall and assess disease activity with a sensitivity of 93% and a specificity of 93%.
7. CT scanning. Multiplanar imaging can visualise overlapping bowel loops, and complications such as fistulae or abscesses. Traditional CT uses a contrast agent such as barium or iodine, which highlights intraluminal filling defects such as masses. CT enterography (CTE) uses neutral (low-density) oral contrast and intravenous contrast to accentuate the distinction between the enhancing small bowel wall and the low-attenuation gut lumen, to visualise the bowel wall and mucosa. CT enteroclysis comprises placement of a nasojejunal tube, and infusing a contrast agent into the proximal small bowel. CTE and CT enteroclysis may detect segmental mural enhancement, and wall thickening, consistent with active inflammation; reactive mesenteric adenopathy also may be found, supporting the diagnosis of active inflammation. For any CT study, the benefits must be weighed against the cumulative deleterious effects of ionising radiation, especially in younger children.
8. MR enterography (MRE) and MR enteroclysis both have a sensitivity of 88% for detecting IBD. MR enterography has a specificity of 92–100% for detection of IBD. Gadolinium-enhanced magnetic resonance imaging (MRI) has improved intestinal image resolution: CD shows transmural enhancement of the colon, and bowel wall thickening of the terminal ileum or proximal small bowel; UC shows mucosal enhancement and submucosal sparing extending proximally from the rectum. The technique is cheaper and more pleasant than endoscopy. MR provides detailed images of perianal fistulae and is the dominant imaging technique for assessing perianal disease in CD. MRE is preferable to CTE because of lack of radiation, and because of superior images that can distinguish active inflammation from fibrosis.
9. Positron emission tomography (PET) scanning can identify areas of active inflammation, in UC. PET uses [F18] fluoro-2-deoxy-D-glucose to spot areas of increased metabolism.
10. Radio-labelled white blood cell scan (useful if mid-small bowel pathology suspected but not able to be found using conventional evaluations).
Management
Short-term aims include induction and maintenance of clinical and histological remission, improvement in overall quality of life and prevention of complications. The main goals are now mucosal healing, and transmural healing, rather than just clinical healing; patients in clinical remission with steroids may have active endoscopically identified mucosal lesions in up to 70% of cases. Risk stratification based on genotype, phenotype and serology is another useful tool. The only factor that predicts prolonged remission in CD (persisting 3–4 years after initiating treatment) is complete mucosal healing. Longer-term aims include preventing relapses, normalising growth and pubertal development, maintaining bone mass and minimising the need for surgery. These therapeutic aims are based on early aggressive therapy, including the use of newer biological agents. Earlier use of potent agents, previously considered ‘third line’, may be more effective than awaiting resistance to treatment. When evaluating new therapies, remember that a placebo response rate over 30% has been noted in IBD.
Crohn’s disease (CD)
Induction therapy: exclusive enteral nutrition (EEN) (first line), corticosteroids (second line), infliximab
After EEN, the second-line agents for remission in CD are prednisolone (for moderate to severe disease), budesonide (a potent steroid control-released in the small intestine and right colon) and the biological agent infliximab (for refractory disease, steroid dependency or fistulising disease).
Maintenance: Immunomodulators AZA, 6MP and MTX; infliximab
Agents such as 6-MP and AZA (which is metabolised to 6-MP, the active agent) can be used for maintenance therapy for CD, prophylaxis after surgery in CD and perianal CD. In particular, 6-MP has been shown useful as initial treatment in children with moderate to severe CD. AZA and 6-MP have a slow onset of action (3–4 months) and so need to be combined with nutritional therapy or steroids until their effect is seen. Duration should be over years, as there is a high relapse rate if AZA or 6-MP are stopped within the first year. Bone marrow suppression can occur (in 2–5% of patients) at any time. If using AZA or 6-MP, it is useful to know the patient’s thiopurinemethyltransferase (TPMT) status. TPMT is the enzyme that catalyses the conversion of 6-MP to 6-methylmercaptopurine (6-MMP). If there is a deficiency of TPMT, then 6-MP can be metabolised along an alternate pathway to 6-thioguanine (6-TG), which is toxic and can cause bone marrow depression; that is, low TPMT means high 6-TG levels and a high risk of leukopenia. Higher 6-MMP levels correlate with hepatotoxicity in adults. Aminosalicylates and methotrexate inhibit TPMT activity and can increase 6-TG levels.
Ulcerative colitis (UC)
Mild disease
5-ASAs can treat the acute situation and are also valuable prophylactically. Treatment is usually lifelong (for the life of the colon). Sulphasalazine comprises sulphapyridine linked to 5-aminosalicylate (5-ASA) by an azo bond, which is broken by colonic bacteria; 5-ASA, the active component, is released directly to the colon. The 5-ASAs are enteric-coated and deliver the drug to the small and large bowel. The most common problems encountered with sulphasalazine are related to the sulpha part (rashes, bone marrow depression), whereas 5-ASAs can cause nephrotoxicity.
Growth and pubertal delay
Children with CD have delayed puberty and decreased final adult height, and often do not show catch-up growth after diagnosis. No particular treatment regime has shown superiority in improving growth. Biological agents, immunomodulators and surgical resection all can improve growth in IBD. Management of poor growth and pubertal delay involves obtaining adequate remission and maintaining this. Reduction of intestinal inflammation and caloric supplementation to reverse malnutrition are important. It is well described that surgical intervention, in UC particularly, can be associated with the onset of puberty. Pubertal delay is thus a relative indication for surgery. Escalating therapy using biological agents or immunomodulators can be considered to maintain remission through puberty. Many adolescents are psychologically upset by pubertal delay, and many units use sex steroid therapy in these patients. In boys with IBD, testosterone for 3–6 months can have very positive effects on virilisation, growth and psychological well-being, although there are no controlled trials of testosterone use. In girls with IBD, ethinylestradiol can be used for a similar period of time.
Chronic liver disease (CLD)
The causes of CLD can be divided into three groups: cholestatic diseases, metabolic diseases and chronic hepatitis (various forms).
Cholestatic diseases
Metabolic disease
WATCH this is not missed (mnemonic).
Wilson’s disease (WD)
This is the most common cause of fulminant liver failure in children over 3 years. It is an autosomal recessive disorder of copper metabolism in which the liver cannot excrete this metal into the bile. The WD gene ATP7B is located on 13q14.3–q21.1, and encodes a copper-transporting P-type ATPase; there are 40 normal allelic variants, and more than 400 disease-causing mutations of this gene—this gene is needed to enable copper to attach to caeruloplasmin and to excrete copper into the bile. Initially, there is copper accumulation in the liver, then excess copper spills over into the brain (basal ganglia), kidneys, bones and cornea (Kayser–Fleischer rings represent copper deposition in Descemet’s membrane of the cornea, and indicate significant copper storage in the body); it also, less commonly, spills over into the lens (sunflower cataracts), kidneys (proteinuria, microscopic haematuria, Fanconi syndrome), joints (arthritis), heart (cardiomyopathy, cardiac arrhythmias) and skeletal muscle (rhabomyolysis). Investigation findings include low-serum copper and caeruloplasmin, and raised urinary copper. It is rare for WD to present before 3 years. Neurological features (movement disorders, or rigid dystonia) present in WD in adolescence. WD is managed with copper chelating agents (penicillamine [given with pyridoxine], trientine) and zinc (enterocyte metallothionein inducer, interferes with absorption of copper from gut); chelated copper is excreted in urine. Foods high in copper content are restricted: liver, brain, shellfish, mushrooms, chocolate and nuts. Vitamin E may be used along with chelator or zinc to consume free radicals made by excess copper. One third of patients die if untreated. LTx is required if the patient is unresponsive to penicillamine, or has advanced liver failure with coagulopathy and encephalopathy. WD liver disease does not recur after LTx, which provides an effective phenotypic cure, converting the copper kinetics of a homozygous child to that of a heterozygote.
Tyrosinaemia type 1
Management of chronic tyrosinaemia includes a low-protein diet (to reduce tyrosine), vitamin D and 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexenedione (NTBC), which prevents the formation of toxic metabolites, reverses biochemical and clinical effects, allows hepatic regeneration and almost eliminates the risk of hepatocellular carcinoma (HCC). If NTBC fails, LTx is required. Also, LTx is required if HCC (rising alpha-fetoprotein, liver nodularity on ultrasound, CT or MRI) is suspected.
Chronic hepatitis
Liver function tests can be divided into four categories:
(a) tests for liver synthetic function (albumin—synthesised exclusively in the liver; PT/international normalised ratio (INR)—clotting cascade proteins that are needed to have a normal PT are synthesised in the liver);
(b) tests for biliary excretion (total and direct bilirubin);
(c) tests for cholestasis (GGT, ALP—cholestasis means reduced or absent/stopped bile flow); and
(d) tests for hepatocellular damage (AST, ALT—hepatic enzymes released from damaged hepatocytes into the blood stream).
Ultrasound may show echogenic liver, splenomegaly and oesophageal varices, and endoscopy may show gastric and oesophageal varices.
Complications of cirrhosis are as follows (mnemonic: HEPATIC):
History for CLD
Past history (including initial presentation)
Examination for CLD
Figure 8.2 shows complications (only) of CLD. For the approach to the examination for the aetiology (e.g. KF rings and neurological assessment for WD), refer to the short-case section.
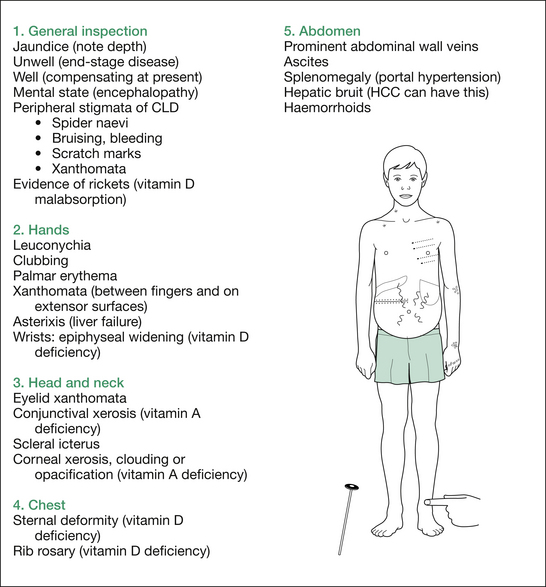
Principles of management of CLD
General
Monitor weight, serum albumin, serum bilirubin (total and conjugated), prothrombin time. Psychosocial support is important.
Portal hypertension, varices and variceal haemorrhage
Avoid splenectomy if possible. As well as the risk of infection after splenectomy, it may lead to increased bleeding from removal of good collateral vessels from the splenic capsule (azygos system) that bypass the lower oesophageal junction vessels. Haemorrhage can be exacerbated by deficiency of vitamin K dependent factors, thrombocytopenia secondary to hypersplenism and circulating fibrinolysins.
Liver transplantation (LTx)
Timing of transplantation
The timing of LTx depends upon a variety of factors and may be hastened by any of the following:
1. A history of life-threatening variceal haemorrhage.
2. The development of: (a) a hepatorenal state; (b) hepatic encephalopathy; (c) refractory ascites; (d) reduction in psychosocial development; (e) intractable pruritus; (f) severe metabolic bone disease; or (g) a diminished quality of life.
Assessment occurs in specialist liver units, with preparatory education and counselling of the child and family, a multidisciplinary team that may include a psychologist and a play therapist (especially for children under 2 years), intensive nutritional support, completion of routine immunisation (especially hepatitis A and B) before commencement of immunosuppression, and management of CLD complications.
Indications for LTx
Specific diseases requiring LTx, in order of decreasing frequency, include the following:
1. Extrahepatic biliary atresia (EHBA): over 50% of LTx.
2. IEMs (10–15% of LTx) include AT deficiency, CF, galactosaemia, glycogen storage diseases (GSDs) type IA and IV, mitochondrial functional defects (e.g. defects of fatty acid beta-oxidation), some organic acidaemias, tyrosinaemia, urea cycle defects and WD.
3. Acute hepatic necrosis (around 10%).
4. Cirrhosis from chronic active hepatitis or primary biliary cirrhosis (under 10%).
The following requirements must be met for a cadaveric donor for LTx:
1. Brain death prior to circulatory arrest.
2. The absence of sepsis, HBsAg and HBeAg, HIV, malignancy, drug abuse, liver or gallbladder disease, chronic hypertension.
3. Liver enzymes, serum bilirubin within three times normal values.
4. No period of hypoxia (PaO2 < 60 mmHg) or hypotension (over 2 hours).
5. The absence of more than minimal pressor support to maintain normal blood pressure and peripheral perfusion.
6. To match a recipient, the suitable donor must have ABO compatibility with the recipient. Sex and HLA type compatibility are not necessary.
Transplant surgery procedure
The procedure itself has three main phases: (a) recipient hepatectomy, which can be complicated because of coexistent coagulopathy and portal hypertension (bleeding) and previous Kasai (adhesions); (b) the anhepatic phase, when the patient has the vascular anastomoses performed (vena cava, portal vein, hepatic artery), and then placement of the graft; (c) the neohepatic phase, where the liver graft is reperfused (cardiovascular instability can occur here, as the liver has been exposed to cold preservation techniques) and the biliary anastomosis performed—in patients with EHBA, the donor duct will be implanted into a Roux-en-Y loop, as there is no recipient biliary tree. This latter technique is also useful in small children who receive a segmental graft.
Complications of LTx
Graft rejection (60%)
• Rejection is the main cause of graft dysfunction following LTx.
• Late acute rejection can occur anywhere from 1 month to several years post-LTx. It can be associated with immunosuppressant non-compliance. It presents with increased transaminases, fever, malaise or jaundice; liver biopsy shows acute cellular rejection (ACR); it responds to pulse steroids, increased calcineurin inhibitors, and addition of another immunosuppressant such as mycophenolate mofetil; if unresponsive to these, treat with anti-T cell antibody preparations.
• Chronic rejection (also known as ‘ductopaenic rejection’), with the loss of over 50% of bile ducts, occurs 6 weeks to 6 months after LTx and can lead to biliary strictures, cholangitis and cholestasis. Treatment may include optimising immunosuppression, and dilatation of strictures. Severe allograft dysfunction could require re-transplantation.
• Autoimmune hepatitis can develop irrespective of initial disease process. It can occur months to years after LTx. It is treated with the addition of azathioprine and prednisolone to the immunosuppressive regimen.
Infection (50%)
• Earlier infections (up to 1 month after LTx) tend to be bacterial or fungal. Intra-abdominal and line infections are the most common early causes. Bacterial and fungal infections can be secondary to technical complications such as vascular thrombosis, bile leaks and strictures. Fungal sepsis, especially aspergillosis, can be seen with re-operation, bowel perforation and long stays in intensive care: prophylaxis is with antifungal agents such as itraconazole and fluconazole, or newer drugs (e.g. the caspofungins) and the lipid complex formulation of amphotericin (less nephrotoxic).
• Later infections (2–6 months after LTx) tend to be viral (e.g. cytomegalovirus [CMV], Epstein–Barr virus [EBV], herpes varicella-zoster virus [HVZ]) and have been reduced by prophylactic courses of ganciclovir, acyclovir and CMV hyper-immunoglobulin. Still later infections (>6 months) may be viral or fungal. Common respiratory viruses (adenovirus, parainfluenza, influenza) can produce necrotising pneumonitis.
• Fever in a young child in the first 12 months after LTx deserves assessment for sepsis, and depending on degree of immunosuppression, may need hospitalisation and intravenous broad-spectrum antibiotic cover.
Recurrence of primary disease (uncommon)
Malabsorption/maldigestion
Aetiology
The common causes in the developed world include:
3. Post-gastroenteritis syndrome.
5. Bacterial overgrowth (usually associated with congenital gut anomalies or prior gastrointestinal surgery).
Other causes in the developed world include the following:
1. Inflammatory bowel disease (predominantly Crohn’s disease [CD]).
2. Syndromic causes of exocrine pancreatic insufficiency:
3. Short bowel syndrome (SBS): residual small bowel length less than 100 cm (e.g. post-necrotising enterocolitis).
4. Chronic liver disease (CLD) with cholestasis.
5. Others: immunodeficiency syndromes, intestinal lymphangiectasia, abetalipo-proteinaemia.
Infants may have congenital disorders of specific nutrient digestion or absorption/transport.
These include disorders involving the following:
1. Aminoacids: for example, enterokinase deficiency.
2. Fats: for example, chylomicron retention disease, intestinal lymphangiectasia; abetalipoproteinaemia.
3. Carbohydrates: congenital intestinal enterocyte brush border enzyme deficiencies or transport defects (e.g. glucose–galactose transporter deficiency, sucrase–isomaltase deficiency, lactase deficiency, microvillus inclusion disease).
4. Electrolytes, vitamins, trace elements: for example, congenital chloride diarrhoea, acrodermatitis enteropathica (zinc deficiency), selective B12 deficiency.
Mechanisms of malabsorption
Absorptive factors
1. Disordered mucosal villus function:
2. Defective intracellular lipid transport: for example, abetalipoproteinaemia.
3. Inadequate lymphatic circulation of chylomicrons: for example, lymphangiectasia (primary, or secondary to CD), mycobacterial gut infection, radiation enteritis, forms of congenital heart disease with chronically elevated right-sided heart pressure (impeded flow from lymphatics [thoracic duct] into systemic venous circulation).
4. Abnormal water and electrolyte transport (e.g. giardiasis).
History
Feeding
Dietary milestones, initial feeding, age at introduction of cow’s milk (CMP intolerance), age at introduction of first solids, age at introduction of first cereals and the type of cereal (gluten-containing [wheat, rye, barley] in coeliac disease), age at introduction of first fruits (sucrase–isomaltase deficiency), appetite, poor feeding with irritability (iron deficiency associated with coeliac disease), parent-devised dietary restriction (attempting to avoid diarrhoea), iatrogenic dietary manipulation (decreased caloric intake and chronic non-specific diarrhoea).
Past medical history
Necrotising enterocolitis (NEC) as neonate, past bowel resections or other surgery.
Investigations
Stool
1. Pus cells (colonic disease, e.g. CD).
2. Eosinophils (CMP intolerance).
4. Cysts or trophozoites (e.g. Giardia lamblia).
5. Fat globules (CF or SDS-maldigestion [luminal] rather than malabsorption. (Note: lubricants or ointments can produce false-positive results.)
6. Fatty acid crystals (coeliac disease—malabsorption [mucosal]).
7. Reducing substances (carbohydrate maldigestion or malabsorption).
8. Faecal alpha-1-antitrypsin excretion test (FA1AT). This screens for PLE. An elevated FA1AT result suggests a mucosal disorder, as inflamed mucosa allows transudation of serum proteins. This is common in CD and occurs in coeliac disease, but not in CF or CLD, where there is an intraluminal maldigestive defect, which does not involve any mucosal damage.
9. Culture for pathogens (Cryptosporidium, Yersinia).
10. Three-day faecal fat assessment (more accurate than a qualitative stool stain).
11. Bile acids (in SBS with 40–80% ileal resection).
12. Colonic hydroxy fatty acids (in SBS with 80–100% ileal resection).
13. Ostomy output: electrolytes, osmolality (children with stomas).
Blood
1. Full blood count and film (anaemia, neutropenia [SDS], lymphopenia [lymphangiectasia], acanthocytosis [abetalipoproteinaemia, hypobetalipoproteinaemia, chylomicron retention disease]), megaloblastic anaemia (B12, folate malabsorption).
2. Erythrocyte sedimentation rate, ESR (chronic infection, IBD).
3. Liver function tests (albumin, total protein, transaminases) (CLD, CD with PLE).
4. Vitamins: folate (serum and red cell), vitamin B12 (ileal disease), vitamins A, 25-OH D and E, and prothrombin time for vitamin K (fat malabsorption).
5. Minerals: calcium, magnesium, phosphate, iron, ferritin, zinc, copper, selenium.
6. Electrolytes, urea and creatinine (hydration, associated renal disease).
7. Fetal haemoglobin, HbF (SDS).
8. Pancreatic isoamylase (SDS).
9. IgA and coeliac disease serology (anti-tissue transglutaminase [tTG] IgA antibodies, EMA IgA). In addition, molecular genetic testing can be carried out: targeted mutation analysis can determine HLA-DQA1 and HLA-DQB1 genotypes to detect the presence or absence of coeliac disease associated alleles: HLA-DQA1∗0501, HLA-DQA1∗0505, HLA-DQB1∗0201, HLA-DQB1∗0202 and HLA-DQB1∗0302. Over 99.9% of these alleles will be detected by this test.
10. IBD serology screening tests. None are routine, but they are included for completeness. Perinuclear anticytoplasmic antibodies (pANCAs) to neutrophil proteins may be elevated in UC. Anti-Saccharomyces cerevisiae (ASCA) antibodies may be present in CD (more sensitive and specific with elevated pANCA). Antibodies to E. coli (outer membrane porin C [anti-ompC] antibodies) may correlate with diagnosis of IBD.
11. Immunoglobulins (severe combined immunodeficiency syndrome and other primary immune defects).
12. Human immunodeficiency virus (HIV) serology in high-risk groups.
Imaging
1. Bone age (delay in skeletal development; abnormal in SDS syndrome with metaphyseal dysostosis; film will also show rickets).
2. Bowel contrast studies (anatomical lesions, CD).
3. Motility studies (nuclear medicine gastric emptying scan, upper gastrointestinal barium contrast study).
4. Liver scan (causes of CLD).
5. CT scan of liver and pancreas.
6. Radiolabelled Tc albumin lymphatic scan (lymphangiectasia).
Short Cases
Gastrointestinal system
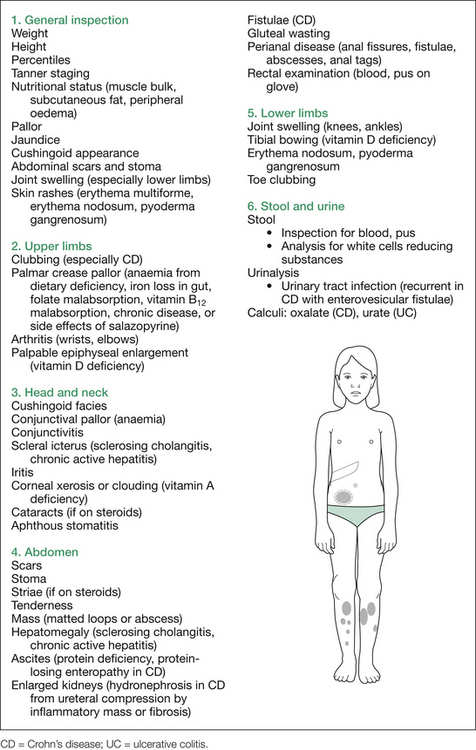
The relevant findings sought are outlined in Figure 8.3. A more detailed listing of possible examination findings is given in Table 8.1.
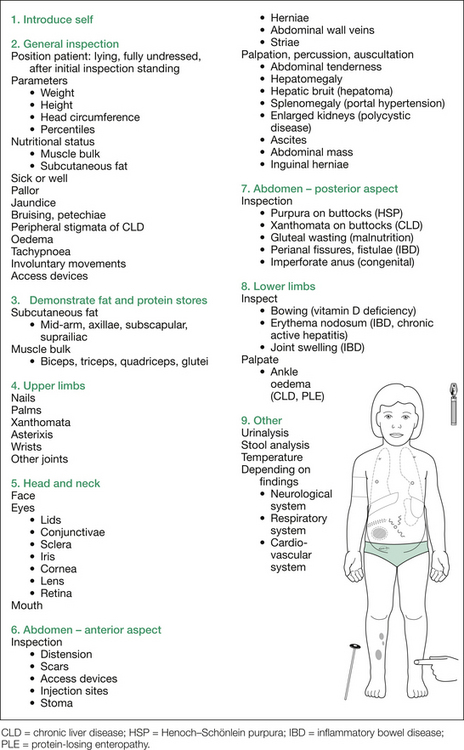
Table 8.1 Additional information: details of possible findings on gastrointestinal examination
| General inspection |
| Pallor (GIT blood loss, CLD, nutritional deficiencies in iron, folate, various vitamins) |
| Jaundice (CLD, vitamin B12 deficiency with ileal resection or disease) |
| Bruising (CLD, thrombocytopenia in hypersplenism, HSP) |
| Petechiae (hypersplenism in portal hypertension) |
| Peripheral stigmata of CLD: spider naevi, scratch marks (pruritis with cholestasis), xanthomata (cholestasis) |
| Oedema (CLD, PLE) |
| Tachypnoea, cyanosis, cough, barrel chest (CF) |
| Irritability (iron deficiency, coeliac disease) |
| Mental state (hepatic encephalopathy) |
| Dysarthria (Wilson’s) |
| Involuntary movements: athetosis, chorea, tremor, dystonia or myoclonus (Wilson’s) |
| Access devices (venous ports used in CF for administering antibiotics; intravenous access for total parenteral nutrition, or hyperalimentation) |
| Nasogastric tube |
| Dysmorphic features |
• Pigmentation around eyes (2–3 mm brown–black spots in Peutz–Jeghers)
• Lids: xanthelasma (hyperlipidaemia)
• Conjunctivae: pallor (anaemias, with nutritional deficiencies, GIT blood loss [CLD, Wilson’s]), xerosis (vitamin A deficiency)
• Sclera: jaundice (note depth thereof)
• Cornea: xerosis, clouding, opacification (vitamin A deficiency), Kayser–Fleischer rings (brown/green pigmentation in limbic region, occur in Wilson’s disease)
• Lens: cataracts (congenital TORCH infections, steroid-treated IBD or chronic active hepatitis, Wilson’s)
• Retina: retinal pigmentation (abetalipoproteinaemia, Alagille), chorioretinitis (TORCH), retinopathy of prematurity (ex-premature with NEC)
CF = cystic fibrosis; CLD = chronic liver disease; GIT = gastrointestinal tract; HSP = Henoch–Schönlein purpura; NEC = necrotising enterocolitis; PLE = protein-losing enteropathy; TORCH = toxoplasmosis, other (e.g. HIV, syphilis), rubella, cytomegalovirus, herpes (both simplex and varicella).
Now, take 20 seconds or so to visually scan for those features outlined in the figure. After this, the child can be systematically examined, commencing with the hands, noting any clubbing, leuconychia or other peripheral stigmata of chronic liver disease (CLD). Move on to the head and neck, examining in particular the eyes and mouth. When examining the eyes, mention the relevance of examining the retinae, but defer actually doing it until you have completed the rest of your examination, as it is too time-consuming at this stage. This is followed by a thorough abdominal examination.
Neurological assessment
A neurological examination is appropriate in the following circumstances:
1. Any jaundice or other suggestion of liver disease in a child over 5 years (e.g. vitamin E deficiency causing cerebellar ataxia and/or peripheral neuropathy, Wilson’s disease causing ataxia, dysarthria, dystonia).
2. Any malabsorption or pigmentary retinopathy (e.g. abetalipoproteinaemia with vitamin E deficiency).
3. Dysmorphism in an infant (marked hypotonia in Zellweger syndrome).
4. Microcephaly (congenital TORCH infections, with developmental delay).
The abdomen
Next, ballot for the kidneys, noting their size, and percuss for resonance above them. Percuss for ascites; if resonant to percussion to the flanks, there is no need to test for shifting dullness. If not, then check for the latter by rolling the patient away from you, waiting 20 seconds for any fluid to settle, and percussing again. Note any change in the site where the percussion note became dull. Measure this distance. Also check for a fluid thrill if there is evidence of any shifting dullness. An assistant (e.g. an examiner) is required to test for a fluid thrill.
Common findings in the examination setting include hepatosplenomegaly, ascites and enlarged kidneys.
In view of the large number of possibilities in this case, they are best enumerated in list form. Figure 8.4 shows several of the findings sought. For further details on the many possible signs on inspection, see Table 8.2.
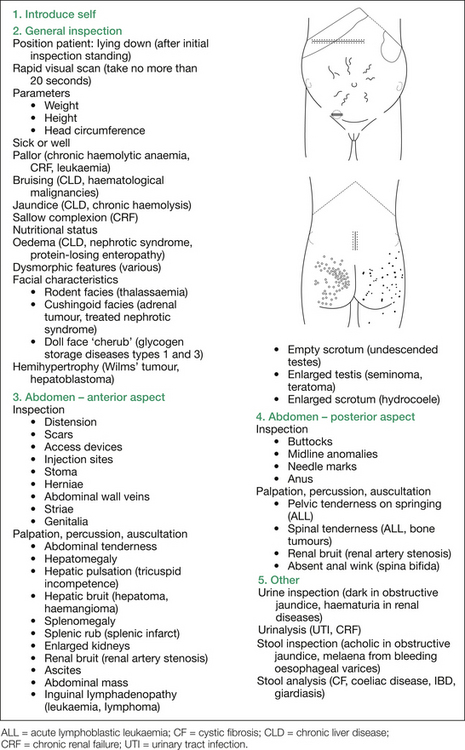
Table 8.2 Additional information: details of possible findings on inspection of abdomen
| Inspection |
| Anterior aspect of abdomen |
| Distension, best assessed standing (ascites, coeliac disease, PCM, organomegaly) |
| Operative scars (e.g. Kasai, renal transplant, peritoneal dialysis, exomphalos repair) |
| Access devices (e.g. Tenckhoff catheter, subcutaneous venous port) |
| Injection sites (e.g. insulin, desferrioxamine) |
| Stoma (colostomy, ileostomy, ileal conduit, gastrostomy) |
| Herniae (umbilical, paraumbilical, inguinal, incisional) |
| Prominent abdominal wall veins (portal hypertension) |
| Striae (Cushing) |
| Visible peristalsis (pyloric stenosis) |
| Inguinal lymphadenopathy (leukaemia, lymphoma) |
| Pubertal status (precocity) |
| External genitalia (ambiguous) |
| Enlarged testis (seminoma, teratoma) |
| Posterior aspect of abdomen |
| Purpura on buttocks (HSP) |
| Xanthomata on buttocks (cholestasis) |
| Buttock asymmetry (sacral tumour) |
| Gluteal wasting (malnutrition) |
| Midline anomalies related to spinal dysraphism |
HSP = Henoch–Schönlein purpura; IBD = inflammatory bowel disease; PCM = protein calorie malnutrition.
The abdominal findings will determine the remainder of the general physical examination.
• If the only finding is minimal tenderness, proceed to a gastrointestinal or renal examination, depending on the site of tenderness.
• If the finding is hepatosplenomegaly, proceed to a gastrointestinal and haematological assessment.
• If the finding is enlarged kidneys, proceed with a renal examination, after requesting the blood pressure and urinalysis.
• If the finding is lymphadenopathy and splenomegaly, examine the haematological system.
• If ascites is the finding, proceed as outlined in the short-case approach to the child with oedema.
• If ambiguous genitalia is the finding, request the blood pressure, look for hyperpigmentation and then proceed as in the short-case approach to ambiguous genitalia in Chapter 7.
Common findings on abdominal examination and their causes are outlined in the following sections.
Hepatomegaly
The following two lists give different classifications of hepatomegaly. The first is a mnemonic, SHIRT, which the author has found useful. The second describes the more common findings found in three different age groups: infant, preschool (less than 5 years) and school age (5 years and over), further subdivided into jaundiced or not jaundiced. This second classification is more practical than comprehensive.
Causes of hepatomegaly (mnemonic SHIRT)
S Structural
Storage/metabolic
1. Defective lipid metabolism—Gaucher disease, Niemann–Pick disease, hyperlipoproteinaemias, cholesteryl ester storage disease, carnitine deficiency, mucolipidoses.
2. Defective carbohydrate metabolism—diabetes mellitus, glycogen storage diseases (types 1, 3, 4 and 6), hereditary fructose intolerance (HFI), galactosaemia, Cushing’s syndrome, mucopolysaccharidoses.
3. Defective amino acid/protein metabolism—tyrosinaemia (type 1), urea cycle enzyme disorders.
4. Defective mineral metabolism—Wilson’s disease.
5. Defective electrolyte transport—CF.
6. Defective nutrition—protein calorie malnutrition (PCM), total parenteral nutrition (TPN).
I Infection
1. Viral infections—congenital rubella, cytomegalovirus infection, coxsackie virus, echovirus, hepatitis (A, B, C, D and E) viruses, infectious mononucleosis.
2. Bacterial infections—neonatal septicaemia, E. coli urinary tract infections, tuberculosis, congenital syphilis.
3. Parasitic infections—hydatid disease, malaria, schistosomiasis, toxoplasmosis, visceral larva migrans.
CAH, chronic persistent hepatitis, IBD associated liver disease.
Splenomegaly
A mnemonic (CHIMPS) is helpful to remember the causes of splenomegaly.
Renal enlargement
Bilateral flank masses
1. Polycystic kidney disease (infantile—autosomal recessive, adult type—autosomal dominant).
2. Hydronephrosis—posterior urethral valves, vesico-ureteral reflux, neurogenic bladder.
3. Tumour—Wilms’, leukaemia, lymphoma, tuberous sclerosis.
4. Metabolic—glycogen storage disease type 1 (A and B), tyrosinaemia type 1.
Ascites
1. Hepatic—cirrhosis and portal hypertension.
3. Gastrointestinal—protein-losing enteropathy (e.g. coeliac disease, inflammatory bowel disease), nutritional (PCM, beri-beri), intestinal lymphangiectasia.
4. Cardiovascular—right ventricular failure, constrictive pericarditis, IVC obstruction, hepatic vein obstruction (Budd–Chiari syndrome).
5. Lymphatic—acquired chylous ascites (thoracic duct obstruction by enlarged lymph nodes or neoplasms).
Jaundice
The infant
The systematic general examination of the infant commences with the hands, followed by the head and neck, the abdomen, a neurological assessment, cardiac and chest examinations and, finally, interpretation of the urinalysis, stool analysis and temperature chart. This approach is outlined in Figure 8.5. Note that the sequence outlined also includes assessing for complications of (unconjugated) hyperbilirubi-naemia itself (that is, kernicterus) or for bilirubin encephalopathy, and also examines for complications of cholestasis (if that is the underlying mechanism suspected); namely, deficiencies in the fat soluble vitamins A, D, E and K. All but vitamin E deficiency can manifest themselves clinically in this age group.
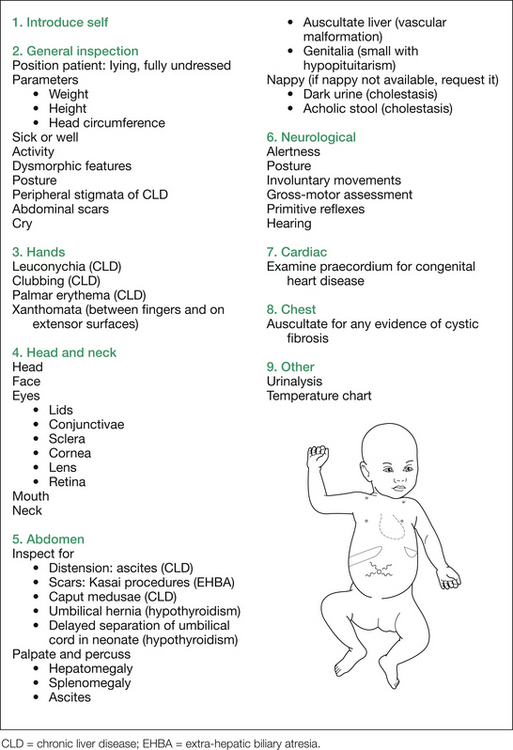
The other possibilities at this age include the following:
1. Metabolic—galactosaemia, tyrosinaemia, hereditary fructose intolerance (HFI), cystic fibrosis (CF).
1. Polysplenia/asplenia syndromes, in which biliary atresia indistinguishable from EHBA can occur in association with dextrocardia and congenital heart disease.
2. Arteriovenous malformation of the liver, which can lead to heart failure.
3. Congestive cardiac failure, which can cause hepatomegaly and jaundice.
1. Stool inspection—EHBA causes acholic stools.
2. Ultrasound of abdomen—EHBA is associated with a gall bladder less than 2.5 cm in diameter. Ultrasound can also identify choledochal cysts and exclude calculi.
3. Nuclear DISIDA scan—extrahepatic or intrahepatic biliary atresia and severe cholestasis can be indistinguishable. All are associated with lack of tracer in the gastrointestinal tract. For this test the patient should be primed with choleretic therapy with phenobarbitone and cholestyramine.
(c) Sweat: iontophoresis (CF).
(d) Radiology: chest X-ray to identify butterfly vertebrae (Alagille).
(e) Cardiac assessment: to exclude pulmonary artery stenosis (Alagille).
If all the above are normal, proceed as follows.
4. Liver biopsy (percutaneous)—EHBA shows extrahepatic biliary obstruction, and bile duct proliferation (discriminates EHBA from other diagnoses in 80% of cases).
5. Percutaneous transhepatic cholangiography (PTC), usually done with biopsy.
6. Operative cholangiogram—the ‘gold standard’ for diagnosing EHBA.
Table 8.3 gives details of the possible findings on examination of an infant with jaundice.
Table 8.3 Additional information: details of possible findings on jaundice examination (infant)
| Inspection |
| Dysmorphic features |
CLD = chronic liver disease; TORCH = toxoplasmosis, other (e.g. HIV, syphilis), rubella, cytomegalovirus, herpes (both simplex and varicella); UTI = urinary tract infection.
The older child
Systematic examination commences with the hands, followed by the head and neck, abdomen, heart and chest, neurological assessment and then interpretation of the urinalysis, stool analysis and temperature chart. This is outlined in Figure 8.6.
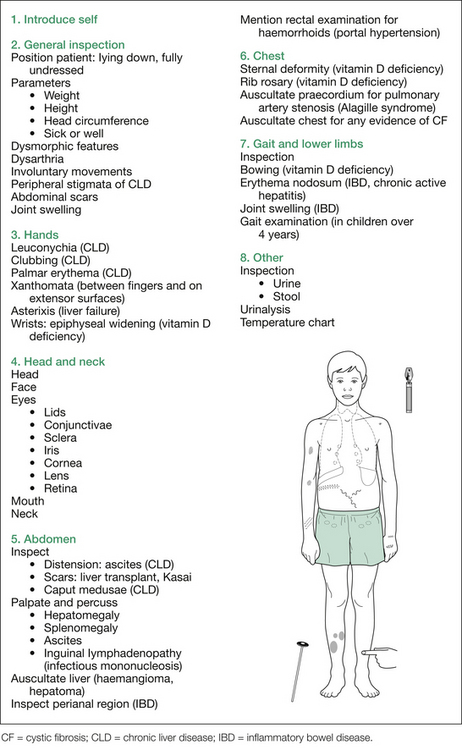
Important points regarding Wilson’s disease are as follows:
1. Neurological problems are extremely rare in children (although they are listed here for completeness).
2. If there is neurological disease, Kayser–Fleischer rings are usually present.
3. Cystic fibrosis often presents when screening an asymptomatic sibling of a child with Wilson’s disease.
4. It may present as a psychiatric problem or with deteriorating school performance.
5. It can present early in childhood, around 5 years, as a haemolytic anaemia, which can be associated with jaundice.
6. It is crucial not to miss the diagnosis, as it is one of the few curable causes of cirrhosis.
Investigations
Liver function tests (LFTs)
1. Serum bilirubin (direct and indirect): total indicates severity of jaundice; fractional determination (conjugated or unconjugated) valuable in diagnosis.
3. Serum proteins: albumin—index of liver capacity for protein synthesis.
Assessment for deficiency of fat-soluble vitamins
After these have been discussed, mention appropriate investigations to clarify the diagnosis.
Table 8.4 gives an incomplete list of some of the more important possible investigations. Table 8.5 details the possible findings on examination of an older child for jaundice.
Table 8.4 Diagnostic investigations in the older child with jaundice
| Disease | Investigations |
|---|---|
| Wilson’s disease | Reduced serum copper (usually) and caeruloplasmin |
| Elevated urinary copper excretion | |
| Markedly elevated liver copper content on liver biopsy (most reliable) | |
| Chronic active hepatitis | |
| 1. HBsAg negative | Markedly elevated serum gammaglobulin (over 90% of cases) |
| Positive for antinuclear antibody (75%) | |
| High-titre smooth muscle antibody | |
| Positive direct Coomb’s test (75%) | |
| 2. HBsAg positive | Mildly elevated serum gammaglobulin |
| Negative for antinuclear antibody (95%) | |
| Low-titre smooth muscle antibody | |
| Negative direct Coomb’s test | |
| Positive (often) HbsAb | |
| 3. HCAb positive | |
| Alpha-1-antitrypsin (A1AT) deficiency | Pi phenotyping: |
Table 8.5 Additional information: details of possible findings on jaundice examination (older child)
| Inspection |
| Dysmorphic features (Alagille) |
| Dysarthria (Wilson’s) |
| Involuntary movements: athetosis, chorea, tremor, dystonia or myoclonus (Wilson’s) |
| Peripheral stigmata of CLD: bruising, bleeding, spider naevi |
| Scratch marks (pruritis with cholestasis) |
| Abdominal scars (liver transplant, Kasai) |
| Joint swelling (IBD) |
| Head and neck |
| Head |
| Facial features of Alagille syndrome |
| Coarse facial features (hypothyroid) |
| Frontal and parietal bossing (thalassaemia) |
| Craniotabes (vitamin D deficiency) |
| Eyes |
| Lids: xanthelasma |
| Conjunctivae |
| Sclerae: depth of jaundice |
| Cornea |
| Lens: cataracts (steroid-treated IBD) |
| Mouth |
| Poor state of dentition (vitamin D deficiency) |
| Palatal petechiae (infectious mononucleosis) |
| Tonsillar exudate (infectious mononucleosis) |
| Neck |
| Cervical lymphadenopathy (infectious mononucleosis) |
| Goitre (hypothyroidism) |
| Gait and lower limbs |
| Most of this section is only applicable to a child over 4 years of age, as vitamin E deficiency and Wilson’s disease do not exhibit neurological effects until later in childhood |
| Gait examination |
| Romberg’s sign (vitamin E deficiency) |
| Hold arms horizontally: wing beating tremor (Wilson’s) |
| Knee and ankle reflexes |
| Sensation: diminished (vitamin E deficiency) |
| Other |
| Request inspection of: |
| Urinalysis |
| Temperature chart (hepatitis, UTI, chronic active hepatitis) |
CLD = chronic liver disease; IBD = inflammatory bowel disease: UTI = urinary tract infection.
Nutritional assessment
The simplest approach to this case comprises three successive components:
First, introduce yourself to the child and the parent. Ensure that the child is fully undressed, then stand back and inspect the child carefully. Visually scan for subcutaneous tissue and muscle bulk. Comment on the child’s height and weight, request the percentile charts and interpret these. If only one measurement is given, request previous measurements to observe their progression. Work out the weight age and height age; compare these and comment. Next, if the child is underweight, work out the weight for height, to quantitate the difference in kilograms between this value and the child’s actual weight.
The next step is a systematic general examination directed at detection of various deficiencies. It commences at the hands, continues up to the head, and then essentially moves from head to toe. Figure 8.7 outlines the order of examination, and Table 8.6 at the end of this section gives additional information. Each deficiency sought is given in parentheses after the relevant physical sign.
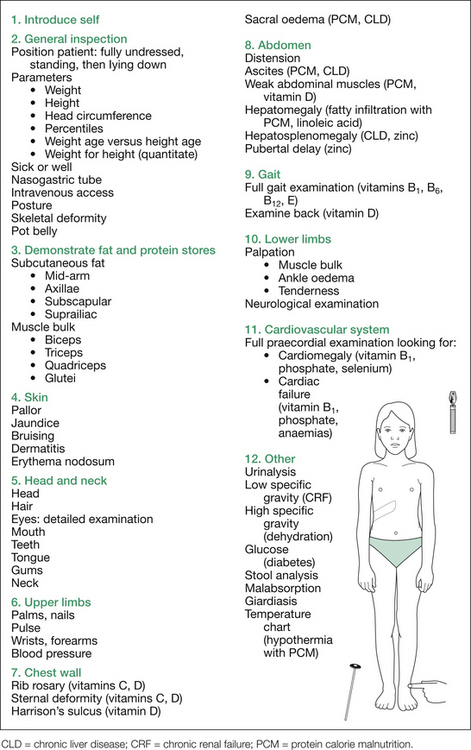
Table 8.6 Additional information: details of possible findings on nutritional assessment
| Inspection |
| Activity, awareness (PCM) |
| Irritability (vitamin C, iron, coeliac) |
| Nasogastric tube |
| Intravenous access for total parenteral nutrition |
| Posture |
CHD = congenital heart disease; CLD = chronic liver disease; IBD = inflammatory bowel disease; PCM = protein calorie malnutrition.
Next, examine the head and neck. Look for thinning of hair or areas of alopecia (linoleic acid, zinc), and for dyspigmentation of hair (PCM), and feel the hair for dryness (iodine) or excessive pluckability (PCM).
The eyes are the next area on which to focus, and many signs are possible here (see Table 8.6). In particular, look at the conjunctivae for pallor (iron, copper, B group vitamins, folate), or dryness and wrinkling (vitamin A), or Bitot’s spots (silver plaques of desquamated epithelial cells and mucus) on the bulbar aspect (vitamin A). Look for scleral icterus (vitamin B12, CLD), corneal dryness, wrinkling or clouding (vitamin A) or opacification (vitamin A, zinc). Quickly assess the external ocular movement (vitamin E) and check for photophobia (riboflavin, zinc). Offer fundoscopy for optic nerve inflammation (vitamin B12) or atrophy (vitamin B1); usually this will not be required.
Examine the chest for sternal deformity (vitamins C, D), or any ‘rib rosary’ (vitamins C, D).
Failure to thrive
Commence with general inspection for obvious abnormalities, such as recognisable dysmorphic syndromes, central nervous system disease (e.g. cerebral palsy [CP]), neuromuscular disease (congenital myopathies, spinal muscular atrophy), tachypnoea (cardiac, respiratory, or renal—metabolic acidosis—in origin), cyanosis (congenital heart disease [CHD]) and any findings related to nutritional status. Next, request the child’s parameters. Failure to thrive as a term is used to describe failure of weight gain in particular, but—particularly if long-standing—may include lack of linear growth as well. If the head circumference is significantly affected, this suggests an intrauterine onset.
1. If all percentiles are equally affected, the possibilities include intrauterine TORCH infections or chromosomal abnormalities.
2. If height is most affected, possibilities include endocrinopathies and skeletal dysplasias.
3. The common pattern for malnutrition is that the weight is most affected, the height less affected and the head circumference relatively normal.
Request the urinalysis for specific gravity (low with chronic kidney disease [CKD], diabetes insipidus), glucose (diabetes), pH (renal tubular acidosis), protein (structural kidney disease, proximal tubular disease), blood (structural kidney disease, urinary tract infection), nitrites (urinary tract infection) and bilirubin (CLD). Request stool analysis, for evidence of steatorrhoea, fat crystals (coeliac disease) or globules (CF), low pH and reducing substances (carbohydrate intolerance), or giardia (cysts or vegetative forms). Mention inspection of any vomitus for bile (obstructive bowel lesions) or blood (portal hypertension) and the temperature chart for infection, as well as watching the mother feeding the child, noting their interaction, the feeding technique and any maternal anxiety.
Weight loss—older child/adolescent
This is very similar in approach to failure to thrive, but there are numerous disorders that occur in older children that are not relevant to the infant or younger child in whom the term ‘failure to thrive’ is used. The commonest cause of weight loss in adolescent girls in Australia and the USA is anorexia nervosa (AN). Other conditions that tend to affect older children need to be considered, including inflammatory bowel disease (IBD), thyrotoxicosis, Addison’s disease, Wilson’s disease and various malignancies. The usual lead-in will be ‘This child is thin’ or ‘This child has lost weight over the past [specified time period]’.
1. Lack of caloric intake: voluntary (e.g. by desire in AN, to avoid nausea in Crohn’s disease) or involuntary (e.g. protein calorie malnutrition [PCM] in the third world, neglect, abuse, Münchausen’s syndrome by proxy [a specific form of abuse], local structural craniofacial problems), or neuromuscular and neuro-developmental disorders, making intake difficult.
2. Lack of calorie utilisation, from lack of food digestion (e.g. cystic fibrosis [CF], Shwachman–Diamond syndrome [SDS]) or food absorption (e.g. coeliac disease, severe Crohn’s disease, short bowel syndrome [SBS]).
3. Excessive use of calories in metabolic processes. Any child with ‘chronic [fill in the organ system here—any organ] failure’ can lose weight. Hence, numerous causes can be remembered easily: chronic kidney failure/disease (CKD), chronic liver failure (CLD), chronic respiratory failure (as in CF), congestive cardiac failure (various forms of congenital and acquired heart disease), chronic immune failure (immunodeficiencies such as HIV), chronic atopic dermatitis (‘skin failure’), chronic adrenocortical failure (Addison’s) or chronic pancreatic endocrine failure (type 1 diabetes mellitus [T1DM]). Any child with cancer will lose weight. Any child with an active inflammatory process can lose weight (e.g. juvenile idiopathic arthritis, IBD).
4. Excessive loss of calories/nutrients/water: various causes of diarrhoea (coeliac disease), various causes of polyuria (CKD, T1DM, diabetes inspidus), various causes of vomiting (AN patients with bulimic features, subacute bowel obstruction, intracranial pathology, malignancy).
Depending on the lead-in given, as alluded to above, malignancy may need to be considered. Leukaemia, intracranial tumours and solid tumours (lymphoma, neuroblastoma, Wilms’) thus all come into the differential diagnosis. The approach given in Table 8.7 includes selected findings only; the list is by no means complete. While it is unlikely that a child with a malignancy would be a short-case subject in an examination format, it is still worth learning a comprehensive approach that will be useful in day-to-day practice.
Table 8.7 Weight loss in the older child: list of possible findings on examination
| 1. General inspection |
| Position patient: fully undressed, standing then lying down |
| Parameters |
• Exophthalmos (Graves’, neuroblastoma, orbital tumour)
• Thyroid stare (hyperthyroidism)
• Lid retraction (hyperthyroidism)
• Ptosis (intracranial tumour)
• Horner’s syndrome: miosis, partial ptosis, anhydrosis (neuroblastoma)
• Heterochromia (associated malignancy)
• Conjunctival pallor (anaemias)
• Papilloedema (intracranial tumour)
• Extraocular movements: paresis of inferior oblique; muscles of convergence—medial, superior, inferior recti; lateral rectus (intracranial tumours)
• Full examination of all aspects of a mass: location (site), size (measure), if crosses midline, consistency, surface, tenderness, mobility, relations to adjacent structures, associated bruit, venous dilatation, enlarged draining lymph nodes, percussion note, associated ascites, extension into pelvis
AIDS = acquired immune deficiency syndrome; ALL = acute lymphoblastic leukaemia; AML = acute myeloid leukaemia; AN = anorexia nervosa; AP = antero-posterior; BSL = blood sugar level; CF = cystic fibrosis; CHD = congenital heart disease; CLD = chronic liver disease; CLL = chronic lymphocytic leukaemia; CKD = chronic kidney disease; DFO = desferrioxamine; HPOA = hypertrophic pulmonary osteoarthropathy; IBD = inflammatory bowel disease; JA = juvenile arthritis; NF-1 = neurofibromatosis type 1; PCM = protein calorie malnutrition; SVC = superior vena cava; TPN = total parenteral nutrition.
For more detailed findings in the examinations for the various systems and their disorders, refer to the relevant sections (e.g. CF, IBD, CLD, CKD, oncology). Wherever a vitamin or trace element is noted in brackets, this refers to a deficiency of this, in the context of malnutrition.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 8 Gastroenterology
Long Cases
Inflammatory bowel disease
In the last few years, there has been an increase in the understanding of genetic susceptibility to IBD, suggesting that Crohn’s disease (CD) and ulcerative colitis (UC) may represent a continuum of disease. Recently, there has been a shift in the IBD management paradigm. Mucosal healing is now used as an end point rather than a clinical indicator of remission, as it is realised that endoscopic lesions and symptoms may not correlate. Exclusive enteral nutrition (EEN) has emerged as the ideal induction therapy in CD. There is a risk stratification evolving, with those at higher risk of severe CD—predictors of more aggressive disease including younger age of onset, extensive small bowel disease, deep colonic ulcers, perianal disease and early need for corticosteroids—receiving more aggressive therapy, with earlier use of immunosuppressive and biological agents; this is termed ‘top-down’ therapy (as opposed to the traditional approach of escalating, or ‘step up’ therapy). In the case of UC, the best evidence for prevention of dysplasia is for 5-aminosalicylates, not infliximab, so there is no reason for top-down therapy in UC.
History
Extraintestinal symptoms
Interval/current: rashes (e.g. erythema nodosum [more in CD], pyoderma gangrenosum [more in ulcerative colitis, UC]), mouth involvement (e.g. aphthous ulcers), liver involvement (e.g. chronic active hepatitis), joint disease (e.g. arthralgias, spondylo-arthropathies), visual problems (e.g. uveitis), vascular complications (e.g. vasculitis), renal tract involvement (e.g. nephrolithiasis), hypertension, myocarditis, pericarditis, fever, pubertal delay, neurological problems (e.g. peripheral neuropathy), haematological disease (e.g. anaemia), musculoskeletal weakness (e.g. vasculitic myositis, steroid-induced myopathy), pancreatitis, extraintestinal neoplasia (e.g. lymphoma), amyloidosis, thromboembolic disease (e.g. cerebral, retinal).
Investigations
Stool
Mainly to differentiate other causes of symptoms:
1. Microscopy (fresh specimen) for cysts, ova, parasites.
2. Culture for bacteria (e.g. Campylobacter, enteropathogenic E. coli, Shigella).
3. Clostridium difficile toxin.
4. Alpha-1-antitrypsin level (screen for protein loss from bowel).
5. Neutrophil-derived proteins: (a) calprotectin—95% sensitivity, 91% specificity for diagnosis of IBD; (b) lactoferrin—80% sensitivity, 82% specificity for diagnosis of IBD.
Blood
1. Full blood count (leucocytosis found in two thirds of CD; anaemia plus high ESR has 96% specificity in diagnosing IBD in a referred patient population with suspected IBD).
2. CRP (elevated in 70–100% of CD and 50–60% of UC; also useful to prognosticate disease course)/ESR (elevated in 80–90% IBD patients)
3. Liver function tests (liver dysfunction as part of IBD; low albumin, total protein from protein-losing enteropathy).
4. Vitamins: folate (serum and red cell), vitamin B12 (ileal disease), vitamins A, D, E and prothrombin time for vitamin K (fat malabsorption).
5. Minerals: calcium, magnesium, phosphate, iron, ferritin, zinc, copper, selenium.
6. Electrolytes, urea and creatinine (associated renal disease).
7. Serology for Yersinia species (enteropathica and pseudotuberculosis) to exclude this as a diagnosis. Yersinia colitis can present with erythema nodosum and arthralgia, as can CD.
8. Perinuclear antineutrophil cytoplasmic antibodies (pANCAs) to neutrophil proteins, with an atypical staining pattern, may be elevated in UC (in 40–80%) or CD (in 5–25%); for diagnosing UC, sensitivity 55%, specificity 89%. Not tested routinely.
9. Anti-Saccharomyces cerevisiae antibodies (ASCAs) are present in 50–60% of patients with CD, 20% of healthy first-degree relatives of those with CD, 10–15% of patients with UC and 0–5% of controls. Not tested routinely.
10. Antibodies to E. coli (outer membrane porin C protein antibodies), anti-ompC antibodies. These are present in 55% of patients with CD, 5–11% those with UC and 5% of controls. Not tested routinely.
11. Antibodies to I2, a bacterial sequence derived from Pseudomonas fluorescens, are present in 30–50% of CD patients (with a sensitivity of only 42%, and a specificity of 76%), 10% of UC patients, 19% of other inflammatory conditions and 5% of controls. Not tested routinely.
12. Antibodies to CBir1 flagellin, an antigen of enteric microbial flora, are present in 50% of CD patients, but only 6% of UC patients and 8% of controls. Not tested routinely.
13. Three different carbohydrate (glycan) antibodies are highly specific, but not sensitive, for CD. These comprise ALCA, ACCA and AMCA, which stand for: antiaminaribioside carbohydrate IgG antibodies (ALCA); antichitobioside carbohydrate IgA antibodies (ACCA); and antimannobioside antibodies (AMCA). These are present in 44–50% of ASCA-negative CD patients, and have been shown to be associated with the NOD2/CARD15 genotype in CD. Not tested routinely.
14. Two other anticarbohydrate antibodies, anti-chitin IgA (anti-C) and anti-laminarin (anti-L), are associated with complicated CD, and improve differentiation between CD and UC.
Imaging
1. Plain abdominal X-ray, erect and supine (may see evidence of incomplete bowel obstruction with distended bowel loops and air/fluid levels in CD).
2. Fluoroscopic barium studies: small bowel follow through (SBFT), and small bowel enteroclysis (SBE) can detect mucosal ulceration, or irregularities, narrowing or distension of the gut lumen, and the presence of fistulous communications, with a sensitivity of 85–95% and a specificity of 89–94% for SBFT in patients with terminal ileal disease in CD.
3. Double-contrast barium enema (may identify rectal and colonic disease in CD and UC).
4. Chest X-ray (to help detect tuberculosis, a well-known cause of chronic bowel inflammation).
5. Ultrasound of abdomen and pelvis. Ultrasound identifies inflammation by increased bowel wall diameter and altered pattern of mural stratification, and Doppler studies can detect increased blood flow. Ultrasound may be useful in delineating intra-abdominal masses (e.g. solid lesions or bowel loops), including abdominal or pelvic abscesses.
Ultrasound for the initial diagnosis of IBD has a sensitivity of 75–94%, with a specificity of 67–100%, can identify bowel wall thickening and can tell fibrosis from oedema.
6. Contrast-enhanced ultrasound (using the new microbubble contrast agent that lasts a few minutes in the bloodstream) can evaluate the bowel wall and assess disease activity with a sensitivity of 93% and a specificity of 93%.
7. CT scanning. Multiplanar imaging can visualise overlapping bowel loops, and complications such as fistulae or abscesses. Traditional CT uses a contrast agent such as barium or iodine, which highlights intraluminal filling defects such as masses. CT enterography (CTE) uses neutral (low-density) oral contrast and intravenous contrast to accentuate the distinction between the enhancing small bowel wall and the low-attenuation gut lumen, to visualise the bowel wall and mucosa. CT enteroclysis comprises placement of a nasojejunal tube, and infusing a contrast agent into the proximal small bowel. CTE and CT enteroclysis may detect segmental mural enhancement, and wall thickening, consistent with active inflammation; reactive mesenteric adenopathy also may be found, supporting the diagnosis of active inflammation. For any CT study, the benefits must be weighed against the cumulative deleterious effects of ionising radiation, especially in younger children.
8. MR enterography (MRE) and MR enteroclysis both have a sensitivity of 88% for detecting IBD. MR enterography has a specificity of 92–100% for detection of IBD. Gadolinium-enhanced magnetic resonance imaging (MRI) has improved intestinal image resolution: CD shows transmural enhancement of the colon, and bowel wall thickening of the terminal ileum or proximal small bowel; UC shows mucosal enhancement and submucosal sparing extending proximally from the rectum. The technique is cheaper and more pleasant than endoscopy. MR provides detailed images of perianal fistulae and is the dominant imaging technique for assessing perianal disease in CD. MRE is preferable to CTE because of lack of radiation, and because of superior images that can distinguish active inflammation from fibrosis.
9. Positron emission tomography (PET) scanning can identify areas of active inflammation, in UC. PET uses [F18] fluoro-2-deoxy-D-glucose to spot areas of increased metabolism.
10. Radio-labelled white blood cell scan (useful if mid-small bowel pathology suspected but not able to be found using conventional evaluations).
Management
Short-term aims include induction and maintenance of clinical and histological remission, improvement in overall quality of life and prevention of complications. The main goals are now mucosal healing, and transmural healing, rather than just clinical healing; patients in clinical remission with steroids may have active endoscopically identified mucosal lesions in up to 70% of cases. Risk stratification based on genotype, phenotype and serology is another useful tool. The only factor that predicts prolonged remission in CD (persisting 3–4 years after initiating treatment) is complete mucosal healing. Longer-term aims include preventing relapses, normalising growth and pubertal development, maintaining bone mass and minimising the need for surgery. These therapeutic aims are based on early aggressive therapy, including the use of newer biological agents. Earlier use of potent agents, previously considered ‘third line’, may be more effective than awaiting resistance to treatment. When evaluating new therapies, remember that a placebo response rate over 30% has been noted in IBD.
Crohn’s disease (CD)
Induction therapy: exclusive enteral nutrition (EEN) (first line), corticosteroids (second line), infliximab
After EEN, the second-line agents for remission in CD are prednisolone (for moderate to severe disease), budesonide (a potent steroid control-released in the small intestine and right colon) and the biological agent infliximab (for refractory disease, steroid dependency or fistulising disease).
Maintenance: Immunomodulators AZA, 6MP and MTX; infliximab
Agents such as 6-MP and AZA (which is metabolised to 6-MP, the active agent) can be used for maintenance therapy for CD, prophylaxis after surgery in CD and perianal CD. In particular, 6-MP has been shown useful as initial treatment in children with moderate to severe CD. AZA and 6-MP have a slow onset of action (3–4 months) and so need to be combined with nutritional therapy or steroids until their effect is seen. Duration should be over years, as there is a high relapse rate if AZA or 6-MP are stopped within the first year. Bone marrow suppression can occur (in 2–5% of patients) at any time. If using AZA or 6-MP, it is useful to know the patient’s thiopurinemethyltransferase (TPMT) status. TPMT is the enzyme that catalyses the conversion of 6-MP to 6-methylmercaptopurine (6-MMP). If there is a deficiency of TPMT, then 6-MP can be metabolised along an alternate pathway to 6-thioguanine (6-TG), which is toxic and can cause bone marrow depression; that is, low TPMT means high 6-TG levels and a high risk of leukopenia. Higher 6-MMP levels correlate with hepatotoxicity in adults. Aminosalicylates and methotrexate inhibit TPMT activity and can increase 6-TG levels.
Ulcerative colitis (UC)
Mild disease
5-ASAs can treat the acute situation and are also valuable prophylactically. Treatment is usually lifelong (for the life of the colon). Sulphasalazine comprises sulphapyridine linked to 5-aminosalicylate (5-ASA) by an azo bond, which is broken by colonic bacteria; 5-ASA, the active component, is released directly to the colon. The 5-ASAs are enteric-coated and deliver the drug to the small and large bowel. The most common problems encountered with sulphasalazine are related to the sulpha part (rashes, bone marrow depression), whereas 5-ASAs can cause nephrotoxicity.
Growth and pubertal delay
Children with CD have delayed puberty and decreased final adult height, and often do not show catch-up growth after diagnosis. No particular treatment regime has shown superiority in improving growth. Biological agents, immunomodulators and surgical resection all can improve growth in IBD. Management of poor growth and pubertal delay involves obtaining adequate remission and maintaining this. Reduction of intestinal inflammation and caloric supplementation to reverse malnutrition are important. It is well described that surgical intervention, in UC particularly, can be associated with the onset of puberty. Pubertal delay is thus a relative indication for surgery. Escalating therapy using biological agents or immunomodulators can be considered to maintain remission through puberty. Many adolescents are psychologically upset by pubertal delay, and many units use sex steroid therapy in these patients. In boys with IBD, testosterone for 3–6 months can have very positive effects on virilisation, growth and psychological well-being, although there are no controlled trials of testosterone use. In girls with IBD, ethinylestradiol can be used for a similar period of time.
Chronic liver disease (CLD)
The causes of CLD can be divided into three groups: cholestatic diseases, metabolic diseases and chronic hepatitis (various forms).
Cholestatic diseases
Metabolic disease
WATCH this is not missed (mnemonic).
Wilson’s disease (WD)
This is the most common cause of fulminant liver failure in children over 3 years. It is an autosomal recessive disorder of copper metabolism in which the liver cannot excrete this metal into the bile. The WD gene ATP7B is located on 13q14.3–q21.1, and encodes a copper-transporting P-type ATPase; there are 40 normal allelic variants, and more than 400 disease-causing mutations of this gene—this gene is needed to enable copper to attach to caeruloplasmin and to excrete copper into the bile. Initially, there is copper accumulation in the liver, then excess copper spills over into the brain (basal ganglia), kidneys, bones and cornea (Kayser–Fleischer rings represent copper deposition in Descemet’s membrane of the cornea, and indicate significant copper storage in the body); it also, less commonly, spills over into the lens (sunflower cataracts), kidneys (proteinuria, microscopic haematuria, Fanconi syndrome), joints (arthritis), heart (cardiomyopathy, cardiac arrhythmias) and skeletal muscle (rhabomyolysis). Investigation findings include low-serum copper and caeruloplasmin, and raised urinary copper. It is rare for WD to present before 3 years. Neurological features (movement disorders, or rigid dystonia) present in WD in adolescence. WD is managed with copper chelating agents (penicillamine [given with pyridoxine], trientine) and zinc (enterocyte metallothionein inducer, interferes with absorption of copper from gut); chelated copper is excreted in urine. Foods high in copper content are restricted: liver, brain, shellfish, mushrooms, chocolate and nuts. Vitamin E may be used along with chelator or zinc to consume free radicals made by excess copper. One third of patients die if untreated. LTx is required if the patient is unresponsive to penicillamine, or has advanced liver failure with coagulopathy and encephalopathy. WD liver disease does not recur after LTx, which provides an effective phenotypic cure, converting the copper kinetics of a homozygous child to that of a heterozygote.
Tyrosinaemia type 1
Management of chronic tyrosinaemia includes a low-protein diet (to reduce tyrosine), vitamin D and 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexenedione (NTBC), which prevents the formation of toxic metabolites, reverses biochemical and clinical effects, allows hepatic regeneration and almost eliminates the risk of hepatocellular carcinoma (HCC). If NTBC fails, LTx is required. Also, LTx is required if HCC (rising alpha-fetoprotein, liver nodularity on ultrasound, CT or MRI) is suspected.
Chronic hepatitis
Liver function tests can be divided into four categories:
(a) tests for liver synthetic function (albumin—synthesised exclusively in the liver; PT/international normalised ratio (INR)—clotting cascade proteins that are needed to have a normal PT are synthesised in the liver);
(b) tests for biliary excretion (total and direct bilirubin);
(c) tests for cholestasis (GGT, ALP—cholestasis means reduced or absent/stopped bile flow); and
(d) tests for hepatocellular damage (AST, ALT—hepatic enzymes released from damaged hepatocytes into the blood stream).
Ultrasound may show echogenic liver, splenomegaly and oesophageal varices, and endoscopy may show gastric and oesophageal varices.
Complications of cirrhosis are as follows (mnemonic: HEPATIC):
History for CLD
Past history (including initial presentation)
Examination for CLD
Figure 8.2 shows complications (only) of CLD. For the approach to the examination for the aetiology (e.g. KF rings and neurological assessment for WD), refer to the short-case section.
Principles of management of CLD
General
Monitor weight, serum albumin, serum bilirubin (total and conjugated), prothrombin time. Psychosocial support is important.
Portal hypertension, varices and variceal haemorrhage
Avoid splenectomy if possible. As well as the risk of infection after splenectomy, it may lead to increased bleeding from removal of good collateral vessels from the splenic capsule (azygos system) that bypass the lower oesophageal junction vessels. Haemorrhage can be exacerbated by deficiency of vitamin K dependent factors, thrombocytopenia secondary to hypersplenism and circulating fibrinolysins.
Liver transplantation (LTx)
Timing of transplantation
The timing of LTx depends upon a variety of factors and may be hastened by any of the following:
1. A history of life-threatening variceal haemorrhage.
2. The development of: (a) a hepatorenal state; (b) hepatic encephalopathy; (c) refractory ascites; (d) reduction in psychosocial development; (e) intractable pruritus; (f) severe metabolic bone disease; or (g) a diminished quality of life.
Assessment occurs in specialist liver units, with preparatory education and counselling of the child and family, a multidisciplinary team that may include a psychologist and a play therapist (especially for children under 2 years), intensive nutritional support, completion of routine immunisation (especially hepatitis A and B) before commencement of immunosuppression, and management of CLD complications.
Indications for LTx
Specific diseases requiring LTx, in order of decreasing frequency, include the following:
1. Extrahepatic biliary atresia (EHBA): over 50% of LTx.
2. IEMs (10–15% of LTx) include AT deficiency, CF, galactosaemia, glycogen storage diseases (GSDs) type IA and IV, mitochondrial functional defects (e.g. defects of fatty acid beta-oxidation), some organic acidaemias, tyrosinaemia, urea cycle defects and WD.
3. Acute hepatic necrosis (around 10%).
4. Cirrhosis from chronic active hepatitis or primary biliary cirrhosis (under 10%).
The following requirements must be met for a cadaveric donor for LTx:
1. Brain death prior to circulatory arrest.
2. The absence of sepsis, HBsAg and HBeAg, HIV, malignancy, drug abuse, liver or gallbladder disease, chronic hypertension.
3. Liver enzymes, serum bilirubin within three times normal values.
4. No period of hypoxia (PaO2 < 60 mmHg) or hypotension (over 2 hours).
5. The absence of more than minimal pressor support to maintain normal blood pressure and peripheral perfusion.
6. To match a recipient, the suitable donor must have ABO compatibility with the recipient. Sex and HLA type compatibility are not necessary.
Transplant surgery procedure
The procedure itself has three main phases: (a) recipient hepatectomy, which can be complicated because of coexistent coagulopathy and portal hypertension (bleeding) and previous Kasai (adhesions); (b) the anhepatic phase, when the patient has the vascular anastomoses performed (vena cava, portal vein, hepatic artery), and then placement of the graft; (c) the neohepatic phase, where the liver graft is reperfused (cardiovascular instability can occur here, as the liver has been exposed to cold preservation techniques) and the biliary anastomosis performed—in patients with EHBA, the donor duct will be implanted into a Roux-en-Y loop, as there is no recipient biliary tree. This latter technique is also useful in small children who receive a segmental graft.
