[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 49 Gastric Secretion
The stomach is an active reservoir that stores, grinds, and slowly dispenses partially digested food to the intestine for further digestion and absorption. Its main secretory function is the production of hydrochloric acid. Gastric acid secretion is present on the first day of life and increases as infants become more mature.1 By two years of age, acid secretion is similar to that of adults, when corrected for body weight.2 Most studies indicate that the rate of acid secretion changes little after the second decade of life unless there is coexisting disease of the acid-secreting glandular mucosa such as infection with Helicobacter pylori (HP) or atrophic gastritis.3–5
Acid facilitates the digestion of protein by converting the proenzyme pepsinogen to the active proteolytic enzyme pepsin. It also facilitates the absorption of iron, calcium, vitamin B12, and certain medications (e.g., thyroxin) as well as prevents bacterial overgrowth, enteric infection, and possibly community-acquired pneumonia.6–18
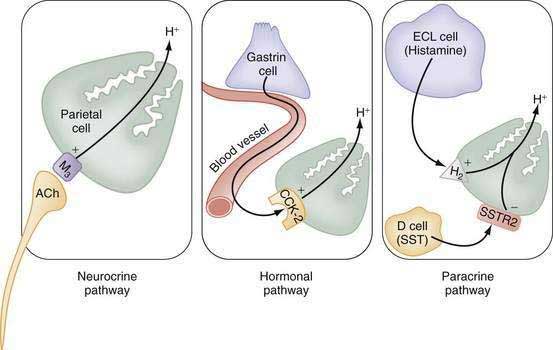
The stomach also secretes lipase, intrinsic factor, electrolytes (e.g., HCO3−, K+, and Cl−), and mucins in addition to a variety of neurocrine, paracrine, and hormonal agents (Fig. 49-1). Neurocrine agents are released from nerve terminals and reach their targets via synaptic diffusion (e.g., acetylcholine [ACh], gastrin-releasing peptide [GRP], and vasoactive intestinal peptide [VIP]). Paracrine agents are released in proximity to their targets and reach them via diffusion (e.g., histamine and somatostatin). Hormones are released into the circulation and reach their targets via the bloodstream (e.g., gastrin).
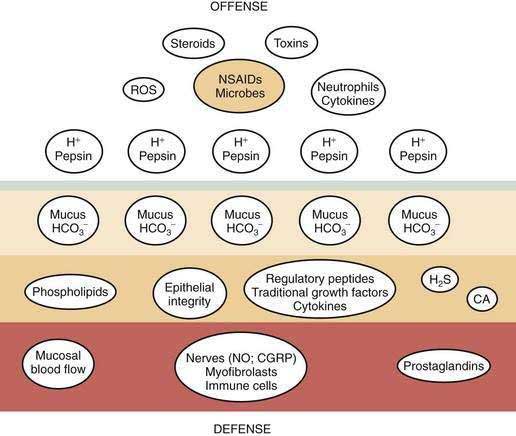
Gastric mucosal integrity depends on a delicate balance between secretion of aggressive (e.g., acid and pepsin) and defensive (e.g. bicarbonate and mucin) substances (Fig. 49-2).19 When mucosal defense mechanisms are overwhelmed, ulceration may occur. In order to reap the benefits of acid without untoward effects, gastric exocrine and endocrine secretion must be precisely regulated. This is accomplished by a highly coordinated interaction among a multitude of neural, paracrine, and hormonal pathways.
FUNCTIONAL ANATOMY
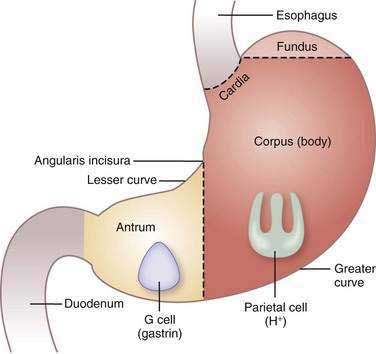
The stomach consists of three anatomic (fundus, corpus or body, and antrum) and two functional (oxyntic and pyloric gland) areas (Fig. 49-3). The oxyntic gland area, the hallmark of which is the oxyntic (oxys, Greek for acid) or parietal cell, comprises 80% of the organ (fundus and corpus). The pyloric gland area, the hallmark of which is the G or gastrin cell, comprises 20% of the organ (antrum). The human stomach contains approximately 1 × 109 parietal cells and 9 × 106 gastrin cells.20 There is debate as to whether the cardia, a transition zone of 0 to 9 mm between the squamous mucosa of the esophagus and the oxyntic mucosa of the stomach, exists as a normal anatomic structure or develops as a result of abnormal reflux. Autopsy and endoscopic studies suggest that cardiac mucosa is absent in more than 50% of the general population.21 Gastric anatomy is discussed in greater detail in Chapter 47.
The glandular area is organized in vertical tubular units that consist of an apical pit region, an isthmus, and the actual gland region that forms the lower part of the unit; the latter consists of a neck and a base (Fig. 49-4). The progenitor cell of the gastric unit, located in the isthmus, gives rise to all gastric epithelial cells. In the oxyntic gland area, the mucus-producing pit cells migrate upward from the progenitor cell toward the gastric lumen. Acid-secreting parietal cells migrate downward to the middle and lower regions of the gland; those at the bases are less active acid secretors. The turnover time for parietal cells is 54 days in mice and 164 days in rats.20 Chief (zymogenic) cells predominate at the base and secrete pepsinogen and leptin22; the latter is also present in parietal cells.23 Several distinct neuroendocrine cell types are contained within the gland but only some of their products have been assigned physiologic functions (see Chapter 1). The cells include enterochromaffin (EC) cells (atrial natriuretic peptide [ANP], serotonin, and adrenomedullin), enterochromaffin-like (ECL) cells (histamine), D cells (somatostatin and amylin), and A-like or Gr cells (ghrelin and obestatin).24–29 ECL cells constitute 66% of the neuroendocrine cell population in rats and 30% in humans. Somatostatin-containing D cells possess neural-like cytoplasmic processes that terminate in the vicinity of parietal and ECL cells (see Fig. 49-4). The functional correlate of this anatomic coupling is a tonic paracrine restraint exerted by somatostatin on acid secretion directly as well as indirectly by inhibiting histamine secretion (Fig. 49-5).30–32
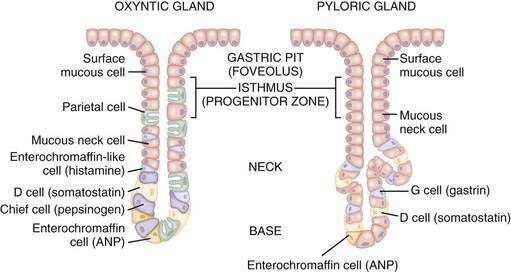
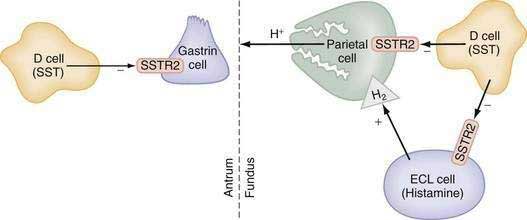
Somatostatin-containing D cells are also present in the pyloric gland area (see Figs. 49-4 and 49-5); in this region they exert a tonic paracrine restraint on gastrin secretion from G cells.33,34 The pyloric gland also contains EC cells (ANP and serotonin), A-like or Gr cells (ghrelin and obestatin), and endocrine cells containing orexin.26,35,36
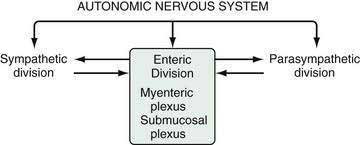
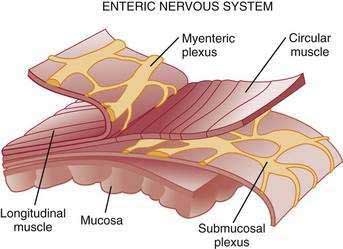
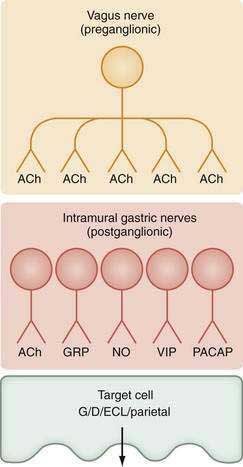
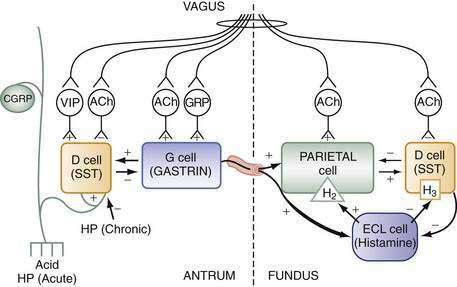
The stomach is innervated by a neural network, the enteric nervous system (ENS), that contains intrinsic neurons, that is, neurons whose cell bodies are contained within the gastric wall (Figs. 49-6 to 49-8). The ENS, the third division of the autonomic nervous system (the other two being the sympathetic and parasympathetic), is often referred to as the “little brain” because it contains as many neurons as the spinal cord, approximately 106, and can function autonomous of central input (see Fig. 49-6).37 Nevertheless, the ENS receives input from and sends input to the central nervous system via sympathetic and parasympathetic neurons. In rats and guinea pigs, most of the intrinsic neural innervation of the stomach originates in the myenteric plexus, located between the circular and longitudinal muscle layers; the submucosal plexus in these species, adjacent to the mucosal layer, contains only a small number of neurons. Humans, in contrast, have a clearly defined submucosal plexus that regulates gastric secretion and contains a variety of neurotransmitters (see Figs. 49-7 and 49-8). It should be noted that the vagus nerve is predominantly afferent, containing 80% to 90% afferent fibers and 10% to 20% efferent fibers. The efferent fibers arise from the dorsal motor nucleus of the brainstem. They are preganglionic and do not directly innervate parietal or neuroendocrine cells but rather synapse with postganglionic neurons of the ENS. The postganglionic neurons contain a variety of transmitters including ACh, GRP, nitric oxide (NO), VIP, and pituitary adenylate cyclase–activating polypeptide (PACAP) (see Fig. 49-8).38 In the stomach, afferent nerve fibers containing calcitonin gene–related peptide (CGRP) are of extrinsic origin, that is, the cell bodies are located outside the stomach wall.39 Postganglionic neurons of the ENS regulate acid secretion directly, as is the case for ACh, and/or indirectly by modulating the secretion of gastrin from G cells, somatostatin from D cells, and possibly histamine from ECL cells (see Fig. 49-8).
ACID SECRETION: PARACRINE, HORMONAL, NEURAL, AND INTRACELLULAR REGULATION
Parietal cells secrete hydrochloric acid at a concentration of approximately 160 mM or pH 0.8. Acid is thought to gain access to the lumen via channels in the mucus layer created by the relatively high intraglandular hydrostatic pressures generated during secretion, about 17 mm Hg.40
Acid facilitates the digestion of protein and absorption of iron, calcium, and vitamin B12 as well as prevents bacterial overgrowth, enteric infection, and possibly community acquired pneumonia.6–14,18,41 However, when levels of acid (and pepsin) overwhelm mucosal defense mechanisms, ulcers occur. To prevent such damage, gastric acid must be precisely regulated and produced according to need. This is accomplished by a highly coordinated interaction among a number of neural, hormonal, and paracrine pathways. These pathways can be activated directly by stimuli originating in the brain or reflexively by stimuli originating in the stomach after ingestion of a meal such as mechanical stimulation (e.g., distention) or chemical stimulation (e.g., protein and acid).
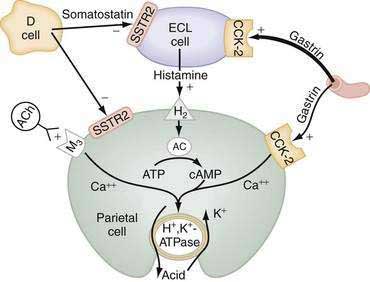
The principal stimulants of acid secretion are (1) ACh, released from postganglionic enteric neurons (neurocrine), (2) gastrin, released from antral G cells (hormonal), and (3) histamine, released from oxyntic ECL cells (paracrine) (see Fig. 49-1; Fig. 49-9). These agents interact with specific G protein–binding receptors (M3, CCK-2, and H2, respectively) that are coupled to two major signal transduction pathways: intracellular calcium in the case of gastrin and ACh, and adenylate cyclase or adenosine 3′,5′-cyclic monophosphate (cAMP) in the case of histamine (see Fig. 49-9). There is evidence for potentiation (or synergism) between histamine and either ACh or gastrin, probably as a result of postreceptor interaction between the two signaling pathways.42 The main inhibitor of acid secretion is somatostatin, released from oxyntic and antral D cells (paracrine) (see Figs. 49-1, 49-5, and 49-9). Each of these agents acts directly on the parietal cell as well as indirectly by modulating the secretion of neuroendocrine cells (Fig. 49-10).
HISTAMINE
Histamine, produced in ECL cells by decarboxylation of l-histidine by histidine decarboxylase (HDC), stimulates the parietal cell directly by binding to H2 receptors coupled to activation of adenylate cyclase and generation of cAMP (see Fig. 49-9).43 Histamine also stimulates acid secretion indirectly by binding to H3 receptors coupled to inhibition of somatostatin release from oxyntic D cells, thus resulting in stimulation of histamine release and acid secretion (see Fig. 49-10).44,45 Gastrin, PACAP, VIP, and ghrelin stimulate, whereas somatostatin, CGRP, prostaglandins, peptide YY (PYY), and galanin inhibit histamine secretion.46,47 As discussed following, gastrin also exerts a direct proliferative effect on ECL cells. ACh has no direct effect on histamine secretion.48–50
GASTRIN
Gastrin, the main stimulant of acid secretion during meal ingestion, is produced in G cells of the gastric antrum and, in much lower and variable amounts, in the proximal small intestine, colon, and pancreas. Gastrin is synthesized as a large precursor molecule of 101 amino acids, which is converted to progastrin (80 amino acids) by cleavage of the N-terminal signal peptide. Progastrin is further processed to yield peptides with C-terminal glycine, that is, G34gly and G17gly. The final processing step involves amidation to yield G34amide and G17amide. The plasma half-life of G34amide is 30 minutes and that of G17amide is three to seven minutes; they are metabolized primarily by the kidney and, in addition, by the intestine and liver.51,52 Consequently, most gastrin in the circulation during fasting is G34, whereas after a meal it is G17. In patients with renal insuffiency as well as massive small bowel resection, fasting blood levels of G17 and G34 are elevated.53,54 It should be noted that the commercially available test substance pentagastrin is not a naturally occurring peptide but rather is a manufactured analog that contains the biologically active C-terminus sequence Trp-Met-Asp-Phe-NH2.
Gastrin and cholecystokinin (CCK) belong to the same family of peptides and possess an identical carboxyl-terminal pentapeptide sequence (-Gly-Trp-Met-Asp-Phe-NH2). Two main classes of gastrin/CCK receptors have been characterized: CCK1 (formerly CCK-A) and CCK-2 (formerly CCKB or CCKB/gastrin). CCK1 receptors are specific for CCK, whereas CCK-2 receptors recognize both CCK and gastrin with high affinity. Gastrin, acting via CCK-2 receptors that activate phospholipase C to induce release of intracellular calcium, stimulates the parietal cell directly and, more importantly, indirectly by releasing histamine from ECL cells (see Figs. 49-9 and 49-10).55,56 Gastrin regulates the secretion and synthesis of histamine in a biphasic manner. The first phase involves release of stored histamine. The second phase relates to the replenishment of histamine stores and involves an increase in HDC activity followed by an increase in HDC gene transcription.57 H2 receptor, HDC, and CCK-2 receptor knockout mice manifest decreased acid secretion, especially in response to gastrin.58–60
Although amidated gastrins had been thought to be the only forms with biological activity, glycine-extended gastrins may regulate the capacity of the parietal cell to respond to secretagogues, release histamine from ECL cells, and stimulate proliferation of colonic mucosa and colorectal cancers.61,62 ACh, GRP, secretin, β2/β3-adrenergic agonists, calcium, protein, and alcoholic beverages produced by fermentation stimulate, whereas somatostatin, galanin, and adenosine inhibit gastrin secretion. In addition, at least two negative-feedback pathways, mediated via release of somatostatin, regulate gastrin secretion. The first is activated by luminal acidity and involves sensory CGRP neurons (see Fig. 49-10). Low intragastric pH (high intragastric acidity) activates CGRP neurons that, via an axon reflex, stimulate somatostatin and thus inhibit gastrin secretion.63–65 Conversely, when intragastric pH rises (low intragastric acidity), for example, by administering antisecretory medications such as proton pump inhibitors (PPIs) or by developing gastric atrophy, somatostatin secretion is inhibited and patients develop hypergastrinemia. There is some evidence that bacterial overgrowth induced by hypochlorhydria may also contribute to hypergastrinemia.66 The second negative-feedback pathway involves a paracrine action whereby gastrin directly stimulates somatostatin and thus attenuates its own secretion (see Fig. 49-10).67
Gastrin also functions as a trophic hormone to stimulate mucosal proliferation. CCK-2 receptors have been localized to the progenitor zone in oxyntic glands, and chronic hypergastrinemia induces proliferation of ECL and parietal cells directly as well as indirectly via the autocrine or paracrine action of growth factors such as heparin-binding epidermal growth factor, amphiregulin, transforming growth factor-α, metalloproteinases, and regenerating islet-derived 1.68,69 Rats rendered hypergastrinemic by a PPI demonstrate a five-fold increase in the number of ECL cells and a 1.5-fold increase in the number of parietal cells.70 Gastrin acts directly on ECL cells to induce hyperplasia, dysplasia, and eventually neoplasia (carcinoids).71 In contrast to rodents, humans rarely develop carcinoid tumors in response to hypergastrinemia unless other factors are present such as chronic active gastritis or gastrinoma associated with multiple endocrine neoplasia type 1 (see Chapter 31).72 Because ECL cells contain somatostatin subtype 2 receptors (SSTR2), somatostatin scintigraphy with 111indium-diethylenetriamine pentaacetic acid [111In-DTPA]octreotide is the preferred imaging method to detect carcinoid tumors (see Chapter 31).73,74
ACETYLCHOLINE
ACh, released from postganglionic neurons whose cell bodies are located primarily in the submucosal (Meissner’s) plexus, stimulates the parietal cell directly as well as indirectly by inhibiting somatostatin secretion (see Fig. 49-10). The parietal cell muscarinic receptor is of the M3 subtype.75,76 Like CCK-2 receptors, M3 receptors are coupled to activation of phospholipase C with generation of inositol trisphosphate and release of intracellular calcium (see Fig. 49-9).77 Alcoholic beverages produced by fermentation stimulate gastric acid secretion and the effect may be mediated via activation of M3 receptors.78 ACh also stimulates acid secretion indirectly by activating M2 and M4 receptors on D cells coupled to inhibition of somatostatin secretion, thus removing the tonic restraint exerted by this peptide on gastrin, ECL, and parietal cells (see Fig. 49-10).
SOMATOSTATIN
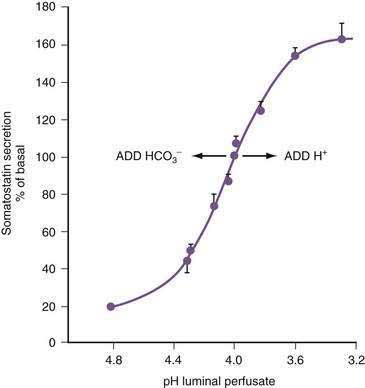
In the stomach, somatostatin cells are closely coupled to their target cells (gastrin cells in the antrum, or parietal and ECL cells in the fundus/body) either directly via cytoplasmic processes or indirectly via the local circulation.28,79 The functional correlate of this anatomic coupling is a tonic restraint exerted by somatostatin on acid secretion from the parietal cell, histamine secretion from the ECL cell, and gastrin secretion from the G cell (see Figs. 49-5 and 49-10).30,31,34,80,81 Removing this restraint (i.e., disinhibition or elimination of the influence of an inhibitor), by activation of cholinergic neurons, is an important physiologic mechanism for stimulating acid secretion (see Fig. 49-10). In the stomach, the actions of somatostatin are mediated primarily via the somatostatin subtype 2 receptor (SSTR2).82–84 Gastrin, GRP, VIP, PACAP, β2/β3-adrenergic agonists, secretin, ANP, adrenomedullin, amylin, adenosine, and CGRP stimulate, whereas ACh and interferon-γ inhibit somatostatin secretion. As mentioned, an increase in luminal acidity acts to attenuate acid secretion via a pathway involving release of somatostatin in the antrum and the fundus. The change in gastric somatostatin secretion can be demonstrated over a range of pH 3 to pH 5, which is within the range observed after ingestion of a meal (Fig. 49-11).85
MISCELLANEOUS PEPTIDES
Ghrelin, the natural ligand for the growth hormone secretagogue receptor, is present in greatest concentrations in gastric oxyntic mucosa and is localized to A-like (or Gr) cells.86,87 Lesser amounts are present in the antrum, small intestine, and colon (see Chapter 1). Plasma ghrelin concentrations increase before meals and decrease postprandially.88 It is postulated that ghrelin triggers premeal hunger and promotes feeding. Its suppression after Roux-en-Y gastric bypass may, in part, contribute to weight loss.89 Most studies report that exogenously administered ghrelin stimulates acid secretion.90,91 The stimulatory effect appears to involve the vagus nerve and histamine release because it is abolished by vagotomy and is associated with an increase in HDC messenger ribonucleic acid (mRNA).92,93
Orexin-A, derived from propro-orexin by post-translational processing, is co-localized with gastrin in human pyloric mucosa.91,94 Intracerebroventricular and peripherally administered orexin-A stimulate gastric acid secretion.95 In rats equipped with gastric fistulas, an orexin receptor 1 antagonist inhibits basal and pentagastrin-stimulated acid secretion, implying that endogenous orexin-A stimulates acid secretion.94,95
ANP, CCK, secretin, neurotensin, glucagon-like peptide 1 (GLP-1), glicentin, oxyntomodulin, peptide YY, adrenomedullin, amylin, glucose-dependent insulinotropic polypeptide (GIP), leptin, epidermal growth factor, and interleukin-1β (IL-1β) inhibit acid secretion. The effect of each, except perhaps for IL-1β, is mediated via release of somatostatin.24–2696 The term enterogastrone has been used to describe the intestinal factor or factors responsible for inhibiting acid secretion in response to nutrients in the intestine. Prime candidates include CCK, secretin, neurotensin, GLP-1, glicentin, and oxyntomodulin because they are present in intestinal mucosa, released into the circulation in response to luminal nutrients, and capable of inhibiting acid secretion at “physiologic” concentrations.97–101 Although it is likely that enterogastrone activity represents the combined influence of several of these peptides, the strongest evidence favors CCK. CCK, produced in I cells in the proximal small intestine, is released by luminal protein and fat. The acid-inhibitory response to intraduodenal fat is abolished by pretreatment with a CCK-1 receptor antagonist in dogs, and the response is blocked in CCK-1 receptor knockout mice as well.102–105
PARIETAL CELL INTRACELLULAR PATHWAYS
In parietal cells, acid secretion is increased by activation of intracellular cAMP- and calcium-dependent signaling pathways that activate downstream protein kinases, ultimately leading to fusion and activation of H+,K+-ATPase (the proton pump) with concomitant activation of luminal membrane conductances for K+ and Cl− (see Fig. 49-9; Fig. 49-12). The H+,K+-ATPase actively pumps out H+ against a tremendous concentration gradient (cell interior pH 7.4 or 40 nM; acid secreted at pH 0.8 or 160 million nM) in exchange for luminal K+. The energy required comes from adenosine triphosphate (ATP) produced by the parietal cell’s extensive mitochondrial network. H+,K+-ATPase consists of an α–subunit that carries out the catalytic and transport function of the enzyme and contains sequences responsible for apical membrane localization106 as well as a β-subunit, which is heavily glycosylated, and protects the enzyme from degradation and is necessary for trafficking to and from the luminal membrane.107
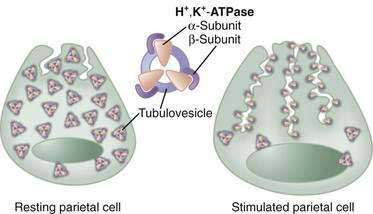
In the resting unstimulated state, H+,K+-ATPase activity is sequestered within cytoplasmic tubulovesicles. On stimulation, there is a dramatic morphologic transformation as these vesicles fuse with the apical plasma membrane, resulting in a 6- to 10-fold increase in the membrane and the formation of the canalicular system (Fig. 49-13). Translocation of the H+,K+-ATPase into the canalicular membrane together with the presence of luminal K+ activates the enzyme.108 On cessation of secretion, the H+,K+-ATPase is retrieved from the apical membrane and the tubulovesicular compartment is reestablished. The precise mechanisms regulating trafficking are not known, but data suggest that it involves actin-based microfilaments, small GTPases, docking/fusion proteins, ezrin, and clathrin.109–111
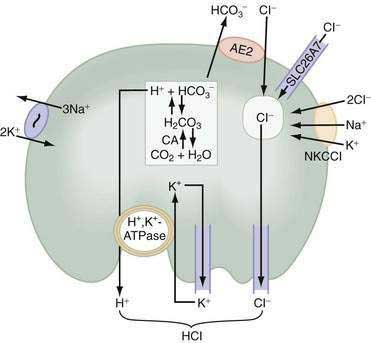
Acid secretion requires not only a functional H+,K+-ATPase but also apical K+ and Cl− channels and basolateral HCO3− and Cl− exchangers. Acid is produced from the hydration of CO2 to form H+ and HCO3−, a reaction catalyzed by cytoplasmic carbonic anhydrase (see Fig. 49-12). Because the H+,K+-ATPase is unable to pump H+ into the lumen without a parallel uptake of K+, sufficient quantities of K+ must be delivered to the lumen. This K+ recycling is accomplished by luminal KCNE2/KCNQ1 and ROMK (KCNJ1) potassium channels. KCNQ1 is a voltage-activated K+ channel, which, when modified by the small regulatory subunit, KCNE2, becomes voltage insensitive, constitutively open, and acid activated.108,112,113 ROMK may be regulated by the cystic fibrosis transmembrane conductance regulator (CFTR).108,112–114 The concentration of K+ in gastric juice (8 to 12 mM) exceeds plasma K+ by two- to four-fold.
For each H+ secreted, an HCO3− ion exits the cell across the basolateral membrane via the anion exchanger 2 (AE2), Slc4a2 (see Fig. 49-12).115 As a result of this HCO3−/Cl− exchange, the pH within the parietal cell remains only slightly alkaline during acid secretion.116 Rapid entry of HCO3− from parietal cells into blood has been referred to as the alkaline tide. Some of this HCO3− may be taken up and secreted by surface epithelial cells.
Concurrently with H+, Cl− is extruded across the luminal membrane via an apical chloride channel, the precise identity of which is not known. The sources of intracellular Cl− are the basolateral anion exchanger 2 (AE2), sodium-2 chloride potassium-cotransporter-1 (NKCC1), and the SLC26A7 channel (see Fig. 49-12).117,118
PROTON PUMP INHIBITORS
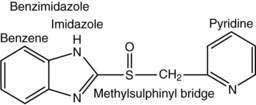
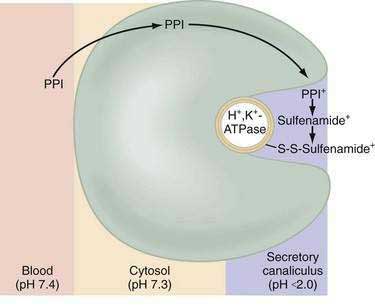
Current PPIs (e.g., omeprazole, lansoprazole, rabeprazole, pantoprazole, and esomeprazole) consist of two heterocyclic moieties, a pyridine and a benzimidazole ring, connected by a methylsulphinyl group (Fig. 49-14). They are weak bases (pKa 4 or 5) that concentrate in acidic spaces within the body that have a pH less than 4. The pKa of a molecule refers to the degree of willingness of the compound to accept or donate a proton and is based on a logarithmic scale such that a compound with a pKa of 5 is 10-fold more basic than a compound with a pKa of 4. When a compound is in an environment with a pH equal to its pKa, half the molecules will be protonated and half will be non-protonated. If a compound with a pKa of 4 is placed in an environment with a pH less than 1, more than 99.9% of the molecules will be protonated. PPIs are membrane permeable in the nonprotonated form and relatively impermeable in the protonated form. In blood (pH 7.4), PPIs are predominantly nonprotonated and thus pass readily into cells (time to reach peak plasma concentration, ≈2 hours; elimination half-life, ≈1 hour), diffuse through the cytoplasm, and then become protonated and trapped, probably as sulfenamides or sulfenic acid, in the acid environment of the secretory canaliculus (Fig. 49-15).119 As a consequence of the basic pKa of PPIs and “ion trapping,” the concentration of PPIs in the secretory canaliculus is 100,000- to 1,000,000-fold higher than in the blood. The sulfenamide rapidly reacts with cysteines on the luminally exposed α-subunit of the H+,K+-ATPase to form a covalent (electrochemical) disulfide bond (see Fig. 49-15).120 Whereas all PPIs bind to cysteine 813, omeprazole also binds to cysteine 892, lansoprazole to cysteine 321, and pantoprazole to cysteine 822. Because only the apical-membrane inserted H+,K+-ATPase is susceptible to blockade by PPIs and an acid environment (pH < 4) is necessary for trapping and converting the PPI to its reactive species, the potency of PPIs is decreased when administered during the basal (fasting) state or when acid secretion is inhibited.121,122 Because most pumps are inserted with breakfast, it is recommended that PPIs be taken 30 minutes to 1 hour before the first meal. If greater inhibition is needed, an additional dose should be taken before dinner. Recovery from inhibition of acid secretion occurs by de novo synthesis of pump protein (54 hours in rat). It has been postulated that reduction of the cysteine disulfide bonds by reducing agents such as glutathione (15 hours in rat) could also play a role.123
INTEGRATED RESPONSE TO A MEAL: INTERPLAY OF NEURAL, PARACRINE, AND HORMONAL PATHWAYS
Stimuli originating inside and outside the stomach converge on gastric intramural efferent neurons that are the primary regulators of acid secretion. The effector neurons comprise cholinergic and peptidergic (i.e., GRP and VIP) neurons. Although nitric oxide and PACAP neurons are present in gastric mucosa their precise physiologic roles as regulators of acid secretion are not known. The effector neurons act on target cells directly as well as indirectly by regulating secretion of gastrin, somatostatin, and possibly histamine (see Figs. 49-8 and 49-10).
During the basal state, acid secretion is maintained at an economically low level by the continuous inhibitory restraint exerted by somatostatin on the ECL cell (histamine) and parietal cell (acid) in the fundus/body and on the G cell (gastrin) in the antrum (see Figs. 49-5, 49-9, and 49-10). During ingestion of a meal, maximal secretion may be achieved by removing the inhibitory influence of somatostatin while at the same time directly stimulating acid and gastrin secretion. This is accomplished, in large part, by activation of intramural cholinergic neurons (see Fig. 49-10). The thought, sight, smell, and taste of food contributes up to 50% of total postprandial acid secretion.124,125 Anticipation of a meal activates central neurons whose input is relayed via the vagus nerve to gastric intramural cholinergic neurons in oxyntic as well as pyloric mucosa. The components of the central nervous system include the dorsal motor nucleus of the vagus, the nucleus tractus solitarius, and the hypothalamus. In the fundus/body, ACh, released from cholinergic neurons, stimulates the parietal cell directly, as well as indirectly, by eliminating the inhibitory paracrine influence of somatostatin on parietal and ECL cells.44,126 The resultant increase in histamine stimulates acid secretion directly via H2 receptors on the parietal cell and indirectly via H3 receptors that mediate suppression of somatostatin secretion (see Fig. 49-10).44,127 Thus, histamine, acting via H3 receptors, amplifies the ability of secretagogues to stimulate acid secretion by suppressing somatostatin secretion. The net effect of cholinergic neurons is suppression of all paracrine inhibitory influence (i.e., somatostatin) and enhancement of paracrine stimulatory influences (i.e., histamine acting via H2 receptors) on parietal cells. There is some evidence that PACAP, a member of the glucagon/VIP superfamily of regulatory peptides, may participate in the regulation of acid secretion, but its precise physiologic role is not known.26,128,129 PACAP is present in gastric mucosal nerves and capable of releasing histamine from ECL cells and somatostatin from D cells. The net effect of exogenous PACAP on acid secretion has been reported to be either stimulation or inhibition, depending on the relative contributions of released histamine and somatostatin in each preparation.128,130,131
In the antrum, cholinergic neurons activated by anticipation of the meal stimulate gastrin secretion directly as well as indirectly by suppressing somatostatin secretion (see Fig. 49-10).30,31,34,132–144 In physiologic concentrations, gastrin stimulates parietal cells directly and, more importantly, indirectly by enhancing the secretion of histamine.145,146
As the meal enters the stomach, the same cholinergic neurons are further activated mechanically by high distention and chemically by protein components of the food.141,142,147,148 Initially, the ingested meal acts as a buffer of secreted acid. The decrease in acidity (i.e., increase in pH) further inhibits somatostatin secretion and thus increases gastrin secretion. Luminal protein activates GRP neurons that directly stimulate gastrin secretion (see Fig. 49-10).79,142 It should be noted that suppression of somatostatin secretion permits an optimal gastrin response.
As the meal empties the stomach, a number of paracrine and neural pathways are activated to restore the inhibitory influence of somatostatin in the fundus/body and antrum and hence restrain acid secretion (see Fig. 49-10). First, a stimulatory paracrine pathway linking gastrin to antral somatostatin cells is activated that acts to restore antral somatostatin secretion after release of gastrin.67 Second, there is less activation of cholinergic neurons by anticipation of the meal as well as by protein and distention. Third, as distention decreases, VIP neurons are preferentially activated that stimulate somatostatin secretion.141 Fourth, as the buffering capacity of the meal is lost, antral and fundic somatostatin cells (via sensory CGRP neurons) are exposed to the full stimulatory effect of luminal acid. Fifth, enterogastrones released from the small intestine, such as CCK, stimulate somatostatin secretion. The resultant increase in antral and fundic somatostatin secretion attenuates gastrin and acid secretion and restores the basal interdigestive state. This state is marked by the continuous restraint exerted on G (gastrin), ECL (histamine), and parietal (acid) cells by contiguous somatostatin cells (see Figs. 49-9 and 49-10). A decrease in this restraint is sufficient to again initiate acid secretion.
HELICOBACTER PYLORI–INDUCED PERTURBATIONS IN ACID SECRETION (see also Chapter 50)
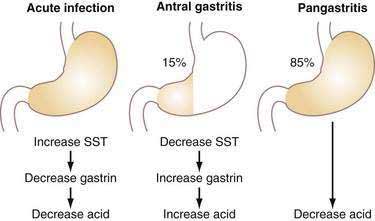
Acute infection with H. pylori (HP) results in hypochlorhydria,149–152 whereas chronic infection results in either hypo- or hyperchlorhydria (Fig. 49-16). Appreciation of the pathways discussed earlier provides some insight into the mechanisms whereby HP colonizes the stomach and may lead to ulceration.
The decrease in acid secretion during acute HP infection is thought to facilitate survival of the organism and its colonization of the stomach.153 The mechanism whereby HP inhibits acid secretion is multifactorial and includes (1) direct inhibition of the parietal cell (and perhaps ECL cell) by a constituent of the bacterium (e.g., vacuolating cytotoxin, lipopolysaccharide, or acid-inhibitory factor) and (2) indirect inhibition of parietal cell function as a result of changes in cytokines as well as hormonal, paracrine, and neural regulatory mechanisms.154–157 HP itself inhibits human H+,K+-ATPase α-subunit gene expression.158 It also elicits secretion of at least two cytokines, IL-1β and tumor necrosis factor-α, that directly inhibit parietal cell secretion.159 In preliminary studies we have shown that HP activates CGRP sensory neurons coupled to stimulation of somatostatin and thus inhibition of gastrin, histamine, and acid secretion.160
Chronic infection with HP may be associated with either decreased or increased acid secretion depending on the severity and distribution of gastritis (see Fig. 49-16).161 Most patients chronically infected with HP manifest a pangastritis and produce less than normal amounts of acid.162 Reduced acid secretion, initially, is due to functional inhibition of parietal cells by either products of HP itself or, more likely, products of the inflammatory process, as discussed earlier for acute infection163,164; this is usually reversible on eradication of the organism.165–167 In such patients, HP may be protective against gastroesophageal reflux disease (GERD), Barrett’s esophagus, and esophageal adenocarcinoma, as well as augment the antisecretory effect of PPIs.168,169 Conversely, rebound acid hypersecretion may occur in HP-eradicated patients when long-term (i.e., >8 weeks) use of PPIs is discontinued; this may unleash or exacerbate GERD, particularly in patients with large hiatal hernias.170,171 Acid hypersecretion persists at least eight weeks and appears to be due to hypergastrinemia-induced increases in parietal and ECL cell masses.172 The reason the phenomenon does not occur in HP-positive individuals who stop taking PPIs may be due to the fact that HP as well as the cytokines produced by the inflammatory infiltrate inhibit acid secretion and thus mask the rebound. With time, atrophy of oxyntic glands with loss of parietal cells may occur in patients chronically infected with HP, resulting in irreversible achlorhydria.
Autoimmune gastritis is an inflammatory disorder of the oxyntic mucosa often associated with antiparietal cell autoantibodies directed against H+,K+-ATPase with subsequent loss of parietal cells.173 H+,K+-ATPase is a major autoantigen in a subset of patients infected with HP and these antibodies may play a role in the subsequent development of atrophic gastritis. It is postulated that antibodies are acquired due to molecular mimicry between HP lipopolysaccharide and H+,K+-ATPase, both of which contain Lewis epitopes.174
About 10% to 15% of patients chronically infected with HP have antral-predominant inflammation. These patients, who are predisposed to duodenal ulcer (see Chapters 50, 51, and 52), produce increased amounts of acid as a result of reduced antral somatostatin content and elevated basal and stimulated gastrin secretion (see Fig. 49-16).175–177 Gastrin stimulates histamine secretion from fundic/body ECL cells and induces ECL hyperplasia.178 The mechanism by which somatostatin secretion is decreased is not known but may involve cytokines induced by the inflammation and/or the production of Na-methyl histamine, a selective H3-receptor agonist, by HP.179,180 One may speculate that the H3-receptor agonist could diffuse across the antral mucosa to interact with H3 receptors on antral somatostatin cells, causing inhibition of somatostatin secretion, and, thus, stimulation of gastrin secretion.45 In addition, IL-8 and platelet activating factor are upregulated in HP-infected mucosa and are capable of stimulating gastrin release from isolated G cells.181,182
MEASUREMENT OF GASTRIC ACID SECRETION
INDICATIONS FOR SECRETORY TESTING
Gastric secretory testing assesses the basal and maximal capacity of the stomach to produce acid. Clinically, its utility has diminished but it may assist in the diagnosis and management of patients with hypergastrinemia (e.g., gastrinoma) and in the diagnosis of incomplete vagotomy in patients with postoperative recurrent ulcer. Demonstrating fasting acid secretion or an acidic fasting gastric pH excludes achlorhydria as a cause of elevated fasting serum gastrin concentration. Patients with gastrinoma (Zollinger-Ellison syndrome; ZES) demonstrate hypergastrinemia with elevated basal acid output (see Chapter 32).
METHODS FOR MEASURING ACID SECRETION
Aspiration of gastric juice is the most widely used method for measuring acid secretion in humans. Traditionally, this is performed by positioning a nasogastric tube into the most dependent portion of the stomach of a fasted individual. Proper positioning may be verified fluoroscopically or by recovery of more than 90 mL after injection of 100 mL water. Gastric juice is collected by suction. When the tube is properly positioned, only 5% to 10% of gastric juice escapes collection and enters the duodenum. Neutralization by bicarbonate and diffusion of tiny amounts of acid back into the mucosa result in a small underestimation of the true rate of secretion. More recently, an endoscopic technique has been described to measure acid secretion in patients with gastrinoma. In this technique, all gastric contents are aspirated and discarded and then a single 15-minute sample of gastric juice is collected under direct endoscopic visualization.183
The H+ concentration in a sample of gastric juice can be determined by one of two methods. First, the specimen can be titrated in vitro with a base (e.g., NaOH). The millimoles (mmol) of base needed to titrate a volume of gastric juice to an arbitrary pH endpoint (e.g., 7) represents the “titratable” acidity in mmol per liter of the sample. The other method is to measure the pH of the sample with an electrode. Because pH electrodes measure H+ activity and not concentration, it is necessary to convert activity to concentration using a table of activity coefficients for H+ in gastric juice.184 Once the H+ concentration of the sample in mmol per liter is determined by either of these methods, it is multiplied by the volume of the sample in liters to determine the acid output during the collection period (e.g., mmol per hour or mmol per kilogram of body weight per hour).
BASAL ACID OUTPUT
Basal acid output (BAO) estimates resting acid secretion in the absence of intentional and avoidable stimulation. It is expressed as the sum of the measured acid output, expressed as mmol H+ per hour, for four consecutive 15-minute periods. The upper limit of normal for BAO is about 10 mmol H+ per hour in men and 5 mmol H+ per hour in women (Table 49-1).185 BAO fluctuates from hour to hour in the same person. The lowest BAO occurs between 6 and 11 am and the highest occurs between 2 and 11 pm. Variation is also related to cyclic gastric motor activity with increased BAO in late gastric phase III (migrating motor complex).186
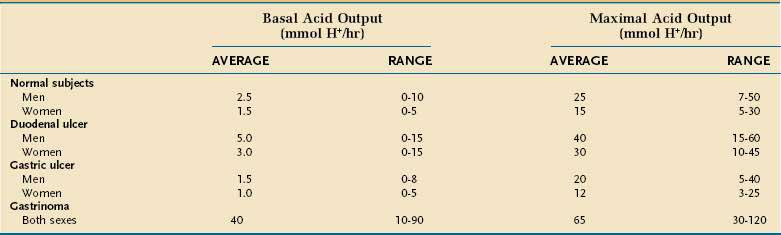
MAXIMAL ACID OUTPUT AND PEAK ACID OUTPUT
Maximal acid output (MAO) and peak acid output (PAO) estimate the acid secretory response to an exogenous secretagogue, usually pentagastrin (6 µg/kg subcutaneous or intramuscular or 6 µg/kg/hr continuous intravenous infusion). Pentagastrin is a manufactured analog of gastrin that contains its biologically active C-terminus sequence. Possible side effects include flushing, nausea, abdominal pain, dizziness, and palpitations. MAO is the sum of acid output of four consecutive 15-minute collection periods, and PAO is calculated by multiplying by two the sum of the two highest outputs recorded in the four test periods. The expected range for MAO is 5 to 50 mmol H+ per hour and for PAO is 10 to 60 mmol H+ per hour. MAO and PAO are higher in men and in smokers; they correlate with parietal cell mass (i.e., the total number of parietal cells). Typical results for MAO in healthy subjects and in disease are shown in Table 49-1.
SHAM FEEDING–STIMULATED ACID OUTPUT
The cephalic phase of acid secretion whereby the smell, sight, and thought of appetizing food, transmitted via the vagus nerve, stimulates acid secretion can be studied by sham feeding. Sham feeding, in which foods are chewed then spit out, increases acid secretion to about 50% of PAO. Thought and taste appear to play more important roles than sight and smell. Cholinergic and GRP neurons are involved because the response can be abolished by atropine or a selective GRP antagonist.187
MEAL-STIMULATED ACID OUTPUT
Continuous intragastric titration is primarily a research tool used to measure acid secretion in response to food in the stomach.188,189 It measures the cephalic and gastric phases of acid secretion. A double-lumen tube is placed in the most dependent part of the stomach and a homogenized meal buffered to pH 5.5 or 5 is infused into the stomach. Small volumes of gastric contents are sampled from one lumen, the pH is measured, and the contents are returned to the stomach. The second lumen is used to infuse sodium bicarbonate to maintain gastric pH at the meal pH. The amount of bicarbonate required to keep the pH of gastric contents constant is a measure of the postprandial acid secretory response. Rates of gastric acid secretion after eating increase rapidly and approach the PAO.
DISEASES ASSOCIATED WITH INCREASED GASTRIC ACID SECRETION
Duodenal ulcer patients, as a group, manifest increased basal and stimulated gastrin and acid production (see Table 49-1).190 It is recognized that most cases of duodenal ulcer are due to infection with HP (Chapter 52) and that this infection is responsible for the perturbations in acid secretion observed in these patients. Pentagastrin-stimulated PAO, an indicator of functional parietal cell mass, is increased in HP-infected duodenal ulcer patients as is GRP-stimulated peak acid output, an indicator of the stomach’s functional response to endogenous gastrin.177,191,192 Suppression of somatostatin secretion by the infection may be the root cause for these changes (see Fig. 49-10; Fig. 49-16). Eradication of HP restores somatostatin as well as basal and stimulated gastrin and acid secretion, over time, to normal in most individuals, thus providing a permanent cure for duodenal ulcer disease.176,177,191,193–195
In contrast to duodenal ulcer, gastric ulcer patients, as a group, exhibit normal or decreased basal and stimulated acid production (see Table 49-1), even though they too are often infected with HP. This suggests that altered gastric mucosal defense may be of primary pathophysiologic importance. Gastric ulcers have been classified according to their location and concomitant association with duodenal ulcer.196 Type I ulcers occur in the gastric body and are generally characterized by low acid secretion. These findings may reflect a greater degree and more generalized mucosal inflammation of the oxyntic mucosa with reduced functional parietal cell mass. Type II ulcers occur in the antrum and are characterized by low, normal, or high acid secretion. Type III ulcers occur within 3 cm of the pylorus, commonly accompany duodenal ulcer, and are characterized by high acid output. Type IV ulcers occur in the gastric cardia and are characterized by low acid secretion.197 Accordingly, the more distant a gastric ulcer is from the pylorus the more likely acid secretion will be low.
A number of uncommon conditions are marked by gastric acid hypersecretion and subsequent ulceration (see Chapter 52). In patients with systemic mastocytosis, high histamine levels, as a consequence of increased numbers of mast cells, continuously stimulate parietal cells to secrete acid.198 When a portion of gastric antrum is retained in the afferent remnant after antrectomy with Billroth II anastomosis, it is bathed in alkaline secretions leading to decreased somatostatin secretion, hypergastrinemia, increased acid production, and anastomotic ulceration.65,85,199 Acid hypersecretion also can result from chronic hypercalcemia of any cause because calcium directly stimulates gastrin secretion from G cells and acid secretion from parietal cells.200,201
The best characterized acid hypersecretory condition is ZES,202–204 as discussed in Chapter 32. The BAO is almost always higher than 15 mmol/hr and the BAO/PAO ratio is usually 0.6 or greater (see Table 49-1). Gastrin, synthesized by the tumor, is secreted into the bloodstream, where it binds to CCK-2 receptors on acid-producing parietal and histamine-containing ECL cells to induce secretion as well as proliferation. The clinical correlate of the proliferation is rugal hypertrophy with prominent gastric folds.
Diagnosis and treatment of gastrinoma are discussed in detail in Chapter 32. The basis of the secretin test to diagnose gastrinoma is that normally somatostatin cells in the antrum tonically restrain gastrin secretion from G cells. Secretin stimulates the G cell directly and, at the same time, inhibits the G cell indirectly by stimulating somatostatin secretion; the effect of the latter usually dominates and gastrin is not stimulated to a significant degree (Fig. 49-17). Because the gastrinoma does not contain functionally coupled somatostatin cells, the effect of secretin is solely stimulation of gastrin secretion from the tumor.205–207 Almost all gastrinomas contain somatostatin receptors and somatostatin receptor scintigraphy (SRS) using [111In-DPTA-Dphe1]-octreotide is considered the initial localization study of choice, with a 71% sensitivity and 86% specificity for primary tumors and 92% detection for metastatic disease.208,209 PPIs are the antisecretory therapies of choice and are able to control acid secretion and prevent complications in most patients with ZES.210
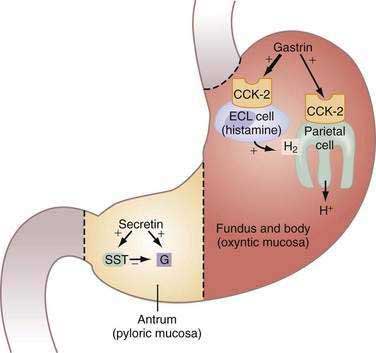
Figure 49-17. Zollinger-Ellison syndrome (ZES). Model illustrating the action of gastrin in oxyntic mucosa and secretin in pyloric mucosa of stomach in patients with ZES. Gastrin, synthesized and secreted by the gastrinoma into the bloodstream, acts via cholecystokinin-2 (CCK-2) receptors on acid-secreting parietal and histamine-secreting enterochromaffin-like (ECL) cells to increase acid secretion and induce cell proliferation. In the antrum, exogenous secretin (i.e., secretin stimulation test [see text and Chapter 32]) stimulates gastrin secretion directly and concomitantly inhibits gastrin secretion by stimulating somatostatin (SST) secretion resulting in little or no gastrin release. Because the gastrinoma does not contain functionally coupled SST cells, the effect of secretin in ZES patients is solely to stimulate gastrin secretion from the tumor.
(From Hung PD, Schubert ML, Mihas AA. Zollinger-Ellison syndrome. Curr Treat Options Gastroenterol 2003; 6:163-70.)
PEPSINOGEN SECRETION
Pepsinogens, which belong to a family of enzymes called gastric aspartic proteases, are inactive polypeptide proenzymes known as zymogens. They are synthesized primarily in chief cells but also in mucous neck cells. Pepsinogens are converted in the gastric lumen by gastric acid to pepsins, which contain two active-site aspartate residues. Once this reaction begins, pepsins can autocatalyze the conversion of pepsinogens to pepsins.211 Pepsins are optimally active at pH 1.8 to 3.5, reversibly inactivated at pH 5, and irreversibly denatured at pH 7. Gastric acid not only provides an optimum pH for peptic activity but itself denatures dietary protein, making it more susceptible to peptic hydrolysis. Thus, acid and pepsin work in concert to promote digestion of dietary protein. As discussed, partially digested protein stimulates gastrin and thus acid secretion.142 More recent data suggest that pepsins may also be important for killing ingested bacteria.17,212
Pepsinogens have been electrophoretically separated into seven isozymogens. The five fractions (pepsinogen 1 to pepsinogen 5) that migrate toward the anode most rapidly at pH 5 are similar immunologically and are referred to as group I pepsinogens (PGI; old term, pepsinogen A).213 PGI is expressed in chief and mucous cells of the oxyntic mucosa. Migrating slightly behind the PGIs are two immunologically similar isozymogens, pepsinogen 6 and pepsinogen 7, that are referred to as group II pepsinogens (PGII; old term, pepsinogen C). PGII, which represents approximately 20% of total pepsin content, is expressed in oxyntic and pyloric mucosa as well as in duodenal Brunner’s glands.
The most important physiologic stimulant for pepsinogen secretion is ACh released from intramural cholinergic neurons. ACh, acting via both M1 and M3 muscarinic receptors on chief cells, induces an increase in cytosolic calcium.214 Calcium, in turn, activates cytosolic kinases, phosphatases, and nitric oxide synthase that induce pepsinogen secretion.215 Other agents capable of stimulating pepsinogen secretion from chief cells via the calcium signaling pathway include CCK, gastrin, and GRP.216–219 Agents that increase cAMP within chief cells, such as isoproterenol, secretin, VIP, and histamine also augment pepsinogen secretion as do agents that activate tyrosine kinase such as epidermal growth factor and transforming growth factor-α.218 Inhibitors of pepsinogen secretion include somatostatin, neuropeptide Y, PYY, and IL-1β. Optimal pepsinogen secretion requires a functional sodium-2 chloride potassium cotransporter-1 (NKCC1), a basolateral cotransporter responsible for chloride uptake into secretory epithelia,118 including the chief cell (and the parietal cell; see Fig. 49-12). It has been postulated that loss of the flushing action of neutral gastric secretion may impair pepsinogen secretion.
Serum levels of PGI correlate with maximal acid output. A linear correlation exists between the loss of chief cells in patients with oxyntic atrophy and serum PGI220,221; a serum PGI/PGII ratio of 2.5 or less has been used as a noninvasive test to detect gastric mucosal atrophy.5,222 Serum PGI is also increased in humans treated with PPIs, but the precise mechanism is not known.223 Both PGI and PGII are filtered and metabolized by the kidney, but serum PGI concentration is increased more than PGII concentration in patients with renal insufficiency.224,225
GASTRIC LIPASE SECRETION
Gastric lipase, secreted by chief cells of the oxyntic mucosa, helps initiate the digestion of dietary triglycerides by hydrolyzing them to free fatty acids, diglycerides, and 2-monoglycerides. The properties of gastric lipase are quite distinct from those of pancreatic lipase. Gastric lipase has a pH optimum of 4.5 to 5.5 (compared with 6.5 to 7.4 for pancreatic lipase) and does not require colipase. Furthermore, protection from peptic proteolysis by an N-glycosylated asparagine at residue 308 permits gastric lipase to retains its full activity in acidic gastric juice (pH 2) despite a high gastric juice peptic activity.226,227
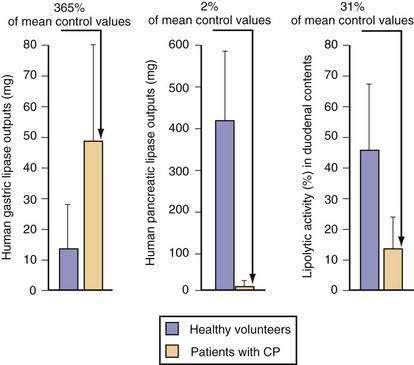
Stimulants and inhibitors of gastric lipase are similar to those for pepsinogen secretion. Aging has been reported to decrease gastric lipase secretion, but data are controversial.228 In humans, increasing the amount of lipid in the diet causes a corresponding increase in gastric lipase secretion into gastric juice.229 The amount of gastric lipase secreted after a meal is small relative to the amount of pancreatic lipase. However, the specific activity of gastric lipase is equal to or greater than that of pancreatic lipase. Thus, gastric lipase is capable of digesting 10% to 25% of dietary triglyceride.230 In patients with chronic pancreatitis, gastric lipase secretion is increased three- to four-fold and can partly, but incompletely, compensate for loss of pancreatic lipase (Fig. 49-18).231 A feedback mechanism exists whereby fat in the small intestine inhibits gastric lipase secretion by a humoral mechanism, with GLP-1 a prime candidate to be the mediator.232
Orlistat (tetrahydrolipstatin) is a lipophilic compound derived from lipstatin produced by Streptomyces toxitricini and is a potent inhibitor of all lipases.233 Orlistat reacts covalently with the catalytic serine residue of lipases.234 Orlistat decreases lipid digestion by about 35% and has been used clinically to induce modest weight loss.235 Adverse gastrointestinal events include loss of lipid-soluble vitamins, accelerated gastric emptying, fecal urgency, abdominal discomfort, flatus with discharge, and fecal urgency (see Chapter 6).236,237
INTRINSIC FACTOR SECRETION
Intrinsic factor (IF), a 50-kd glycoprotein secreted by human parietal cells and, to a lesser degree, chief cells, is necessary for the absorption of cobalamin (vitamin B12).238
Although all stimulants (e.g., gastrin, histamine, and acetylcholine) and inhibitors (e.g., somatostatin) of gastric acid secretion discussed previously have similar effects on IF secretion, the secretion of IF is not coupled to acid secretion. PPIs, for example, inhibit acid but have no significant effect on basal or stimulated IF secretion.239 Several reports indicate, however, that chronic use of PPIs may result in low serum levels of cobalamin, probably as a result of impaired acid-facilitated release of cobalamin from food.240,241 The recommended daily allowance of cobalamin is 2 µg/day and total body stores are 2.5 mg. Cobalamin deficiency due to antisecretory therapy is therefore rare because acid secretion is not completely suppressed and because the body contains relatively large stores.10,242,243
The delivery of cobalamin from food to tissues begins with release of cobalamin from dietary protein by the pH-dependent activity of pepsin, followed by binding of cobalamin to two binding proteins that are secreted into gastric juice: IF and haptocorrin (R binder).244,245 Haptocorrin is also secreted in saliva and bile. Haptocorrin binds cobalamin more avidly than IF in the acidic stomach and, therefore, most cobalamin initially becomes attached to haptocorrin. In the duodenum, cobalamin is released from haptocorrin by pancreatic trypsin and the free cobalamin then binds to IF. IF-cobalamin complexes are resistant to pancreatic proteolysis and eventually bind to a specific receptor on the distal ileal mucosa. This receptor, cubilin, is expressed in clefts between microvilli and mediates endocytosis of the IF-cobalamin complex.246 Once within the ileal enterocyte, IF is degraded by lysosomal enzymes and cobalamin binds to transcobalamin. The cobalamin-transcobalamin complex is released into the circulation and from there enters cells by receptor-mediated endocytosis. Once within cells, cobalamin is dissociated from its transport protein and converted to its active forms, methylcobalamin and 5-deoxyadenosyl cobalamin. The active forms serve as coenzymes for methionine synthase and methylmalonyl-coenzyme A mutase, enzymes involved in methylation of homocysteine to methionine and the catabolism of branched-chain amino acids and odd-chain fatty acids in mitochondria, respectively.247
When radiolabeled cobalamin is administered orally after a large dose of nonradioactive cobalamin is given parenterally, patients with IF deficiency excrete much lower amounts of radioactive cobalamin in a 24-hour urine collection than do normal controls (Schilling test, part I). If IF is administered orally together with radioactive cobalamin to IF-deficient patients, urinary radioactive cobalamin excretion normalizes (Schilling test, part II). In addition to IF deficiency, cobalamin malabsorption may result from achlorhydria or hypochlorhydria (reduced peptic hydrolysis of cobalamin from food protein), bacterial overgrowth (cobalamin competed for by bacteria), pancreatic insufficiency (impaired tryptic cleavage of haptocorrin-cobalamin complex), ileal receptor defect (cubilin mutation), and ileitis or ileal resection (absent IF-cobalamin absorptive site).248–250
Secretion of IF far exceeds the amount necessary for cobalamin absorption. Thus, in most patients with hypochlorhydria, continued IF secretion in low amounts is sufficient to prevent cobalamin deficiency. Patients with pernicious anemia, however, which affects 2% of individuals older than the age of 60, do develop cobalamin deficiency.251 The pathology involves a chronic inflammatory, mainly lymphocytic, infiltrate of the oxyntic mucosa accompanied by loss of parietal and chief cells. The pathogenesis involves proinflammatory TH1 CD4 T cells directed toward the α- and β-subunits of the parietal cell H+,K+-ATPase, as well as circulating antibodies directed against H+,K+-ATPase or IF.252 Chronic infection with HP may play a primary role in the immunologic response. It is proposed that HP-induced inflammation results in breakdown of tolerance for self antigens such as H+,K+-ATPase in genetically susceptible individuals or that antibodies are acquired due to molecular mimicry between HP lipopolysaccharide and H+,K+-ATPase, both of which contain Lewis epitopes.173,174 Antibodies directed against H+,K+-ATPase are found in 90% of patients with pernicious anemia but the incidence of these antibodies may decrease to about 55% to 80% with progression of autoimmune gastritis presumably because of disappearance of HP and the loss of antigenic drive.252–254
BICARBONATE SECRETION
Regulation of gastric bicarbonate (HCO3−) secretion has been studied much less intensively than has duodenal HCO3− secretion. The precise function of gastric HCO3− secretion is uncertain given the overwhelming simultaneous secretion of H+. Nevertheless, HCO3− secretion has been implicated in the formation of a protective pre-epithelial alkaline layer (see Fig. 49-2).
Measurement of gastric HCO3− secretion has been impeded by the presence of considerable H+ secretion, necessitating the use of potent antisecretory compounds or measurement methods unaffected by the presence of acid. One means of measuring bulk HCO3− secretion is with the use of inline pH and CO2 electrodes, in which HCO3− concentration is calculated using the Henderson-Hasselbach equation.255,256 An electrophysiologic method has been used to measure alkaline secretion in the lumen of individual gastric glands isolated from the frog Rana esculenta.257
Gastric HCO3− secretion is an energy-dependent process. The finding that there is virtually no change in gastric electrical potential difference during HCO3− secretion suggests that HCO3− transport takes place via an electroneutral ion exchange mechanism, probably an exchange of HCO3− for Cl− at the luminal surface.258 Although several candidate anion exchangers, solute carriers, and anion transporters have been localized to gastric surface cells including AE2, AE4, Slc4a2, Slc4a4, PAT1, and Slc26a6, there is little evidence that gastric surface cells actually secrete HCO3−.259–263 The cell responsible for HCO3− secretion is not known. The source of some of the HCO3− secreted during H+ secretion may actually be the parietal cell. As discussed previously, for each H+ secreted, a HCO3− ion exits the parietal cell across the basolateral membrane via the anion exchanger 2 (AE2), Slc4a2 (see Fig. 49-12).115 This HCO3− may alkalinize the blood that perfuses surface epithelial cells, be taken up by sodium bicarbonate cotransporters (NBC1 and NBC2), and then be secreted by epithelial cells in an effort to protect them from luminal acid.264 The parietal cell, however, may not be the only source of HCO3− for surface cells, as marked inhibition of gastric H+ secretion by PPIs does not significantly diminish gastric HCO3− secretion in patients with duodenal ulcer.265
Prostaglandin E2 analogs stimulate gastric HCO3− and mucus secretion266,267 and blockade of endogenous prostaglandin synthesis reduces gastric HCO3− secretion.268 In rats and mice, the prostaglandin E receptor subtype involved in stimulation of gastric HCO3− secretion is EP1.269,270 Gastric mucosal prostaglandin synthesis and HCO3− secretion decline in older adults.271,272
MUCUS SECRETION
A firmly adherent viscous mucus gel overlies the gastric surface. It is composed of 95% water and 5% extensively cross-linked mucin glycoproteins that are products of MUC genes.273,274 Ultrastructural studies reveal alternating layers of two distinct mucin classes, MUC5AC (secreted by surface and pit area epithelial cells) and MUC6 (secreted from neck and gland cells).275–278 Observations that the gastroprotective compound, geranylgeranylacetone (GGA), increases MUC6 expression in rat gastric mucosa279 and that lafutidine, a gastroprotective compound used clinically in Japan, increases mucus thickness and mucin content in humans280 suggest that the mucus gel may contribute to mucosal defense.273,281,282 Furthermore, electron microscopy studies demonstrate that HP accumulates within and disrupts the MUC5AC-enriched gel layer.277,283,284 MUC5AC secretion is increased in first-degree relatives of subjects with gastric cancer infected with HP, suggesting that mucus secretion may be a marker for a more severe inflammatory response to the organism.285
Mucus gel thickness can be measured continuously and noninvasively in living rodents by alternately focusing between fluorescently labeled surface epithelial cells and the mucus gel surface, as delineated by carbon particles or by fluorescent microspheres.286–288
A pH gradient at the gastric mucosal surface has been observed in a variety of species including humans.289–292 In most cases, the gradient is relatively alkaline at the tissue surface and gradually more acidic at distances further from the surface. The gradient is due to active bicarbonate secretion and is considered a defense against luminal acid.290,293 Although most measurements using microelectrodes have reported values near neutrality at the gastric mucosal surface, more recent measurements using pH-sensitive fluorescent dyes with ex vivo confocal microscopy report a surface pH near pH 4 in guinea pigs and frogs.294,295 The observation that the steady-state surface pH gradient extends beyond the thickness of the mucus gel layer suggests that the unstirred layer formed at the interface between the mucus and the aqueous lumen or the interface between the epithelial surface and the luminal contents may also play a role in mucosal defense by restricting mixing of molecules within the unstirred layer.283,293,296
Although it has been suggested that gastric mucus may physically retard diffusion of protons, the evidence for this is equivocal. In vitro measurements of proton and bicarbonate diffusion have yielded diffusion coefficients that are up to 10 times slower in isolated mucus compared with saline solution (≈0.5 − 2 × 10−5 cm2/sec).287,297 However, these diffusion coefficients in mucus are comparable to those found in saline for ions such as Na+, K+, and Cl−. Even accepting the slowest reported diffusion of protons, one-third of protons will theoretically diffuse 80 µm or greater within 10 seconds, a distance greater than the average mucus thickness in rats and mice.281,290,296 There are also concerns regarding diffusion measurements performed in vitro as nondestructive removal of the tightly adherent mucus is extremely difficult, mucus structure and function is sensitive to environmental conditions, and measurement instruments may produce artifactual unstirred layers.298,299
Trefoil factor (TFF) peptides are cosecreted with mucins.300 These are 7- to 12-kd peptides sharing a common structure of three internal disulfide bonds that yield the signature “trefoil” structure of three internal loops and are designated TFF1, TFF2, and TFF3. TFFs are markedly pepsin resistant and able to survive intact in the gastric lumen301; the concentration of TFF2 in rat gastric mucus has been estimated at 10 µm.302,303 The distribution of each trefoil peptide in the normal GI tract is distinct. TFF1 is stored in gastric pit and surface mucous cells, TFF2 is present in gastric gland mucous cells, and TFF3 is stored in intestinal goblet cells.303–307 The relative abundance of TFF1 and TFF2 expression is reciprocally regulated by gastrin, H+,K+-ATPase, and inflammation.308
The localization and coordinated secretion of trefoil peptides with mucins suggest that they too may be involved in mucosal defense.305,309,310 In support of this notion (1) increased TFF1 expression is observed after administration of gastroprotective agents311; (2) addition of TFF2 to mucin solutions significantly increases viscosity and elasticity in vitro and in vivo312–313; (3) chronic treatment with PPIs increases TFF2 concentration in gastric secretions coincident with promoting repair and preventing injury in response to luminal noxious agents314; and (4) TFF2 -/- knockout mice exhibit shortened gastric glands with decreased epithelial migration, increased net acid secretion, and a four-fold increase in number of lesions after a 12 hour-exposure to the nonselective cyclooxygenase (COX) inhibitor indomethacin.315
Carrière F, Grandval P, Renou C, et al. Quantitative study of digestive enzyme secretion and gastrointestinal lipolysis in chronic pancreatitis. Clin Gastroenterol Hepatol. 2005;3:28-38. (Ref 231.)
Flemström G, Isenberg JI. Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol Sci. 2001;16:23-8. (Ref 258.)
Fossmark R, Johnsen G, Johanessen E, Waldum HL. Rebound hypersecretion after long-term inhibition of gastric acid secretion. Aliment Pharmacol Therapeut. 2005;21:149-54. (Ref 172.)
Gillen D, Wirz AA, Neithercut WD, et al. Helicobacter pylori infection potentiates the inhibition of gastric acid secretion by omeprazole. Gut. 1999;44:468-75. (Ref 169.)
Hatlebakk JG, Katz PO, Camacho-Lobato L, Castell DO. Proton pump inhibitors: Better acid suppression when taken before a meal than without a meal. Aliment Pharmacol Therapeut. 2000;14:1267-72. (Ref 122.)
Heitzmann D, Warth R. No potassium, no acid: K+ channels and gastric acid secretion. Physiology. 2007;22:335-41. (Ref 108.)
Jain RN, Samuelson LC. Differentiation of the gastric mucosa: Role of gastrin in gastric epithelial cell proliferation and maturation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G762-5. (Ref 68.)
Prinz C, Zanner R, Gratzl M. Physiology of gastric enterochromaffin-like cells. Ann Rev Physiol. 2003;65:371-82. (Ref 46.)
Manela FD, Ren J, Gao J, et al. Calcitonin gene-related peptide modulates acid-mediated regulation of somatostatin and gastrin release from rat antrum. Gastroenterology. 1995;109:701-6. (Ref 65.)
Moss SF, Legon S, Bishop AE, et al. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet. 1992;340:930-2. (Ref 176.)
Schubert ML, Edwards NF, Makhlouf GM. Regulation of gastric somatostatin secretion in the mouse by luminal acid: A local feedback mechanism. Gastroenterology. 1988;94:317-22. (Ref 85.)
Wang TC, Dockray GJ. Lessons from genetically engineered animal models I. Physiological studies with gastrin in transgenic mice. Am J Physiol Gastrointest Liver Physiol. 1999;277:G6-11. (Ref 60.)
Whited KL, Thao D, Lloyd KCK, et al. Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. Am J Physiol Gastrointest Liver Physiol. 2006;291:G156-62. (Ref 104.)
1. Kelly EJ, Newell SJ, Brownlee KG, et al. Gastric acid secretion in preterm infants. Early Human Dev. 1993;35:215-20.
2. Agunod M, Yamaguchi N, Lopez R, et al. Correlative study of hydrochloric acid, pepsin, and intrinsic factor secretion in newborns and infants. Am J Dig Dis. 1969;14:400-14.
3. Hurwitz A, Brady DA, Schaal SE, et al. Gastric acidity in older adults. JAMA. 1997;278:659-62.
4. Trey G, Marks IN, Louw JA, et al. Changes in acid secretion over the years—A 30-year longitudinal study. J Clin Gastroenterol. 1997;25:499-502.
5. Nakamura K, Ogoshi K, Makuuchi H. Influence of aging, gastric mucosal atrophy, and dietary habits on gastric secretion. Hepatogastroenterology. 2006;53:624-8.
6. Hutchinson C, Geissler CA, Powell JJ, Bomford A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut. 2007;56:1291-5.
7. Sharma VR, Brannon MA, Carlson HE I. Effect of omeprazole on oral iron replacement in patients with iron deficiency anemia. South Med J. 2004;97:887-9.
8. O’Connell MB, Madden DM, Murray AM, et al. Effects of proton pump inhibitors on calcium carbonate absorption in women: A randomized crossover trial. Am J Med. 2005;118:778-81.
9. Yang YX, Lewis JD, Epstein S, Metz DC. Long-term proton pump inhibitor therapy and risk of hip fracture. JAMA. 2007;297:470.
10. Den Elzen WPJ, Groeneveld Y, De Ruijter W, et al. Long-term use of proton pump inhibitors and vitamin B12 status in elderly individuals. Aliment Pharmacol Therapeut. 2008;27:491-7.
11. Williams C, McColl KE. Review article: Proton pump inhibitors and bacterial overgrowth. Aliment Pharmacol Therapeut. 2006;23:3-10.
12. Leonard J, Marshall JK, Moayyedi P. Systemic review of the risk of enteric infection in patients taking acid suppression. Am J Gastroenterol. 2007;102:2047-56.
13. Dial S, Delaney JA, Barkun AN, Suissa S. Use of gastric acid-suppressive agents and the risk of community-acquired Clostridium difficile–associated disease. JAMA. 2005;294:2989-95.
14. Gulmez SE, Holm A, Frederiksen H, et al. Use of proton pump inhibitors and the risk of community-acquired pneumonia—A population-based case-control study. Arch Intern Med. 2007;167:950-5.
15. Checchi S, Montanaro A, Pasqui L, et al. L-thyroxine requirement in patients with autoimmune hypothyroidism and parietal cell antibodies. J Clin Endocrinol Metab. 2008;93:465-9.
16. Centanni M, Gargano L, Canettieri G, et al. Thyroxin in goiter, Helicobacter pylori infection, and chronic gastritis. N Engl J Med. 2006;354:1787-95.
17. Zhu H, Hart CA, Sales D, Roberts NB. Bacterial killing in gastric juice—Effect of pH and pepsin on Escherichia coli and Helicobacter pylori. J Med Microbiol. 2006;55:1265-70.
18. Sarkar M, Hennessy S, Yang Y-X. Proton-pump inhibitor use and the risk for community-acquired pneumonia. Ann Intern Med. 2008;149:391-8.
19. Dimaline R, Varro A. Attack and defence in the gastric epithelium—A delicate balance. Exp Physiol. 2007;92:591-601.
20. Joseph IMP, Zavros Y, Merchant JL, Kirschner D. A model for integrative study of human gastric acid secretion. J Appl Physiol. 2003;94:1602-18.
21. Chandrasoma P. Controversies of the cardiac mucosa and Barrett’s oesophagus. Histopathology. 2005;46:361-73.
22. Bado A, Levasseur S, Attoub S, et al. The stomach is a source of leptin. Nature. 1998;394:790-3.
23. Mix H, Widjaja A, Jandl O, et al. Expression of leptin and leptin receptor isoforms in the human stomach. Gut. 2000;47:481-6.
24. Hirsch AB, McCuen RW, Arimura A, Schubert ML. Adrenomedulin stimulates somatostatin and thus inhibits histamine and acid secretion in the fundus of the stomach. Regul Pept. 2003;110:189-95.
25. Zaki M, Koduru S, McCuen RW, et al. Amylin, released from the gastric fundus, stimulates somatostatin and thus inhibits histamine and acid secretion in mice. Gastroenterology. 2002;123:247-55.
26. Gower WR, Premaratne S, McCuen RW, et al. Gastric atrial natriuretic peptide regulated endocrine secretion in antrum and fundus of human and rat stomach. Am J Physiol Gastrointest Liver Physiol. 2003;284:G638-45.
27. Asakawa A, Inui A, Kaga T, et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology. 2001;120:337-45.
28. Larsson L-I, Goltermann N, DeMagistris L, et al. Somatostatin cell processes as pathways for paracrine secretion. Science. 1979;205:1393-5.
29. Zhao C-M, Furnes MW, Stenstrom B, et al. Characterization of obestatin- and ghrelin-producing cells in the gastrointestinal tract and pancreas of rats: An immunohistochemical and electron-microscopic study. Cell Tissue Res. 2008;331:575-87.
30. Schubert ML, Edwards NF, Arimura A, Makhlouf GM. Paracrine regulation of gastric acid secretion by fundic somatostatin. Am J Physiol Gastrointest Liver Physiol. 1987;252:G485-90.
31. Schubert ML, Hightower J, Makhlouf GM. Linkage between somatostatin and acid secretion: Evidence from use of pertussis toxin. Am J Physiol Gastrointest Liver Physiol. 1989;256:G418-22.
32. Makhlouf GM, Schubert ML. Gastric somatostatin: A paracrine regulator of acid secretion. Metabolism. 1990;39:138-42.
33. Schubert ML. Gastric somatostatin: A paracrine regulator of gastrin and acid secretion. Reg Pep Letter. 1991;3:7-11.
34. Saffouri B, Weir GC, Bitar KN, Makhlouf GM. Stimulation of gastrin secretion from the perfused rat stomach by somatostatin antiserum. Life Sci. 1979;25:1749-54.
35. Yu PL, Fujimura M, Hayashi N, et al. Mechanisms in regulating the release of serotonin from the perfused rat stomach. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1099-1105.
36. De la Cour CD, Björkqvist M, Sandvik AK, et al. A-like cells in the rat stomach contain ghrelin and do not operate under gastrin control. Regulatory Peptides. 2001;99:141-50.
37. Furness JB, Costa M. Types of nerves in the enteric nervous system. Neuroscience. 1980;5:1-20.
38. Smith VC, Dhatt N, Buchan AMJ. The innervation of the human antro-pyloric region: Organization and composition. Can J Physiol Pharmacol. 2001;79:905-18.
39. Green T, Dockray GJ. Characterization of the peptidergic afferent innervation of the stomach in the rat. Neuroscience. 1988;25:181-93.
40. Johansson M, Synnerstad I, Holm L. Acid transport through channels in the mucous layer of rat stomach. Gastroenterology. 2001;119:1297-304.
41. Schubert ML, Shamburek R. Control of acid secretion. Gastroenterol Clin North Am. 1990;19:1-25.
42. Soll AH. Potentiating interactions of gastric stimulants on (14C)-aminopyrine accumulation by isolated canine parietal cells. Gastroenterology. 1982;83:216-23.
43. Soll AH, Wollin A. Histamine and cyclic AMP in isolated canine parietal cells. Am J Physiol Endocrinol Metab. 1979;237:E444-50.
44. Vuyyuru L, Harrington L, Arimura A, Schubert ML. Reciprocal inhibitory paracrine pathways link histamine and somatostatin secretion in the fundus of the stomach. Am J Physiol Gastrointest Liver Physiol. 1997;273:G106-11.
45. Vuyyuru L, Schubert ML, Harrington L, et al. Dual inhibitory pathways link antral somatostatin and histamine secretion in human, dog, and rat stomach. Gastroenterology. 1995;109:1566-74.
46. Prinz C, Zanner R, Gratzl M. Physiology of gastric enterochromaffin-like cells. Annu Rev Physiol. 2003;65:371-82.
47. Lindström E, Håkanson R. Prostaglandins inhibit secretion of histamine and pancreastatin from isolated rat stomach ECL cells. Br J Pharmacol. 1998;124:1307-13.
48. Sachs G, Zeng NX, Prinz C. Physiology of isolated gastric endocrine cells. Annu Rev Physiol. 1997;59:243-56.
49. Sandvik AK, Mårvik R, Dimaline R, Waldum HL. Carbachol stimulation of gastric acid secretion and its effects on the parietal cell. Br J Pharmacol. 1998;124:69-74.
50. Zeng NX, Walsh JH, Kang T, et al. Selective ligand-induced intracellular calcium changes in a population of rat isolated gastric endocrine cells. Gastroenterology. 1996;110:1835-46.
51. Hansen CP, Stadil F, Yucun L, Rehfeld JF. Pharmacokinetics and organ metabolism of carboxyamidated and glycine-extended gastrins in pigs. Am J Physiol Gastrointest Liver Physiol. 1996;271:G156-63.
52. Hansen CP, Stadil F, Rehfeld JF. Hepatic and intestinal elimination of endogenous gastrin in pigs. Digestion. 1996;57:356-61.
53. Hansen CP, Stadil F, Rehfeld JF. Renal tubular transport and metabolism of carboxyamidated and glycine-extended gastrins in pigs. Acta Physiol Scand. 1998;164:29-38.
54. Ciccotosto GD, Dawborn JK, Hardy KJ, Shulkes A. Gastrin processing and secretion in patients with end-stage renal failure. J Clin Endocrinol Metab. 1996;81:3231-8.
55. Schmitz F, Göke MN, Otte JM, et al. Cellular expression of CCK-A and CCK-B/gastrin receptors in human gastric mucosa. Regulatory Peptides. 2001;102:101-10.
56. Tari A, Kamiyasu T, Yonei Y, et al. Role of gastrin/CCK-B receptor in the regulation of gastric acid secretion in rat. Dig Dis Sci. 1997;42:1901-7.
57. Zanner R, Hapfelmeier G, Gratzl M, Prinz C. Intracellular signal transduction during gastrin-induced histamine secretion in rat gastric ECL cells. Am J Physiol Cell Physiol. 2002;282:C374-82.
58. Kobayashi T, Tonai S, Ishihara Y, et al. Abnormal functional and morphological regulation of the gastric mucosa in histamine H2 receptor-deficient mice. J Clin Invest. 2000;105:1741-9.
59. Tanaka S, Hamada K, Yamada N, et al. Gastric acid secretion in L-histidine decarboxylase-deficient mice. Gastroenterology. 2002;122:145-55.
60. Wang TC, Dockray GJ. Lessons from genetically engineered animal models I. Physiological studies with gastrin in transgenic mice. Am J Physiol Gastrointest Liver Physiol. 1999;277:G6-11.
61. Cui GL, Sandvik AK, Munkvold B, Waldum HL. Glycine-extended gastrin-17 stimulates acid secretion only via CCK-2 receptor-induced histamine release in the totally isolated vascularly perfused rat stomach. Acta Physiol Scand. 2002;174:125-30.
62. Paterson AC, Lockhart SM, Baker J, et al. Identity and regulation of stored and secreted progastrin-derived peptides in sheep. Endocrinology. 2004;145:5129-40.
63. Brand S, Stone D. Reciprocal regulation of antral gastrin and somatostatin gene expression by omeprazole-induced achlorhydria. J Clin Invest. 1988;82:1059-66.
64. Wu S, Giraud A, Mogard M, et al. Effects of inhibition of gastric secretion on antral gastrin and somatostatin gene expression in rat. Am J Physiol Gastrointest Liver Physiol. 1990;258:G788-93.
65. Manela FD, Ren J, Gao J, et al. Calcitonin gene-related peptide modulates acid-mediated regulation of somatostatin and gastrin release from rat antrum. Gastroenterology. 1995;109:701-6.
66. Zavros Y, Rieder G, Ferguson A, et al. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology. 2002;122:119-33.
67. Schubert ML, Jong MJ, Makhlouf GM. Bombesin/GRP-stimulated somatostatin secretion is mediated by gastrin in the antrum and intrinsic neurons in the fundus. Am J Physiol Gastrointest Liver Physiol. 1991;261:G885-9.
68. Jain RN, Samuelson LC. Differentiation of the gastric mucosa: role of gastrin in gastric epithelial cell proliferation and maturation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G762-5.
69. Tommerås K, Hammer P, Sundler F, et al. Immunolocalization of cholecystokinin-2 receptors in rat gastric mucosa. Scand J Gastroenterol. 2002;37:1017-24.
70. Bakke I, Qvigstad G, Brenna E, et al. Gastrin has a specific proliferative effect on the rat enterochromaffin-like cell, but not on the parietal cell: A study by elutriation centrifugation. Acta Physiol Scand. 2000;169:29-37.
71. Hou W, Schubert ML. Treatment of gastric carcinoids. Curr Treat Options Gastroenterol. 2007;10:123-33.
72. Peghini PL, Annibale B, Azzoni C, et al. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: Insights from patients with sporadic gastrinomas. Gastroenterology. 2002;123:68-85.
73. Modlin IM, Kidd M, Latich I, et al. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128:1717-51.
74. Gibril F, Reynolds JC, Lubensky IA, et al. Ability of somatostatin receptor scintigraphy to identify patients with gastric carcinoids: a prospective study. J Nucl Med. 2000;41:1646-56.
75. Leonard A, Cuq P, Magous R, Bali J-P. M3-subtype muscarinic receptor that controls intracellular calcium release and inositol phosphate accumulation in gastric parietal cells. Biochem Pharmacol. 1991;42:839-45.
76. Wilkes JM, Kajimura M, Scott DR, et al. Muscarinic responses of gastric parietal cells. J Membr Biol. 1991;122:97-110.
77. Kajimura M, Reuben MA, Sachs G. The muscarinic receptor gene expressed in rabbit parietal cells is the m3 subtype. Gastroenterology. 1992;103:870-5.
78. Yamaji N, Yokoo Y, Iwashita T, et al. Structural determination of two active compounds that bind to the muscarinic M-3 receptor in beer. Alcohol Clin Exp Res. 2007;31:9-14.
79. Schubert ML, Saffouri B, Walsh JH, Makhlouf GM. Inhibition of neurally mediated gastrin secretion by bombesin antiserum. Am J Physiol Gastrointest Liver Physiol. 1985;248:G546-62.
80. Nylander O, Bergqvist E, Obrink K. Dual inhibitory actions of somatostatin on isolated gastric glands. Acta Physiol Scand. 1985;125:111-19.
81. Athmann C, Zeng NX, Scott DR, Sachs G. Regulation of parietal cell calcium signaling in gastric glands. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1048-58.
82. Allen JP, Canty AJ, Schulz S, et al. Identification of cells expressing somatostatin receptor 2 in the gastrointestinal tract of Sstr2 knockout/lacZ knockin mice. J Comp Neurol. 2002;454:329-40.
83. Martinez V, Curi AP, Torkian B, et al. High basal gastric acid secretion in somatostatin receptor subtype 2 knockout mice. Gastroenterology. 1998;114:1125-32.
84. Zaki M, Harrington L, McCuen R, et al. Somatostatin receptor subtype 2 mediates inhibition of gastrin and histamine secretion from human, dog, and rat antrum. Gastroenterology. 1996;111:919-24.
85. Schubert ML, Edwards NF, Makhlouf GM. Regulation of gastric somatostatin secretion in the mouse by luminal acid: A local feedback mechanism. Gastroenterology. 1988;94:317-22.
86. Kojima M. The discovery of ghrelin—A personal memory. Regul Peptides. 2008;145:2-6.
87. Zhao CM, Furnes MW, Stenstrom B, et al. Characterization of obestatin- and ghrelin-producing cells in the gastrointestinal tract and pancreas of rats: An immunohistochemical and electron-microscopic study. Cell Tissue Res. 2008;331:575-87.
88. Sonmez MF, Ozan E. Determination of ghrelin immunoreactivity in the rat stomach after fasting and refeeding. Acta Histochem. 2007;109:193-9.
89. Lin E, Gletsu N, Fugate K, et al. The effects of gastric surgery on systemic ghrelin levels in the morbidly obese. Arch Surg. 2004;139:780-4.
90. Brzozowski T, Konturek PC, Konturek SJ, et al. Exogenous and endogenous ghrelin in gastroprotection against stress-induced gastric damage. Regul Pept. 2004;120:39-51.
91. Levin F, Edholm T, Ehrstrom M, et al. Effect of peripherally administered ghrelin on gastric emptying and acid secretion in the rat. Regul. Peptides. 2005;131:59-65.
92. Mori M, Suzuki H, Masaoka T, et al. Intravenous ghrelin administration enhances gastric acid secretion—Evaluation using wireless pH capsule. Aliment Pharmacol Ther. 2006;24:96-103.
93. Yakabi K, Ro S, Onouhi T, et al. Histamine mediates the stimulatory action of ghrelin on acid secretion in rat stomach. Dig Dis Sci. 2006;51:1313-21.
94. Ehrstrom M, Gustafsson T, Finn A, et al. Inhibitory effect of exogenous orexin A on gastric emptying, plasma leptin, and the distribution of orexin and orexin receptors in the gut and pancreas in man. J Clin Endocrinol Metab. 2005;90:2370-7.
95. Heinonen MV, Purhonen AK, Makela KA, Herzig KH. Functions of orexins in peripheral tissues. Acta Physiol. 2008;192:471-85.
96. Rossowski WJ, Cheng BL, Jiang NY, Coy DH. Examination of somatostatin involvement in the inhibitory action of GIP, GLP-1, amylin and adrenomedullin on gastric acid release using a new SRIF antagonist analogue. Br J Pharmacol. 1998;125:1081-7.
97. Lloyd KCK, Amirmoazzami S, Friedik F, et al. Candidate canine enterogastrones: acid inhibition before and after vagotomy. Am J Physiol Gastrointest Liver Physiol. 1997;272:G1236-42.
98. Wettergren A, Wojdemann M, Meisner S, et al. The inhibitory effect of glucagon-like peptide-1 (GLP-1) 7-36 amide on gastric acid secretion in humans depends on an intact vagal innervation. Gut. 1997;40:597-601.
99. Fung LC, Chisholm C, Greenberg GR. Glucagon-like peptide-1-(7-36) amide and peptide YY mediate intraduodenal fat-induced inhibition of acid secretion in dogs. Endocrinology. 1998;139:189-94.
100. Chey WY, Kim MS, Lee KY, Chang TM. Secretin is an enterogastrone in the dog. Am J Physiol Gastrointest Liver Physiol. 1981;240:G239-44.
101. Seal A, Liu E, Buchan A, Brown J. Immunoneutralization of somatostatin and neurotensin: Effect on gastric acid secretion. Am J Physiol Gastrointest Liver Physiol. 1988;255:G40-5.
102. Fung L, Pokol-Daniel S, Greenberg GR. Cholecystokinin type A receptors mediate intestinal fat-induced inhibition of acid secretion through somatostatin-14 in dogs. Endocrinology. 1994;134:2376-82.
103. Lloyd KCK, Wang JF, Solomon TE. Acid inhibition by intestinal nutrients mediated by CCK-A receptors but not plasma CCK. Am J Physiol Gastrointest Liver Physiol. 2001;281:G924-30.
104. Whited KL, Thao D, Lloyd KCK, et al. Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. Am J Physiol Gastrointest Liver Physiol. 2006;291:G156-62.
105. Zhao XT, Walsh JH, Wong H, et al. Intestinal fat-induced inhibition of meal-stimulated gastric acid secretion depends on CCK but not peptide YY. Am J Physiol Gastrointest Liver Physiol. 1999;276:G550-5.
106. Spicer Z, Miller ML, Andringa A, et al. Stomachs of mice lacking the gastric H,K-ATPase α-subunit have achlorhydria, abnormal parietal cells, and ciliated metaplasia. J Biol Chem. 2000;275:21555-65.
107. Asano S, Kawada H, Kimura T, et al. The roles of carbohydrate chains of the β-subunit on the functional expression of gastric H+,K+-ATPase. J Biol Chem. 2000;275:8324-30.
108. Heitzmann D, Warth R. No potassium, no acid: K+ channels and gastric acid secretion. Physiology. 2007;22:335-41.
109. Forte JG, Ly B, Rong QF, et al. State of actin in gastric parietal cells. Am J Physiol Cell Physiol. 1998;274:C97-104.
110. Karvar S, Yao X, Duman JG, et al. Intracellular distribution and functional importance of vesicle-associated membrane protein 2 in gastric parietal cells. Gastroenterology. 2002;123:281-90.
111. Okamoto CT, Duman JG, Tyagarajan K, et al. Clathrin in gastric acid secretory (parietal) cells: Biochemical characterization and subcellular localization. Am J Physiol Cell Physiol. 2000;279:C833-51.
112. Roepke TK, Anantharam A, Kirchhoff P, et al. The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J Biol Chem. 2006;281:23740-7.
113. Kaufhold M-A, Krabbenhoft A, Song P, et al. Localization, trafficking, and significance for acid secretion of parietal cell Kir4.1 and KCNQ1 K+ channels. Gastroenterology. 2008;134:1058-69.
114. Sidani SM, Kirchhoff P, Socrates T, et al. Delta F508 mutation results in impaired gastric acid secretion. J Biol Chem. 2007;282:6068-74.
115. Rossmann H, Bachmann O, Wang Z, et al. Differential expression and regulation of AE2 anion exchanger subtypes in rabbit parietal and mucous cells. J Physiol (Lond). 2001;534:837-48.
116. Schreiber S, Nguyen TH, Stuben M, Scheid P. Demonstration of a pH gradient in the gastric gland of the acid-secreting guinea pig mucosa. Am J Physiol Gastrointest Liver Physiol. 2000;279:G597-604.
117. Kosiek O, Busque SM, Foller M, et al. SLC26A7 Can function as a chloride-loading mechanism in parietal cells. Pflugers Arch Eur J Physiol. 2007;454:989-98.
118. McDaniel N, Pace AJ, Spiegel S, et al. Role of Na-K-2Cl cotransporter-1 in gastric secretion of nonacidic fluid and pepsinogen. Am J Physiol Gastrointest Liver Physiol. 2005;289:G550-60.
119. Fock KM, Ang TL, Bee LC, Lee EJD. Proton pump inhibitors: Do differences in pharmacokinetics translate into differences in clinical outcomes? Clin Pharmacokinet. 2008;47:1-6.
120. Lorentzon P, Jackson R, Wallmark B, Sachs G. Inhibition of H+,K+-ATPase by omeprazole in isolated gastric vesicles requires proton transport. Biochim Biophys Acta. 1987;897:41-51.
121. Lindberg P, Brändström A, Wallmark B, et al. Omeprazole: The first proton pump inhibitor. Med Res Rev. 1990;10:1-54.
122. Hatlebakk JG, Katz PO, Camacho-Lobato L, Castell DO. Proton pump inhibitors: better acid suppression when taken before a meal than without a meal. Aliment Pharmacol Therapeut. 2000;14:1267-72.
123. Shin JM, Sachs G. Restoration of acid secretion following treatment with proton pump inhibitors. Gastroenterology. 2002;123:281-90.
124. Feldman M, Richardson CT. Role of thought, sight, smell and taste of food in the cephalic phase of gastric acid secretion in humans. Gastroenterology. 1986;90:428-33.
125. Richardson CT, Walsh JH, Cooper KA, et al. Studies of the role of cephalic-vagal stimulation in the acid secretory response to eating in normal human subjects. J Clin Invest. 1977;60:435-41.
126. Chuang C-N, Tanner M, Lloyd KCK, et al. Endogenous somatostatin inhibits histamine release from canine gastric mucosal cells in primary culture. Am J Physiol Gastrointest Liver Physiol. 1993;265:G521-5.
127. Vuyyuru L, Schubert ML. Histamine, acting via H3 receptors, inhibits somatostatin and stimulates acid secretion in isolated mouse stomach. Gastroenterology. 1997;113:1545-52.
128. Sandvik AK, Cui GL, Bakke I, et al. PACAP stimulates gastric acid secretion in the rat by inducing histamine release. Am J Physiol Gastrointest Liver Physiol. 2001;281:G997-1003.
129. Gower WR, Dietz JR, McCuen RW, et al. Regulation of atrial natriuretic peptide secretion by cholinergic and PACAP neurons of the gastric antrum. Am J Physiol Gastrointest Liver Physiol. 2003;284:G68-74.
130. Li P, Chang TM, Coy D, Chey WY. Inhibition of gastric acid secretion in rat stomach by PACAP is mediated by secretin, somatostatin, and PGE2. Am J Physiol Gastrointest Liver Physiol. 2000;278:G121-7.
131. Pisegna JR, Ohning GV, Athmann C, et al. Role of PACAP1 receptor in regulation of ECL cells and gastric acid secretion by pituitary adenylate cyclase activating peptide. Ann N Y Acad Sci. 2000;921:233-41.
132. Chiba T, Kadowaki S, Taminato T, et al. Effect of antisomatostatin gamma-globulin on gastrin release in rats. Gastroenterology. 1981;81:321-6.
133. Karnik PS, Wolfe MM. Somatostatin stimulates gastrin mRNA turnover in dog antral mucosa. J Biol Chem. 1990;265:2550-5.
134. Yang H, Wong H, Wu V, et al. Somatostatin monoclonal antibody immunoneutralization increases gastrin and gastric acid secretion in urethane-anesthetized rats. Gastroenterology. 1990;99:659-65.
135. Martindale R, Kauffman GL, Levin S, et al. Differential regulation of gastrin and somatostatin secretion from isolated perfused rat stomach. Gastroenterology. 1982;83:240-4.
136. Saffouri B, Weir GC, Bitar KN, Makhlouf GM. Gastrin and somatostatin secretion by perfused rat stomach: functional linkage of antral peptides. Am J Physiol Gastrointest Liver Physiol. 1980;238:G495-501.
137. Schubert ML, Hightower J. Functionally distinct muscarinic receptors on gastric somatostatin cells. Am J Physiol Gastrointest Liver Physiol. 1990;258:G982-7.
138. Schubert ML, Saffouri B, Makhlouf GM. Identical patterns of somatostatin secretion from isolated antrum and fundus of rat stomach. Am J Physiol Gastrointest Liver Physiol. 1988;254:G20-4.
139. Wolfe MM, Jain DK, Reel GM, McGuigan JE. Effects of carbachol on gastrin and somatostatin release in rat antral tissue culture. Gastroenterology. 1984;87:86-93.
140. Schubert ML, Bitar KN, Makhlouf GM. Regulation of gastrin and somatostatin secretion by cholinergic and noncholinergic intramural neurons. Am J Physiol Gastrointest Liver Physiol. 1982;243:G442-7.
141. Schubert ML, Makhlouf GM. Gastrin secretion induced by distension is regulated by cholinergic and vasoactive intestinal peptide neurons in rats. Gastroenterology. 1993;104:834-9.
142. Schubert ML, Coy DH, Makhlouf GM. Peptone stimulates gastrin secretion from the stomach by activating bombesin/GRP and cholinergic neurons. Am J Physiol Gastrointest Liver Physiol. 1992;262:G685-9.
143. Short GM, Doyle W, Wolfe MM. Effect of antibodies to somatostatin on acid secretion and gastrin release by isolated perfused rat stomach. Gastroenterology. 1985;88:984-8.
144. Kovacs TOG, Lloyd KCK, Lawson DC, et al. Inhibition of sham feeding-stimulated acid secretion in dogs by immunoneutralization of gastrin. Am J Physiol Gastrointest Liver Physiol. 1997;273:G399-403.
145. Sandvik AK, Waldum HL. CCK-B (gastrin) receptor regulates gastric histamine release and acid secretion. Am J Physiol Gastrointest Liver Physiol. 1991;260:G925-8.
146. Chuang C-N, Tanner M, Chen MCY, et al. Gastrin induction of histamine release from primary cultures of canine oxyntic mucosal cells. Am J Physiol Gastrointest Liver Physiol. 1992;263:G460-5.
147. Schiller LR, Walsh JH, Feldman M. Distension-induced gastrin release. Gastroenterology. 1980;78:912-17.
148. Lichtenberger LM, Delansorne R, Graziani LA. Importance of amino acid uptake and decarboxylation in gastrin release from isolated G cells. Nature. 1982;295:698-700.
149. Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. 1987;82:192-9.
150. Graham DY, Alpert LC, Smith JL, Yoshimura HH. Iatrogenic Campylobacter pylori infection is a cause of epidemic achlorhydria. Am J Gastroenterol. 1988;83:974-80.
151. Ramsey EJ, Carey KV, Peterson WL, et al. Epidemic gastritis and achlorhydria. Gastroenterology. 1979;76:1449-57.
152. Harford WV, Barnett C, Lee E, et al. Acute gastritis with hypochlorhydria: Report of 35 cases with long term follow up. Gut. 2000;47:467-72.
153. Meyer-Rosberg K, Scott DR, Rex D, et al. The effect of environmental pH on the proton motive force of Helicobacter pylori. Gastroenterology. 1996;111:886-900.
154. Kobayashi H, Kamiya S, Suzuki T, et al. The effect of Helicobacter pylori on gastric acid secretion by isolated parietal cells from a guinea pig—Association with production of vacuolating toxin by H. pylori. Scand J Gastroenterol. 1996;31:428-33.
155. Hoffman JS, King WW, Fox JG, et al. Rabbit and ferret parietal cell inhibition by Helicobacter species. Dig Dis Sci. 1995;40:147-52.
156. Cave DR, King WW, Hoffman JS. Production of two chemically distinct acid inhibitory factors by Helicobacter pylori. Eur J Gastroenterol Hepatol. 1993;5:S23-7.
157. Konturek P, Brzozowski T, Karczewska E, et al. Water extracts of Helicobacter pylori suppress the expression of histidine decarboxylase and reduce histamine content in the rat gastric mucosa. Digestion. 2000;62:100-9.
158. Göoz M, Hammond CE, Larsen K, et al. Inhibition of human gastric H+-K+-ATPase α-subunit gene expression by Helicobacter pylori. Am J Physiol Gastrointest Liver Physiol. 2000;278:G981-91.
159. Beales ILP, Calam J. Inhibition of carbachol stimulated acid secretion by interleukin 1β in rabbit parietal cells requires protein kinase C. Gut. 2001;48:782-9.
160. Zaki M, Coudron PE, McCuen R, et al. Helicobacter pylori, acting via neural pathways, stimulates somatostatin and thus inhibits histamine and acid secretion in the fundus of rat stomach. Gastroenterology. 1998;114:A344.
161. El-Omar EM. Mechanisms of increased acid secretion after eradication of Helicobacter pylori infection. Gut. 2006;55:144-6.
162. Derakhshan MH, El Omar E, Oien K, et al. Gastric histology, serological markers and age as predictors of gastric acid secretion in patients infected with Helicobacter pylori. J Clin Pathol. 2006;59:1293-9.
163. Saha A, Hammond CE, Gooz M, Smolka AJ. IL-1B modulation of H,K-ATPase α-subunit gene transcription in Helicobacter pylori infection. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1055-61.
164. Kato S, Matsukura N, Matsuda N, et al. Helicobacter pylori eradication therapy modulates acidity and interleukin-1B mRNA levels in un-operated stomach and in remnant stomach after gastrectomy in gastric cancer patients. Aliment Pharmacol Ther. 2006;24:278-84.
165. El-Omar EM, Oien K, El-Nujumi A, et al. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15-24.
166. Feldman M, Cryer B, Lee E. Effects of Helicobacter pylori gastritis on gastric secretion in healthy human beings. Am J Physiol Gastrointest Liver Physiol. 1998;274:G1011-17.
167. Shimatini T, Inoue M, Iwamoto K, et al. Gastric acidity in patients with follicular gastritis is significantly reduced, but can be normalized after eradication for Helicobacter pylori. Helicobacter. 2005;10:256-65.
168. Loffeld RJ, Werdmuller BF, Kuster JG, et al. Colonization with cagA-positive Helicobacter pylori strains inversely associated with reflux esophagitis and Barrett’s esophagus. Digestion. 2000;62:95-9.
169. Gillen D, Wirz AA, Neithercut WD, et al. Helicobacter pylori infection potentiates the inhibition of gastric acid secretion by omeprazole. Gut. 1999;44:468-75.
170. Gillen D, Wirz A, Ardill JE, McColl KE. Rebound hypersecretion after omeprazole and its relation to on-treatment acid suppression and Helicobacter pylori status. Gastroenterology. 1999;116:239-47.
171. Inoue H, Imoto I, Taguchi Y, et al. Reflux esophagitis after eradication of Helicobacter pylori is associated with the degree of hiatal hernia. Scand J Gastroenterol. 2004;39:1061-5.
172. Fossmark R, Johnsen G, Johanessen E, et al. Rebound hypersecretion after long-term inhibition of gastric acid secretion. Aliment Pharmacol Therapeut. 2005;21:149-54.
173. D’Elios MM, Bergman MP, Azzurri A, et al. H+,K+-ATPase (proton pump) is the target autoantigen of Th1-type cytotoxic T cells in autoimmune gastritis. Gastroenterology. 2001;120:377-86.
174. Claeys D, Faller G, Appelmelk BJ, et al. The gastric H+,K+-ATPase is a major autoantigen in chronic Helicobacter pylori gastritis with body mucosa atrophy. Gastroenterology. 1998;115:340-7.
175. Gillen D, El-Omar EM, Wirz AA, et al. The acid response to gastrin distinguishes duodenal ulcer patients from Helicobacter pylori–infected healthy subjects. Gastroenterology. 1998;114:50-7.
176. Moss SF, Legon S, Bishop AE, et al. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet. 1992;340:930-2.
177. El-Omar EM, Penman ID, Ardill JES, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology. 1995;109:681-91.
178. Bechi P, Romagnoli P, Bacci S, et al. Helicobacter pylori and duodenal ulcer: Evidence for a histamine pathways-involving link. Am J Gastroenterol. 1996;91:2338-43.
179. Courillon-Mallet A, Launay J, Roucayrol A, et al. Helicobacter pylori infection: physiopathologic implication of Nα-methyl histamine. Gastroenterology. 1995;108:959-66.
180. Zavros Y, Rieder G, Ferguson A, et al. Hypergastrinemia in response to gastric inflammation suppresses somatostatin. Am J Physiol Gastrointest Liver Physiol. 2002;282:G175-83.
181. Beales I, Blaser MJ, Srinivasan S, et al. Effect of Helicobacter pylori products and recombinant cytokines on gastrin release from cultures canine G cells. Gastroenterology. 1997;113:465-71.
182. Beales ILP. Effect of platelet-activating factor on gastrin release from cultured rabbit G-cells. Dig Dis Sci. 2001;46:301-6.
183. Oh DS, Wang HS, Ohning GV, Pisegna JR. Validation of a new endoscopic technique to assess acid output in Zollinger-Ellison syndrome. Clin Gastroenterol Hepatol. 2006;4:1467-73.
184. Moore EW, Scarlata RW. The determination of gastric acidity by the glass electrode. Gastroenterology. 1965;49:178-88.
185. Klein KB. Gastric secretory testing. In: Drossman DA, editor. Manual of gastroenterologic procedures. 3rd ed. New York: Raven Press; 1993:61-7.
186. Dalenbäck J, Fändriks L, Olbe L, Sjövall H. Mechanisms behind changes in gastric acid and bicarbonate outputs during the human interdigestive motility cycle. Am J Physiol Gastrointest Liver Physiol. 1996;270:G113-22.
187. Hildebrand P, Lehmann FS, Ketterer S, et al. Regulation of gastric function by endogenous gastrin releasing peptide in humans: studies with a specific gastrin releasing peptide receptor antagonist. Gut. 2001;49:23-8.
188. Gardner JD, Ciociola AA, Robinson M. Measurement of meal-stimulated gastric acid secretion by in vivo gastric autotitration. J Appl Physiol. 2002;92:427-34.
189. Feldman M. Comparison of the effects of over-the-counter famotidine and calcium carbonate antacid on postprandial gastric acid—A randomized controlled trial. JAMA. 1996;275:1428-31.
190. Blair AJ, Feldman M, Barnett C, et al. Detailed comparison of basal and food-stimulated gastric acid secretion rates and serum gastrin concentrations in duodenal ulcer patients and normal subjects. J Clin Invest. 1987;79:582-7.
191. El-Omar E, Penman I, Dorrian CA, et al. Eradicating Helicobacter pylori infection lowers gastrin mediated acid secretion by two thirds in patients with duodenal ulcer. Gut. 1993;34:1060-5.
192. Jacobson K, Chiba N, Chen Y, et al. Gastric acid secretory response in Helicobacter pylori–positive patients with duodenal ulcer disease. Can J Gastroenterol. 2001;15:29-39.
193. Moss SF, Calam J. Acid secretion and sensitivity to gastrin in patients with duodenal ulcer: Effect of eradication of Helicobacter pylori. Gut. 1993;34:888-92.
194. Peterson WL, Barnett CC, Evans DJJr., Feldman M, et al. Acid secretion and serum gastrin in normal subjects and patients with duodenal ulcer: The role of Helicobacter pylori. Am J Gastroenterol. 1993;88:2038-43.
195. Peterson WL. Gastrin and acid in relation to Helicobacter pylori. Aliment Pharmacol Therapeut. 1996;10:97-102.
196. Johnson HD, Love AH, Rogers NC, Wyatt AP. Gastric ulcers, blood groups, and acid secretion. Gut. 1964;5:402-11.
197. Csendes A, Braghetto I, Smok G. Type IV gastric ulcer: A new hypothesis. Surgery. 1987;101:361-6.
198. Jensen RT. Gastrointestinal abnormalities and involvement in systemic mastocytosis. Hematol Oncol Clin North Amer. 2000;14:579-623.
199. Webster MW, Barnes EL, Stremple JF. Serum gastrin levels in the differential diagnosis of recurrent peptic ulceration due to retained gastric antrum. Am J Surg. 1978;135:248-52.
200. Dent RI, James JH, Want CA, et al. Hyperparathyroidism: Gastric acid secretion and gastrin. Ann Surg. 1972;176:360-9.
201. Buchan AMJ, Squires PE, Ring M, Meloche RM. Mechanism of action of the calcium-sensing receptor in human antral gastrin cells. Gastroenterology. 2001;120:1128-39.
202. Jensen RT, Niederle B, Mitry E, et al. Gastrinoma (duodenal and pancreatic). Neuroendocrinology. 2006;84:173-82.
203. Hung PD, Schubert ML, Mihas AA. Zollinger-Ellison syndrome. Curr Treat Options Gastroenterol. 2003;6:163-70.
204. Meko JB, Norton JA. Management of patients with Zollinger-Ellison syndrome. Annu Rev Med. 1995;46:395-411.
205. Wolfe MM, Reel GM, McGuigan JE. Inhibition of gastrin release by secretin is mediated by somatostatin in cultured rat antral mucosa. J Clin Invest. 1983;72:1586-93.
206. Chiba T, Taminato T, Kadowaki S, et al. Effects of glucagon, secretin and vasoactive intestinal polypeptide on gastric somatostatin and gastrin release from isolated perfused rat stomach. Gastroenterology. 1980;79:67-71.
207. Chiba T, Yamatani T, Yamaguchi A, et al. Mechanism for increase of gastrin release by secretin in Zollinger-Ellison syndrome. Gastroenterology. 1989;96:1439-44.
208. Gibril F, Reynolds JC, Doppman JL, et al. Somatostatin receptor scintigraphy: Its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas—A prospective study. Ann Intern Med. 1996;125:26-34.
209. Gibril F, Reynolds JC, Chen CC, et al. Specificity of somatostatin receptor scintigraphy: A prospective study and effects of false-positive localizations on management in patients with gastrinomas. J Nucl Med. 1999;40:539-53.
210. Metz DC, Pisegna JR, Fishbeyn VA, et al. Control of gastric acid hypersecretion in the management of patients with Zollinger-Ellison syndrome. World J Surg. 1993;17:468-80.
211. Richter C, Tanaka T, Yada RY. Pepsinogen, progastricsin, and prochymosin. Biochem J. 1998;335:481-90.
212. Schreiber S, Bucker R, Groll C, et al. Gastric antibacterial efficiency is different for pepsin A and C. Arch Microbiol. 2006;184:335-40.
213. Etherington DJ, Taylor WH. Nomenclature of the pepsins. Nature. 1967;216:279-80.
214. Xie G, Drachenberg C, Yamada M, et al. Cholinergic agonist-induced pepsinogen secretion from murine gastric chief cells is mediated by M1 and M3 muscarinic receptors. Am J Physiol Gastrointest Liver Physiol. 2005;289:G521-9.
215. Fiorucci S, Distrutti E, Chiorean M, et al. Nitric oxide modulates pepsinogen secretion induced by calcium-mediated agonist in guinea pig gastric chief cells. Gastroenterology. 1995;109:1214-23.
216. Roberts NB, Sheers R, Taylor WH. Secretion of total pepsin and pepsin 1 in healthy volunteers in response to pentagastrin and to insulin-induced hypoglycemia. Scand J Gastroenterol. 2007;42:555-61.
217. Skak-Nielsen T, Holst JJ, Nielsen OV. Role of gastrin-releasing peptide in the neural control of pepsinogen secretion from the pig stomach. Gastroenterology. 1988;95:1216-20.
218. Kasbekar DK, Jensen RT, Gardner JD. Pepsinogen secretion from dispersed glands from rabbit stomach. Am J Physiol Gastrointest Liver Physiol. 1983;244:G392-6.
219. Kitsukawa Y, Felley C, Metz DC, Jensen RT. Thapsigargin defines roles of Ca2+ in initial, sustained, and potentiated stimulation of pepsinogen secretion. Am J Physiol Gastrointest Liver Physiol. 1994;266:G613-23.
220. Sipponen P, Ranta P, Helske T, et al. Serum levels of amidated gastrin-17 and pepsinogen I in atrophic gastritis: An observational case-control study. Scand J Gastroenterol. 2002;37:785-91.
221. Vaananen H, Vauhkonen M, Helske T, et al. Non-endoscopic diagnosis of atrophic gastritis with a blood test. Correlation between gastric histology and serum levels of gastrin-17 and pepsinogen I: A multicentre study. Eur J Gastroenterol Hepatol. 2003;15:885-91.
222. Miki K, Ichinose M, Shimizu A, et al. Serum pepsinogens as a screening test of extensive chronic gastritis. Gastroenterol Jpn. 1987;22:133-41.
223. Di Mario F, Ingegnoli A, Altavilla N, et al. Influence of antisecretory treatment with proton pump inhibitors on serum pepsinogen I levels. Fund Clin Pharmacol. 2005;19:497-501.
224. ten Kate RW, Pals G, Eriksson AW, et al. The renal metabolism of pepsinogen A and C in man. Clin Nephrol. 1989;31:103-6.
225. Tamura H, Tokushima H, Murakawa M, et al. Influences of Helicobacter pylori on serum pepsinogen concentrations in dialysis patients. Nephrol Dial Transplant. 1999;14:113-17.
226. Ville E, Carrière F, Renou C, Laugier R. Physiological study of pH stability and sensitivity to pepsin of human gastric lipase. Digestion. 2002;67:73-81.
227. Wicker-Planquart C, Canaan S, Riviere M, Dupuis L. Site-directed removal of N-glycosylation sites in human gastric lipase. Eur J Biochem. 1999;262:644-51.
228. Armand M. Lipases and lipolysis in the human digestive tract: where do we stand? Curr Opin Clin Nutr Metab Care. 2008;10:156-64.
229. Armand M, Hamosh M, Dipalma JS, et al. Dietary fat modulates gastric lipase activity in healthy humans. Am J Clin Nutr. 1995;62:74-80.
230. Aloulou A, Carrière F. Gastric lipase: An extremophilic interfacial enzyme with medical applications. Cell Mol Life Sci. 2008;65:851-4.
231. Carrière F, Grandval P, Renou C, et al. Quantitative study of digestive enzyme secretion and gastrointestinal lipolysis in chronic pancreatitis. Clin Gastroenterol Hepatol. 2005;3:28-38.
232. Wojdemann M, Riber C, Bisgaard T, et al. Inhibition of human gastric lipase by intraduodenal fat involves glucagon-like peptide-1 and cholecystokinin. Regul Pept. 1999;80:101-6.
233. Sternby B, Hartmann D, Borgstrom B, Nilsson A. Degree of in vivo inhibition of human gastric and pancreatic lipases by orlistat (tetrahydrolipstatin, THL) in the stomach and small intestine. Clin Nutr. 2002;21:395-402.
234. Hadvary P, Sidler W, Meister W, et al. The lipase inhibitor tetrahydrolipostatin binds covalently to the putative active site serine of pancreatic lipase. J Biol Chem. 1991;266:2021-7.
235. Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. Xenical in the prevention of diabetes in obese subjects (XENDOS) study. Diabetes Care. 2004;27:155-61.
236. O’Meara S, Riemsma R, Shirran L, et al. A rapid and systematic review of the clinical effectiveness and cost-effectiveness of orlistat in the management of obesity. Health Technol Assess. 2001;5:1-81.
237. O’Donovan D, Horowitz M, Russo A, et al. Effects of lipase inhibition on gastric emptying of, and on the glycaemic, insulin and cardiovascular responses to, a high-fat/carbohydrate meal in type 2 diabetes. Diabetologia. 2004;47:2208-14.
238. Howard TA, Misra DN, Grove M, et al. Human gastric intrinsic factor expression is not restricted to parietal cells. J Anat. 1996;189:303-13.
239. Kittang E, Aadland E, Schjowsby H. Effect of omeprazole on secretion of intrinsic factor, gastric acid, and pepsin in man. Gut. 1985;26:594-8.
240. Carmel R. In vitro studies of gastric juice in patients with food-cobalamin malabsorption. Dig Dis Sci. 1994;39:2516-22.
241. Lewerin C, Jacobbson S, Lindstedt G, Nilsson-Ehle H. Serum biomarkers for atrophic gastritis and antibodies against Helicobacter pylori in the elderly: Implications for vitamin B12, folic acid and iron status and response to oral vitamin therapy. Scand J Gastroenterol. 2008;43:1050-6.
242. Miner P, Katz PO, Chen Y, Sostek M. Gastric acid control with esomeprazole, lansoprazole, omeprazole, pantoprazole, and rabeprazole: A five-way crossover study. Am J Gastroenterol. 2003;98:2616-20.
243. Hirschowitz BI, Worthington J, Mohnen J. Vitamin B12 deficiency in hypersecretors during long-term acid suppression with proton pump inhibitors. Aliment Pharmacol Therapeut. 2008;27:1110-21.
244. Gueant JL, Djalali M, Aouadj R, et al. In vitro and in vivo evidences that the malabsorption of cobalamin is related to its binding on haptocorrin (R binder) in chronic pancreatitis. Am J Clin Nutr. 1986;44:265-77.
245. Moestrup SK. New insights into carrier binding and epithelial uptake of the erythropoietic nutrients cobalamin and folate. Curr Opin Hematol. 2006;13:119-23.
246. Xu D, Fyfe JC. Cubilin expression and posttranslational modification in the canine gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2000;279:G748-56.
247. Scott JM. Folate and vitamin B-12. Proc Nutri Soc. 1999;58:441-8.
248. Kristiansen M, Aminoff M, Jacobsen C, et al. Cubilin P1297L mutation associated with hereditary megaoblastic anemia 1 causes impaired recognition of intrinsic factor-vitamin B(12) by cubilin. Blood. 2000;96:405-9.
249. Aimone-Gastin I, Pierson H, Jeandel C, et al. Prospective evaluation of protein bound vitamin B12 (cobalamin) malabsorption in the elderly using trout flesh labelled in vivo with 57CO-cobalamin. Gut. 1997;41:475-9.
250. Suter PM, Goldner BB, Goldin BR, et al. Reversal of protein-bound vitamin B12 malabsorption with antibiotics in atrophic gastritis. Gastroenterology. 1991;101:1039-45.
251. Carmel R. Prevalence of undignosed pernicious anemia in the elderly. Arch Intern Med. 1996;156:1097-100.
252. Toh B-H, Alderuccio F. Pernicious anaemia. Autoimmunity. 2004;37:357-61.
253. Davidson RJ, Atrah HI, Sewell HF. Longitudinal study of circulating gastric antibodies in pernicious anaemia. J Clin Pathol. 1989;42:1092-5.
254. Carmel R. Reassessment of the relative prevalences of antibodies to gastric parietal cell and to intrinsic factor in patients with pernicious anaemia: Influence of patient age and race. Clin Exp Immunol. 1992;89:74-7.
255. Dalenback J, Fandriks L, Olbe L, Sjövall H. The pH/PCO2 method for continuous determination of human gastric acid and bicarbonate secretion. A validation study. Scand J Gastroenterol. 1995;30:861-71.
256. Forssell H, Olbe L. Continuous computerized determination of gastric bicarbonate secretion in man. Scand J Gastroenterol. 1985;20:767-74.
257. Debellis L, Caroppo R, Fromter E, Curci S. Alkaline secretion by frog gastric glands measured with pH microelectrodes in the gland lumen. J Physiol. 1998;513:235-41.
258. Flemström G, Isenberg JI. Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol Sci. 2001;16:23-8.
259. Xu J, Barone S, Petrovic S, et al. Identification of an apical Cl−/HCO3− exchanger in gastric surface mucous and duodenal villus cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1225-34.
260. Cox KH, ir-Kirk TL, Cox JV. Variant AE2 anion exchanger transcripts accumulate in multiple cell types in the chicken gastric epithelium. J Biol Chem. 1996;271:8895-902.
261. Stuarttilley A, Sardet C, Pouyssegur J, et al. Immunolocalization of anion exchanger AE2 and cation exchanger NHE-1 in distinct adjacent cells of gastric mucosa. Am J Physiol. 1994;266:C559-68.
262. Wang Z, Petrovic S, Mann E, Soleimani M. Identification of an apical Cl−/HCO3− exchanger in the small intestine. Am J Physiol Gastrointest Liver Physiol. 2002;282:G573-9.
263. Kraniak J, Koyanagi H, Fromm D. Do isolated gastric mucosal surface cells from rabbits secrete HCO3−. J Surg Res. 1995;58:211-17.
264. De Beus AM, Fabry TL, Lacker HM. A gastric acid secretion model. Biophys J. 1993;65:362-78.
265. Singh K, Nain CK, Singh V. Effect of omeprazole on gastric bicarbonate secretion in patients with duodenal ulcer. Indian J Gastroenterol. 1998;17:136-7.
266. Feldman M. Gastric bicarbonate secretion in humans: effect of pentagastrin, bethanechol, and 11,16,16-trimethyl prostaglandin E2. J Clin Invest. 1983;72:295-303.
267. Johansson C, Kolberg B. Stimulation by intragastrically adminstered E2 prostaglandins of human gastric mucus output. Eur J Clin Invest. 1979;9:229-32.
268. Mertz-Nielsen A, Hillingso J, Bukhave K, Rask-Madsen J. Indomethacin decreases gastroduodenal mucosal bicarbonate secretion in humans. Scand J Gastroenterol. 1995;30:1160-5.
269. Takeuchi K, Yagi K, Kato S, Ukawa H. Roles of prostaglandin E-receptor subtypes in gastric and duodenal bicarbonate secretion in rats. Gastroenterology. 1997;113:1553-9.
270. Takeuchi K, Ukawa H, Furukawa O, et al. Prostaglandin E receptor subtypes involved in stimulation of gastroduodenal bicarbonate secretion in rats and mice. J Physiol Pharmacol. 1999;50:155-67.
271. Cryer B, Lee E, Feldman M. Factors influencing gastroduodenal mucosal prostaglandin concentrations: Roles of smoking and aging. Ann Intern Med. 1992;116:636-40.
272. Feldman M, Cryer B. Effects of age on gastric alkaline and nonparietal fluid secretion in humans. Gerontology. 1998;44:222-7.
273. Allen A, Flemström G, Garner A, Kivilaakso E. Gastroduodenal mucosal protection. Physiol Rev. 1993;73:823-57.
274. Corfield AP, Carroll D, Myerscough N, Probert CS. Mucins in the gastrointestinal tract in health and disease. Front Biosci. 2001;6:D1321-57.
275. Shimizu T, Akamatsu T, Sugiyama A, et al. Helicobacter pylori and the surface mucous gel layer of the human stomach. Helicobacter. 1996;1:207-18.
276. Morgenstern S, Koren R, Fraser G, et al. Gastric corpus mucin expression after partial gastrectomy, in relation to colonization with Helicobacter pylori. J Clin Gastroenterol. 2001;32:218-21.
277. Van den Brink GR, Tytgat KN, Van der Hulst RW, et al. H. pylori colocalises with MUC5AC in the human stomach. Gut. 2000;46:601-7.
278. Ota H, Katsuyama T. Alternating laminated array of two types of mucin in the human gastric surface mucous layer. Histochem J. 1992;24:86-92.
279. Nam SY, Kim N, Lee CS, et al. Gastric mucosal protection via enhancement of MUC5AC and MUC6 by geranylgeranylacetone. Dig Dis Sci. 2005;50:2110-20.
280. Ichikawa T, Ota H, Sugiyama A, et al. Effects of a novel histamine H2-receptor antagonist, lafutidine, on the mucus barrier of human gastric mucosa. J Gastroenterol Hepatol. 2007;22:1800-5.
281. Atuma C, Strugala V, Allen A, Holm L. The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;280:G922-9.
282. Wallace JL, Granger DN. The cellular and molecular basis of gastric mucosal defense. FASEB J. 1996;10:731-40.
283. Williams SE, Turnberg LA. Retardation of acid diffusion by pig gastric mucosa: a potential role in mucosal protection. Gastroenterology. 1980;79:299-304.
284. Kubota S, Yamauchi K, Kumagai T, et al. Quantitative determination of gland mucous cells-type mucin using a monoclonal antibody, HIK1083: Its pathophysiological changes in human gastric juice. Clin Chim Acta. 2007;377:261-7.
285. Vilkin A, Levi Z, Morgenstern S, et al. Higher gastric mucin secretion and lower gastric acid output in first-degree relatives of gastric cancer patients. J Clin Gastroenterol. 2008;42:36-41.
286. Kaunitz JD, Nishizaki Y, Kaneko K, Guth PH. Effect of orogastric nicotine on rat gastric mucosal gel thickness, surface cell viability, and intracellular pH. J Pharmacol Exp Ther. 1993;265:948-54.
287. Engel E, Guth PH, Nishizaki Y, Kaunitz JD. Barrier function of the gastric mucus gel. Am J Physiol Gastrointest Liver Physiol. 1995;269:G994-9.
288. Tanaka S, Podolsky DK, Engel E, et al. Human spasmolytic polypeptide decreases proton permeation through gastric mucus in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol. 1997;272:G1473-80.
289. Williams SE, Turnberg LA. Demonstration of a pH gradient across mucus adherent to rabbit gastric mucosa: Evidence for a “mucus-bicarbonate” barrier. Gut. 1981;22:94-6.
290. Chu S, Tanaka S, Kaunitz JD, Montrose MH. Dynamic regulation of gastric surface pH by luminal pH. J Clin Invest. 1999;103:605-12.
291. Schade C, Flemström G, Holm L. Hydrogen ion concentration in the mucus layer on top of acid-stimulated and -inhibited rat gastric mucosa. Gastroenterology. 1994;107:180-8.
292. Bahari HMM, Ross IN, Turnberg LA. Demonstration of a pH gradient across the mucus layer on the surface of human gastric mucosa in vitro. Gut. 1982;23:513-16.
293. Baumgartner HK, Montrose MH. Regulated alkali secretion acts in tandem with unstirred layers to regulate mouse gastric surface pH. Gastroenterology. 2004;126:774-83.
294. Schreiber S, Scheid P. Gastric mucus of the guinea pig: proton carrier and diffusion barrier. Am J Physiol. 1997;272:G63-70.
295. Nagano F, Masaki H, Fujimoto M, et al. Effects of H+ and HCO3− secretion on mucus gel pH in isolated antral mucosa of bullfrog stomach. Bull Osaka Med College. 1990;36:13-25.
296. Allen A, Hutton D, McQueen S, Garner A. Dimensions of gastroduodenal surface pH gradients exceed those of adherent mucus gel layers. Gastroenterology. 1983;85:463-76.
297. Tanaka S, Meiselman HHJ, Engel E, et al. Regional differences of H+, HCO3−, and CO2 diffusion through native porcine gastroduodenal mucus. Dig Dis Sci. 2002;47:967-73.
298. Phillipson M, Atuma C, Henriksnas J, Holm L. The importance of mucus layers and bicarbonate transport in preservation of gastric juxtamucosal pH. Am J Physiol Gastrointest Liver Physiol. 2002;282:G211-19.
299. Cao X, Bansil R, Bhaskar KR, et al. pH-dependent conformational change of gastric mucin leads to sol-gel transition. Biophys J. 1999;76:1250-8.
300. Taupin D, Podolsky DK. Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol. 2003;4:721-32.
301. Kinoshita K, Taupin DR, Itoh H, Podolsky DK. Distinct pathways of cell migration and antiapoptotic response to epithelial injury: Structure-function analysis of human intestinal trefoil factor. Mol Cell Biol. 2000;20:4680-90.
302. Ota H, Hayama M, Momose M, et al. Co-localization of TFF2 with gland mucous cell mucin in gastric mucous cells and in extracellular mucous gel adherent to normal and damaged gastric mucosa. Histochem Cell Biol. 2006;126:617-25.
303. Jeffrey GP, Oates PS, Wang TC, et al. Spasmolytic polypeptide: A trefoil peptide secreted by rat gastric mucous cells. Gastroenterology. 1994;106:336-45.
304. Hanby AM, Poulsom R, Singh S, et al. Spasmolytic polypeptide is a major antral peptide: Distribution of the trefoil peptides human spasmolytic polypeptide and pS2 in the stomach. Gastroenterology. 1993;105:1110-16.
305. Longman RJ, Douthwaite J, Sylvester PA, et al. Coordinated localisation of mucins and trefoil peptides in the ulcer associated cell lineage and the gastrointestinal mucosa. Gut. 2000;47:792-800.
306. Sarraf CE, Alison MR, Ansari TW, Wright NA. Subcellular distribution of peptides associated with gastric mucosal healing and neoplasia. Microsc Res Tech. 1995;31:234-47.
307. Ren JL, Luo JY, Lu YP, et al. Molecular forms of trefoil factor 1 in normal gastric mucosa and its expression in normal and abnormal gastric tissues. World J Gastroenterol. 2006;12:7361-4.
308. Franic TV, van Driel IR, Gleeson PA, et al. Reciprocal changes in trefoil 1 and 2 expression in stomachs of mice with gastric unit hypertrophy and inflammation. J Pathol. 2005;207:43-52.
309. Saitoh T, Mochizuki T, Suda T, et al. Elevation of TFF1 gene expression during healing of gastric ulcer at non-ulcerated sites in the stomach: semiquantification using the single tube method of polymerase chain reaction. J Gastroenterol Hepatol. 2000;15:604-9.
310. Hoffmann W. Trefoil factors TFF (trefoil factor family) peptide-triggered signals promoting mucosal restitution. Cell Mol Life Sci. 2005;62:2932-8.
311. Young OT, Ok AB, Jung JE, et al. Accelerated ulcer healing and resistance to ulcer recurrence with gastroprotectants in rat model of acetic acid-induced gastric ulcer. J Clin Biochem Nutr. 2008;42:204-14.
312. Thim L, Madsen F, Poulsen SS. Effect of trefoil factors on the viscoelastic properties of mucus gels. Eur J Clin Invest. 2002;32:519-27.
313. Kjellev S, Nexo E, Thim L, Poulsen SS. Systemically administered trefoil factors are secreted into the gastric lumen and increase the viscosity of gastric contents. Br J Pharmacol. 2006;149:92-9.
314. Kang B, Alderman BM, Nicoll AJ, et al. Effect of omeprazole-induced achlorhydria on trefoil peptide expression in the rat stomach. J Gastroenterol Hepatol. 2001;16:1222-7.
315. Farrell JJ, Taupin D, Koh TJ, et al. TFF2/SP-deficient mice show decreased gastric proliferation, increased acid secretion, and increased susceptibility to NSAID injury. J Clin Invest. 2002;109:193-204.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 49 Gastric Secretion
The stomach is an active reservoir that stores, grinds, and slowly dispenses partially digested food to the intestine for further digestion and absorption. Its main secretory function is the production of hydrochloric acid. Gastric acid secretion is present on the first day of life and increases as infants become more mature.1 By two years of age, acid secretion is similar to that of adults, when corrected for body weight.2 Most studies indicate that the rate of acid secretion changes little after the second decade of life unless there is coexisting disease of the acid-secreting glandular mucosa such as infection with Helicobacter pylori (HP) or atrophic gastritis.3–5
Acid facilitates the digestion of protein by converting the proenzyme pepsinogen to the active proteolytic enzyme pepsin. It also facilitates the absorption of iron, calcium, vitamin B12, and certain medications (e.g., thyroxin) as well as prevents bacterial overgrowth, enteric infection, and possibly community-acquired pneumonia.6–18
The stomach also secretes lipase, intrinsic factor, electrolytes (e.g., HCO3−, K+, and Cl−), and mucins in addition to a variety of neurocrine, paracrine, and hormonal agents (Fig. 49-1). Neurocrine agents are released from nerve terminals and reach their targets via synaptic diffusion (e.g., acetylcholine [ACh], gastrin-releasing peptide [GRP], and vasoactive intestinal peptide [VIP]). Paracrine agents are released in proximity to their targets and reach them via diffusion (e.g., histamine and somatostatin). Hormones are released into the circulation and reach their targets via the bloodstream (e.g., gastrin).
Gastric mucosal integrity depends on a delicate balance between secretion of aggressive (e.g., acid and pepsin) and defensive (e.g. bicarbonate and mucin) substances (Fig. 49-2).19 When mucosal defense mechanisms are overwhelmed, ulceration may occur. In order to reap the benefits of acid without untoward effects, gastric exocrine and endocrine secretion must be precisely regulated. This is accomplished by a highly coordinated interaction among a multitude of neural, paracrine, and hormonal pathways.
FUNCTIONAL ANATOMY
The stomach consists of three anatomic (fundus, corpus or body, and antrum) and two functional (oxyntic and pyloric gland) areas (Fig. 49-3). The oxyntic gland area, the hallmark of which is the oxyntic (oxys, Greek for acid) or parietal cell, comprises 80% of the organ (fundus and corpus). The pyloric gland area, the hallmark of which is the G or gastrin cell, comprises 20% of the organ (antrum). The human stomach contains approximately 1 × 109 parietal cells and 9 × 106 gastrin cells.20 There is debate as to whether the cardia, a transition zone of 0 to 9 mm between the squamous mucosa of the esophagus and the oxyntic mucosa of the stomach, exists as a normal anatomic structure or develops as a result of abnormal reflux. Autopsy and endoscopic studies suggest that cardiac mucosa is absent in more than 50% of the general population.21 Gastric anatomy is discussed in greater detail in Chapter 47.
The glandular area is organized in vertical tubular units that consist of an apical pit region, an isthmus, and the actual gland region that forms the lower part of the unit; the latter consists of a neck and a base (Fig. 49-4). The progenitor cell of the gastric unit, located in the isthmus, gives rise to all gastric epithelial cells. In the oxyntic gland area, the mucus-producing pit cells migrate upward from the progenitor cell toward the gastric lumen. Acid-secreting parietal cells migrate downward to the middle and lower regions of the gland; those at the bases are less active acid secretors. The turnover time for parietal cells is 54 days in mice and 164 days in rats.20 Chief (zymogenic) cells predominate at the base and secrete pepsinogen and leptin22; the latter is also present in parietal cells.23 Several distinct neuroendocrine cell types are contained within the gland but only some of their products have been assigned physiologic functions (see Chapter 1). The cells include enterochromaffin (EC) cells (atrial natriuretic peptide [ANP], serotonin, and adrenomedullin), enterochromaffin-like (ECL) cells (histamine), D cells (somatostatin and amylin), and A-like or Gr cells (ghrelin and obestatin).24–29 ECL cells constitute 66% of the neuroendocrine cell population in rats and 30% in humans. Somatostatin-containing D cells possess neural-like cytoplasmic processes that terminate in the vicinity of parietal and ECL cells (see Fig. 49-4). The functional correlate of this anatomic coupling is a tonic paracrine restraint exerted by somatostatin on acid secretion directly as well as indirectly by inhibiting histamine secretion (Fig. 49-5).30–32
Somatostatin-containing D cells are also present in the pyloric gland area (see Figs. 49-4 and 49-5); in this region they exert a tonic paracrine restraint on gastrin secretion from G cells.33,34 The pyloric gland also contains EC cells (ANP and serotonin), A-like or Gr cells (ghrelin and obestatin), and endocrine cells containing orexin.26,35,36
The stomach is innervated by a neural network, the enteric nervous system (ENS), that contains intrinsic neurons, that is, neurons whose cell bodies are contained within the gastric wall (Figs. 49-6 to 49-8). The ENS, the third division of the autonomic nervous system (the other two being the sympathetic and parasympathetic), is often referred to as the “little brain” because it contains as many neurons as the spinal cord, approximately 106, and can function autonomous of central input (see Fig. 49-6).37 Nevertheless, the ENS receives input from and sends input to the central nervous system via sympathetic and parasympathetic neurons. In rats and guinea pigs, most of the intrinsic neural innervation of the stomach originates in the myenteric plexus, located between the circular and longitudinal muscle layers; the submucosal plexus in these species, adjacent to the mucosal layer, contains only a small number of neurons. Humans, in contrast, have a clearly defined submucosal plexus that regulates gastric secretion and contains a variety of neurotransmitters (see Figs. 49-7 and 49-8). It should be noted that the vagus nerve is predominantly afferent, containing 80% to 90% afferent fibers and 10% to 20% efferent fibers. The efferent fibers arise from the dorsal motor nucleus of the brainstem. They are preganglionic and do not directly innervate parietal or neuroendocrine cells but rather synapse with postganglionic neurons of the ENS. The postganglionic neurons contain a variety of transmitters including ACh, GRP, nitric oxide (NO), VIP, and pituitary adenylate cyclase–activating polypeptide (PACAP) (see Fig. 49-8).38 In the stomach, afferent nerve fibers containing calcitonin gene–related peptide (CGRP) are of extrinsic origin, that is, the cell bodies are located outside the stomach wall.39 Postganglionic neurons of the ENS regulate acid secretion directly, as is the case for ACh, and/or indirectly by modulating the secretion of gastrin from G cells, somatostatin from D cells, and possibly histamine from ECL cells (see Fig. 49-8).
ACID SECRETION: PARACRINE, HORMONAL, NEURAL, AND INTRACELLULAR REGULATION
Parietal cells secrete hydrochloric acid at a concentration of approximately 160 mM or pH 0.8. Acid is thought to gain access to the lumen via channels in the mucus layer created by the relatively high intraglandular hydrostatic pressures generated during secretion, about 17 mm Hg.40
Acid facilitates the digestion of protein and absorption of iron, calcium, and vitamin B12 as well as prevents bacterial overgrowth, enteric infection, and possibly community acquired pneumonia.6–14,18,41 However, when levels of acid (and pepsin) overwhelm mucosal defense mechanisms, ulcers occur. To prevent such damage, gastric acid must be precisely regulated and produced according to need. This is accomplished by a highly coordinated interaction among a number of neural, hormonal, and paracrine pathways. These pathways can be activated directly by stimuli originating in the brain or reflexively by stimuli originating in the stomach after ingestion of a meal such as mechanical stimulation (e.g., distention) or chemical stimulation (e.g., protein and acid).
The principal stimulants of acid secretion are (1) ACh, released from postganglionic enteric neurons (neurocrine), (2) gastrin, released from antral G cells (hormonal), and (3) histamine, released from oxyntic ECL cells (paracrine) (see Fig. 49-1; Fig. 49-9). These agents interact with specific G protein–binding receptors (M3, CCK-2, and H2, respectively) that are coupled to two major signal transduction pathways: intracellular calcium in the case of gastrin and ACh, and adenylate cyclase or adenosine 3′,5′-cyclic monophosphate (cAMP) in the case of histamine (see Fig. 49-9). There is evidence for potentiation (or synergism) between histamine and either ACh or gastrin, probably as a result of postreceptor interaction between the two signaling pathways.42 The main inhibitor of acid secretion is somatostatin, released from oxyntic and antral D cells (paracrine) (see Figs. 49-1, 49-5, and 49-9). Each of these agents acts directly on the parietal cell as well as indirectly by modulating the secretion of neuroendocrine cells (Fig. 49-10).
HISTAMINE
Histamine, produced in ECL cells by decarboxylation of l-histidine by histidine decarboxylase (HDC), stimulates the parietal cell directly by binding to H2 receptors coupled to activation of adenylate cyclase and generation of cAMP (see Fig. 49-9).43 Histamine also stimulates acid secretion indirectly by binding to H3 receptors coupled to inhibition of somatostatin release from oxyntic D cells, thus resulting in stimulation of histamine release and acid secretion (see Fig. 49-10).44,45 Gastrin, PACAP, VIP, and ghrelin stimulate, whereas somatostatin, CGRP, prostaglandins, peptide YY (PYY), and galanin inhibit histamine secretion.46,47 As discussed following, gastrin also exerts a direct proliferative effect on ECL cells. ACh has no direct effect on histamine secretion.48–50
GASTRIN
Gastrin, the main stimulant of acid secretion during meal ingestion, is produced in G cells of the gastric antrum and, in much lower and variable amounts, in the proximal small intestine, colon, and pancreas. Gastrin is synthesized as a large precursor molecule of 101 amino acids, which is converted to progastrin (80 amino acids) by cleavage of the N-terminal signal peptide. Progastrin is further processed to yield peptides with C-terminal glycine, that is, G34gly and G17gly. The final processing step involves amidation to yield G34amide and G17amide. The plasma half-life of G34amide is 30 minutes and that of G17amide is three to seven minutes; they are metabolized primarily by the kidney and, in addition, by the intestine and liver.51,52 Consequently, most gastrin in the circulation during fasting is G34, whereas after a meal it is G17. In patients with renal insuffiency as well as massive small bowel resection, fasting blood levels of G17 and G34 are elevated.53,54 It should be noted that the commercially available test substance pentagastrin is not a naturally occurring peptide but rather is a manufactured analog that contains the biologically active C-terminus sequence Trp-Met-Asp-Phe-NH2.
Gastrin and cholecystokinin (CCK) belong to the same family of peptides and possess an identical carboxyl-terminal pentapeptide sequence (-Gly-Trp-Met-Asp-Phe-NH2). Two main classes of gastrin/CCK receptors have been characterized: CCK1 (formerly CCK-A) and CCK-2 (formerly CCKB or CCKB/gastrin). CCK1 receptors are specific for CCK, whereas CCK-2 receptors recognize both CCK and gastrin with high affinity. Gastrin, acting via CCK-2 receptors that activate phospholipase C to induce release of intracellular calcium, stimulates the parietal cell directly and, more importantly, indirectly by releasing histamine from ECL cells (see Figs. 49-9 and 49-10).55,56 Gastrin regulates the secretion and synthesis of histamine in a biphasic manner. The first phase involves release of stored histamine. The second phase relates to the replenishment of histamine stores and involves an increase in HDC activity followed by an increase in HDC gene transcription.57 H2 receptor, HDC, and CCK-2 receptor knockout mice manifest decreased acid secretion, especially in response to gastrin.58–60
Although amidated gastrins had been thought to be the only forms with biological activity, glycine-extended gastrins may regulate the capacity of the parietal cell to respond to secretagogues, release histamine from ECL cells, and stimulate proliferation of colonic mucosa and colorectal cancers.61,62 ACh, GRP, secretin, β2/β3-adrenergic agonists, calcium, protein, and alcoholic beverages produced by fermentation stimulate, whereas somatostatin, galanin, and adenosine inhibit gastrin secretion. In addition, at least two negative-feedback pathways, mediated via release of somatostatin, regulate gastrin secretion. The first is activated by luminal acidity and involves sensory CGRP neurons (see Fig. 49-10). Low intragastric pH (high intragastric acidity) activates CGRP neurons that, via an axon reflex, stimulate somatostatin and thus inhibit gastrin secretion.63–65 Conversely, when intragastric pH rises (low intragastric acidity), for example, by administering antisecretory medications such as proton pump inhibitors (PPIs) or by developing gastric atrophy, somatostatin secretion is inhibited and patients develop hypergastrinemia. There is some evidence that bacterial overgrowth induced by hypochlorhydria may also contribute to hypergastrinemia.66 The second negative-feedback pathway involves a paracrine action whereby gastrin directly stimulates somatostatin and thus attenuates its own secretion (see Fig. 49-10).67
Gastrin also functions as a trophic hormone to stimulate mucosal proliferation. CCK-2 receptors have been localized to the progenitor zone in oxyntic glands, and chronic hypergastrinemia induces proliferation of ECL and parietal cells directly as well as indirectly via the autocrine or paracrine action of growth factors such as heparin-binding epidermal growth factor, amphiregulin, transforming growth factor-α, metalloproteinases, and regenerating islet-derived 1.68,69 Rats rendered hypergastrinemic by a PPI demonstrate a five-fold increase in the number of ECL cells and a 1.5-fold increase in the number of parietal cells.70 Gastrin acts directly on ECL cells to induce hyperplasia, dysplasia, and eventually neoplasia (carcinoids).71 In contrast to rodents, humans rarely develop carcinoid tumors in response to hypergastrinemia unless other factors are present such as chronic active gastritis or gastrinoma associated with multiple endocrine neoplasia type 1 (see Chapter 31).72 Because ECL cells contain somatostatin subtype 2 receptors (SSTR2), somatostatin scintigraphy with 111indium-diethylenetriamine pentaacetic acid [111In-DTPA]octreotide is the preferred imaging method to detect carcinoid tumors (see Chapter 31).73,74
ACETYLCHOLINE
ACh, released from postganglionic neurons whose cell bodies are located primarily in the submucosal (Meissner’s) plexus, stimulates the parietal cell directly as well as indirectly by inhibiting somatostatin secretion (see Fig. 49-10). The parietal cell muscarinic receptor is of the M3 subtype.75,76 Like CCK-2 receptors, M3 receptors are coupled to activation of phospholipase C with generation of inositol trisphosphate and release of intracellular calcium (see Fig. 49-9).77 Alcoholic beverages produced by fermentation stimulate gastric acid secretion and the effect may be mediated via activation of M3 receptors.78 ACh also stimulates acid secretion indirectly by activating M2 and M4 receptors on D cells coupled to inhibition of somatostatin secretion, thus removing the tonic restraint exerted by this peptide on gastrin, ECL, and parietal cells (see Fig. 49-10).
SOMATOSTATIN
In the stomach, somatostatin cells are closely coupled to their target cells (gastrin cells in the antrum, or parietal and ECL cells in the fundus/body) either directly via cytoplasmic processes or indirectly via the local circulation.28,79 The functional correlate of this anatomic coupling is a tonic restraint exerted by somatostatin on acid secretion from the parietal cell, histamine secretion from the ECL cell, and gastrin secretion from the G cell (see Figs. 49-5 and 49-10).30,31,34,80,81 Removing this restraint (i.e., disinhibition or elimination of the influence of an inhibitor), by activation of cholinergic neurons, is an important physiologic mechanism for stimulating acid secretion (see Fig. 49-10). In the stomach, the actions of somatostatin are mediated primarily via the somatostatin subtype 2 receptor (SSTR2).82–84 Gastrin, GRP, VIP, PACAP, β2/β3-adrenergic agonists, secretin, ANP, adrenomedullin, amylin, adenosine, and CGRP stimulate, whereas ACh and interferon-γ inhibit somatostatin secretion. As mentioned, an increase in luminal acidity acts to attenuate acid secretion via a pathway involving release of somatostatin in the antrum and the fundus. The change in gastric somatostatin secretion can be demonstrated over a range of pH 3 to pH 5, which is within the range observed after ingestion of a meal (Fig. 49-11).85
MISCELLANEOUS PEPTIDES
Ghrelin, the natural ligand for the growth hormone secretagogue receptor, is present in greatest concentrations in gastric oxyntic mucosa and is localized to A-like (or Gr) cells.86,87 Lesser amounts are present in the antrum, small intestine, and colon (see Chapter 1). Plasma ghrelin concentrations increase before meals and decrease postprandially.88 It is postulated that ghrelin triggers premeal hunger and promotes feeding. Its suppression after Roux-en-Y gastric bypass may, in part, contribute to weight loss.89 Most studies report that exogenously administered ghrelin stimulates acid secretion.90,91 The stimulatory effect appears to involve the vagus nerve and histamine release because it is abolished by vagotomy and is associated with an increase in HDC messenger ribonucleic acid (mRNA).92,93
Orexin-A, derived from propro-orexin by post-translational processing, is co-localized with gastrin in human pyloric mucosa.91,94 Intracerebroventricular and peripherally administered orexin-A stimulate gastric acid secretion.95 In rats equipped with gastric fistulas, an orexin receptor 1 antagonist inhibits basal and pentagastrin-stimulated acid secretion, implying that endogenous orexin-A stimulates acid secretion.94,95
ANP, CCK, secretin, neurotensin, glucagon-like peptide 1 (GLP-1), glicentin, oxyntomodulin, peptide YY, adrenomedullin, amylin, glucose-dependent insulinotropic polypeptide (GIP), leptin, epidermal growth factor, and interleukin-1β (IL-1β) inhibit acid secretion. The effect of each, except perhaps for IL-1β, is mediated via release of somatostatin.24–2696 The term enterogastrone has been used to describe the intestinal factor or factors responsible for inhibiting acid secretion in response to nutrients in the intestine. Prime candidates include CCK, secretin, neurotensin, GLP-1, glicentin, and oxyntomodulin because they are present in intestinal mucosa, released into the circulation in response to luminal nutrients, and capable of inhibiting acid secretion at “physiologic” concentrations.97–101 Although it is likely that enterogastrone activity represents the combined influence of several of these peptides, the strongest evidence favors CCK. CCK, produced in I cells in the proximal small intestine, is released by luminal protein and fat. The acid-inhibitory response to intraduodenal fat is abolished by pretreatment with a CCK-1 receptor antagonist in dogs, and the response is blocked in CCK-1 receptor knockout mice as well.102–105
PARIETAL CELL INTRACELLULAR PATHWAYS
In parietal cells, acid secretion is increased by activation of intracellular cAMP- and calcium-dependent signaling pathways that activate downstream protein kinases, ultimately leading to fusion and activation of H+,K+-ATPase (the proton pump) with concomitant activation of luminal membrane conductances for K+ and Cl− (see Fig. 49-9; Fig. 49-12). The H+,K+-ATPase actively pumps out H+ against a tremendous concentration gradient (cell interior pH 7.4 or 40 nM; acid secreted at pH 0.8 or 160 million nM) in exchange for luminal K+. The energy required comes from adenosine triphosphate (ATP) produced by the parietal cell’s extensive mitochondrial network. H+,K+-ATPase consists of an α–subunit that carries out the catalytic and transport function of the enzyme and contains sequences responsible for apical membrane localization106 as well as a β-subunit, which is heavily glycosylated, and protects the enzyme from degradation and is necessary for trafficking to and from the luminal membrane.107
In the resting unstimulated state, H+,K+-ATPase activity is sequestered within cytoplasmic tubulovesicles. On stimulation, there is a dramatic morphologic transformation as these vesicles fuse with the apical plasma membrane, resulting in a 6- to 10-fold increase in the membrane and the formation of the canalicular system (Fig. 49-13). Translocation of the H+,K+-ATPase into the canalicular membrane together with the presence of luminal K+ activates the enzyme.108 On cessation of secretion, the H+,K+-ATPase is retrieved from the apical membrane and the tubulovesicular compartment is reestablished. The precise mechanisms regulating trafficking are not known, but data suggest that it involves actin-based microfilaments, small GTPases, docking/fusion proteins, ezrin, and clathrin.109–111
Acid secretion requires not only a functional H+,K+-ATPase but also apical K+ and Cl− channels and basolateral HCO3− and Cl− exchangers. Acid is produced from the hydration of CO2 to form H+ and HCO3−, a reaction catalyzed by cytoplasmic carbonic anhydrase (see Fig. 49-12). Because the H+,K+-ATPase is unable to pump H+ into the lumen without a parallel uptake of K+, sufficient quantities of K+ must be delivered to the lumen. This K+ recycling is accomplished by luminal KCNE2/KCNQ1 and ROMK (KCNJ1) potassium channels. KCNQ1 is a voltage-activated K+ channel, which, when modified by the small regulatory subunit, KCNE2, becomes voltage insensitive, constitutively open, and acid activated.108,112,113 ROMK may be regulated by the cystic fibrosis transmembrane conductance regulator (CFTR).108,112–114 The concentration of K+ in gastric juice (8 to 12 mM) exceeds plasma K+ by two- to four-fold.
For each H+ secreted, an HCO3− ion exits the cell across the basolateral membrane via the anion exchanger 2 (AE2), Slc4a2 (see Fig. 49-12).115 As a result of this HCO3−/Cl− exchange, the pH within the parietal cell remains only slightly alkaline during acid secretion.116 Rapid entry of HCO3− from parietal cells into blood has been referred to as the alkaline tide. Some of this HCO3− may be taken up and secreted by surface epithelial cells.
Concurrently with H+, Cl− is extruded across the luminal membrane via an apical chloride channel, the precise identity of which is not known. The sources of intracellular Cl− are the basolateral anion exchanger 2 (AE2), sodium-2 chloride potassium-cotransporter-1 (NKCC1), and the SLC26A7 channel (see Fig. 49-12).117,118
PROTON PUMP INHIBITORS
Current PPIs (e.g., omeprazole, lansoprazole, rabeprazole, pantoprazole, and esomeprazole) consist of two heterocyclic moieties, a pyridine and a benzimidazole ring, connected by a methylsulphinyl group (Fig. 49-14). They are weak bases (pKa 4 or 5) that concentrate in acidic spaces within the body that have a pH less than 4. The pKa of a molecule refers to the degree of willingness of the compound to accept or donate a proton and is based on a logarithmic scale such that a compound with a pKa of 5 is 10-fold more basic than a compound with a pKa of 4. When a compound is in an environment with a pH equal to its pKa, half the molecules will be protonated and half will be non-protonated. If a compound with a pKa of 4 is placed in an environment with a pH less than 1, more than 99.9% of the molecules will be protonated. PPIs are membrane permeable in the nonprotonated form and relatively impermeable in the protonated form. In blood (pH 7.4), PPIs are predominantly nonprotonated and thus pass readily into cells (time to reach peak plasma concentration, ≈2 hours; elimination half-life, ≈1 hour), diffuse through the cytoplasm, and then become protonated and trapped, probably as sulfenamides or sulfenic acid, in the acid environment of the secretory canaliculus (Fig. 49-15).119 As a consequence of the basic pKa of PPIs and “ion trapping,” the concentration of PPIs in the secretory canaliculus is 100,000- to 1,000,000-fold higher than in the blood. The sulfenamide rapidly reacts with cysteines on the luminally exposed α-subunit of the H+,K+-ATPase to form a covalent (electrochemical) disulfide bond (see Fig. 49-15).120 Whereas all PPIs bind to cysteine 813, omeprazole also binds to cysteine 892, lansoprazole to cysteine 321, and pantoprazole to cysteine 822. Because only the apical-membrane inserted H+,K+-ATPase is susceptible to blockade by PPIs and an acid environment (pH < 4) is necessary for trapping and converting the PPI to its reactive species, the potency of PPIs is decreased when administered during the basal (fasting) state or when acid secretion is inhibited.121,122 Because most pumps are inserted with breakfast, it is recommended that PPIs be taken 30 minutes to 1 hour before the first meal. If greater inhibition is needed, an additional dose should be taken before dinner. Recovery from inhibition of acid secretion occurs by de novo synthesis of pump protein (54 hours in rat). It has been postulated that reduction of the cysteine disulfide bonds by reducing agents such as glutathione (15 hours in rat) could also play a role.123
INTEGRATED RESPONSE TO A MEAL: INTERPLAY OF NEURAL, PARACRINE, AND HORMONAL PATHWAYS
Stimuli originating inside and outside the stomach converge on gastric intramural efferent neurons that are the primary regulators of acid secretion. The effector neurons comprise cholinergic and peptidergic (i.e., GRP and VIP) neurons. Although nitric oxide and PACAP neurons are present in gastric mucosa their precise physiologic roles as regulators of acid secretion are not known. The effector neurons act on target cells directly as well as indirectly by regulating secretion of gastrin, somatostatin, and possibly histamine (see Figs. 49-8 and 49-10).
During the basal state, acid secretion is maintained at an economically low level by the continuous inhibitory restraint exerted by somatostatin on the ECL cell (histamine) and parietal cell (acid) in the fundus/body and on the G cell (gastrin) in the antrum (see Figs. 49-5, 49-9, and 49-10). During ingestion of a meal, maximal secretion may be achieved by removing the inhibitory influence of somatostatin while at the same time directly stimulating acid and gastrin secretion. This is accomplished, in large part, by activation of intramural cholinergic neurons (see Fig. 49-10). The thought, sight, smell, and taste of food contributes up to 50% of total postprandial acid secretion.124,125 Anticipation of a meal activates central neurons whose input is relayed via the vagus nerve to gastric intramural cholinergic neurons in oxyntic as well as pyloric mucosa. The components of the central nervous system include the dorsal motor nucleus of the vagus, the nucleus tractus solitarius, and the hypothalamus. In the fundus/body, ACh, released from cholinergic neurons, stimulates the parietal cell directly, as well as indirectly, by eliminating the inhibitory paracrine influence of somatostatin on parietal and ECL cells.44,126 The resultant increase in histamine stimulates acid secretion directly via H2 receptors on the parietal cell and indirectly via H3 receptors that mediate suppression of somatostatin secretion (see Fig. 49-10).44,127 Thus, histamine, acting via H3 receptors, amplifies the ability of secretagogues to stimulate acid secretion by suppressing somatostatin secretion. The net effect of cholinergic neurons is suppression of all paracrine inhibitory influence (i.e., somatostatin) and enhancement of paracrine stimulatory influences (i.e., histamine acting via H2 receptors) on parietal cells. There is some evidence that PACAP, a member of the glucagon/VIP superfamily of regulatory peptides, may participate in the regulation of acid secretion, but its precise physiologic role is not known.26,128,129 PACAP is present in gastric mucosal nerves and capable of releasing histamine from ECL cells and somatostatin from D cells. The net effect of exogenous PACAP on acid secretion has been reported to be either stimulation or inhibition, depending on the relative contributions of released histamine and somatostatin in each preparation.128,130,131
In the antrum, cholinergic neurons activated by anticipation of the meal stimulate gastrin secretion directly as well as indirectly by suppressing somatostatin secretion (see Fig. 49-10).30,31,34,132–144 In physiologic concentrations, gastrin stimulates parietal cells directly and, more importantly, indirectly by enhancing the secretion of histamine.145,146
As the meal enters the stomach, the same cholinergic neurons are further activated mechanically by high distention and chemically by protein components of the food.141,142,147,148 Initially, the ingested meal acts as a buffer of secreted acid. The decrease in acidity (i.e., increase in pH) further inhibits somatostatin secretion and thus increases gastrin secretion. Luminal protein activates GRP neurons that directly stimulate gastrin secretion (see Fig. 49-10).79,142 It should be noted that suppression of somatostatin secretion permits an optimal gastrin response.
As the meal empties the stomach, a number of paracrine and neural pathways are activated to restore the inhibitory influence of somatostatin in the fundus/body and antrum and hence restrain acid secretion (see Fig. 49-10). First, a stimulatory paracrine pathway linking gastrin to antral somatostatin cells is activated that acts to restore antral somatostatin secretion after release of gastrin.67 Second, there is less activation of cholinergic neurons by anticipation of the meal as well as by protein and distention. Third, as distention decreases, VIP neurons are preferentially activated that stimulate somatostatin secretion.141 Fourth, as the buffering capacity of the meal is lost, antral and fundic somatostatin cells (via sensory CGRP neurons) are exposed to the full stimulatory effect of luminal acid. Fifth, enterogastrones released from the small intestine, such as CCK, stimulate somatostatin secretion. The resultant increase in antral and fundic somatostatin secretion attenuates gastrin and acid secretion and restores the basal interdigestive state. This state is marked by the continuous restraint exerted on G (gastrin), ECL (histamine), and parietal (acid) cells by contiguous somatostatin cells (see Figs. 49-9 and 49-10). A decrease in this restraint is sufficient to again initiate acid secretion.
HELICOBACTER PYLORI–INDUCED PERTURBATIONS IN ACID SECRETION (see also Chapter 50)
Acute infection with H. pylori (HP) results in hypochlorhydria,149–152 whereas chronic infection results in either hypo- or hyperchlorhydria (Fig. 49-16). Appreciation of the pathways discussed earlier provides some insight into the mechanisms whereby HP colonizes the stomach and may lead to ulceration.
The decrease in acid secretion during acute HP infection is thought to facilitate survival of the organism and its colonization of the stomach.153 The mechanism whereby HP inhibits acid secretion is multifactorial and includes (1) direct inhibition of the parietal cell (and perhaps ECL cell) by a constituent of the bacterium (e.g., vacuolating cytotoxin, lipopolysaccharide, or acid-inhibitory factor) and (2) indirect inhibition of parietal cell function as a result of changes in cytokines as well as hormonal, paracrine, and neural regulatory mechanisms.154–157 HP itself inhibits human H+,K+-ATPase α-subunit gene expression.158
[/not-level-membership-for-gastroenterology-and-hepatology-category]
