[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 65 Gallstone Disease
DEFINITION AND TYPES OF GALLSTONES
Cholesterol cholelithiasis is one of the most prevalent and most costly digestive diseases in western countries. At least 20 million Americans (12% of adults) have gallstones.1–4 The prevalence of gallstones appears to be rising, and each year approximately one million new cases are discovered.5–7 Although many gallstones are “silent,” approximately one third eventually cause symptoms and complications.8 An estimated 700,000 cholecystectomies are performed for gallstone disease each year, and medical expenses for the treatment of gallstones exceeded $6 billion in the year 2000.2 In addition, complications of gallstones result in 3000 deaths (0.12% of all deaths) every year.1
On the basis of chemical composition and macroscopic appearance, gallstones are divided into three types: cholesterol, pigment, and rare stones.3,4,9 The majority (∼75%) of gallstones in the United States and Europe are cholesterol stones,8 which consist of cholesterol monohydrate crystals and precipitates of amorphous calcium bilirubinate, often with calcium carbonate or phosphate in one of the crystalline polymorphs. These stones are usually subclassified as either pure cholesterol or mixed stones that contain at least 50% cholesterol by weight. The remaining gallstones are pigment stones that contain mostly calcium bilirubinate, which are subclassifed into two groups: black pigment stones (∼20%) and brown pigment stones (∼4.5%). Rare stones (∼0.5%) include calcium carbonate stones and fatty acid–calcium stones. Gallstones also are classified by their location into intrahepatic stones, gallbladder stones, and choledocholithiasis (bile duct stones). Intrahepatic stones are predominantly brown pigment stones. Gallbladder gallstones are mainly cholesterol stones, with a small group of black pigment stones. Bile duct stones are composed mostly of mixed cholesterol stones.
EPIDEMIOLOGY
Although determining the true incidence of gallstones in a given population is not easy, a large study of the incidence of gallstones in the Danish population has been reported.10 The five-year incidence of gallstones was 0.3%, 2.9%, 2.5%, and 3.3% for Danish men, and 1.4%, 3.6%, 3.1% and 3.7% for Danish women ages 30, 40, 50, and 60, respectively. Women have a higher incidence than men at age 30 and 40, but the difference declines with increasing age. These incidence rates could reflect genetic and environmental factors in the specific populations studied because they are in accordance with estimated prevalence rates reported for Denmark and other populations.11 In a major Italian study, the incidence of gallstones was obtained at ten years’ follow-up in an originally gallstone-free cohort in the town of Sirmione.12 This study revealed that new cases of gallstones developed at a rate of 0.5% per year. Although age, female gender, parity, obesity, and hypertriglyceridemia were associated with gallstones in the cross-sectional prevalence study of Sirmione, multivariate analyses of risk factors for the formation of gallstones in the longitudinal study identified only age and obesity to be risk factors.
Ultrasonographic screening or necropsy data are often used to estimate the prevalence of gallstone disease in different populations, as illustrated in Figure 65-1. Although ultrasonographic screening cannot be used to distinguish cholesterol from pigment stones, 70% to 80% of detected gallbladder gallstones are assumed to be cholesterol stones.
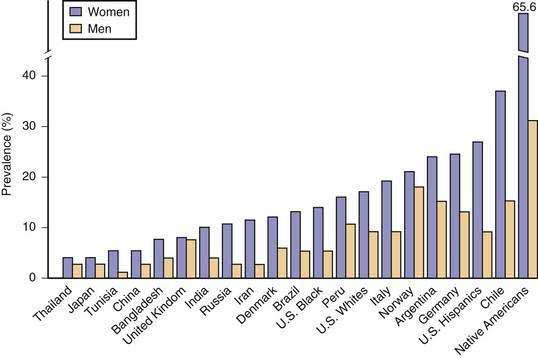
In older studies of American Pima Indians, the prevalence of gallstones was investigated by oral cholecystography.13 The well-studied Pima Indians in southern Arizona display a high prevalence of gallstones, which occur in 70% of the women after the age of 25 years. Subsequently, real-time ultrasonography has been used for screening in nationally representative samples of civilian Mexicans, Hispanic White Americans, non-Hispanic white Americans, and non-Hispanic black Americans of both genders ages 20 to 74. The cross-sectional prevalence rates of gallstones have been found to be highest in certain tribes of Native Americans (e.g., Pima Indians), higher in Hispanic Americans than in whites, and lowest in black Americans.7
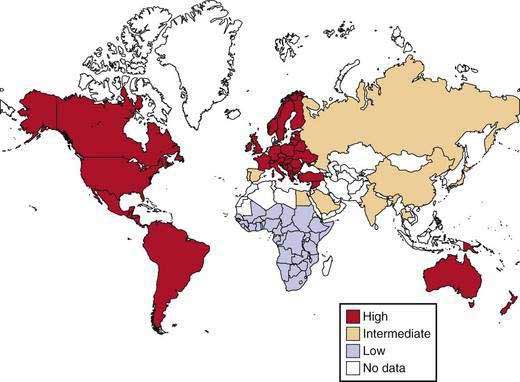
Figure 65-2 displays the world distribution of cholesterol gallstones. American Pima Indians are an extremely high-risk population. Other high-risk populations include Native American groups in North and South America and Scandinavians, of whom 50% develop gallstones by age 50. By contrast, African and Asian populations show the lowest risk of gallstones. Within a given population, first-degree relatives of index cases of persons with gallstones are 4.5 times as likely to form gallstones as matched controls. These epidemiologic investigations underscore the likely role of genetic predisposition in gallstone disease.
RISK FACTORS
Age and Gender
Epidemiologic and clinical studies have reported that cholesterol gallstones occur infrequently in childhood and adolescence, and the prevalence of cholesterol gallstones increases linearly with age in both genders and approaches 50% at age 70 in women.14,15 Furthermore, elderly persons are at higher risk for complications of gallstones, and mortality from surgery is often unacceptably high in patients older than age 65. Cholesterol saturation of bile is significantly higher in elderly Swedes and Chilean women than in younger controls, and age correlates positively with an increased hepatic secretion rate of biliary cholesterol.16,17 In animals, aging has been shown to be associated with increased cholesterol gallstone formation as a result of increased biliary secretion and intestinal absorption of cholesterol, decreased hepatic synthesis and secretion of bile salts, and reduced gallbladder contractility (see later).18
Epidemiologic investigations have found, and clinical studies have confirmed, that at all ages, women are twice as likely as men to form cholesterol gallstones. The difference between women and men begins during puberty and continues through the childbearing years because of the effects of female sex hormones8 and differences between the sexes in how the liver metabolizes cholesterol in response to estrogen. Human and animal studies have suggested that estrogen increases the risk of cholesterol gallstones by augmenting the hepatic secretion of biliary cholesterol, thereby leading to an increase in cholesterol saturation of bile.19–22
Diet
Epidemiologic investigations have shown that cholesterol cholelithiasis is prevalent in populations that consume a Western diet that consists of high amounts of total calories, cholesterol, saturated fatty acids, refined carbohydrates, proteins, and salt, as well as a low amount of fiber. The incidence of cholesterol gallstones is significantly higher in North and South American as well as European populations than in Asian and African populations.3,23 Several clinical studies have found an association between the increased incidence of cholesterol gallstones in China and westernization of the traditional Chinese diet.24 In Japan, cholesterol cholelithiasis was once rare, but since the 1970s, the adoption of Western-type dietary habits has led to a markedly increased incidence.25
Pregnancy and Parity
Pregnancy is a risk factor for the development of biliary sludge and gallstones.26 During pregnancy, bile becomes more lithogenic as a result of increased estrogen levels, which result in increased cholesterol secretion and supersaturated bile. In addition, gallbladder motility is reduced, with a resulting increase in gallbladder volume and bile stasis. These alterations promote sludge and stone formation.27 Increased plasma levels of progestogen also reduce gallbladder motility. Because plasma hormone concentrations increase linearly with duration of gestation, the risk of gallstone formation is especially hazardous in the third trimester of pregnancy. Increasing parity is probably a risk factor for gallstones, especially in younger women.
Rapid Weight Loss
Rapid weight loss is a well-known risk factor for the formation of cholesterol gallstones.28 As many as 50% of obese patients who undergo gastric bypass surgery form biliary sludge and eventually gallstones within six months after surgery. Gallstones also develop in 25% of patients who undergo strict dietary restriction. Furthermore, 40% of these patients display symptoms related to gallstones within the same six-month period. The mechanisms by which rapid weight loss causes gallstone formation include increased hepatic secretion of biliary cholesterol during caloric restriction, increased production of mucin by the gallbladder, and impaired gallbladder motility. Gallstones could be prevented in this high-risk population by prophylactic administration of ursodeoxycholic acid (UDCA). UDCA in a dose of 600 mg/day has been reported to reduce the frequency of gallstones from 28% to 3% in obese patients on a very-low-calorie diet.29
Total Parenteral Nutrition
Total parenteral nutrition (TPN) is associated with the development of cholelithiasis and of acalculous cholecystitis. As early as three weeks after the initiation of TPN, biliary sludge often forms in the gallbladder because of prolonged fasting. In addition, the sphincter of Oddi may fail to relax, leading to preferential flow of bile into the gallbladder. Finally, approximately 45% of adults and 43% of children form gallstones after three to four months of TPN.30,31 Because patients receiving TPN often have serious medical problems and are not good candidates for abdominal surgery, prophylactic treatment to prevent gallstones should be prescribed if no contraindication exists. Cholecystokinin (CCK) octapeptide administered twice daily via an intravenous line to patients on long-term TPN has proved to be safe and cost effective32 and should be used routinely in TPN-treated patients.
Biliary Sludge
Biliary sludge is a crucial intermediate stage in the pathogenesis of both cholesterol and pigment gallstones because it facilitates crystallization and agglomeration of solid cholesterol crystals, as well as precipitation of calcium bilirubinate, and leads ultimately to the development into macroscopic stones.33,34 In addition, biliary sludge can induce acute cholecystitis, cholangitis, and acute pancreatitis. Furthermore, biliary sludge is associated with many conditions that predispose to gallstone formation, including pregnancy, rapid weight loss, spinal cord injury, long-term TPN, and treatment with octreotide.3 Although biliary sludge is reversible in most cases, it persists or disappears and reappears in 12% to 20% of affected persons and eventually leads to gallstones.35 Treatment of patients with persistent biliary sludge with UDCA decreases the frequency of clinical complications of biliary sludge.
Drugs
Estrogens
Most, but not all, relevant studies have shown that the use of oral contraceptive steroids and conjugated estrogens in premenopausal women doubles the frequency of cholesterol gallstones.8,36 The administration of estrogen to postmenopausal women and of estrogen therapy to men with prostatic carcinoma have similar lithogenic effects.36,37 Therefore, estrogen has been proposed to be an important risk factor for the formation of cholesterol gallstones. In mice, the hepatic estrogen receptor α, but not β, plays a crucial role in the formation of cholesterol gallstones in response to estrogen.21 The hepatic estrogen receptor α, which is activated by estrogen, interferes with the negative feedback regulation of cholesterol biosynthesis by stimulating sterol-regulatory element binding protein-2 (SREBP-2), with the resulting activation of the SREBP-2–responsive genes in the cholesterol biosynthetic pathway.22 These alterations result in increased secretion of newly synthesized cholesterol and supersaturation of bile, thereby predisposing to precipitation of cholesterol levels and formation of gallstones. In addition, estrogen leads to a decrease in plasma low-density lipoprotein (LDL) cholesterol levels and an increase in plasma high-density lipoprotein (HDL) cholesterol levels. The decrease in plasma LDL levels is a result of increased expression of the hepatic LDL receptor, which increases the clearance of plasma LDL. The increased uptake of LDL by the liver may also result in increased secretion of cholesterol into the bile. High levels of estrogen may impair gallbladder motility and consequently induce gallbladder hypomotility.
Lipid-Lowering Drugs
Lipid-lowering drugs may influence the formation of gallstones because they regulate key pathways in cholesterol and bile salt metabolism. Clofibrate is a lipid-lowering drug that has a significant association with gallstone formation. Clofibrate induces cholesterol supersaturation in bile and diminishes bile salt concentrations by reducing the activity of cholesterol 7α-hydroxylase (the rate-limiting enzyme in bile salt synthesis).38 The 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) reduce the biliary cholesterol saturation index, but their role in the prevention or therapy of gallstone disease needs to be further investigated.39 The potent cholesterol absorption inhibitor ezetimibe prevents the formation of cholesterol gallstones and facilitate the dissolution of gallstones in gallstone-susceptible C57L mice. Ezetimibe also may act as a potent biliary cholesterol-desaturating agent in patients with gallstones.40 Cholestyramine and nicotinic acid have no statistical association with gallstone formation.
Octreotide
The somatostatin analog octreotide increases the frequency of gallstones when administered to patients as treatment for acromegaly, with approximately 28% of treated acromegalic patients forming gallstones. Acromegalic patients who are treated with octreotide display dysfunctional gallbladder motility, sluggish intestinal transit, and increased colonic deoxycholic acid formation and absorption41; all of these physiologic effects facilitate the formation of cholesterol gallstones.
Ceftriaxone
The third-generation cephalosporin ceftriaxone has a long duration of action, with much of the drug excreted in the urine. Approximately 40% of the drug, however, is secreted in an unmetabolized form into bile, where its concentration reaches 100 to 200 times that of the concentration in plasma and exceeds its saturation level in bile. Once the saturation level of ceftriaxone is exceeded, it complexes with calcium to form insoluble salts, thereby resulting in the formation of biliary sludge. Up to 43% of children who receive high-dose ceftriaxone (60 to 100 mg/kg/day) have been reported to form biliary sludge, and 19% of these patients experience biliary symptoms.42 The sludge usually disappears spontaneously after ceftriaxone is discontinued.
Lipid Abnormalities
Epidemiologic investigations have shown that plasma HDL cholesterol levels are inversely correlated with the frequency of cholesterol gallstones.43 By contrast, hypertriglyceridemia is positively associated with an increased frequency of gallstones.44 These seemingly independent variables are actually interrelated because high plasma triglyceride levels tend to increase with increasing body mass and are inversely correlated with plasma HDL levels. Interestingly, high plasma total and LDL cholesterol levels are not likely to be risk factors for the formation of gallstones.
Systemic Diseases
Obesity
Obesity is a well-known risk factor for cholelithiasis. A large prospective study of obese women demonstrated a strong linear association between the body mass index (BMI) and frequency of cholelithiasis.45 In this study,45 the risk of gallstones was seven-fold higher in women with the highest BMI (>45 kg/m2) than in nonobese control women.47 Obesity is associated with increased hepatic secretion of cholesterol into bile, possibly because of higher levels of HMG-CoA reductase activity (the rate-limiting enzyme in cholesterol synthesis), which lead to high levels of cholesterol biosynthesis in the liver. Studies that compare the amounts of pronucleating and antinucleating factors in the gallbladder bile of obese and nonobese subjects have not been performed, nor have studies of gallbladder motility in obese persons.
Diabetes Mellitus
Patients with diabetes mellitus have long been assumed to be at an increased risk of gallstones because hypertriglyceridemia and obesity are risk factors for gallstones and are associated with diabetes mellitus and because gallbladder motility is often impaired in patients with diabetes mellitus. Proving that diabetes mellitus is an independent risk factor for gallstones has been difficult, however. Mice with hepatic insulin resistance induced by liver-specific disruption of the insulin receptor are markedly predisposed toward the formation of cholesterol gallstones because of increased expression of the biliary cholesterol transporters Abcg5 and Abcg8, with a resulting increase in hepatic sterol secretion, and decreased expression of the bile acid synthetic enzymes, particularly Cyp7b1, with a resulting lithogenic bile salt profile.46
Diseases of the Ileum
Disease or resection of the terminal ileum is considered a risk factor for gallstone formation. For example, intestinal bile salt absorption is often impaired in patients with Crohn’s disease, who are at increased risk of gallstones.47 The loss of specific bile salt transporters (e.g., ileal apical sodium/bile salt transporter) in the terminal ileum may result in excessive bile salt excretion in feces and a diminished bile salt pool size, presumably with a consequent increase in the risk of cholesterol gallstones. These changes may lead, however, to the formation of pigment gallstones because increased bile salt delivery to the colon enhances solubilization of unconjugated bilirubin, thereby increasing bilirubin concentrations in bile.48
COMPOSITION AND ABNORMALITIES OF BILE
PHYSICAL CHEMISTRY OF BILE
Chemical Composition of Bile
The primary bile salts are hepatic catabolic products of cholesterol and are composed of cholate (a trihydroxy bile salt) and chenodeoxycholate (a dihydroxy bile salt) (see Chapter 64). The secondary bile salts are derived from the primary bile salt species by the action of intestinal bacteria in the ileum and colon and include deoxycholate, ursodeoxycholate, and lithocholate. The most important of the conversion reactions is 7α-dehydroxylation of primary bile salts to produce deoxycholate from cholate and lithocholate from chenodoxycholate. Another important conversion reaction is the 7α-dehydrogenation of chenodeoxycholate to form 7α-oxo-lithocholate. This bile salt does not accumulate in bile but is metabolized by hepatic or bacterial reduction to form the tertiary bile salt chenodeoxycholate (mainly in the liver) or its 7β-epimer ursodeoxycholate (primarily by bacteria in the colon).
Phase Diagrams and Cholesterol Solubility in Bile
In the 1960s, Small and colleagues defined the maximal solubility (saturation) limits for cholesterol in model quaternary bile systems that consisted of varying proportions of cholesterol, phospholipids, bile salts, and water.49,50 The relative proportions (as molar percentages) of the three lipids in bile play a critical role in determining the maximal solubility of cholesterol. When the relative proportions of the three lipids at a fixed total lipid concentration are plotted in a triangular coordinate, the solubility of cholesterol for any given solute concentration can be determined.51 The triangular coordinate diagram also illustrates the physical phases of cholesterol in bile. For example, the phase diagram shown in Figure 65-3 is specific for a total lipid concentration of 7.5 g/dL, which is typical of human gallbladder bile.52,53 For hepatic bile, with a typical total lipid concentration of 3 g/dL, the phase boundaries would be different, with a smaller micellar zone, all phase boundaries shifted to the left, and an expanded two-phase zone on the right (i.e., region E in Figure 65-3). The effect of the total lipid content on cholesterol solubilization in the micellar zone explains why hepatic bile tends to be more saturated with cholesterol than is gallbladder bile in the same subject. Because hepatic bile contains cholesterol-phospholipid vesicles that are relatively stable, solid cholesterol monohydrate crystals never occur in hepatic bile.
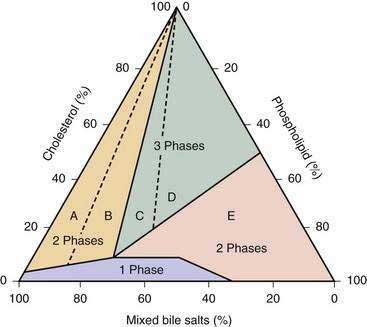
Within the micellar zone (see Fig. 65-3), bile is a visually clear, stable solution that is considered unsaturated because all cholesterol can be solubilized in thermodynamically stable simple and mixed micelles. At the boundary line of the micellar zone, bile is saturated because all the solubilizing capacity for cholesterol is utilized and no further cholesterol can be carried in micelles. Outside the micellar zone, bile is supersaturated because excess cholesterol cannot be solubilized by micelles51,54 and exists in more than one phase (micelles, liquid crystals, and solid monohydrate crystals); the solution is visually cloudy. Obviously, relatively stable unilamellar cholesterol-phospholipid vesicles solubilize a significant proportion of cholesterol outside the micellar zone. The term metastable zone refers to the area in the phase diagram (above but near the micellar zone) in which bile is supersaturated with cholesterol but may not form solid cholesterol crystals even after many days. The diagram also suggests that when the quantity of cholesterol in bile exceeds that which can be solubilized by the available bile salts and phospholipids, solid cholesterol crystals precipitate in bile. Furthermore, the proportional distance outside the micellar zone directed along an axis joined to the cholesterol apex is often calculated as the cholesterol saturation index (CSI) (or lithogenic index).54 Therefore, the degree of saturation of bile with cholesterol can be quantitated. A CSI for a sample of bile can be estimated directly from the diagram or calculated by using a formula. The CSI is the ratio of the actual amount of cholesterol present in a bile sample to the maximal amount of cholesterol that can be dissolved in it. Bile that has a CSI of 1 is saturated; bile with a saturation index less than 1 is unsaturated; and bile with a saturation index greater than 1 is supersaturated. The degree of saturation can also be expressed as percent saturation by multiplying the saturation index by 100. For example, at the boundary of the micellar zone, bile is saturated, and the CSI is 100%. Supersaturated bile has a CSI above 100%, and unsaturated bile has a CSI below 100%. The CSI values are also useful for predicting the proportion of lipid particles and the metastable and equilibrium physical-chemical states in bile.
HEPATIC SECRETION OF BILIARY LIPIDS
Source of Lipids Secreted in Bile
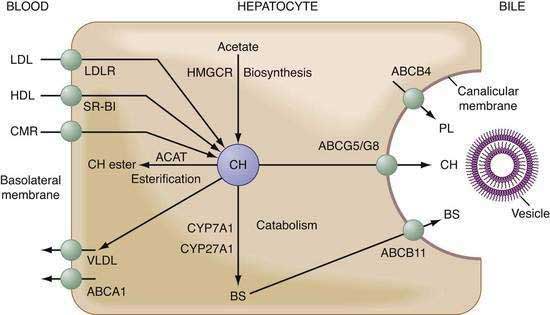
The supply of hepatic cholesterol molecules that can be recruited for biliary secretion depends on the balance of input and output of cholesterol and its catabolism in the liver (see also Chapter 72) (Fig. 65-4). Input is related to the amount of cholesterol (both unesterified and esterified) taken up by the liver from plasma lipoproteins (LDL > HDL > chylomicron remnants) plus de novo hepatic cholesterol synthesis. Output is related to the amount of cholesterol disposed of within the liver by conversion to cholesteryl ester (to form new very-low-density lipoprotein [VLDL] and for storage) minus the amount of cholesterol converted to primary bile salts. An appreciable fraction of cholesterol in bile also may be derived from the diet via apolipoprotein E–dependent delivery of chylomicron remnants to the liver. Under low or no dietary cholesterol conditions, bile contains newly synthesized cholesterol from the liver and preformed cholesterol that reaches the liver in several different ways. Approximately 20% of the cholesterol in bile comes from de novo hepatic biosynthesis, and 80% is from pools of preformed cholesterol within the liver. De novo cholesterol synthesis in the liver uses acetate as a substrate and is regulated mainly by the rate-limited enzyme HMG-CoA reductase. This enzyme can be up- or down-regulated depending on the overall cholesterol balance in the liver. An increase in the activity of this rate-limiting enzyme leads to excessive cholesterol secretion in bile. The major sources of preformed cholesterol are hepatic uptake of plasma lipoproteins (mainly HDL and LDL through their receptors on the basolateral membrane of hepatocytes). Consistent with their central role in reverse cholesterol transport, HDL particles are the main lipoprotein source of cholesterol that is targeted for biliary secretion. Under conditions of a diet high in cholesterol, dietary cholesterol reaches the liver through the intestinal lymphatic pathways as chylomicrons and then chylomicron remnants, after chylomicrons are hydrolyzed by plasma lipoprotein lipase and hepatic lipase. The synthesis of new cholesterol in the liver is reduced and comprises only approximately 5% of biliary cholesterol. Overall, the liver can systematically regulate the total amount of cholesterol within it, and any excess cholesterol is handled efficiently.
More than 95% of bile salt molecules, after secretion into bile, return to the liver through the enterohepatic circulation by absorption mostly from the distal ileum via an active transport system (see also Chapter 64). Consequently, newly synthesized bile salts in the liver contribute only a small fraction (<5%) to biliary secretion and compensate for bile salts that escape intestinal absorption and are lost in feces. The fecal excretion of bile salts is increased when the enterohepatic circulation of bile salts is partially or completely interrupted by surgery, disease states, or drugs (e.g., bile salt-binding resins such as cholestyramine). Complete interruption of the enterohepatic circulation results in up-regulation of bile salt synthesis in the liver, which restores bile salt secretion rates to approximately 25% of their usual values. Cholesterol from two sources serves as substrate for de novo bile salt synthesis—cholesterol that is newly synthesized in the smooth endoplasmic reticulum and cholesterol that is preformed outside the smooth endoplasmic reticulum. The first step in this process is catalyzed by cholesterol 7α-hydroxylase. In the basal state, de novo bile salt synthesis uses principally newly synthesized cholesterol as substrate. When de novo cholesterol biosynthesis is suppressed by long-term therapy with a HMG-CoA reductase inhibitor, preformed cholesterol originating from plasma lipoprotein substitutes for newly synthesized cholesterol.
Biliary Lipid Secretion
Bile salts have been shown to stimulate secretion of vesicles, which are always detected in freshly collected hepatic bile.55,56 When cultured under specified conditions, rat hepatocytes form couplets with isolated “bile canaliculi” at the interface between adjoining cells. With the use of laser light-scattering techniques, vesicle formation can be observed within these bile canaliculi after exposure to bile salts. In addition, rapid fixation techniques and electronic microscopy have provided direct morphologic evidence of vesicle formation at the outer surface of the canalicular membrane.57,58 Most, if not all, bile salts are thought to enter canalicular spaces as monomers, whereas biliary phospholipids and cholesterol enter as unilamellar vesicles (see Fig. 65-4). A study of the molecular genetics of sitosterolemia (see Chapter 64) has shown that efflux of biliary cholesterol from the canalicular membrane is protein mediated. Two plasma membrane proteins—adenosine triphosphate (ATP)-binding cassette (ABC) transporters ABCG5 and ABCG8—promote cellular efflux of cholesterol. The significance of this process for bile formation has been examined in genetically modified mice, in which overexpression of abcg5 and abcg8 in the liver was shown to increase the cholesterol content of gallbladder bile.59–63 Despite a reduced frequency of gallstones, the formation of gallstones is still observed in abcg5/g8 double-knockout mice, as well as in abcg5 or abcg8 single-knockout mice, challenged with a lithogenic diet.59–63 These findings strongly suggest the existence of an ABCG5/G8-independent pathway for hepatic secretion of biliary cholesterol and its role in the formation of cholesterol gallstones. In addition, scavenger receptor class B type I (SR-BI) is localized in sinusoidal, and possibly, canalicular, membranes of the hepatocyte, and in transgenic and knockout mice fed a chow diet, biliary secretion of cholesterol varies in proportion to hepatic expression of SR-BI and to the contribution of SR-BI to sinusoidal uptake of HDL cholesterol destined for secretion into bile.64,65
Deletion of the Abcb4 gene completely inhibits hepatic secretion of biliary phospholipids in mice,66 suggesting that ABCB4 could be responsible for the translocation, or “flip,” of phosphatidylcholine from the endoplasmic (inner) to ectoplasmic (outer) leaflet of the canalicular membrane bilayer and that the action of ABCB4 may form phosphatidylcholine-rich microdomains within the outer membrane leaflet. Although the ectoplasmic leaflet of the canalicular membrane is cholesterol- and sphingomyelin-rich and is relatively resistant to penetration by bile salts, bile salts may promote vesicular secretion of biliary cholesterol and phosphatidylcholine. Bile salts may partition preferentially into these areas to destabilize the membrane and release phosphatidylcholine-rich vesicles because detergent-like bile salt molecules within the canalicular space could interact with canalicular membrane. Mutations of the ABCB4 gene in humans result in the molecular defect underlying type 3 progressive familial intrahepatic cholestasis (see Chapter 76).67
Biliary bile salts include those that are newly synthesized in the liver and those that undergo enterohepatic cycling. The precise molecular mechanism of bile salt secretion is not known, although it probably involves ABCB11, a bile salt export pump (see Chapter 64).68–70 Although hepatic secretion of biliary bile salts directly affects cholesterol-phospholipid vesicle secretion, whether bile salt secretion is coupled to cholesterol and phospholipid secretion at a molecular level is not known. The relationship between bile salt secretion and cholesterol secretion is curvilinear: At low bile salt secretion rates (usually less than 10 µmol/hr/kg), more cholesterol is secreted per molecule of bile salt than at higher rates. Although bile salt secretion rates are not low in normal subjects, they could diminish during prolonged fasting, during the overnight period, and with substantial bile salt losses, as occur with a biliary fistula or ileal resection when the liver cannot compensate sufficiently by increasing bile salt synthesis. At high bile salt secretion rates, for example, during and after eating, biliary cholesterol saturation is less than that during interprandial periods. In laboratory animals, biliary secretion of organic anions does not influence bile salt secretion but does inhibit secretion of phospholipid and cholesterol into bile because organic anions bind bile salts within bile canaliculi and prevent interactions with the canalicular membrane.
PATHOPHYSIOLOGY
As shown in Figure 65-5, at least five primary defects must be present simultaneously for cholesterol gallstone formation: certain genetic factors, including LITH genes (see later); hepatic hypersecretion; gallbladder hypomotility; rapid phase transitions; and certain intestinal factors.
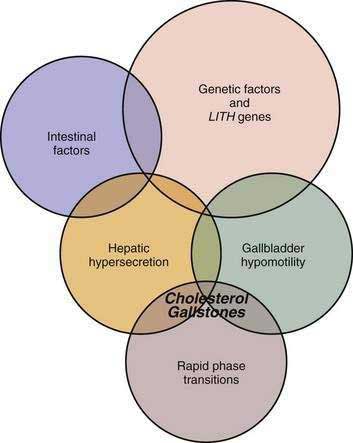
Figure 65-5. Venn diagram of five primary defects that must be present simultaneously in gallbladder bile for cholesterol crystallization to take place. The five defects are genetic factors and LITH (gallstone) genes, hepatic hypersecretion of cholesterol, gallbladder hypomotility, rapid phase transitions, and intestinal factors. The hypothesis proposed is that hepatic cholesterol hypersecretion into bile is the primary defect and is the outcome in part of a complex genetic predisposition. The downstream effects include gallbladder hypomotility and rapid phase transitions (see Fig. 65-3). A major result of gallbladder hypomotility is alteration in the kinetics of the enterohepatic circulation of bile salts (intestinal factors). The alterations in the intestinal factors result in increased cholesterol absorption as well as reduced bile salt absorption that lead to abnormal enterohepatic circulation of bile salts and diminished biliary bile salt pool size. Not only does gallbladder hypomotility facilitate nucleation, but it also allows the gallbladder to retain cholesterol monohydrate crystals. Although a large number of candidate Lith genes have been identified in mouse models, the identification of human LITH genes and their contributions to gallstones require further investigation.
RAPID CHOLESTEROL NUCLEATION AND CRYSTALLIZATION
Cholesterol nucleation and crystallization is a process by which solid plate-like cholesterol crystals precipitate from supersaturated bile. The crystals can be detected by polarizing light microscopy in a sample of bile previously rendered crystal-free (“isotropic”).71 Bile from patients with cholesterol gallstones and from controls is supersaturated with cholesterol, and the degree of cholesterol supersaturation is not a reliable predictor of gallstones. On the other hand, rapid in vitro nucleation and crystallization of cholesterol monohydrate crystals from the isotropic phase of gallbladder bile distinguishes the lithogenic bile of patients with cholesterol gallstones from cholesterol-supersaturated bile of non-gallstone control subjects.71 The phase diagram of cholesterol, phospholipids, and bile salts discussed earlier (see Fig. 65-3) is often used to study the phase transitions where metastable intermediates form. Five crystallization pathways can be identified on the basis of the bile salt-to-phospholipid ratio, total lipid concentration, bile salt species (hydrophilic and hydrophobic properties), temperature, and CSI.52,72 Furthermore, these crystallization pathways have been confirmed in fresh human and mouse gallbladder bile.52,72,73 In Figure 65-3, for the cholesterol-phospholipid-mixed bile salt model system, the five distinct crystallization pathways are designated A to E, with each representing a different sequence of phase transitions, including an anhydrous cholesterol pathway and a liquid crystalline pathway leading to the formation of solid plate-like cholesterol monohydrate crystals.52,72 Transient arc-like crystals appear in some of the pathways and are consistent with crystalline anhydrous cholesterol (see later).74,75 Why anhydrous cholesterol crystals should precipitate in an aqueous environment is unknown, but they are characteristic of the pathways that seem to originate from unilamellar, as opposed to multilamellar, vesicles. In these pathways, the critical nucleus may be a unilamellar vesicle that contains liquid anhydrous cholesterol molecules in its core, possibly reflecting internal nucleation. In essence, these early vesicular “nuclei” may already have initiated the nucleation cascade by the time bile enters the gallbladder. The current paradigm for nucleation and crystallization, based principally on observations from video-enhanced polarized light microscopy, suggests that biliary vesicles must fuse or at least aggregate to form crystalline cholesterol monohydrate. Because cholesterol nucleation and crystallization are apparently initiated in vesicles, the stability of the vesicle determines the stability of bile. Unstable vesicles can fuse, aggregate, and grow into multilamellar liquid crystalline structures (liposomes) in which cholesterol crystallizes out of solution. Furthermore, evidence from quasi-elastic light-scattering spectroscopy shows that nucleation of solid cholesterol crystals may occur directly from supersaturated micelles in conjugated deoxycholate-rich bile in vitro without an intervening vesicle or liquid crystalline phase.
In bile with the lowest phospholipid contents (region A in Fig. 65-3), arc-like crystals with a density (d = 1.030 g/mL) consistent with anhydrous cholesterol appear first and evolve via helical and tubular crystals to form plate-like cholesterol monohydrate crystals (d = 1.045 g/mL).52,74,75 With higher phospholipid contents (region B), cholesterol monohydrate crystals appear earlier than arc-like crystals and other transitional crystals. With typical physiologic phospholipid contents (region C), early liquid crystals (d = 1.020 g/mL) are followed by cholesterol monohydrate crystals; subsequently, arc-like and other intermediate crystals appear. With still higher phospholipid contents (region D), liquid crystals are followed by cholesterol monohydrate crystals only. At the highest phospholipid mole fractions (region E), liquid crystals are stable and no solid crystals form. Decreases in temperature (37°C→4°C), total lipid concentration (7.5 g/dL → 2.5 g/dL), and bile salt hydrophobicity (3α,12α→3α,7α→3α,7α,12α→3α,7β hydroxylated taurine conjugates) progressively shift all crystallization pathways to lower phospholipid contents, reduce micellar cholesterol solubilization, and retard crystallization.52,72
Cholesterol crystallization pathways and sequences in human gallbladder bile are identical to those of model bile samples matched for appropriate physical-chemical conditions, and in a physiologic state, three of the five sequences observed in model bile samples are found in human and mouse gallbladder bile.72 Most notably, the kinetics of all these phase transitions are faster in lithogenic human bile than in identically patterned model bile samples, most likely a result, in part, of the combined influences of increased levels of cholesterol, secondary bile salts, and mucin glycoproteins.53 In addition, biliary lipid, electrolyte, and protein factors may be important in stabilizing supersaturated bile. Nonprotein factors that retard cholesterol nucleation and crystallization include (1) a total lipid concentration less than 3 g/dL; (2) reduced hydrophobicity of the bile salt pool; (3) low bile salt-to-lecithin ratios; (4) low cholesterol-to-lecithin ratios in vesicles; and (5) low total calcium ion concentrations. The states opposite to these conditions accelerate nucleation and crystallization.76
IMBALANCE OF PRONUCLEATING AND ANTINUCLEATING FACTORS
More rapid crystallization of cholesterol in the bile of patients with gallstones implies that lithogenic bile may contain pronucleating agents that accelerate crystallization or that normal bile may contain antinucleating agents that inhibit crystallization. Furthermore, bile may contain both accelerators and inhibitors of crystallization, and imbalances between them can induce rapid crystallization in gallbladder bile in patients with cholesterol gallstones.77,78
Mucin was the first biliary protein shown to promote cholesterol crystallization.79 The epithelial cells of the gallbladder secrete mucin that normally serves as a protective layer over the mucosa. Mucin or mucin glycoproteins are large molecules that consist of a protein core and many carbohydrate side chains.80 An important property of mucin is its ability to form a gel phase in higher concentrations, and the gel has greatly increased viscosity compared with the sol (soluble) phase. Gallbladder mucins, a heterogeneous family of O-linked glycoproteins, are divided into two classes: epithelial and gel-forming mucins.81 The epithelial mucins, which are produced by mucin gene 1 (MUC1), MUC3, and MUC4, do not seem to form aggregates and are integral membrane glycoproteins located on the apical surface of epithelial cells.82–85 The gel-forming mucins, MUC2, MUC5AC, and MUC5B, which are secreted by specialized gallbladder mucin-producing cells, provide a protective coating on the underlying mucosa.82–85 They form disulfide-stabilized oligomers or polymers, a phenomenon that accounts for their viscoelastic properties. Mucins from different organs vary in carbohydrate side chain, protein composition, and charge but generally have similar properties. Mucins have hydrophilic domains to which many water molecules bind. They have an overall charge and are capable of binding other charged species such as calcium. Hydrophobic domains in the mucin molecule (on the nonglycosylated regions of the polypeptide core) allow binding of lipids such as cholesterol, phospholipids, and bilirubin. Evidence suggests that gallbladder mucins play an important role in the early stages of gallstone formation and are a potent pronucleating agent for accelerating cholesterol crystallization in native and model biles. Indeed, hypersecretion of gallbladder mucins is a prerequisite for gallstone formation, and increased amounts of gallbladder mucins are consistently observed in gallbladder bile of several animal models of gallstones.73,79,86 Mucins are also found within gallstones, where they act as a matrix for stone growth.87 The mucins in gallstones have been found to extend from the amorphous center to the periphery in either a radial or laminated fashion. Mucins are also a major component of sludge in the gallbladder, and sludge has been suggested to be a precursor of gallstones. Therefore, two roles in the formation of gallstones have been proposed for mucins (1) a pronucleating agent for the nucleation and crystallization of cholesterol from saturated bile and (2) a scaffolding for the deposition of crystals during the growth of stones.
The synthesis of mucin glycoproteins that are secreted by the epithelium of the gallbladder and biliary ducts may be regulated by mucosal prostaglandins that are derived from arachidonic acid–containing biliary phospholipids.80 During the formation of gallstones, the gallbladder hypersecretes mucins, mostly as a result of stimulation by some components of saturated bile. Then, the carbohydrate groups of the polymers of mucins avidly bind water to form gels. The hydrophobic polypeptides in the core of mucin glycoproteins also can bind bilirubin and calcium in bile. The resulting water-insoluble complex of mucin glycoproteins and calcium bilirubinate provides a surface for nucleation of cholesterol monohydrate crystals and a matrix for the growth of stones.
Secretion and accumulation of mucin in the gallbladder is controlled by multiple mucin genes. MUC1 mucin in the gallbladders of mice has been shown to be reduced with disruption of the Muc1 gene, with a consequent decrease in susceptibility to cholesterol gallstone formation.88 Also, gene expression of the gallbladder Muc5ac gel-forming mucin gene is significantly reduced in Muc1-knockout mice in response to a lithogenic diet. As a result, cholesterol crystallization and the development of gallstone formation are significantly retarded. These findings suggest that gene-gene interactions between the Muc1 and Muc5ac genes might affect mucin secretion and accumulation in the gallbladder. Furthermore, increased gallbladder epithelial Muc1 mucin enhances cholelithogenesis mostly by promoting gallbladder cholesterol absorption and impairing gallbladder motility in mice that are transgenic for the human MUC1 gene; this lithogenic mechanism is completely different from that associated with the gel-forming mucins.89 Collectively, these findings support the concept that inhibition of the secretion and accumulation of not only the gel-forming mucins, but also the epithelial mucins in the gallbladder may completely prevent the formation of cholesterol gallstones.
Many glycoproteins that bind reversibly to concanavalin A-Sepharose also speed up cholesterol crystallization.90 These glycoproteins include aminopeptidase N, immunoglobulins, α1-acid glycoprotein, phospholipase C, fibronectin, and haptoglobin. Other pronucleating agents are the amphipathic anionic polypeptide fraction/calcium-binding protein, albumin-lipid complexes, and group II phospholipase A2. Nonprotein components of bile also expedite cholesterol crystallization. Calcium bound to micelles and vesicles in bile may accelerate cholesterol crystallization by promoting fusion of cholesterol-rich vesicles. The precipitation of calcium salts in bile that is supersaturated with calcium salts and cholesterol may lead to rapid crystallization of cholesterol, an effect that is enhanced by the presence of mucins. The rapidity of cholesterol crystal formation also varies in proportion to the deoxycholate content of bile and is related to the effect of deoxycholate on the equilibrium phase relationships of biliary lipids. The degree of cholesterol supersaturation of bile may also be a determinant of rapid crystallization of cholesterol.
Several inhibitors of cholesterol crystallization have been identified, including apolipoproteins AI and AII, a 120-kd glycoprotein, a 15-kd protein, and secretory immunoglobulin A and its heavy and light chains.91–93 Apolipoproteins AI and AII may prolong crystal detection time of supersaturated model bile. Apolipoproteins AI and AII are present in a fraction of human bile that may inhibit cholesterol nucleation and crystallization. Precholecystectomy treatment with UDCA for three months prolongs the crystal detection time of bile from patients with cholesterol gallstones, suggesting also that UDCA could be an antinucleating factor.52,94–96 UDCA may exert its effect by stabilizing vesicles, perhaps by enhancing the incorporation of apolipoprotein AI into (or onto) the vesicles. In addition, a potential antinucleating factor from normal human gallbladder bile is detected by lectin affinity chromatography and high performance liquid ion-exchange chromatography and found to be a slightly acidic glycoprotein with an apparent molecular size of 120 kd. The protein may inhibit crystal growth by attaching to the most rapidly growing microdomains on a crystal face and interfering with further solute attachment. Whether only one or several antinucleating factors exist and how they may inhibit the initiation of cholesterol crystal formation are uncertain, but unilamellar vesicles have been proposed to be the key sites of action.
GALLBLADDER DYSFUNCTION
Under normal physiologic conditions, frequent gallbladder contractions occur throughout the day. Between meals, the gallbladder stores hepatic bile (with an average fasting volume of 25 to 30 mL in healthy subjects). Following a meal, depending on the degree of neurohormonal response, the gallbladder discharges a variable amount of bile.97 Use of a combined method of cholescintigraphy and ultrasonography has demonstrated that after a meal, the gallbladder empties immediately and refills repeatedly.97 By contrast, an increased fasting gallbladder volume as well as incomplete emptying and high residual gallbladder volume are often observed in patients with cholesterol gallstones, whether they have tiny or large stones or simply lithogenic bile. In this group of patients with cholesterol gallstones and gallbladder motility abnormalities, gallbladder wall inflammation is usually mild and cannot account for the impaired dynamics of the gallbladder. Furthermore, the poor interdigestive gallbladder filling is consistent with delivery of a greater percentage of lithogenic bile from the liver directly into the small intestine, with augmentation of the enterohepatic effects of increased recycling and bile salt hydrophobicity. This observation suggests that emptying and filling of the gallbladder are affected in patients with gallbladder hypomotility.97,98 Clinical investigations confirm that gallbladder hypomotility is associated principally with the formation of cholesterol gallstones, although a milder degree of gallbladder dysmotility, in the absence of an enlarged gallbladder in the fasting state and any gallbladder inflammation, is also found in patients with pigment gallstones.99 In patients with cholesterol gallstones, impaired gallbladder motility persists in the stone-free gallbladder following successful extracorporeal shock-wave lithotripsy and oral bile acid dissolution therapy.100,101 The degree of impairment of gallbladder emptying has been found to increase in proportion to the cholesterol content of gallbladder bile, even in healthy subjects without gallstones. These findings suggest that excess cholesterol molecules in the gallbladder wall may act as myotoxic agents.
In vitro studies in which gallbladder function was compared in patients with cholesterol gallstones and control subjects have shown abnormalities in the binding of agonists such as CCK to plasma membrane CCK-1 receptors, alterations in contraction of isolated smooth muscle cells, and decreased contractility of isolated smooth muscle strips and whole gallbladder preparations. In particular, signal transduction in response to binding of agonists is impaired. Defects in contractility associated with cholesterol gallstones are reversible at an early stage and are attributable primarily to excess accumulation of biliary cholesterol in the membranes of gallbladder smooth muscle cells. This mechanism appears to explain why gallbladder emptying is impaired before gallstones are formed in animal models at a time when bile is supersaturated with cholesterol. In addition, the intracellular mechanisms of smooth muscle contraction in human gallbladder muscle cells from patients with cholesterol gallstones seem to be intact. These findings support the hypothesis that the absorption of cholesterol from the gallbladder lumen is associated with gallbladder smooth muscle dysfunction. This alteration may induce stiffening of sarcoplasmic membranes secondary to an increase in cholesterol content of the membranes. As a result, when CCK binds to its receptor on smooth muscle cells of the lithogenic gallbladder, G-proteins are not activated and gallbladder motility is impaired.102,103
Gallbladder hypomotility could precede gallstone formation. Gallbladder stasis induced by the hypofunctioning gallbladder could provide the time necessary to accommodate nucleation of cholesterol crystals and growth of gallstones within the mucin gel in the gallbladder.104,105 Furthermore, the viscous mucin gel that forms within the gallbladder may contribute to hypomotility by impairing gallbladder emptying mechanically, possibly at the level of the cystic duct. In particular, sludge contains calcium, pigment, bile salts, and glycoproteins and could serve as a nidus for nucleation and crystallization of cholesterol or precipitation of calcium bilirubinate. The high frequency of cholelithiasis in patients receiving long-term TPN highlights the importance of gallbladder stasis in the formation of gallstones.106 For example, 49% of patients with Crohn’s disease who are on TPN have gallstones, whereas Crohn’s disease alone leads to gallstones in 27% of patients. During TPN, the gallbladder does not empty completely because the stimulus (ingestion of meals) for the release of CCK is eliminated. As a result, bile stagnates and sludge develops in the gallbladder, thereby enhancing the formation of gallstones. Daily intravenous administration of CCK can completely prevent gallbladder dysmotility and eliminate the inevitable risk of biliary sludge and gallstone formation. In addition, slow emptying and increased volume of the gallbladder, as measured by ultrasonography, occur during pregnancy and during administration of oral contraceptives, two conditions that predispose to the formation of gallstones (see earlier).20,21
The concentration of bile by the gallbladder increases cholesterol solubility; however, it also enhances cholesterol nucleation and crystallization in bile, thereby suggesting that increased concentration of bile is a contributing factor for gallstone formation.107,108 In addition to concentrating bile, the normal gallbladder can also acidify bile. Acidification increases the solubility in bile of calcium salts (e.g., bilirubinate and carbonate), which may be promoters of nucleation and crystallization. Therefore, defective acidification may have an effect on the formation of gallstones.
Differential absorption rates of cholesterol, phospholipids, and bile salts by the gallbladder epithelial cells may reduce cholesterol saturation of bile in normal subjects; however, the gallbladder epithelium of patients with cholesterol gallstones loses the capacity for selective absorption of biliary cholesterol and phospholipids.109,110 Impaired lipid absorption by the gallbladder may contribute to gallstone formation by sustaining cholesterol supersaturation of bile during storage.111 The physical-chemical fate of cholesterol absorbed by the gallbladder may be similar to that which occurs during the development of an atherosclerotic plaque. In all likelihood, cholesterol molecules are absorbed continuously by the gallbladder mucosa from supersaturated bile,112 and the unesterified cholesterol molecules diffuse rapidly to the muscularis propria because the gallbladder lacks an intervening muscularis mucosae and submucosa. Because the gallbladder apparently does not synthesize lipoproteins for exporting cholesterol to plasma, excess unesterified cholesterol molecules are removable from gallbladder mucosa and muscle only by esterification and storage or back diffusion into bile.113 In the lithogenic state, back diffusion of cholesterol molecules into bile is blocked because gallbladder bile is continuously saturated. As a result, gallbladder mucosal acyl-coenzyme A:cholesterol acyltransferase (ACAT) esterifies most, but not all, cholesterol molecules. As in an atherosclerotic plaque, mucosal and muscle membranes apparently become saturated with cholesterol and coexist with stored cholesteryl ester droplets. Furthermore, the unesterified cholesterol molecules become intercalated within the membrane bilayer of muscle cells, a process that may alter the physical state of phospholipid molecules, as reflected by their increased rigidity. Consequently, gallbladder motility function is impaired because signal transduction in response to CCK is diminished markedly. In addition, excess cholesterol molecules absorbed from the lithogenic bile may be direct stimulants to proliferative and inflammatory changes in the mucosa and lamina propria of the gallbladder.97
INTESTINAL FACTORS
The high efficiency of intestinal cholesterol absorption correlates positively and significantly with the frequency of cholesterol gallstones in inbred mice, and gallstone-susceptible C57L mice display significantly higher intestinal cholesterol absorption than gallstone-resistant AKR mice.114 These observations suggest that high dietary cholesterol intake and high efficiency of intestinal cholesterol absorption are independent risk factors for cholesterol gallstone formation. Differences in the metabolism of chylomicron remnant cholesterol between C57L and AKR mice may account for lithogenic bile formation in the former, and the cholesterol absorbed from the small intestine provides an important source for biliary cholesterol hypersecretion in mice challenged by a lithogenic diet.115
Altered intestinal motility also may have a role in gallstone formation. Delayed or impaired small intestinal transit is associated with enhanced intestinal cholesterol absorption, biliary cholesterol secretion, and gallstones in CCK-1 receptor-knockout mice.115 The association of impaired colonic motility with increased biliary deoxycholate levels is found in some patients with cholesterol gallstones. Evidence for a causal relation between impaired intestinal motility, deoxycholate formation, and bile lithogenicity comes from studies in humans and mice. Clinical studies have found that acromegalic patients treated with octreotide (a known risk factor for cholesterol gallstone disease [see earlier]) display prolonged colonic transit times, high levels of biliary deoxycholate concentration, and biliary cholesterol precipitation.116–119 Furthermore, higher levels of biliary deoxycholate are associated with increased amounts of gram-positive anaerobic bacteria and increased activity of 7α-dehydroxylase in the cecums of patients with cholesterol gallstones compared with control subjects who have no stones.120 Biliary deoxycholate and cholesterol concentrations can be lowered by antibiotic treatment that reduces fecal 7α-dehydroxylation activity. Compared with resistant mice, gallstone-susceptible mice also have high biliary levels of deoxycholate, which are associated with cholesterol supersaturation and gallstone formation.73,121 Chronic intestinal infection has been proposed to be a potential factor in cholesterol gallstone pathogenesis. A mouse study has shown that distal intestinal infection with a variety of enterohepatic Helicobacter species (but not Helicobacter pylori) is essential for nucleation and crystallization of cholesterol from supersaturated bile.122,123 These Helicobacter species also have been identified in the bile and gallbladder tissue of Chilean patients with chronic cholecystitis.124 Whether chronic intestinal infection has a direct pathogenic role in the formation of cholesterol gallstones requires further investigation.
Patients with Crohn’s disease and those who have undergone intestinal resection or total colectomy have bile that is supersaturated with cholesterol and are prone to precipitation of cholesterol crystals and formation of gallstones.125 The enterohepatic circulation of bile salts is probably impaired in these patients so that biliary bile salt secretion is greatly reduced and the solubilization of cholesterol in bile is decreased. Moreover, Crohn’s disease might lead to impaired enterohepatic cycling of bilirubin so that biliary bilirubin levels and precipitation of calcium bilirubinate are increased, thereby providing a nidus for cholesterol nucleation and crystallization.48,126
GROWTH OF GALLSTONES
Although cholesterol nucleation and crystallization is a critical stage in the formation of cholesterol gallstones, findings in patients who have cholesterol crystals but no gallstones in the gallbladder suggest that growth of cholesterol crystals into gallstones does not always follow crystallization. Stone growth may represent a second critical stage in the formation of gallstones that results from delayed emptying of the gallbladder. When multiple gallstones are found in the gallbladder, they often are equal in size, indicating that cholesterol crystallization for this family of stones occurred simultaneously and the stones grew at the same rate. By contrast, stones of unequal size could represent different generations. The amorphous material in the center of stones contains bilirubin, bile salts, mucin glycoproteins, calcium carbonate, phosphate, copper, and sulfur, which could have provided a required nidus for cholesterol nucleation and crystallization. Cholesterol crystals could assemble about this nidus. The formation of a nidus and subsequent stone growth could be determined by mucins, other biliary proteins, and the cholesterol saturation of bile. The growth of stones is likely a discontinuous process that is punctuated by deposition of rings of calcium bilirubinate and calcium carbonate. Because cholesterol crystals often aggregate randomly in amorphous groupings and layer radially and concentrically, cholesterol stones consist of radially or horizontally oriented cholesterol crystals embedded within an organic matrix. In the outer portion of stones, cholesterol crystals are oriented perpendicularly to the surface.127 Throughout the formation of gallstones, mucins could provide a matrix on which the growth of gallstones occurs. Furthermore, concentric pigmented rings separate layers of cholesterol crystals that have different axial orientations. The chemical composition of these rings often resembles the center of gallstones, and the rings may reflect cyclic deposition of calcium bilirubinate, other calcium salts, and mucin glycoproteins.
GENETICS
The evidence for a genetic component of cholesterol gallstone disease in humans is mostly indirect and based on geographic and ethnic differences, as well as on family and twin studies.13,128–135 A genetic predisposition is clearly present in the Pima and certain other North and South American Indians, who display the highest frequency rates (48%) of gallstones.13,128,129 By contrast, the overall frequency in other American (whites) and European populations is about 20%. The lowest rates (<5%) are observed in African populations and intermediate rates are found in Asian populations (5% to 20%), as shown in Figures 65-1 and 65-2. Although some independent risk factors, such as aging, gender, parity, obesity, some drugs, and rapid weight loss, for gallstone formation have been found (see later),18,22,46,136–138 none of these factors can explain the striking differences in incidence rates of gallstones among different populations, thereby suggesting a genetic contribution to the etiology of the disease.
Gallstones are more frequent by a ratio of 3 : 1 in siblings and other family members of affected persons than in spouses or unrelated controls.130 Using ultrasonography to detect gallstones in first-degree relatives of index patients, Gilat and colleagues132 found a 21% frequency in first-degree relatives compared with 9% in matched controls, and Sarin and coworkers133 also observed a frequency that was five times higher in relatives than in controls. Furthermore, cholesterol supersaturation is higher in fasting duodenal bile of older sisters of patients with cholesterol gallstone than in controls.134 Cholesterol synthesis rates, bile saturation levels, and gallstone frequency rates are also significantly higher on pair-wise correlations in monozygotic than in dizygotic male twins.135 Despite these observations, a mode of inheritance that fits a mendelian pattern cannot be shown in most cases.
To examine the influence of the genetic factors more rigorously, a study of populations with different incidence rates of gallstones but living in the same environment should provide insights into genetic mechanisms of the disease. Unfortunately, intermarriages between two populations result in a rapid loss of the original genetic background within a few generations, thereby making such a study impossible. A large study of 43,141 twin pairs in Sweden, however, has provided conclusive evidence for the role of genetic factors in the pathogenesis of cholesterol gallstones.139 In this study, concordance rates were significantly higher in monozygotic twins than in dizygotic twins, with genetic factors accounting for 25% of the phenotypic variation between the twins.
The first evidence that human gallstones might be caused by a single gene defect came from a study by Lin and colleagues,140 who reported that among 232 Mexican-Americans, a variant of the cholesterol 7α-hydroxylase (CYP7A1) gene was associated with gallstones in men but not in women. CYP7A1 is an attractive candidate gene because it encodes the rate-limiting enzyme in the “neutral” pathway for hepatic bile salt synthesis (see Chapter 64) and because bile salts are essential for forming bile and for keeping cholesterol molecules solubilized in simple and mixed micelles in bile. Furthermore, Pullinger and colleagues found a link between another single gene defect of CYP7A1 and cholesterol gallstones associated with hypercholesterolemia resistant to HMG-CoA reductase inhibitors in two male homoyzgotes.141
Missense mutations in the ABC transporter B4 (ABCB4) gene (formerly named multidrug resistance gene 3, MDR3), which encodes the phosphatidylcholine transporter in the canalicular membrane of hepatocytes, are the basis for a particular type of cholelithiasis.66,142 The disorder is characterized by intrahepatic sludge, gallbladder cholesterol gallstones, mild chronic cholestasis, a high cholesterol-to-phospholipid ratio in bile, and recurrent symptoms after cholecystectomy.143–145 A defect in the ABC transporter B4 gene could constitute the basis for this highly symptomatic and recurrent form of gallstone disease. In patients with hepatolithiasis, a common disease in Asia (see Chapter 68), low expression levels of ABCB4 and phosphatidylcholine transfer protein occur together, with markedly reduced phospholipid concentrations in bile.146 Furthermore, HMG-CoA reductase activity is increased and CYP7A1 activity is reduced in patients with gallstones compared with controls. In this disorder, the formation of cholesterol-rich intrahepatic stones could be induced by decreased biliary secretion of phospholipids in the setting of increased cholesterol synthesis and decreased bile salt synthesis.
Because hypomotility of the gallbladder favors gallstone formation, the genes for CCK and the CCK-1 receptor (CCK-1R), which regulate gallbladder motility, are attractive candidates.115,148 Genetic variation in CCK-1R is associated with gallstone risk, and an aberrant splicing of CCK-1R, which is predicted to result in a nonfunctional receptor, is found in a few obese patients with gallstones.148,149 A search for mutations or polymorphisms in the CCK-1R gene in patients with gallstones has been unsuccessful, however.150
Some studies have reported that certain polymorphisms of the apolipoprotein (APO) E and APOB genes and the cholesteryl ester transfer protein, all of which are involved in carrying cholesterol in the plasma, are associated with gallstones. The APOE polymorphisms are the most extensively studied polymorphisms in patients with gallstones, but reports concerning the protective role of the ε4 allele against gallstones have been inconsistent.151–155 The ε2 allele appears to protect against gallstones, and the degree of dietary cholesterol absorption in the intestine varies with the APOE isoform (ε4>ε3>ε2). Also, the fecal excretion of cholesterol tends to be higher in persons with the APOE2 phenotype than in those with the APOE3 or APOE4 phenotypes.156 In a study of polymorphisms at the APOB, APOAI, and cholesteryl ester transfer protein gene loci in patients with gallbladder disease, a polymorphism of the cholesteryl ester transfer protein gene, in relation to another HDL lowering factor, was found to be associated with cholesterol gallstones.157 Also, a link was found between the X+ allele of the APOB gene and an increased risk of cholesterol gallstones.158 More recently, a genome-wide association study in a large cohort of patients with gallstones from Germany159 and a linkage study in affected sibling pairs160 identified a common variant (D19H) of the sterol transporters ABCG5 and ABCG8 on the canalicular membrane of hepatocytes as a risk factor for gallstones. This variant is also a susceptibility factor for gallstones in Chilean Hispanics,159 and other ABCG8 variants (T400K, D19H, A632V, M429V, and C54Y) as well as ABCG5 variants (Q604E) may be important risk factors for gallstone formation in Chinese and Canadian white populations.161–163
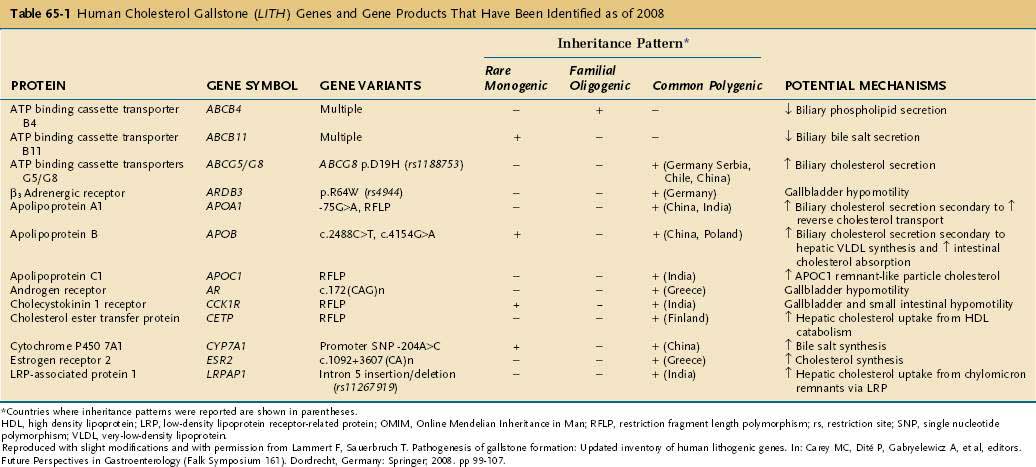
Table 65-1 summarizes the major classes of candidate genes for cholesterol gallstones (or Lith genes).23 Some candidate genes have not yet been identified in humans, and their roles in cholelithogenesis need to be investigated further. In general, genes that contribute to cholesterol gallstone formation include those that encode (1) hepatic and intestinal membrane lipid transporters; (2) hepatic and intestinal lipid regulatory enzymes; (3) hepatic and intestinal intracellular lipid transporters; (4) hepatic and intestinal lipid regulatory transcription factors; (5) hepatic lipoprotein receptors and related proteins; (6) hormone receptors in the gallbladder; and (7) biliary mucins. Human analogs of these genes could be involved in human gallstone disease.
Table 65-1 Human Cholesterol Gallstone (LITH) Genes and Gene Products That Have Been Identified as of 2008
The factors that regulate intestinal membrane lipid transporters, lipid regulatory enzymes, intracellular lipid transporters, and lipid regulatory transcription factors may influence the amount of cholesterol of intestinal origin contributing to the liver for biliary secretion. Direct evidence for the role of intestinal factors in mouse gallstones comes from a study of ACAT gene 2-knockout mice in which the lack of cholesteryl ester synthesis in the intestine significantly reduces intestinal cholesterol absorption and causes complete resistance to diet-induced cholesterol gallstones.164 One study found that the potent cholesterol absorption inhibitor ezetimibe prevents gallstones by effectively reducing intestinal cholesterol absorption and biliary cholesterol secretion and protects gallbladder motility function by desaturating bile in mice.165 Therefore, reduced cholesterol absorption or hepatic chylomicron remnant uptake may induce a decrease in biliary cholesterol secretion and saturation.
PIGMENT STONES
Although the pathogeneses of black and brown pigment gallstones are not as well understood as that of cholesterol gallstones and each type of stone probably has a distinctive pathogenesis, both types of pigment stone result from abnormalities in the metabolism of bilirubin and are pigmented as a result of bilirubin precipitation.166–168 In general, the bile of patients with both types of pigment stones contains an excess of unconjugated bilirubin, analogous to the saturation of bile with cholesterol in patients with cholesterol stones.169 Also, both types of pigment stones are composed primarily of bile pigment and contain a matrix of mucin glycoproteins. In black stones, however, the pigment is predominantly an insoluble, highly cross-linked polymer of calcium bilirubinate, whereas in brown stones, the main pigment is monomeric calcium bilirubinate. The two types of pigment stones also differ in radiodensity, location within the biliary system, and geographic distribution.
BLACK PIGMENT STONES
Black pigment stones are formed in uninfected gallbladders, particularly in patients with chronic hemolytic anemia (e.g., β-thalassemia, hereditary spherocytosis, and sickle cell disease) and liver cirrhosis. The unconjugated bilirubin produced in increased amounts precipitates as calcium bilirubinate to form stones.170 This type of stone is composed of either pure calcium bilirubinate or polymer-like complexes consisting of unconjugated bilirubin, calcium bilirubinate, calcium, and copper. Mucin glycoproteins account for as much as 20% of the weight of black stones. A regular crystalline structure is not present.
Under normal physiologic conditions, unconjugated bilirubin is not secreted into bile. Bilirubin glucuronides are hydrolyzed by endogenous β-glucuronidase, and unconjugated bilirubin constitutes less than 1% of total bile pigment, mostly because the activity of the enzyme is inhibited by β-glucaro-1,4-lactone in the biliary system.171,172 The unifying predisposing factor in the formation of black pigment stones is the hypersecretion of bilirubin conjugates (especially monoglucuronides) into bile. In the presence of hemolysis, secretion of these bilirubin conjugates increases ten-fold. Unconjugated monohydrogenated bilirubin is formed by the action of endogenous β-glucuronidase, which coprecipitates with calcium as a result of supersaturation. A 1% hydrolysis rate could give rise to high concentrations of unconjugated bilirubin that often greatly exceed the solubility of bilirubin in bile. A defect in acidification of bile also may be induced by gallbladder inflammation or the reduced buffering capacity of sialic acid and sulfate moieties in the mucin gel. The reduction in buffering capacity facilitates the supersaturation of calcium carbonate and phosphate that would not occur at a more acidic pH. Gallbladder motility defects are not observed in patients with black pigment stones, as inferred from in vitro experiments of human gallbladder muscles.
BROWN PIGMENT STONES
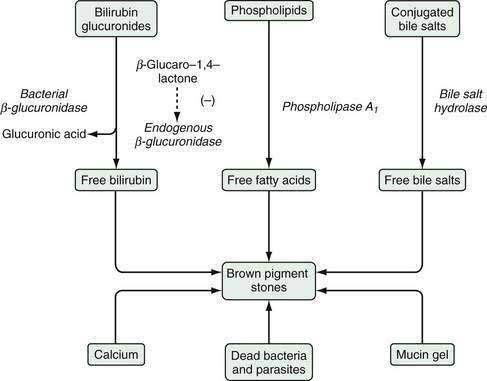
Brown pigment stones are formed not only in the gallbladder, but also commonly in other portions of the biliary tree, especially in intrahepatic bile ducts. The formation of brown pigment stones requires the presence of structural or functional stasis of bile associated with biliary infection, especially with Escherichia coli.173 These stones are more common in areas such as Asia, where Clonorchis sinensis and roundworm infestations are prevalent, and parasitic elements have been considered to be kernels of brown pigment stone formation (see Chapters 68 and 82).174 Bile stasis predisposes to the bacterial infection as well as the accumulation of mucins and bacterial cytoskeletons in the bile ducts. Bile stasis may be caused by bile duct stenosis and bacterial infection caused by infestation by parasites such as Clonorchis sinensis, roundworms, and their ova.175 Additionally, bacterial infection and colonization in bile ducts by enteric bacteria are found commonly in patients with brown pigment stones. As the incidence of biliary infections has decreased in Asian populations prone to development of brown pigment stones, the ratio of cholesterol stones to pigment stones also has changed in these populations. The percentage of brown pigment stones in Japan has fallen from 60% to 24% since the 1950s, and similar changes have been reported from other Asian countries.176–178
Enteric bacteria produce β-glucuronidase, phospholipase A1, and conjugated bile acid hydrolase. Activity of β-glucuronidase results in the production of unconjugated bilirubin from bilirubin glucuronide; phospholipase A1 liberates palmitic and stearic acids from phospholipids; and bile acid hydrolases produce unconjugated bile salts from glycine or taurine-conjugated bile salts. Partially ionized saturated fatty acids, unconjugated bilirubin, and unconjugated bile salts may precipitate as calcium salts. Mucin gel can trap these complex precipitates and facilitate their growth into macroscopic stones. Figure 65-6 shows the postulated mechanisms underlying the formation of brown pigment stones. Under normal physiologic conditions, bilirubin in bile exists mainly as bilirubin glucuronide, which is soluble in aqueous media. Bile also contains β-glucuronidase of tissue origin, the activity of which is inhibited by glucaro-1,4-lactone, which is also formed in the liver. If infection with E. coli occurs, the concentration of bacterial β-glucuronidase increases significantly and exceeds the inhibitory power of glucaro-1,4-lactone. As a result, bilirubin glucuronide is hydrolyzed to produce unconjugated bilirubin and glucuronic acid; the former is water-insoluble and combines with calcium to form calcium bilirubin at its carboxyl radical, leading to the formation of brown pigment gallstones.
NATURAL HISTORY
The natural history of gallstones typically is described in two separate groups of patients: those who have symptoms and those who are asymptomatic. Necropsy studies clearly show that the vast majority of patients with gallstones are asymptomatic and remain so. Ascertaining the true frequency of complications in persons with asymptomatic stones (as well as those with symptomatic stones) is critical to providing rational, cost-effective recommendations regarding therapy (see later). Unfortunately, the information available on the natural history of gallstones has been sparse and somewhat varied.179–181
ASYMPTOMATIC STONES
The study that changed our understanding of the course and appropriate therapy of gallstone disease was performed by Gracie and Ransohoff.179 They monitored 123 University of Michigan faculty members for 15 years after they had been found to have gallstones on routine screening ultrasonography. At 5, 10, and 15 years of follow-up, 10%, 15%, and 18% of the patients, respectively, had become symptomatic, and none had experienced serious complications. The investigators suggested that the rate at which biliary pain develops in persons with asymptomatic gallstones is about 2% per year for five years and then decreases over time. Biliary complications developed in only three patients in this study, and all complications were preceded by episodes of biliary pain. Studies have suggested that biliary pain, not a biliary complication, is the initial manifesting symptom in 90% of people with previously asymptomatic gallstones.179 Therefore, in patients with asymptomatic stones, the frequency of complications is low, and prophylactic cholecystectomy is not necessary.
Subsequent studies have reported slightly higher rates of biliary pain and complications in patients with initially asymptomatic gallstones,180 but only one was a long-term and prospective study.181 The Group for Epidemiology and Prevention of Cholelithiasis (GREPCO) in Rome reported the courses of 151 subjects with gallstones, 118 of whom were asymptomatic on entering the study. In those who were initially asymptomatic, the frequency of biliary pain was 12% at 2 years, 17% at 4 years, and 26% at 10 years, and the cumulative rate of biliary complication was 3% at 10 years.181
SYMPTOMATIC STONES
The natural history of symptomatic gallstones has a more aggressive course than that of asymptomatic stones. The U.S. National Cooperative Gallstone Study showed that in persons who had an episode of uncomplicated biliary pain in the year before entering the study, the rate of recurrent biliary pain was 38% per year.182 Other investigators have reported a rate of recurrent biliary pain as high as 50% per year in persons with symptomatic gallstones.183 As noted earlier, biliary complications also are more likely to develop in persons with symptomatic gallstones. The risk of biliary complications is estimated to be 1% to 2% per year and is believed to remain relatively constant over time.184 Therefore, cholecystectomy should be offered to patients only after biliary symptoms develop. Depending on the patient, a reasonable alternative approach may be to observe the pattern of pain before deciding on therapy because up to 30% of patients with one episode of biliary pain do not have a recurrent episode. This approach is particularly useful in patients with a high operative risk.
STONES IN PATIENTS WITH DIABETES MELLITUS
Diabetic patients with incidental cholelithiasis were long considered to have an increased risk of serious complications even when the gallstones were asymptomatic. Subsequent studies have shown that the natural history of gallstones in diabetic patients follows the same pattern observed in nondiabetic persons. A prospective study of patients with insulin-resistant diabetes mellitus showed that after five years of follow-up, symptoms had developed in 15% of the asymptomatic patients.185 This frequency is roughly the same as that reported for nondiabetic patients. Moreover, the complication and mortality rates were comparable to those in studies of nondiabetic patients with gallstones. Therefore, prophylactic cholecystectomy is generally not recommended in patients with insulin-resistant diabetes mellitus and asymptomatic gallstones.
DIAGNOSIS AND CLINICAL DISODERS
The clinical manifestations of gallstones are shown schematically in Figure 65-7 and summarized in more detail in Table 65-2.186–190 Biliary pancreatitis is discussed in Chapter 58. Although the standard approach to asymptomatic gallstones is observation, some patients with asymptomatic gallstones may be at increased risk of complications and may require special consideration.
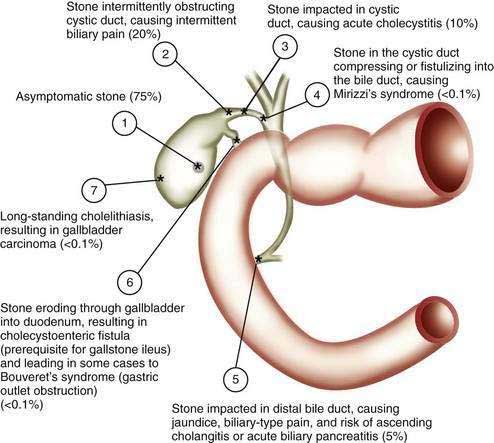
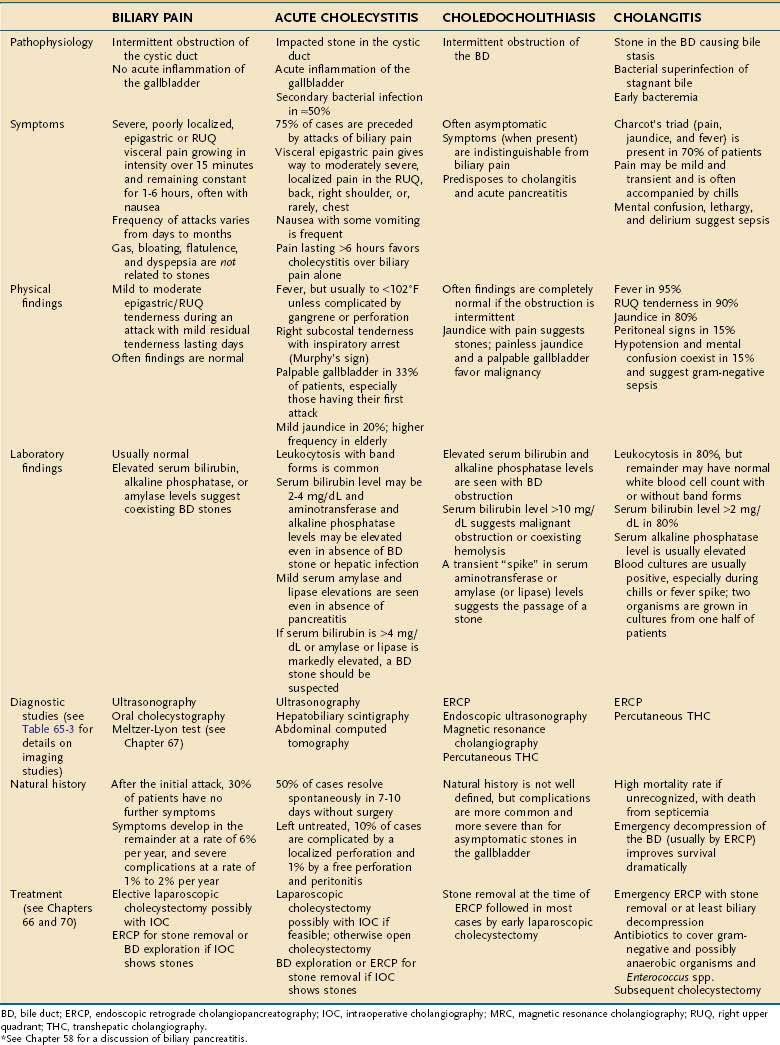
An increased risk of cholangiocarcinoma and gallbladder carcinoma has been associated with certain disorders of the biliary tree and in some ethnic groups (such as Native Americans) (see Chapter 69). Risk factors include choledochal cysts, Caroli’s disease, anomalous pancreatic ductal drainage (in which the pancreatic duct drains into the bile duct), large gallbladder adenomas, and porcelain gallbladder (see Chapters 62 and 67). Patients at increased risk of biliary cancer may benefit from prophylactic cholecystectomy. In particular, if abdominal surgery is planned for another indication, an incidental cholecystectomy should be performed.
Morbidly obese persons who undergo bariatric surgery are at high risk of complications of gallstones (see Chapters 6 and 7). These patients have a frequency of gallstones of greater than 30%. An incidental cholecystectomy is recommended at the time of surgery.
Some investigators have proposed that patients with incidental cholelithiasis who are awaiting heart transplantation undergo a prophylactic cholecystectomy irrespective of the presence or absence of biliary tract symptoms because they are at increased risk of post-transplant gallstone complications.191 A retrospective study that addressed this issue in renal transplant recipients, however, concluded that complications of gallstones could be managed safely after symptoms emerged.192
IMAGING STUDIES
As shown in Table 65-3, a wide array of imaging techniques are available to evaluate the biliary tract.193–196 Each modality has its strengths and limitations, and the methods vary widely in relative cost and risk to the patient. With the possible exception of ultrasonography, none of the modalities should be ordered routinely in the evaluation of a patient with suspected gallstone disease; rather, the diagnostic evaluation should proceed in a rational, stepwise fashion based on the individual patient’s symptoms, signs, and results of laboratory studies.
| TECHNIQUE | CONDITION TESTED FOR | FINDINGS/COMMENTS |
|---|---|---|
| Ultrasonography | Cholelithiasis | Stones manifest as mobile, dependent echogenic foci within the gallbladder lumen with acoustic shadowing |
| Sludge appears as layering echogenic material without shadows | ||
| Sensitivity rate >95% for stones >2 mm | ||
| Specificity rate >95% for stones with acoustic shadows | ||
| Rarely, a stone-filled gallbladder may be contracted and difficult to see, with a “wall-echo-shadow” sign | ||
| Best single test for stones in the gallbladder | ||
| Choledocholithiasis | Stones are seen in BD in only ≈50% of cases but can be inferred from the finding of a dilated BD (>6 mm diameter), with or without gallstones, in another ≈25% of cases | |
| Can confirm, but not exclude, BD stones | ||
| Acute cholecystitis | Ultrasonographic Murphy’s sign (focal gallbladder tenderness under the transducer) has a positive predictive value of >90% in detecting acute cholecystitis when stones are seen | |
| Pericholecystic fluid (in the absence of ascites) and gallbladder wall thickening to >4 mm (in the absence of hypoalbuminemia) are nonspecific findings but are suggestive of acute cholecystitis | ||
| EUS | Choledocholithiasis | Highly accurate for excluding or confirming stones in the BD |
| Concordance of EUS with the ERCP diagnosis ≈95%; many studies suggest slightly higher sensitivity rates for EUS than for ERCP | ||
| Specificity rate ≈97% | ||
| Positive predictive value ≈99%, negative predictive value ≈98%, accuracy rate ≈97% | ||
| With experienced operators, EUS can be used in lieu of ERCP to exclude BD stones, particularly when the clinical suspicion is low or intermediate | ||
| Considered for patients with a low-to-moderate clinical probability of choledocholithiasis | ||
| Oral cholecystography* | Cholelithiasis | Stones manifest as mobile filling defects in an opacified gallbladder |
| Sensitivity and specificity rates exceed 90% when the gallbladder is opacified, but nonvisualization occurs in 25% of studies and can result from multiple causes other than stones | ||
| Opacification of the gallbladder indicates patency of the cystic duct | ||
| May be useful in the evaluation of acalculous gallbladder diseases such as cholesterolosis and adenomyomatosis (see Chapter 67) | ||
| Cholescintigraphy (hepatobiliary scintigraphy; hydroxyiminodiacetic acid or diisopropyl iminodiacetic acid scan) | Acute cholecystitis | Assesses patency of the cystic duct |
| Normal scan shows radioactivity in the gallbladder, BD, and small bowel within 30-60 minutes | ||
| Positive result is defined as nonvisualization of the gallbladder with preserved hepatic excretion of radionuclide into the BD or small bowel | ||
| Sensitivity rate is ≈95% and specificity rate is ≈90%, with false-positive results seen in fasted, critically ill patients | ||
| With cholecystokinin stimulation, gallbladder “ejection fraction” can be determined and may help evaluate patients with acalculous biliary pain (see Chapter 67) | ||
| Normal scan result virtually excludes acute cholecystitis | ||
| ERCP | Choledocholithiasis | ERCP is the standard diagnostic test for stones in the BD, with sensitivity and specificity rates of ≈95% |
| Use of ERCP to extract stones (or at least to drain infected bile) is life-saving in severe cholangitis and reduces the need for BD exploration at the time of cholecystectomy | ||
| Recommended for patients with a high clinical probability of choledocholithiasis | ||
| Cholelithiasis | When contrast agent flows retrograde into the gallbladder, stones appear as filling defects and can be detected with a sensitivity rate of ≈80%, but ultrasonography remains the mainstay for confirming cholelithiasis | |
| MRCP | Choledocholithiasis | Rapid, noninvasive modality that provides detailed bile duct and pancreatic duct images equal to those of ERCP |
| Sensitivity rate ≈93% and specificity rate ≈94%, comparable with those for ERCP | ||
| Useful for examining nondilated ducts, particularly at the distal portion, which often is not well visualized by ultrasonography | ||
| Adjacent structures such as liver and pancreas can be examined at the same time | ||
| Recommended for patients with a low-to-moderate clinical probability of choledocholithiasis | ||
| CT | Complications of gallstones | Not well suited for detecting uncomplicated stones, but excellent for detecting complications, such as abscess, perforation of the gallbladder or BD, and pancreatitis |
| Spiral CT may prove useful as a noninvasive means of excluding BD stones; some studies suggest improved diagnostic accuracy when CT is combined with an oral cholecystographic contrast agent |
BD, bile duct; CT, computed tomography; ERCP, endoscopic retrograde cholangiopancreatography; EUS, endoscopic ultrasonography; MRCP, magnetic resonance cholangiopancreatography.
* Performed infrequently nowadays.
Ultrasonography
Since its introduction in the 1970s, ultrasonographic examination of the biliary tract has become the principal imaging modality for the diagnosis of cholelithiasis. Ultrasonography requires only an overnight or eight-hour fast, involves no ionizing radiation, is simple to perform, and provides accurate anatomic information. It has the additional advantage of being portable and thus available at the bedside of a critically ill patient.194
The diagnosis of gallstones relies on the detection of echogenic objects within the lumen of the gallbladder that produce an acoustic shadow (Fig. 65-8A). The stones are mobile and generally congregate in the dependent portion of the gallbladder. Modern ultrasonography is able to detect stones as small as 2 mm in diameter routinely. Smaller stones may be missed or may be confused with biliary sludge (layering echogenic material that does not cast acoustic shadows).197
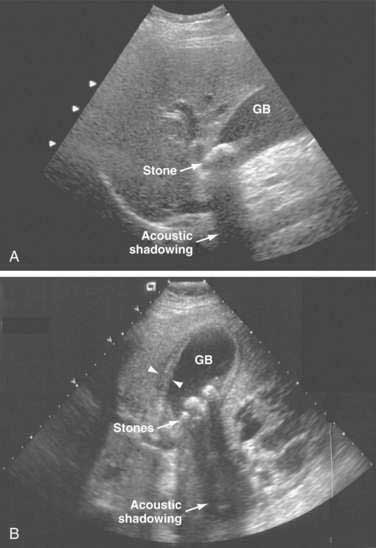
The sensitivity of ultrasonography for the detection of gallstones in the gallbladder is more than 95% for stones larger than 2 mm.198 The specificity is greater than 95% when stones produce acoustic shadows. Rarely, advanced scarring and contraction of the gallbladder around gallstones make it impossible to locate the gallbladder or the stones; this finding should also raise the possibility of gallbladder cancer. The contracted gallbladder filled with stones may give a “double-arc shadow” or “wall-echo shadow” sign, with the gallbladder wall, echogenic stones, and acoustic shadowing seen in immediate proximity. If the gallbladder cannot be identified ultrasonographically, then a complementary imaging modality such as oral cholecystography or abdominal computed tomography (CT) is warranted.
Ultrasonography is the standard for the diagnosis of stones in the gallbladder but is distinctly less sensitive for the detection of stones in the bile duct (previously termed common bile duct).199 Because of the proximity of the distal bile duct to the duodenum, luminal bowel gas often interferes with the ultrasonographic image, and the entire length of the bile duct cannot be examined.200 As a result, only approximately 50% of bile duct stones are actually seen on ultrasonography.194 The presence of an obstructing bile duct stone, however, can be inferred when a dilated duct is found. Now that endoscopic retrograde cholangiopancreatography (ERCP) has uncovered a rising frequency of falsely negative ultrasonograms, the upper limit of normal of the diameter of the bile duct has declined from 10 mm to 6 mm. Even so, inferring choledocholithiasis from a dilated bile duct on ultrasonography has a sensitivity of only 75%.
Finally, ultrasonography is quite useful for diagnosing acute cholecystitis.201 Pericholecystic fluid (in the absence of ascites) and gallbladder wall thickening to more than 4 mm (in the absence of hypoalbuminemia) are suggestive of acute cholecystitis (see Fig. 65-8B). Unfortunately, in the critical care setting, these nonspecific findings are seen frequently in patients with no other evidence of gallbladder disease.201 A more specific finding is the so-called sonographic Murphy’s sign, in which the ultrasonographer elicits focal gallbladder tenderness under the ultrasound transducer. Eliciting a sonographic Murphy’s sign is somewhat operator dependent and requires an alert patient. Presence of the sign has a positive predictive value of greater than 90% for detecting acute cholecystitis if gallstones are present.202
Endoscopic Ultrasonography
Endoscopic ultrasonography (EUS) is highly accurate for detecting choledocholithiasis. Inherently more invasive and more expensive than standard ultrasonography, EUS has the advantage of being able to visualize the bile duct from within the gastrointestinal lumen and is reported to be comparable to ERCP in this respect. Intraluminal imaging provides several advantages over transabdominal ultrasonography, including closer proximity to the bile duct, higher resolution, and lack of interference by bowel gas or abdominal wall layers (Fig. 65-9). In several studies, EUS had a positive predictive value of 99%, a negative predictive value of 98%, and an accuracy rate of 97% for the diagnosis of bile duct stones compared with ERCP.203,204 If bile duct stones are found on EUS, endoscopic removal of the stones is necessary, and it can be argued that ERCP should be the initial study if choledocholithiasis is strongly suspected. Nonetheless, several studies that compared EUS with ERCP have found both techniques to be accurate for confirming or excluding choledocholithiasis, with EUS having advantages in both safety and cost.205–207
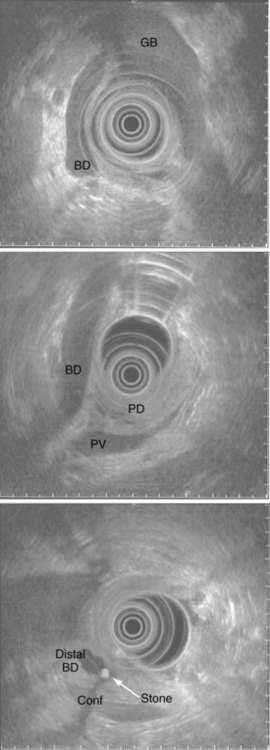
Oral Cholecystography
Once the mainstay of imaging studies of the gallbladder, oral cholecystography (OCG) now has limited application as a secondary approach to identifying stones in the gallbladder.194 The ease, reliability, and rapidity with which it can detect stones, along with the absence of ionizing radiation, have made ultrasonography the imaging study of choice. In unusual cases in which the gallbladder cannot be identified ultrasonographically (as when the gallbladder is contracted and full of stones),208 OCG is complementary to ultrasonography for demonstrating cholelithiasis. Additionally, when medical dissolution of stones or lithotripsy is being considered, visualization of the gallbladder on OCG excludes cystic duct obstruction (see Chapter 66).209 Because of the time required to complete the test (48 hours), OCG is not useful in patients with suspected acute cholecystitis or other complications of gallstone disease. On occasion, OCG may detect unsuspected disease of the gallbladder, such as adenomyomatosis or cholesterolosis (see Chapter 67).
Cholescintigraphy
Cholescintigraphy (hepatobiliary scintigraphy) is a radionuclide imaging test of the gallbladder and biliary tract that is most useful for evaluating patients with suspected acute cholecystitis. By demonstrating patency of the cystic duct, cholescintigraphy can exclude acute cholecystitis rapidly (within 90 minutes) from the differential diagnosis in a patient who presents with abdominal pain.210,211
The procedure can be performed on an emergency basis in a nonfasting patient after intravenous administration of gamma-emitting 99mTc-labeled hydroxyl iminodiacetic acid (HIDA) or diisopropyl iminodiacetic acid (DISIDA), which is taken up rapidly by the liver and secreted into bile. As shown in Figure 65-10, serial scans after injection normally should show radioactivity in the gallbladder, bile duct, and small intestine within 30 to 60 minutes.167 In the past, imaging of jaundiced patients with this technique was limited, but use of DISIDA may allow imaging of the biliary tree in a patient with a serum bilirubin value as high as 20 mg/dL.
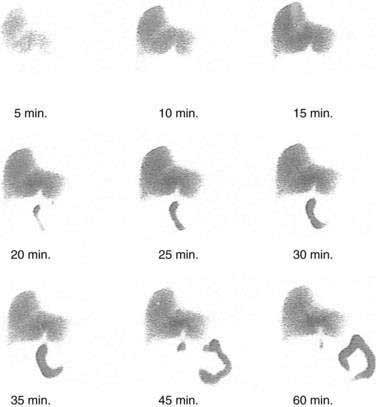
An abnormal or “positive” scan result is defined as nonvisualization of the gallbladder with preserved excretion into the bile duct or small intestine. The accuracy of the test for detecting acute cholecystitis is 92%, superior to that for ultrasonography. False-positive results occur primarily in fasting or critically ill patients, in whom gallbladder motility is decreased. The reduction in gallbladder motility leads to greater water resorption, which results in a gelatinous bile. In critically ill patients, cholestasis and hepatocyte dysfunction result in reduced clearance of radionuclide imaging agents. Although nonvisualization of the gallbladder because of cystic duct obstruction is the hallmark of acute cholecystitis, pericholecystic hepatic uptake of radionuclide is a useful secondary sign.212
Although primarily a tool for evaluating acutely ill patients with suspected acute cholecystitis, cholescintigraphy after administration of CCK may be useful in identifying patients with chronic acalculous biliary pain who are likely to benefit from empirical cholecystectomy (see Chapter 67). An additional important role for cholescintigraphy is the noninvasive and clear detection of bile leakage from the cystic duct as a complication of cholecystectomy (see Chapter 66).213
Endoscopic Retrograde Cholangiopancreatography
Endoscopic retrograde cholangiopancreatography (ERCP) is one of the most effective modalities for detecting choledocholithiasis. The technique of this procedure is discussed in more detail in Chapter 70. Stones within the bile duct appear as filling defects and can be detected with a sensitivity of approximately 95% (Fig. 65-11). Care should be taken to avoid inadvertent injection of air into the biliary tract214 because bubbles may mimic gallstones. The specificity of ERCP for the detection of bile duct stones is approximately 95%.
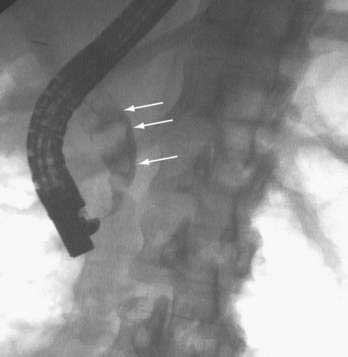
The therapeutic applications of ERCP have revolutionized the treatment of patients with choledocholithiasis215 and other bile duct disorders (see Chapter 70). Because ERCP is invasive and is associated with potential complications such as pancreatitis and postsphincterotomy bleeding, however, the role of ERCP has been challenged by other modalities that exclude choledocholithiasis more safely and accurately in patients in whom the clinical suspicion of choledocholithiasis is not high. As the use of EUS and MRC has increased, the role of ERCP in the diagnosis of choledocholithiasis has changed considerably. A National Institutes of Health consensus conference has recommended the use of ERCP only when the clinical probability for choledocholithiasis is high (i.e., when the need for therapeutic intervention is likely). For diagnosis of choledocholithiasis alone, EUS and MRC are equal in accuracy to ERCP.216
Computed Tomographic Cholangiography and Magnetic Resonance Cholangiography
In patients with cholelithiasis or choledocholithiasis, CT has been used principally for detecting complications such as pericholecystic fluid in acute cholecystitis, gas in the gallbladder wall suggesting emphysematous cholecystitis, gallbladder perforation, and abscesses (Fig. 65-12). Spiral CT cholangiography (CTC) with use of an oral cholecystographic contrast agent has been studied for the detection of choledocholithiasis.217,218 Although CTC is still inferior to ERCP imaging for detecting bile duct stones, it may reveal other surrounding pathologic abnormalities.217
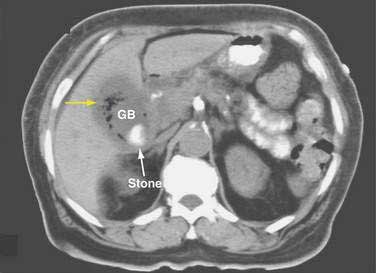
MRC is highly useful for imaging the bile duct and detecting gallstones. This modality is especially useful for detecting abnormalities in the most distal extrahepatic portion of the bile duct when the duct is not dilated; this region often is not well visualized by transabdominal ultrasonography.195 With the advent of laparoscopic cholecystectomy, an easy, quick, and, preferably, noninvasive method of excluding bile duct stones is needed. MRC permits the construction of a three-dimensional image of the bile duct with a high sensitivity for detecting bile duct stones (Fig. 65-13).219,220 In a systematic review that compared MRC with diagnostic ERCP for the detection of choledocholithiasis, MRC had a sensitivity of 93% and a specificity of 94%.221
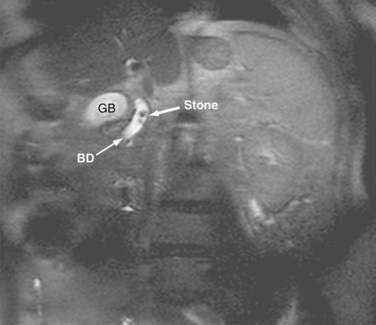
BILIARY PAIN AND CHRONIC CHOLECYSTITIS
Pathogenesis
Biliary pain (conventionally referred to as biliary “colic,” a misnomer) is caused by intermittent obstruction of the cystic duct by one or more gallstones. Biliary pain does not require that inflammation of the gallbladder accompany the obstruction. The term chronic cholecystitis to describe biliary pain should be avoided because it implies the presence of a chronic inflammatory infiltrate that may or may not be present in a given patient. Indeed, the severity and frequency of biliary pain and the pathologic changes in the gallbladder do not correlate significantly.222 The most common histologic changes observed in patients with biliary pain are mild fibrosis of the gallbladder wall with a chronic inflammatory cell infiltrate and an intact mucosa. Recurrent episodes of biliary pain also can be associated with a scarred, shrunken gallbladder and Rokitansky-Aschoff sinuses (intramural diverticula). Bacteria can be cultured from gallbladder bile or gallstones themselves in about 10% of patients with biliary pain, but bacterial infection is not believed to contribute to the symptoms (see also Chapter 67).
Clinical Features
Biliary pain is visceral in nature and, thus, poorly localized.223 In a typical case, the patient experiences episodes of upper abdominal pain, usually in the epigastrium or right upper quadrant (RUQ) but sometimes in other abdominal locations. Ingestion of a meal often can precipitate pain, but more commonly no inciting event is apparent. The onset of biliary pain is more likely to occur during periods of weight reduction and marked physical inactivity such as prolonged bed rest than at other times.
Natural History
Biliary pain is cause for concern but not alarm. Approximately 30% of patients who have an attack of classic biliary pain will experience no additional attacks over the next 24 months. Therefore, a reasonable approach would be to offer cholecystectomy to patients with recurring episodes of biliary pain.224 In the remaining 70%, the frequency of recurrent attacks varies widely from patient to patient, but the pattern remains relatively constant for an individual patient over time. In patients monitored after an initial attack of biliary pain, symptoms sufficient to warrant cholecystectomy develop on average at a rate of approximately 6% per year. The cumulative risk that symptoms that require therapy will develop in asymptomatic persons with gallstones who are followed up for five years is 7.6%.225 The probability that a patient with a history of biliary pain will experience a complication of gallstones that requires urgent surgical intervention is only 1% to 2% per year.224
Diagnosis
In general, the first, and often the only, imaging study recommended in patients with biliary pain is ultrasound of the RUQ. Ultrasonography is rapid, noninvasive, highly sensitive, and highly specific for detecting stones in the gallbladder. Despite the impressive diagnostic accuracy of ultrasonography, a clinically important stone is occasionally missed and the correct diagnosis delayed because of the large number of patients who undergo ultrasonography for any reason.195 Given the relatively benign natural history of biliary pain, patients with suspected gallstones but a negative ultrasonography result can safely be observed, further diagnostic testing being reserved for those in whom symptoms recur.226
Oral cholecystography is generally viewed as a secondary imaging study of the gallbladder and is reserved for patients in whom medical dissolution therapy or lithotripsy of gallstones is planned (see Chapter 66). In such cases, patency of the cystic duct must be confirmed by OCG before therapy. On rare occasions, OCG may demonstrate small floating gallstones that were missed by ultrasonography.
Treatment
Patients with recurrent, uncomplicated biliary pain and documented gallstones are generally treated with elective laparoscopic cholecystectomy, as discussed in Chapter 66. Acute biliary pain improves with administration of meperidine, with or without ketorolac or diclofenac. Aspirin taken prophylactically has been reported to prevent gallstone formation as well as acute attacks of biliary pain in patients with gallstones, but long-term use of NSAIDs does not prevent gallstone formation.227,228
ACUTE CHOLECYSTITIS
Acute cholecystitis is the most common complication of gallstone disease. Inflammation of the gallbladder wall associated with abdominal pain, RUQ tenderness, fever, and leukocytosis is the hallmark of acute cholecystitis. In approximately 90% of cases, the underlying cause is obstruction of the outlet of the gallbladder by a gallstone in the cystic duct, the gallbladder neck, or Hartman’s pouch.229 In the remaining 10% of cases, cholecystitis occurs in the absence of gallstones (acalculous cholecystitis; see Chapter 67). Acute cholecystitis caused by gallstones is a disease of young, otherwise healthy women and generally has a favorable prognosis, whereas acute acalculous cholecystitis occurs more commonly in critically ill elderly men and is associated with high morbidity and mortality rates.
Pathogenesis
Acute cholecystitis generally occurs when a stone becomes embedded in the cystic duct and causes chronic obstruction, rather than transient obstruction as in biliary pain.229 Stasis of bile within the gallbladder lumen results in damage of the gallbladder mucosa with consequent release of intracellular enzymes and activation of a cascade of inflammatory mediators.
In animal studies, if the cystic duct is ligated, the usual result is gradual absorption of the gallbladder contents without the development of inflammation230; the additional instillation of a luminal irritant, such as concentrated bile or lysolecithin, or trauma from an indwelling catheter is required to cause acute cholecystitis in an obstructed gallbladder.
Phospholipase A is believed to be released by gallstone-induced mucosal trauma and converts lecithin to lysolecithin. Although normally absent from gallbladder bile, lysolecithin is present in the gallbladder contents of patients with acute cholecystitis.231 In animal models, installation of lysolecithin into the gallbladder produces acute cholecystitis associated with increased protein secretion, decreased water absorption, and evidence of white blood cell (WBC) invasion associated with elevated production of prostaglandins E and F1α. Administration of indomethacin, a cyclooxygenase inhibitor, has been shown to block this inflammatory response.
Studies of human tissue obtained at cholecystectomy have demonstrated enhanced prostaglandin production in the inflamed gallbladder. Additionally, administration of intravenous indomethacin and oral ibuprofen to patients with acute cholecystitis has been shown to diminish both luminal pressure in the gallbladder and pain.231
Supporting evidence for the role of prostaglandins in the development of acute cholecystitis comes from a prospective study in which patients who presented with biliary pain were randomized to receive diclofenac, a prostaglandin synthetase inhibitor, or placebo.232 Ultimately, acute cholecystitis developed in 9 of 40 patients who received placebo, whereas episodes of biliary pain resolved in all 20 patients who received diclofenac. These data suggest a chain of events in which obstruction of the cystic duct in association with one or more intraluminal factors damages the gallbladder mucosa and stimulates prostaglandin synthetase. The resulting fluid secretion and inflammatory changes promote a cycle of further mucosal damage and inflammation.232
Enteric bacteria can be cultured from gallbladder bile in approximately one half of patients with acute cholecystitis.233 Bacteria are not believed, however, to trigger the actual onset of acute cholecystitis.
Pathology
If examined in the first few days of an attack of acute cholecystitis, the gallbladder usually is distended and contains a stone embedded in the cystic duct.234 After the gallbladder is opened, inflammatory exudate and, rarely, pus are present. Later in the attack, the bile pigments that are normally present are absorbed, having been replaced by thin mucoid fluid, pus, or blood. If the attack of acute cholecystitis is left untreated for a long period but the cystic duct remains obstructed, the lumen of the gallbladder may become distended with clear mucoid fluid, a condition known as hydrops of the gallbladder.
Histologic changes range from mild acute inflammation with edema to necrosis and perforation of the gallbladder wall. Surprisingly, the severity of histologic changes correlates little with the patient’s symptoms.234 If the gallbladder is resected for acute cholecystitis and no stones are found, the specimen should be carefully examined histologically for evidence of vasculitis or cholesterol emboli because these systemic disorders may manifest as acalculous cholecystitis (see Chapter 35).
Clinical Features
Approximately 75% of patients with acute cholecystitis report prior attacks of biliary pain (see Table 65-2).235 Often, such a patient is alerted to the possibility that more than simple biliary pain is occurring by the prolonged duration of the pain. If biliary pain has been constant for more than six hours, uncomplicated biliary pain is increasingly unlikely, and acute cholecystitis should be suspected.
As inflammation in the gallbladder wall progresses, poorly localized visceral pain gives way to moderately severe parietal pain that localizes to the RUQ.235 Less commonly the back or rarely the chest may be the site of maximal pain. Nausea with some vomiting is characteristic of acute cholecystitis, but these symptoms almost invariably follow, rather than precede, the onset of pain. Vomiting is not as persistent or as severe as that with intestinal obstruction or acute pancreatitis.
In contrast to uncomplicated biliary pain, the physical findings can, in many cases, suggest the diagnosis of acute cholecystitis. Fever is common, but body temperature is usually less than 102°F unless the gallbladder has become gangrenous or has perforated (Fig. 65-14). Mild jaundice is present in 20% of patients with acute cholecystitis and 40% of elderly patients. Serum bilirubin levels usually are less than 4 mg/dL.236 Bilirubin levels above this value suggest the possibility of bile duct stones, which may be found in 50% of jaundiced patients with acute cholecystitis. Another cause of pronounced jaundice in patients with acute cholecystitis is Mirizzi’s syndrome, which is associated with inflammatory obstruction of the common hepatic duct (see later).
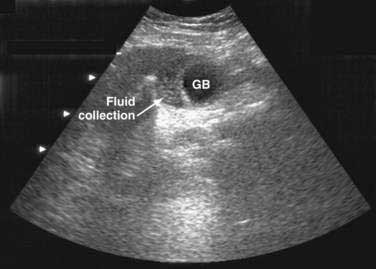
A relatively specific finding of acute cholecystitis is Murphy’s sign.235 During palpation in the right subcostal region, pain and inspiratory arrest may occur when the patient takes a deep breath that brings the inflamed gallbladder into contact with the examiner’s hand. The presence of Murphy’s sign in the appropriate clinical setting is a reliable predictor of acute cholecystitis, although gallstones should still be confirmed by ultrasonography.
Natural History
The pain of untreated acute cholecystitis generally resolves in 7 to 10 days.237 Not uncommonly, symptoms remit within 48 hours of hospitalization. One study has shown that acute cholecystitis resolves without complications in approximately 83% of patients but results in gangrenous cholecystitis in 7%, gallbladder empyema in 6%, perforation in 3%, and emphysematous cholecystitis in fewer than 1%.238
Diagnosis
Table 65-3 details the most common laboratory findings in acute cholecystitis.237 Leukocytosis with a shift to immature neutrophils is common. Because a diagnosis of bile duct stones with cholangitis usually is in the differential diagnosis, attention is directed to results of liver biochemical tests.236 Even without any detectable bile duct obstruction, acute cholecystitis often causes mild elevations in serum aminotransferase and alkaline phosphatase levels. As noted earlier, the serum bilirubin level may also be mildly elevated (2 to 4 mg/dL), and even serum amylase and lipase values may be elevated nonspecifically. A serum bilirubin value greater than 4 mg/dL or amylase value greater than 1000 U/L usually indicates coexisting bile duct obstruction or acute pancreatitis and warrants further evaluation.
Ultrasonography is the single most useful imaging study in acutely ill patients with RUQ pain and tenderness. It accurately establishes the presence or absence of gallstones and serves as an extension of the physical examination. Presence of sonographic Murphy’s sign, defined as focal gallbladder tenderness under the transducer, has a positive predictive value greater than 90% for detecting acute cholecystitis if gallstones are also present, the operator is skillful, and the patient is alert.239 Additionally, ultrasonography can detect nonspecific findings suggestive of acute cholecystitis, such as pericholecystic fluid and gallbladder wall thickening greater than 4 mm. Both findings lose specificity for acute cholecystitis if the patient has ascites or hypoalbuminemia.195,240
Because the prevalence of gallstones is high in the population, many patients with nonbiliary tract diseases that manifest as acute abdominal pain (such as acute pancreatitis and complications of peptic ulcer) may have incidental and clinically irrelevant gallstones. The greatest usefulness of cholescintigraphy in these patients is its ability to exclude acute cholecystitis and allow the clinician to focus on nonbiliary causes of the patient’s acute abdominal pain.188 A normal cholescintigraphy scan result shows radioactivity in the gallbladder, bile duct, and small intestine within 30 to 60 minutes of injection of the isotope. With rare exceptions, a normal result excludes acute cholecystitis caused by gallstones. Several studies have suggested that the sensitivity and specificity of scintigraphy in the setting of acute cholecystitis are approximately 94% each. Its sensitivity and specificity are reduced considerably, however, in patients who have liver disease, are receiving parenteral nutrition, or are fasting. These conditions can lead to a false-positive scan result, defined as the absence of isotope in the gallbladder in a patient who does not have acute cholecystitis. If a positive scan result is defined as the absence of isotope in the gallbladder, then a false-negative scan result would be defined as filling of the gallbladder with isotope in the setting of acute cholecystitis, a situation that virtually never occurs. Therefore, scintigraphy should not be used as the initial imaging study in a patient with suspected cholecystitis but rather should be used as a secondary imaging study in patients who already are known to have gallstones and in whom a nonbiliary cause of acute abdominal pain is possible.241
Differential Diagnosis
Acute appendicitis is the disease most often confused with acute cholecystitis because the initial diagnostic impression is based largely on localized right-sided abdominal tenderness, which may be lower than expected in cholecystitis or higher than expected in appendicitis. In general, fever, leukocytosis, and tenderness progress more inexorably in appendicitis. Abdominal CT usually can distinguish these two entities (see Chapter 116).
Acute pancreatitis also may be difficult to distinguish from acute cholecystitis on the basis of the history and physical examination alone. Generally, vomiting is a more prominent feature of acute pancreatitis, and affected persons are more uncomfortable in a supine position. Hyperamylasemia is more profound in pancreatitis than cholecystitis, and an elevated serum lipase value is more specific (see Chapter 58).
Diseases of the right kidney may produce pain and tenderness that mimic findings in acute cholecystitis, but urinalysis and ultrasonography can usually differentiate renal disease from cholecystitis. The pain of an uncomplicated peptic ulcer is usually chronic and seldom confused with that of acute cholecystitis, but a perforated ulcer, at least initially, may mimic severe acute cholecystitis. Signs of generalized peritonitis or pneumoperitoneum strongly suggest a perforated viscus, necessitating emergency laparotomy (see Chapters 10 and 52).
In some instances, acute hepatitis, especially when caused by alcohol, may manifest as severe RUQ pain and tenderness, fever, and leukocytosis and may be confused diagnostically with acute cholecystitis. In such cases, careful assessment of the liver biochemical values over time in combination with ultrasonography or cholescintigraphy may exclude a diagnosis of acute cholecystitis. Rarely, liver biopsy may be warranted (see Chapters 77 to 81 and 84).
Gonococcal perihepatitis (Fitz-Hugh–Curtis syndrome) produces RUQ pain and tenderness, which often overshadow any pelvic complaints, as well as leukocytosis. Nevertheless, adnexal tenderness is found on physical examination, and a Gram stain of the cervical smear should show gonococci (see Chapter 37).
Hepatic abscesses and tumors usually can be differentiated from acute cholecystitis on the basis of ultra-sonographic findings. Prior undiagnosed gallbladder perforation may manifest with fever from a subhepatic abscess. Pseudolithiasis due to ceftriaxone therapy, most often in children, has caused symptoms resembling those of acute cholecystitis, although the gallbladder is histologically normal (see Chapters 82 and 94).
CHOLEDOCHOLITHIASIS
Choledocholithiasis is defined as the occurrence of stones in the bile ducts. Like stones in the gallbladder, stones in the bile ducts may remain asymptomatic for years, and stones from the bile duct are known to pass silently into the duodenum, perhaps frequently. Unlike stones in the gallbladder, which usually become clinically evident as relatively benign episodes of recurrent biliary pain, stones in the bile duct, when they do cause symptoms, tend to manifest as life-threatening complications such as cholangitis and acute pancreatitis (see Chapter 58). Therefore, discovery of choledocholithiasis generally should be followed by some type of intervention to remove the stones (see Chapter 70).
Etiology
Gallstones may pass from the gallbladder into the bile duct or can form de novo in the duct. Generally, all gallstones from one patient, whether from the gallbladder or bile duct, are of one type, either cholesterol or pigment. Cholesterol stones form only in the gallbladder, and any cholesterol stones found in the bile duct must have migrated there from the gallbladder. Black pigment stones, which are associated with old age, hemolysis, alcoholism, and cirrhosis, also form in the gallbladder and only rarely migrate into the bile duct. The majority of pigment stones in the bile duct are the softer brown pigment stones. These stones form de novo in the bile duct as a result of bacterial action on phospholipid and bilirubin in bile (see earlier).243 They are often found proximal to biliary strictures and are frequently associated with cholangitis. Brown pigment stones are found in patients with hepatolithiasis and recurrent pyogenic cholangitis (see Chapter 68).244
Fifteen percent of patients with gallbladder stones also have bile duct stones. Conversely, of patients with ductal stones, 95% also have gallbladder stones.245 In patients who present with choledocholithiasis months or years after a cholecystectomy, determining whether the stones were overlooked at the earlier operation or have subsequently formed may be impossible. In fact, formation of pigment stones in the bile duct is also a late complication of endoscopic sphincterotomy.246 In a study of the long-term consequences of endoscopic sphincterotomy in more than 400 patients, the cumulative frequency of recurrent bile duct stones was 12%; all the recurrent stones were of the brown pigment type, irrespective of the chemical composition of the original gallstones. This observation suggests that sphincterotomy permits chronic bacterial colonization of the bile duct that results in deconjugation of bilirubin and precipitation of pigment stones.
Clinical Features
The morbidity of choledocholithiasis stems principally from biliary obstruction, which raises biliary pressure and diminishes bile flow. The rate of onset of obstruction, its extent, and the amount of bacterial contamination of the bile are the major factors that determine the resulting symptoms. Acute obstruction usually causes biliary pain and jaundice, whereas obstruction that develops gradually over several months may manifest initially as pruritus or jaundice alone.247 If bacteria proliferate, life-threatening cholangitis may result (see later).
As shown in Table 65-2, results of laboratory studies may be the only clue to the presence of choledocholithiasis.248 With bile duct obstruction, serum bilirubin and alkaline phosphatase levels both increase. Bilirubin accumulates in serum because of blocked excretion, whereas alkaline phosphatase levels rise because of increased synthesis of the enzyme by the canalicular epithelium. The rise in the alkaline phosphatase level is more rapid than and precedes the rise in bilirubin level.249 The absolute height of the serum bilirubin level is proportional to the extent of obstruction, but the height of the alkaline phosphatase level bears no relation to either the extent of obstruction or its cause. In cases of choledocholithiasis, the serum bilirubin level is typically in the range of 2 to 5 mg/dL205 and rarely exceeds 12 mg/dL. Transient “spikes” in serum aminotransferase or amylase levels suggest passage of a bile duct stone into the duodenum. The overall sensitivity of liver biochemical testing for detecting choledocholithiasis is reported to be 94%; serum levels of gamma glutamyl transpeptidase are elevated most commonly but may not be assessed in clinical practice.249
Natural History
Little information is available on the natural history of asymptomatic bile duct stones. In many patients such stones remain asymptomatic for months or years, but available evidence suggests that the natural history of asymptomatic bile duct stones is less benign than that of asymptomatic gallstones.247,250
Diagnosis
Ultrasonography actually visualizes bile duct stones in only about 50% of cases,199 whereas dilatation of the bile duct to a diameter greater than 6 mm is seen in about 75% of cases. Ultrasonography can confirm, or at least suggest, the presence of bile duct stones but cannot exclude choledocholithiasis definitively. EUS, although clearly more invasive than standard ultrasonography, has the advantage of visualizing the bile duct more accurately. In preliminary studies, EUS has excluded or confirmed choledocholithiasis with sensitivity and specificity rates of approximately 98% as compared with ERCP.203
ERCP is the standard method for the diagnosis and therapy of bile duct stones,251 with sensitivity and specificity rates of approximately 95%. When the clinical probability of choledocholithiasis is low, however, less invasive studies, such as EUS and MRCP, should be performed first.216
Laparoscopic ultrasonography may be used in the surgical suite immediately before mobilization of the gallbladder during cholecystectomy. Laparoscopic ultrasonography may be as accurate as surgical cholangiography in detecting bile duct stones and may thereby obviate the need for the latter.252
Differential Diagnosis
In patients who present with jaundice, malignant obstruction of the bile duct or obstruction from a choledochal cyst may be indistinguishable clinically from choledocholithiasis (see Chapters 62 and 69).
Acute passive congestion of the liver, associated with cardiac decompensation, may cause intense RUQ pain, tenderness, and even jaundice with serum bilirubin levels higher than 10 mg/dL (see Chapter 83); however, fever is usually absent, and the WBC count is normal or only slightly elevated. The patient typically has other obvious signs of cardiac decompensation. Constrictive pericarditis and cor pulmonale also may cause acute congestion of the liver with only subtle cardiac findings.
Acquired immunodeficiency syndrome (AIDS)-associated cholangiopathy253 and papillary stenosis must be considered in human immunodeficiency virus–positive patients with RUQ pain and abnormal liver biochemical test results (see Chapter 33).
Treatment
Because of its propensity to result in serious complications such as cholangitis and acute pancreatitis, choledocholithiasis warrants treatment in nearly all cases.254 The optimal therapy for a given patient depends on the severity of symptoms, presence of coexisting medical problems, availability of local expertise, and presence or absence of the gallbladder.
Bile duct stones discovered at the time of a laparoscopic cholecystectomy present a dilemma to the surgeon. Some surgeons may attempt laparoscopic exploration of the bile duct. In other cases, the operation can be converted to an open cholecystectomy with bile duct exploration, but this approach results in greater morbidity and a more prolonged hospital stay. Alternatively, the laparoscopic cholecystectomy can be carried out as planned, and the patient can return for ERCP with removal of the bile duct stones. Such an approach, if successful, cures the disease but runs the risk of necessitating a third procedure, namely a bile duct exploration, if the stones cannot be removed at ERCP. In general, the greater the expertise of the therapeutic endoscopist, the more inclined the surgeon should be to complete the laparoscopic cholecystectomy and have the bile duct stones removed endoscopically.254
In especially high-risk patients, endoscopic removal of bile duct stones may be performed without cholecystectomy. This approach is particularly appropriate for elderly patients with other severe illnesses.255 Cholecystectomy is required subsequently for recurrent symptoms in only 10% of patients. The surgical management and endoscopic treatment of gallstones are discussed in detail in Chapters 66 and 70, respectively.
CHOLANGITIS
Of all the common complications of gallstones, the most serious and the most lethal is acute bacterial cholangitis. Pus under pressure in the bile ducts leads to rapid spread of bacteria via the liver into the blood, with resulting septicemia. Moreover, the diagnosis of cholangitis is often problematic (especially in the critical early phase of the disease) because clinical features that point to the biliary tract as the source of sepsis are often absent.26 Table 65-2 delineates the symptoms, signs, and laboratory findings that can aid in an early diagnosis of cholangitis.
Etiology and Pathophysiology
In approximately 85% of cases, cholangitis is caused by a stone embedded in the bile duct, with resulting bile stasis.257 Other causes of bile duct obstruction that may result in cholangitis are neoplasms (see Chapters 60 and 69), biliary strictures (see Chapters 68 and 70), parasitic infections (see Chapters 68 and 82), and congenital abnormalities of the bile ducts (see Chapter 62). This discussion deals specifically with cholangitis caused by gallstones in the bile duct.
Bile duct obstruction is necessary, but not sufficient, to cause cholangitis. Cholangitis is relatively common in patients with choledocholithiasis and nearly universal in patients with a post-traumatic bile duct stricture but is seen in only 15% of patients with neoplastic obstruction of the bile duct. It is most likely to result when a bile duct that already contains bacteria becomes obstructed, as is the case in most patients with choledocholithiasis and stricture but in few patients with neoplastic obstruction. Malignant obstruction is more often complete than obstruction by a stricture or a bile duct stone and less commonly permits the reflux of bacteria from duodenal contents into the bile ducts.258
The bacterial species most commonly cultured from the bile are E. coli, Klebsiella, Pseudomonas, Proteus, and enterococci. Anaerobic species such as Bacteroides fragilis and Clostridium perfringens are found in about 15% of appropriately cultured bile specimens. Anaerobes usually accompany aerobes, especially E. coli. The shaking chills and fever of cholangitis are caused by bacteremia from bile duct organisms. The degree of regurgitation of bacteria from bile into hepatic venous blood is directly proportional to the biliary pressure and, hence, the degree of obstruction.258 For this reason, decompression alone often effectively treats the illness.
Clinical Features
The hallmark of cholangitis is Charcot’s triad, consisting of RUQ pain, jaundice, and fever (see Table 65-2). The full triad is present in only 70% of patients.258 The pain of cholangitis may be surprisingly mild and transient but is often accompanied by chills and rigors. Elderly patients in particular may present solely with mental confusion, lethargy, and delirium. Altered mental status and hypotension in combination with Charcot’s triad, known commonly as Reynolds’ pentad, occur in severe suppurative cholangitis.
On physical examination, fever is almost universal, occurring in 95% of patients. RUQ tenderness is elicited in approximately 90% of patients, but jaundice is clinically detectable in only 80%. Notably, peritoneal signs are found in only 15% of patients. The combination of hypotension and mental confusion indicates gram-negative septicemia. In overlooked cases of severe cholangitis, intrahepatic abscess may manifest as a late complication (see Chapter 82).
Laboratory study results are often helpful in pointing to the biliary tract as the source of sepsis. In particular, the serum bilirubin level exceeds 2 mg/dL in 80% of patients. When the bilirubin level is normal initially, the diagnosis of cholangitis may not be suspected.249 The WBC count is elevated in 80% of patients. In many patients who have a normal WBC count, examination of the peripheral blood smear reveals a dramatic shift to immature neutrophil forms. The serum alkaline phosphatase level is usually elevated, and the serum amylase level may also be elevated if pancreatitis is also present.
Diagnosis
The principles of radiologic diagnosis of cholangitis are the same as those for choledocholithiasis. As shown in Table 65-3, stones in the bile duct are seen ultrasonographically in only about 50% of cases155 but can be inferred by detection of a dilated bile duct in about 75% of cases. Normal ultrasonography findings do not exclude the possibility of choledocholithiasis in a patient in whom the clinical presentation suggests cholangitis.241
ERCP is the standard test for the diagnosis of bile duct stones and cholangitis. Moreover, the ability of ERCP to establish drainage of infected bile under pressure can be life-saving. If ERCP is unsuccessful, percutaneous THC can be performed (see Chapter 70).
Treatment
In cases of suspected bacterial cholangitis, blood culture specimens should be obtained immediately and therapy started with antibiotics effective against the likely causative organisms.259 In mild cases, initial therapy with a single drug, such as cefoxitin, 2.0 g intravenously every six to eight hours, is usually sufficient. In severe cases, more intensive therapy (e.g., gentamicin, ampicillin, and metronidazole or a broad-spectrum agent such as piperacillin-tazobactam 3.375 g intravenously every six hours or, if resistant organisms are suspected, meropenem 1 g intravenously every eight hours) is indicated.
The patient’s condition should improve within 6 to 12 hours, and in most cases, the infection comes under control within 2 to 3 days, with defervescence, relief of discomfort, and a decline in the WBC count. In these cases, definitive therapy can be planned on an elective basis. If, however, after 6 to 12 hours of careful observation, the patient’s clinical status declines with worsening fever, pain, mental confusion, or hypotension, the bile duct must be decompressed immediately.259 If available, ERCP with stone extraction, or at least decompression of the bile duct with an intrabiliary stent, is the treatment of choice. Controlled studies in which ERCP and decompression of the bile duct were compared with emergency surgery and bile duct exploration have shown dramatically lower morbidity and mortality rates in patients treated endoscopically.254 The surgical treatment and endoscopic management of cholangitis are discussed in detail in Chapters 66 and 70, respectively.
UNCOMMON COMPLICATIONS
Table 65-4 describes the clinical manifestations, diagnosis, and treatment of several uncommon complications of gallstone disease.
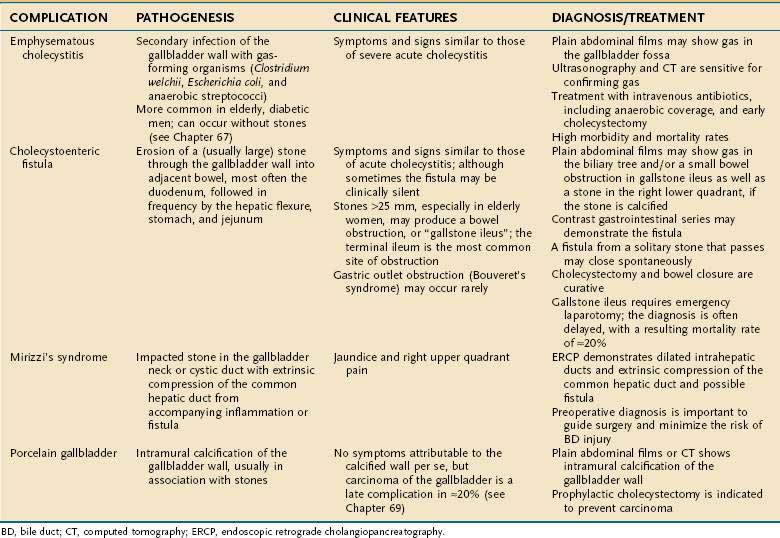
EMPHYSEMATOUS CHOLECYSTITIS
Patients who have emphysematous cholecystitis present with the same clinical manifestations as patients with uncomplicated acute cholecystitis, but in the former, gas-forming organisms have secondarily infected the gallbladder wall. Pockets of gas are evident in the area of the gallbladder fossa on plain abdominal films, ultrasonography, and abdominal CT (see Fig. 65-13).260 Emergency antibiotic therapy with anaerobic coverage and early cholecystectomy are warranted because the risk of gallbladder perforation is high. Emphysematous cholecystitis often occurs in diabetic persons or older men who do not have gallstones, in whom atherosclerosis of the cystic artery with resulting ischemia may be the initiating event (see Chapter 67).
CHOLECYSTOENTERIC FISTULA
A cholecystoenteric fistula occurs when a stone erodes through the gallbladder wall (usually the neck) and into a hollow viscus. The most common entry point into the bowel is the duodenum, followed in frequency by the hepatic flexure of the colon, the stomach, and the jejunum. Symptoms are initially similar to those of acute cholecystitis, although at times the stone may pass into the bowel and may be excreted without causing any symptoms.261 Because the biliary tract is decompressed, cholangitis is not common despite gross seeding of the gallbladder and bile ducts with bacteria. The diagnosis of a cholecystoenteric fistula is suspected from radiographic evidence of pneumobilia and may be confirmed by barium contrast studies of the upper or lower gastrointestinal tract; often the precise anatomic location of the fistula is not identified until surgery.
If the gallstone exceeds 25 mm in diameter, it may manifest, especially in elderly women, as a small intestinal obstruction (gallstone ileus); the ileocecal area is the most common site of obstruction.262 In such cases, a plain abdominal film may show the pathognomonic features of pneumobilia, a dilated small bowel, and a large gallstone in the right lower quadrant. Unfortunately, the diagnosis of a gallstone ileus is often delayed, with a resulting mortality rate of approximately 20%. Bouveret’s syndrome is characterized by gastric outlet obstruction resulting from duodenal impaction of a large gallstone that has migrated through a cholecystoduodenal fistula.263
MIRIZZI’S SYNDROME
Mirizzi’s syndrome is a rare complication in which a stone embedded in the neck of the gallbladder or cystic duct extrinsically compresses the common hepatic duct with resulting jaundice, bile duct obstruction, and, in some cases, a fistula.264,265 Typically the gallbladder contracted and contains stones. ERCP usually demonstrates the characteristic extrinsic compression of the common hepatic duct. Treatment is traditionally by an open cholecystectomy, although endoscopic stenting and laparoscopic cholecystectomy have been performed successfully. Preoperative diagnosis of Mirizzi’s syndrome is important so that bile duct injury can be avoided.266
PORCELAIN GALLBLADDER
Strictly speaking, porcelain gallbladder, defined as intramural calcification of the gallbladder wall, is not a complication of gallstones but is mentioned here because of the remarkable tendency of carcinoma to develop as a late complication of gallbladder calcification (specifically, a gallbladder with focal rather than diffuse wall calcification).267 The diagnosis of a porcelain gallbladder can be made with a plain abdominal film or abdominal CT, which shows intramural calcification of the gallbladder wall. Prophylactic cholecystectomy, preferably through a laparoscopic approach, is indicated to prevent the subsequent development of carcinoma, which may otherwise occur in up to 20% of cases (see Chapter 69).268
Buch S, Schafmayer C, Volzke H, et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet. 2007;39:995-9. (Ref 159.)
Buhman KK, Accad M, Novak S, et al. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med. 2000;6:1341-7. (Ref 164.)
Collins C, Maguire D, Ireland A, et al. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: Natural history of choledocholithiasis revisited. Ann Surg. 2004;239:28-33. (Ref 250.)
Konikoff FM, Chung DS, Donovan JM, et al. Filamentous, helical, and tubular microstructures during cholesterol crystallization from bile. Evidence that cholesterol does not nucleate classic monohydrate plates. J Clin Invest. 1992;90:1155-60. (Ref 74.)
Maurer KJ, Ihrig MM, Rogers AB, et al. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023-33. (Ref 122.)
Paigen B, Carey MC. Gallstones. New York: Oxford University Press; 2002. p 298. (Ref 3.)
Portincasa P, Di Ciaula A, Wang HH, et al. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology. 2008;47:2112-26. (Ref 97.)
Wang DQ, Carey MC. Complete mapping of crystallization pathways during cholesterol precipitation from model bile: Influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J Lipid Res. 1996;37:606-30. (Ref 52.)
Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: Physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395-411. (Ref 73.)
Wang DQ, Schmitz F, Kopin AS, et al. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J Clin Invest. 2004;114:521-8. (Ref 115.)
Wang HH, Portincasa P, Wang DQ. Molecular pathophysiology and physical chemistry of cholesterol gallstones. Front Biosci. 2008;13:401-23. (Ref 4.)
Wang HH, Afdhal NH, Gendler SJ, et al. Evidence that gallbladder epithelial mucin enhances cholesterol cholelithogenesis in MUC1 transgenic mice. Gastroenterology. 2006;131:210-22. (Ref 89.)
Wang HH, Afdhal NH, Wang DQ. Estrogen receptor alpha, but not beta, plays a major role in 17beta-estradiol-induced murine cholesterol gallstones. Gastroenterology. 2004;127:239-49. (Ref 21.)
Wang HH, Afdhal NH, Wang DQ. Overexpression of estrogen receptor alpha increases hepatic cholesterogenesis, leading to biliary hypersecretion in mice. J Lipid Res. 2006;47:778-86. (Ref 22.)
Yu L, Hammer RE, Li-Hawkins J, et al. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237-42. (Ref 60.)
1. Liver Disease Subcommittee of the Digestive Disease Interagency Coordinating Committee. Action Plan for Liver Disease Research. Bethesda, Md.: National Institutes of Health; 2004.
2. Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics, 2004. Gastroenterology. 2004;126:1448-53.
3. Paigen B, Carey MC. Gallstones. New York: Oxford University Press; 2002. p 298
4. Wang HH, Portincasa P, Wang DQ. Molecular pathophysiology and physical chemistry of cholesterol gallstones. Front Biosci. 2008;13:401-23.
5. Everhart JE. Gallstones and ethnicity in the Americas. J Assoc Acad Minor Phys. 2001;12:137-43.
6. Everhart JE, Khare M, Hill M, et al. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology. 1999;117:632-9.
7. Sandler RS, Everhart JE, Donowitz M, et al. The burden of selected digestive diseases in the United States. Gastroenterology. 2002;122:1500-11.
8. Diehl AK. Epidemiology and natural history of gallstone disease. Gastroenterol Clin North Am. 1991;20:1-19.
9. Portincasa P, Moschetta A, Palasciano G. Cholesterol gallstone disease. Lancet. 2006;368:230-9.
10. Jensen KH, Jorgensen T. Incidence of gallstones in a Danish population. Gastroenterology. 1991;100:790-4.
11. Lowenfels AB, Velema JP. Estimating gallstone incidence from prevalence data. Scand J Gastroenterol. 1992;27:984-6.
12. Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: The Sirmione Study. Hepatology. 1987;7:913-17.
13. Sampliner RE, Bennett PH, Comess LJ, et al. Gallbladder disease in Pima Indians. Demonstration of high prevalence and early onset by cholecystography. N Engl J Med. 1970;283:1358-64.
14. Friedman GD, Kannel WB, Dawber TR. The epidemiology of gallbladder disease: Observations in the Framingham Study. J Chronic Dis. 1966;19:273-92.
15. Sama C, Labate AM, Taroni F, et al. Epidemiology and natural history of gallstone disease. Semin ver Dis. 1990;10:149-58.
16. Einarsson K, Nilsell K, Leijd B, et al. Influence of age on secretion of cholesterol and synthesis of bile acids by the liver. N Engl J Med. 1985;313:277-82.
17. Valdivieso V, Palma R, Wunkhaus R, et al. Effect of aging on biliary lipid composition and bile acid metabolism in normal Chilean women. Gastroenterology. 1978;74:871-4.
18. Wang DQ. Aging per se is an independent risk factor for cholesterol gallstone formation in gallstone susceptible mice. J Lipid Res. 2002;43:1950-9.
19. Angelin B, Olivecrona H, Reihner E, et al. Hepatic cholesterol metabolism in estrogen-treated men. Gastroenterology. 1992;103:1657-63.
20. Henriksson P, Einarsson K, Eriksson A, et al. Estrogen-induced gallstone formation in males. Relation to changes in serum and biliary lipids during hormonal treatment of prostatic carcinoma. J Clin Invest. 1989;84:811-16.
21. Wang HH, Afdhal NH, Wang DQ. Estrogen receptor alpha, but not beta, plays a major role in 17beta-estradiol-induced murine cholesterol gallstones. Gastroenterology. 2004;127:239-49.
22. Wang HH, Afdhal NH, Wang DQ. Overexpression of estrogen receptor alpha increases hepatic cholesterogenesis, leading to biliary hypersecretion in mice. J Lipid Res. 2006;47:778-86.
23. Wang DQ, Afdhal NH. Genetic analysis of cholesterol gallstone formation: Searching for Lith (gallstone) genes. Curr Gastroenterol Rep. 2004;6:140-50.
24. Huang YC, Zhang XW, Yang RX. Changes in cholelithiasis in Tianjin in the past 30 years. Chin Med J (Engl). 1984;97:133-5.
25. Nagase M, Tanimura H, Setoyama M, et al. Present features of gallstones in Japan. A collective review of 2,144 cases. Am J Surg. 1978;135:788-90.
26. Valdivieso V, Covarrubias C, Siegel F, et al. Pregnancy and cholelithiasis: Pathogenesis and natural course of gallstones diagnosed in early puerperium. Hepatology. 1993;17:1-4.
27. Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: Incidence, risk factors, and natural history. Ann Intern Med. 1993;119:116-20.
28. Shiffman ML, Sugerman HJ, Kellum JM, et al. Gallstone formation after rapid weight loss: A prospective study in patients undergoing gastric bypass surgery for treatment of morbid obesity. Am J Gastroenterol. 1991;86:1000-5.
29. Shiffman ML, Kaplan GD, Brinkman-Kaplan V, et al. Prophylaxis against gallstone formation with ursodeoxycholic acid in patients participating in a very-low-calorie diet program. Ann Intern Med. 1995;122:899-905.
30. Pitt HA, King W3rd, Mann LL, et al. Increased risk of cholelithiasis with prolonged total parenteral nutrition. Am J Surg. 1983;145:106-12.
31. Roslyn JJ, Berquist WE, Pitt HA, et al. Increased risk of gallstones in children receiving total parenteral nutrition. Pediatrics. 1983;71:784-9.
32. Sitzmann JV, Pitt HA, Steinborn PA, et al. Cholecystokinin prevents parenteral nutrition induced biliary sludge in humans. Surg Gynecol Obstet. 1990;170:25-31.
33. Lee SP, Nicholls JF. Nature and composition of biliary sludge. Gastroenterology. 1986;90:677-86.
34. Lee SP, Maher K, Nicholls JF. Origin and fate of biliary sludge. Gastroenterology. 1988;94:170-6.
35. Ko CW, Beresford SA, Schulte SJ, et al. Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology. 2005;41:359-65.
36. Grodstein F, Colditz GA, Hunter DJ, et al. A prospective study of symptomatic gallstones in women: Relation with oral contraceptives and other risk factors. Obstet Gynecol. 1994;84:207-14.
37. Uhler ML, Marks JW, Judd HL. Estrogen replacement therapy and gallbladder disease in postmenopausal women. Menopause. 2000;7:162-7.
38. Stahlberg D, Reihner E, Rudling M, et al. Influence of bezafibrate on hepatic cholesterol metabolism in gallstone patients: Reduced activity of cholesterol 7 alpha-hydroxylase. Hepatology. 1995;21:1025-30.
39. Chapman BA, Burt MJ, Chisholm RJ, et al. Dissolution of gallstones with simvastatin, an HMG CoA reductase inhibitor. Dig Dis Sci. 1998;43:349-53.
40. Wang HH, Portincasa P, Mendez-Sanchez N, et al. Effect of ezetimibe on the prevention and dissolution of cholesterol gallstones. Gastroenterology. 2008;134:2101-10.
41. Dowling RH, Hussaini SH, Murphy GM, et al. Gallstones during octreotide therapy. Digestion. 1993;54(Suppl 1):107-20.
42. Schaad UB, Wedgwood-Krucko J, Tschaeppeler H. Reversible ceftriaxone-associated biliary pseudolithiasis in children. Lancet. 1988;2:1411-13.
43. Petitti DB, Friedman GD, Klatsky AL. Association of a history of gallbladder disease with a reduced concentration of high-density-lipoprotein cholesterol. N Engl J Med. 1981;304:1396-8.
44. Attili AF, Capocaccia R, Carulli N, et al. Factors associated with gallstone disease in the MICOL experience. Multicenter Italian Study on Epidemiology of Cholelithiasis. Hepatology. 1997;26:809-18.
45. Stampfer MJ, Maclure KM, Colditz GA, et al. Risk of symptomatic gallstones in women with severe obesity. Am J Clin Nutr. 1992;55:652-8.
46. Biddinger SB, Haas JT, Yu BB, et al. Hepatic insulin resistance directly promotes formation of cholesterol gallstones. Nat Med. 2008;14:778-82.
47. Lapidus A, Bangstad M, Astrom M, et al. The prevalence of gallstone disease in a defined cohort of patients with Crohn’s disease. Am J Gastroenterol. 1999;94:1261-6.
48. Brink MA, Slors JF, Keulemans YC, et al. Enterohepatic cycling of bilirubin: A putative mechanism for pigment gallstone formation in ileal Crohn’s disease. Gastroenterology. 1999;116:1420-7.
49. Bourges M, Small DM, Dervichian DG. Biophysics of lipid associations. 3. The quaternary systems lecithin-bile salt-cholesterol-water. Biochim Biophys Acta. 1967;144:189-201.
50. Small DM, Bourges M, Dervichian DG. Ternary and quaternary aqueous systems containing bile salt, lecithin, and cholesterol. Nature. 1966;211:816-18.
51. Carey MC, Small DM. The physical chemistry of cholesterol solubility in bile. Relationship to gallstone formation and dissolution in man. J Clin Invest. 1978;61:998-1026.
52. Wang DQ, Carey MC. Complete mapping of crystallization pathways during cholesterol precipitation from model bile: Influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J Lipid Res. 1996;37:606-30.
53. Wang DQ, Cohen DE, Lammert F, et al. No pathophysiologic relationship of soluble biliary proteins to cholesterol crystallization in human bile. J Lipid Res. 1999;40:415-25.
54. Carey MC. Critical tables for calculating the cholesterol saturation of native bile. J Lipid Res. 1978;19:945-55.
55. Cohen DE, Carey MC. Physical chemistry of biliary lipids during bile formation. Hepatology. 1990;12:143S-7S.
56. Cohen DE, Leighton LS, Carey MC. Bile salt hydrophobicity controls vesicle secretion rates and transformations in native bile. Am J Physiol. 1992;263:G386-95.
57. Crawford JM, Mockel GM, Crawford AR, et al. Imaging biliary lipid secretion in the rat: Ultrastructural evidence for vesiculation of the hepatocyte canalicular membrane. J Lipid Res. 1995;36:2147-63.
58. Crawford AR, Smith AJ, Hatch VC, et al. Hepatic secretion of phospholipid vesicles in the mouse critically depends on mdr2 or MDR3 P-glycoprotein expression. Visualization by electron microscopy. J Clin Invest. 1997;100:2562-7.
59. Graf GA, Yu L, Li WP, et al. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem. 2003;278:48275-82.
60. Yu L, Hammer RE, Li-Hawkins J, et al. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A. 2002;99:16237-42.
61. Yu L, Li-Hawkins J, Hammer RE, et al. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110:671-80.
62. Wang HH, Patel SB, Carey MC, et al. Quantifying anomalous intestinal sterol uptake, lymphatic transport, and biliary secretion in Abcg8(-/-) mice. Hepatology. 2007;45:998-1006.
63. Kosters A, Kunne C, Looije N, et al. The mechanism of ABCG5/ABCG8 in biliary cholesterol secretion in mice. J Lipid Res. 2006;47:1959-66.
64. Wang DQ, Carey MC. Susceptibility to murine cholesterol gallstone formation is not affected by partial disruption of the HDL receptor SR-BI. Biochim Biophys Acta. 2002;1583:141-50.
65. Kozarsky KF, Donahee MH, Rigotti A, et al. Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature. 1997;387:414-17.
66. Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451-62.
67. Degiorgio D, Colombo C, Seia M, et al. Molecular characterization and structural implications of 25 new ABCB4 mutations in progressive familial intrahepatic cholestasis type 3 (PFIC3). Eur J Hum Genet. 2007;15:1230-8.
68. Gerloff T, Stieger B, Hagenbuch B, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046-50.
69. Wang R, Lam P, Liu L, et al. Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatology. 2003;38:1489-99.
70. Wang R, Salem M, Yousef IM, et al. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A. 2001;98:2011-16.
71. Holan KR, Holzbach RT, Hermann RE, et al. Nucleation time: A key factor in the pathogenesis of cholesterol gallstone disease. Gastroenterology. 1979;77:611-17.
72. Wang DQ, Carey MC. Characterization of crystallization pathways during cholesterol precipitation from human gallbladder biles: Identical pathways to corresponding model biles with three predominating sequences. J Lipid Res. 1996;37:2539-49.
73. Wang DQ, Paigen B, Carey MC. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: Physical-chemistry of gallbladder bile. J Lipid Res. 1997;38:1395-411.
74. Konikoff FM, Chung DS, Donovan JM, et al. Filamentous, helical, and tubular microstructures during cholesterol crystallization from bile. Evidence that cholesterol does not nucleate classic monohydrate plates. J Clin Invest. 1992;90:1155-60.
75. Konikoff FM, Cohen DE, Carey MC. Phospholipid molecular species influence crystal habits and transition sequences of metastable intermediates during cholesterol crystallization from bile salt-rich model bile. J Lipid Res. 1994;35:60-70.
76. Carey MC. Pathogenesis of gallstones. Am J Surg. 1993;165:410-19.
77. Holzbach RT. Nucleation of cholesterol crystals in native bile. Hepatology. 1990;12:155S-9S.
78. Holzbach RT. Newer pathogenetic concepts in cholesterol gallstone formation: A unitary hypothesis. Digestion. 1997;58(Suppl 1):29-32.
79. Lee SP, LaMont JT, Carey MC. Role of gallbladder mucus hypersecretion in the evolution of cholesterol gallstones. J Clin Invest. 1981;67:1712-23.
80. Carey MC, Cahalane MJ. Whither biliary sludge? Gastroenterology. 1988;95:508-23.
81. Neutra MR, Forstner JF. Gastrointestinal mucus: Synthesis, secretion and function. New York: Raven Press; 1987. p 975
82. Gendler SJ, Spicer AP. Epithelial mucin genes. Annu Rev Physiol. 1995;57:607-34.
83. Kim YS, Gum JRJr. Diversity of mucin genes, structure, function, and expression. Gastroenterology. 1995;109:999-1001.
84. Verma M, Davidson EA. Mucin genes: Structure, expression and regulation. Glycoconj J. 1994;11:172-9.
85. Ho SB, Niehans GA, Lyftogt C, et al. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53:641-51.
86. Pemsingh RS, MacPherson BR, Scott GW. Mucus hypersecretion in the gallbladder epithelium of ground squirrels fed a lithogenic diet for the induction of cholesterol gallstones. Hepatology. 1987;7:1267-71.
87. Womack NA. The development of gallstones. Surg Gynecol Obstet. 1971;133:937-45.
88. Wang HH, Afdhal NH, Gendler SJ, et al. Targeted disruption of the murine mucin gene 1 decreases susceptibility to cholesterol gallstone formation. J Lipid Res. 2004;45:438-47.
89. Wang HH, Afdhal NH, Gendler SJ, et al. Evidence that gallbladder epithelial mucin enhances cholesterol cholelithogenesis in MUC1 transgenic mice. Gastroenterology. 2006;131:210-22.
90. Groen AK, Noordam C, Drapers JA, et al. Isolation of a potent cholesterol nucleation-promoting activity from human gallbladder bile: Role in the pathogenesis of gallstone disease. Hepatology. 1990;11:525-33.
91. Kibe A, Holzbach RT, LaRusso NF, et al. Inhibition of cholesterol crystal formation by apolipoproteins in supersaturated model bile. Science. 1984;225:514-16.
92. Holzbach RT, Kibe A, Thiel E, et al. Biliary proteins. Unique inhibitors of cholesterol crystal nucleation in human gallbladder bile. J Clin Invest. 1984;73:35-45.
93. Secknus R, Darby GH, Chernosky A, et al. Apolipoprotein A-I in bile inhibits cholesterol crystallization and modifies transcellular lipid transfer through cultured human gallbladder epithelial cells. J Gastroenterol Hepatol. 1999;14:446-56.
94. Stolk MF, van de Heijning BJ, van Erpecum KJ, et al. The effect of bile acid hydrophobicity on nucleation of several types of cholesterol crystals from model bile vesicles. J Hepatol. 1994;20:802-10.
95. van de Heijning BJ, Stolk MF, van Erpecum KJ, et al. The effects of bile salt hydrophobicity on model bile vesicle morphology. Biochim Biophys Acta. 1994;1212:203-10.
96. van Erpecum KJ, Portincasa P, Stolk MF, et al. Effects of bile salt and phospholipid hydrophobicity on lithogenicity of human gallbladder bile. Eur J Clin Invest. 1994;24:744-50.
97. Portincasa P, Di Ciaula A, Wang HH, et al. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology. 2008;47:2112-26.
98. Portincasa P, Di Ciaula A, vanBerge-Henegouwen GP. Smooth muscle function and dysfunction in gallbladder disease. Curr Gastroenterol Rep. 2004;6:151-62.
99. Portincasa P, Di Ciaula A, Vendemiale G, et al. Gallbladder motility and cholesterol crystallization in bile from patients with pigment and cholesterol gallstones. Eur J Clin Invest. 2000;30:317-24.
100. Sackmann M, Niller H, Klueppelberg U, et al. Gallstone recurrence after shock-wave therapy. Gastroenterology. 1994;106:225-30.
101. Pauletzki J, Sailer C, Kluppelberg U, et al. Gallbladder emptying determines early gallstone clearance after shock-wave lithotripsy. Gastroenterology. 1994;107:1496-502.
102. Yu P, Chen Q, Harnett KM, et al. Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am J Physiol. 1995;269:G659-65.
103. Yu P, Chen Q, Xiao Z, et al. Signal transduction pathways mediating CCK-induced gallbladder muscle contraction. Am J Physiol. 1998;275:G203-11.
104. Portincasa P, Stolk MF, van Erpecum KJ, et al. Cholesterol gallstone formation in man and potential treatments of the gallbladder motility defect. Scand J Gastroenterol, Suppl 212, 1995:63-78
105. Stolk MF, van Erpecum KJ, Renooij W, et al. Gallbladder emptying in vivo, bile composition, and nucleation of cholesterol crystals in patients with cholesterol gallstones. Gastroenterology. 1995;108:1882-8.
106. Stolk MF, Van Erpecum KJ, Hiemstra G, et al. Gallbladder motility and cholecystokinin release during long-term enteral nutrition in patients with Crohn’s disease. Scand J Gastroenterol. 1994;29:934-9.
107. Van Erpecum KJ, Stolk MF, van den Broek AM, et al. Bile concentration promotes nucleation of cholesterol monohydrate crystals by increasing the cholesterol concentration in the vesicles. Eur J Clin Invest. 1993;23:283-88.
108. van Erpecum KJ, Wang DQ, Moschetta A, et al. Gallbladder histopathology during murine gallstone formation: Relation to motility and concentrating function. J Lipid Res. 2006;47:32-41.
109. Corradini SG, Elisei W, Giovannelli L, et al. Impaired human gallbladder lipid absorption in cholesterol gallstone disease and its effect on cholesterol solubility in bile. Gastroenterology. 2000;118:912-20.
110. Conter RL, Roslyn JJ, Porter-Fink V, et al. Gallbladder absorption increases during early cholesterol gallstone formation. Am J Surg. 1986;151:184-91.
111. Roslyn JJ, Doty J, Pitt HA, et al. Enhanced gallbladder absorption during gallstone formation: The roles of cholesterol saturated bile and gallbladder stasis. Am J Med Sci. 1986;292:75-80.
112. Einarsson C. Lipid absorption by the human gallbladder. Ital J Gastroenterol Hepatol. 1999;31:571-3.
113. Yu P, Chen Q, Biancani P, et al. Membrane cholesterol alters gallbladder muscle contractility in prairie dogs. Am J Physiol. 1996;271:G56-61.
114. Wang DQ, Zhang L, Wang HH. High cholesterol absorption efficiency and rapid biliary secretion of chylomicron remnant cholesterol enhance cholelithogenesis in gallstone-susceptible mice. Biochim Biophys Acta. 2005;1733:90-9.
115. Wang DQ, Schmitz F, Kopin AS, et al. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J Clin Invest. 2004;114:521-8.
116. Dowling RH, Veysey MJ, Pereira SP, et al. Role of intestinal transit in the pathogenesis of gallbladder stones. Can J Gastroenterol. 1997;11:57-64.
117. Hussaini SH, Pereira SP, Dowling RH, et al. Slow intestinal transit and gallstone formation. Lancet. 1993;341:638.
118. Hussaini SH, Pereira SP, Murphy GM, et al. Deoxycholic acid influences cholesterol solubilization and microcrystal nucleation time in gallbladder bile. Hepatology. 1995;22:1735-44.
119. Hussaini SH, Pereira SP, Veysey MJ, et al. Roles of gall bladder emptying and intestinal transit in the pathogenesis of octreotide induced gall bladder stones. Gut. 1996;38:775-83.
120. Hussaini SH, Pereira SP, Murphy GM, et al. Composition of gall bladder stones associated with octreotide: response to oral ursodeoxycholic acid. Gut. 1995;36:126-32.
121. Wang DQ, Lammert F, Paigen B, et al. Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice. Pathophysiology of biliary lipid secretion. J Lipid Res. 1999;40:2066-79.
122. Maurer KJ, Ihrig MM, Rogers AB, et al. Identification of cholelithogenic enterohepatic helicobacter species and their role in murine cholesterol gallstone formation. Gastroenterology. 2005;128:1023-33.
123. Maurer KJ, Rogers AB, Ge Z, et al. Helicobacter pylori and cholesterol gallstone formation in C57L/J mice: a prospective study. Am J Physiol Gastrointest Liver Physiol. 2006;290:G175-82.
124. Fox JG, Dewhirst FE, Shen Z, et al. Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterology. 1998;114:755-63.
125. Pereira SP, Bain IM, Kumar D, et al. Bile composition in inflammatory bowel disease: Ileal disease and colectomy, but not colitis, induce lithogenic bile. Aliment Pharmacol Ther. 2003;17:923-33.
126. Brink MA, Mendez-Sanchez N, Carey MC. Bilirubin cycles enterohepatically after ileal resection in the rat. Gastroenterology. 1996;110:1945-57.
127. van Den Berg AA, van Buul JD, Ostrow JD, et al. Measurement of cholesterol gallstone growth in vitro. J Lipid Res. 2000;41:189-94.
128. Weiss KM, Ferrell RE, Hanis CL, et al. Genetics and epidemiology of gallbladder disease in New World native peoples. Am J Hum Genet. 1984;36:1259-78.
129. Thistle JL, Schoenfield LJ. Lithogenic bile among young Indian women. N Engl J Med. 1971;284:177-81.
130. van der Linden W, Simonson N. Familial occurrence of gallstone disease. Incidence in parents of young patients. Hum Hered. 1973;23:123-7.
131. van der Linden W, Nakayama F. Gallstone disease in Sweden versus Japan. Clinical and etiologic aspects. Am J Surg. 1973;125:267-72.
132. Gilat T, Feldman C, Halpern Z, et al. An increased familial frequency of gallstones. Gastroenterology. 1983;84:242-6.
133. Sarin SK, Negi VS, Dewan R, et al. High familial prevalence of gallstones in the first-degree relatives of gallstone patients. Hepatology. 1995;22:138-41.
134. Danzinger RG, Gordon H, Schoenfield LJ, et al. Lithogenic bile in siblings of young women with cholelithiasis. Mayo Clin Proc. 1972;47:762-6.
135. Antero Kesaniemi Y, Koskenvuo M, Vuoristo M, et al. Biliary lipid composition in monozygotic and dizygotic pairs of twins. Gut. 1989;30:1750-6.
136. Hyogo H, Roy S, Paigen B, et al. Leptin promotes biliary cholesterol elimination during weight loss in ob/ob mice by regulating the enterohepatic circulation of bile salts. J Biol Chem. 2002;277:34117-24.
137. Everson GT, McKinley C, Kern FJr. Mechanisms of gallstone formation in women. Effects of exogenous estrogen (Premarin) and dietary cholesterol on hepatic lipid metabolism. J Clin Invest. 1991;87:237-46.
138. Yang H, Petersen GM, Roth MP, et al. Risk factors for gallstone formation during rapid loss of weight. Dig Dis Sci. 1992;37:912-18.
139. Katsika D, Grjibovski A, Einarsson C, et al. Genetic and environmental influences on symptomatic gallstone disease: A Swedish study of 43,141 twin pairs. Hepatology. 2005;41:1138-43.
140. Lin JP, Hanis CL, Boerwinkle E. Genetic epidemiology of gallbladder disease in Mexican-Americans and cholesterol 7alpha-hydroxylase gene variation. Am J Hum Genet. 1994;55:A48.
141. Pullinger CR, Eng C, Salen G, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109-17.
142. Oude Elferink RP, Ottenhoff R, van Wijland M, et al. Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest. 1995;95:31-8.
143. Rosmorduc O, Hermelin B, Boelle PY, et al. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology. 2003;125:452-9.
144. Rosmorduc O, Hermelin B, Poupon R. MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology. 2001;120:1459-67.
145. Jacquemin E, De Vree JM, Cresteil D, et al. The wide spectrum of multidrug resistance 3 deficiency: From neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001;120:1448-58.
146. Shoda J, Oda K, Suzuki H, et al. Etiologic significance of defects in cholesterol, phospholipid, and bile acid metabolism in the liver of patients with intrahepatic calculi. Hepatology. 2001;33:1194-205.
147. Hanis CL, Hewett-Emmett D, Kubrusly LF, et al. An ultrasound survey of gallbladder disease among Mexican Americans in Starr County, Texas: Frequencies and risk factors. Ethn Dis. 1993;3:32-43.
148. Miller LJ, Holicky EL, Ulrich CD, et al. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology. 1995;109:1375-80.
149. Schneider H, Sanger P, Hanisch E. In vitro effects of cholecystokinin fragments on human gallbladders. Evidence for an altered CCK-receptor structure in a subgroup of patients with gallstones. J Hepatol. 1997;26:1063-8.
150. Nardone G, Ferber IA, Miller LJ. The integrity of the cholecystokinin receptor gene in gallbladder disease and obesity. Hepatology. 1995;22:1751-3.
151. Juvonen T, Kervinen K, Kairaluoma MI, et al. Gallstone cholesterol content is related to apolipoprotein E polymorphism. Gastroenterology. 1993;104:1806-13.
152. Bertomeu A, Ros E, Zambon D, et al. Apolipoprotein E polymorphism and gallstones. Gastroenterology. 1996;111:1603-10.
153. Van Erpecum KJ, Van Berge-henegouwen GP, Eckhardt ER, et al. Cholesterol crystallization in human gallbladder bile: Relation to gallstone number, bile composition, and apolipoprotein E4 isoform. Hepatology. 1998;27:1508-16.
154. Fischer S, Dolu MH, Zundt B, et al. Apolipoprotein E polymorphism and lithogenic factors in gallbladder bile. Eur J Clin Invest. 2001;31:789-95.
155. Mella JG, Schirin-Sokhan R, Rigotti A, et al. Genetic evidence that apolipoprotein E4 is not a relevant susceptibility factor for cholelithiasis in two high-risk populations. J Lipid Res. 2007;48:1378-85.
156. Kesaniemi YA, Ehnholm C, Miettinen TA. Intestinal cholesterol absorption efficiency in man is related to apoprotein E phenotype. J Clin Invest. 1987;80:578-81.
157. Juvonen T, Savolainen MJ, Kairaluoma MI, et al. Polymorphisms at the apoB, apoA-I, and cholesteryl ester transfer protein gene loci in patients with gallbladder disease. J Lipid Res. 1995;36:804-12.
158. Han T, Jiang Z, Suo G, et al. Apolipoprotein B-100 gene Xba I polymorphism and cholesterol gallstone disease. Clin Genet. 2000;57:304-8.
159. Buch S, Schafmayer C, Volzke H, et al. A genome-wide association scan identifies the hepatic cholesterol transporter ABCG8 as a susceptibility factor for human gallstone disease. Nat Genet. 2007;39:995-9.
160. Grunhage F, Acalovschi M, Tirziu S, et al. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology. 2007;46:793-801.
161. Wang Y, Jiang ZY, Fei J, et al. ATP binding cassette G8 T400K polymorphism may affect the risk of gallstone disease among Chinese males. Clin Chim Acta. 2007;384:80-5.
162. Kuo KK, Shin SJ, Chen ZC, et al. Significant association of ABCG5 604Q and ABCG8 D19H polymorphisms with gallstone disease. Br J Surg. 2008;95:1005-11.
163. Rudkowska I, Jones PJ. Polymorphisms in ABCG5/G8 transporters linked to hypercholesterolemia and gallstone disease. Nutr Rev. 2008;66:343-8.
164. Buhman KK, Accad M, Novak S, et al. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med. 2000;6:1341-7.
165. Zuniga S, Molina H, Azocar L, et al. Ezetimibe prevents cholesterol gallstone formation in mice. Liver Int. 2008;28:935-47.
166. Ostrow JD. The etiology of pigment gallstones. Hepatology. 1984;4:215S-22S.
167. Trotman BW. Pigment gallstone disease. Semin Liver Dis. 1983;3:112-19.
168. Cahalane MJ, Neubrand MW, Carey MC. Physical-chemical pathogenesis of pigment gallstones. Semin Liver Dis. 1988;8:317-28.
169. Ostrow JD. Bilirubin solubility and the etiology of pigment gallstones. Prog Clin Biol Res. 1984;152:53-69.
170. Trotman BW, Bernstein SE, Bove KE, et al. Studies on the pathogenesis of pigment gallstones in hemolytic anemia: Description and characteristics of a mouse model. J Clin Invest. 1980;65:1301-8.
171. Ho KJ, Hsu SC, Chen JS, et al. Human biliary beta-glucuronidase: Correlation of its activity with deconjugation of bilirubin in the bile. Eur J Clin Invest. 1986;16:361-7.
172. Matsushiro T. Identification of glucaro-1,4-lactone in bile as a factor responsible for inhibitory effect of bile on bacterial beta-glucuronidase. Tohoku J Exp Med. 1965;85:330-9.
173. Maki T. Pathogenesis of calcium bilirubinate gallstone: Role of E. coli, beta-glucuronidase and coagulation by inorganic ions, polyelectrolytes and agitation. Ann Surg. 1966;164:90-100.
174. Okuda K, Nakayama F, Wong J. Intrahepatic calculi. New York: Alan R. Liss; 1984. p 1
175. Nakayama F. Intrahepatic calculi: A special problem in East Asia. World J Surg. 1982;6:802-4.
176. Nakayama F, Furusawa T, Nakama T. Hepatolithiasis in Japan: Present status. Am J Surg. 1980;139:216-19.
177. Nakayama F, Soloway RD, Nakama T, et al. Hepatolithiasis in East Asia. Retrospective study. Dig Dis Sci. 1986;31:21-6.
178. Nakayama F, Koga A, Ichimiya H, et al. Hepatolithiasis in East Asia: Comparison between Japan and China. J Gastroenterol Hepatol. 1991;6:155-8.
179. Gracie WA, Ransohoff DF. The natural history of silent gallstones: The innocent gallstone is not a myth. N Engl J Med. 1982;307:798-800.
180. Friedman GD, Raviola CA, Fireman B. Prognosis of gallstones with mild or no symptoms: 25 years of follow-up in a health maintenance organization. J Clin Epidemiol. 1989;42:127-36.
181. Attili AF, De Santis A, Capri R, et al. The natural history of gallstones: The GREPCO experience. The GREPCO Group. Hepatology. 1995;21:655-60.
182. Thistle JL, Cleary PA, Lachin JM, et al. The natural history of cholelithiasis: The National Cooperative Gallstone Study. Ann Intern Med. 1984;101:171-5.
183. Newman HF, Northup JD, Rosenblum M, et al. Complications of cholelithiasis. Am J Gastroenterol. 1968;50:476-96.
184. Ransohoff DF, Gracie WA. Treatment of gallstones. Ann Intern Med. 1993;119:606-19.
185. Del Favero G, Caroli A, Meggiato T, et al. Natural history of gallstones in non-insulin-dependent diabetes mellitus: A prospective 5-year follow-up. Dig Dis Sci. 1994;39:1704-7.
186. Traverso LW. Clinical manifestations and impact of gallstone disease. Am J Surg. 1993;165:405-9.
187. Fenster LF, Lonborg R, Thirlby RC, et al. What symptoms does cholecystectomy cure? Insights from an outcomes measurement project and review of the literature. Am J Surg. 1995;169:533-8.
188. Cox MR, Wilson TG, Luck AJ, et al. Laparoscopic cholecystectomy for acute inflammation of the gallbladder. Ann Surg. 1993;218:630-4.
189. Strasberg SM, Clavien PA. Overview of therapeutic modalities for the treatment of gallstone diseases. Am J Surg. 1993;3165:420-6.
190. Lahmann BE, Adrales G, Schwartz RW. Choledocholithiasis—principles of diagnosis and management. Curr Surg. 2004;61:290-3.
191. Richardson WS, Surowiec WJ, Carter KM, et al. Gallstone disease in heart transplant recipients. Ann Surg. 2003;237:273-6.
192. Melvin WS, Meier DJ, Elkhammas EA, et al. Prophylactic cholecystectomy is not indicated following renal transplantation. Am J Surg. 1998;175:317-19.
193. Houdart R, Perniceni T, Darne B, et al. Predicting common bile duct lithiasis: Determination and prospective validation of a model predicting low risk. Am J Surg. 1995;170:38-43.
194. Barkun AN, Barkun JS, Fried GM, et al. Useful predictors of bile duct stones in patients undergoing laparoscopic cholecystectomy. McGill Gallstone Treatment Group. Ann Surg. 1994;220:32-9.
195. Bortoff GA, Chen MY, Ott DJ, et al. Gallbladder stones: Imaging and intervention. Radiographics. 2000;20:751-66.
196. Rubens DJ. Hepatobiliary imaging and its pitfalls. Radiol Clin North Am. 2004;42:257-78.
197. Jain R. Biliary sludge: When should it not be ignored? Curr Treat Options Gastroenterol. 2004;7:105-9.
198. Shea JA, Berlin JA, Escarce JJ, et al. Revised estimates of diagnostic test sensitivity and specificity in suspected biliary tract disease. Arch Intern Med. 1994;154:2573-81.
199. Einstein DM, Lapin SA, Ralls PW, et al. The insensitivity of sonography in the detection of choledocholithiasis. Am J Roentgenol. 1984;142:725-8.
200. Amouyal P, Amouyal G, Levy P, et al. Diagnosis of choledocholithiasis by endoscopic ultrasonography. Gastroenterology. 1994;106:1062-7.
201. Boland GW, Slater G, Lu DS, et al. Prevalence and significance of gallbladder abnormalities seen on sonography in intensive care unit patients. Am J Roentgenol. 2000;174:973-7.
202. Ralls PW, Colletti PM, Lapin SA. Real-time sonography in suspected acute cholecystitis. Radiology. 1985;155:767-71.
203. Buscarini E, Tansini P, Vallisa D, et al. EUS for suspected choledocholithiasis: Do benefits outweigh costs? A prospective, controlled study. Gastrointest Endosc. 2003;57:510-18.
204. Schwartz DA, Wiersema MJ. The role of endoscopic ultrasound in hepatobiliary disease. Curr Gastroenterol Rep. 2002;4:72-8.
205. Canto MI, Chak A, Stellato T, et al. Endoscopic ultrasonography versus cholangiography for the diagnosis of choledocholithiasis. Gastrointest Endosc. 1998;47:439-48.
206. Scheiman JM, Carlos RC, Barnett JL, et al. Can endoscopic ultrasound or magnetic resonance cholangiopancreatography replace ERCP in patients with suspected biliary disease? A prospective trial and cost analysis. Am J Gastroenterol. 2001;96:2900-4.
207. Meenan J, Tibble J, Prasad P, et al. The substitution of endoscopic ultrasound for endoscopic retrograde cholangio-pancreatography: Implications for service development and training. Eur J Gastroenterol Hepatol. 2004;16:299-303.
208. Beswick JS, Hughes PM, Martin DF. Ultrasonic evaluation of gallbladder function prior to non-surgical treatment of gallstones. Br J Radiol. 1991;64:321-3.
209. Maglinte DD, Torres WE, Laufer I. Oral cholecystography in contemporary gallstone imaging: A review. Radiology. 1991;178:49-58.
210. Iqbal M, Aggarwal S, Kumar R, et al. The role of 99mTc mebrofenin hepatobiliary scanning in predicting common bile duct stones in patients with gallstone disease. Nucl Med Commun. 2004;25:285-9.
211. Marton KI, Doubilet P. How to image the gallbladder in suspected cholecystitis. Ann Intern Med. 1988;110:722-9.
212. Chatziioannou SN, Moore WH, Ford PV, et al. Hepatobiliary scintigraphy is superior to abdominal ultrasonography in suspected acute cholecystitis. Surgery. 2000;127:609-13.
213. Tripathi M, Chandrashekar N, Kumar R, et al. Hepatobiliary scintigraphy: An effective tool in the management of bile leak following laparoscopic cholecystectomy. Clin Imaging. 2004;28:40-3.
214. Braun MA, Collins MB. A simple method to reduce air-bubble artifacts during percutaneous extraction of biliary stones. Am J Roentgenol. 1992;158:309-10.
215. Enns R, Baillie J. Review article: The treatment of acute biliary pancreatitis. Aliment Pharmacol Ther. 1999;13:1379-89.
216. NIH state-of-the-science statement on endoscopic retrograde cholangiopancreatography (ERCP) for diagnosis and therapy. NIH Consens State Sci Statements. 2002;19:1-26.
217. Caoili EM, Paulson EK, Heyneman LE, et al. Helical CT cholangiography with three-dimensional volume rendering using an oral biliary contrast agent: Feasibility of a novel technique. Am J Roentgenol. 2000;174:487-92.
218. Naseem I, Rees J. Oral contrast-enhanced CT cholangiography—an initial experience. J Pak Med Assoc. 2004;54:8-12.
219. Haroun A, Hadidi A, Tarawneh E, et al. Magnetic resonance cholangiopancreatography in patients with upper abdominal pain: A prospective study. Hepatogastroenterology. 2003;50:1236-41.
220. Ke ZW, Zheng CZ, Li JH, et al. Prospective evaluation of magnetic resonance cholangiography in patients with suspected common bile duct stones before laparoscopic cholecystectomy. Hepatobiliary Pancreat Dis Int. 2003;2:576-80.
221. Kaltenthaler E, Vergel YB, Chilcott J, et al. A systematic review and economic evaluation of magnetic resonance cholangiopancreatography compared with diagnostic endoscopic retrograde cholangiopancreatography. Health Technol Assess. 2004;8:1-89.
222. Patel NA, Lamb JJ, Hogle NJ, et al. Therapeutic efficacy of laparoscopic cholecystectomy in the treatment of biliary dyskinesia. Am J Surg. 2004;187:209-12.
223. Middelfart HV, Jensen P, Hojgaard L, et al. Pain patterns after distension of the gallbladder in patients with acute cholecystitis. Scand J Gastroenterol. 1998;33:982-7.
224. Friedman GD. Natural history of asymptomatic and symptomatic gallstones. Am J Surg. 1993;165:399-404.
225. Halldestam I, Enell EL, Kullman E, et al. Development of symptoms and complications in individuals with asymptomatic gallstones. Br J Surg. 2004;91:734-8.
226. Farrell T, Mahon T, Daly L, et al. Identification of inappropriate radiological referrals with suspected gallstones: A prospective audit. Br J Radiol. 1994;67:32-5.
227. Morgan G. Beneficial effects of NSAIDs in the gastrointestinal tract. Eur J Gastroenterol Hepatol. 1999;11:393-400.
228. Pazzi P, Scagliarini R, Sighinolfi D, et al. Nonsteroidal antiinflammatory drug use and gallstone disease prevalence: A case-control study. Am J Gastroenterol. 1998;93:1405-7.
229. Turner MA, Fulcher AS. The cystic duct: Normal anatomy and disease processes. Radiographics. 2001;21:3-22.
230. Roslyn JJ, DenBesten L, Thompson JEJr, et al. Roles of lithogenic bile and cystic duct occlusion in the pathogenesis of acute cholecystitis. Am J Surg. 1980;140:126-30.
231. Kaminski DL, Deshpande Y, Thomas L, et al. Effect of oral ibuprofen on formation of prostaglandins E and F by human gallbladder muscle and mucosa. Dig Dis Sci. 1985;30:93-40.
232. Goldman G, Kahn PJ, Alon R, et al. Biliary colic treatment and acute cholecystitis prevention by prostaglandin inhibitor. Dig Dis Sci. 1989;34:809-11.
233. Claesson BE, Holmlund DE, Matzsch TW. Microflora of the gallbladder related to duration of acute cholecystitis. Surg Gynecol Obstet. 1986;162:531-5.
234. Edulund Y, Zettergren L. Histopathology of the gallbladder in gallstone disease related to clinical data: With a proposal for uniform surgical and clinical terminology. Acta Chir Scand. 1959;116:450-60.
235. Raine PA, Gunn AA. Acute cholecystitis. Br J Surg. 1975;62:697-700.
236. Dumont AE. Significance of hyperbilirubinemia in acute cholecystitis. Surg Gynecol Obstet. 1976;142:855.
237. Edlund Y, Olsson O. Acute cholecystitis: Its aetiology and course, with special reference to the timing of cholecystectomy. Acta Chir Scand. 1961;120:479-94.
238. Bedirli A, Sakrak O, Sozuer EM, et al. Factors effecting the complications in the natural history of acute cholecystitis. Hepatogastroenterology. 2001;48:1275-8.
239. Nino-Murcia M, Jeffrey RBJr. Imaging the patient with right upper quadrant pain. Semin Roentgenol. 2001;36:81-91.
240. Cho KS, Baek SY, Kang BC, et al. Evaluation of preoperative sonography in acute cholecystitis to predict technical difficulties during laparoscopic cholecystectomy. J Clin Ultrasound. 2004;32:115-22.
241. Yusoff IF, Barkun JS, Barkun AN. Diagnosis and management of cholecystitis and cholangitis. Gastroenterol Clin North Am. 2003;32:1145-68.
242. Bove A, Bongarzoni G, Serafini FM, et al. Laparoscopic cholecystectomy in acute cholecystitis: Predictors of conversion to open cholecystectomy and preliminary results. G Chir. 2004;25:75-9.
243. Sandstad O, Osnes T, Urdal P, et al. Brown pigment stones in the common bile duct: Reduced bilirubinate diconjugate in bile. Scand J Gastroenterol. 2000;35:198-203.
244. Jeyarajah DR. Recurrent pyogenic cholangitis. Curr Treat Options Gastroenterol. 2004;7:91-8.
245. Soloway RD, Trotman BW, Ostrow JD. Pigment gallstones. Gastroenterology. 1977;72:167-82.
246. Tanaka M, Takahata S, Konomi H, et al. Long-term consequence of endoscopic sphincterotomy for bile duct stones. Gastrointest Endosc. 1998;48:465-9.
247. Way LW. Retained common duct stones. Surg Clin North Am. 1973;53:1139-47.
248. Goldman DE, Gholson CF. Choledocholithiasis in patients with normal serum liver enzymes. Dig Dis Sci. 1995;40:1065-8.
249. Pereira-Lima JC, Jakobs R, Busnello JV, et al. The role of serum liver enzymes in the diagnosis of choledocholithiasis. Hepatogastroenterology. 2000;47:1522-5.
250. Collins C, Maguire D, Ireland A, et al. A prospective study of common bile duct calculi in patients undergoing laparoscopic cholecystectomy: Natural history of choledocholithiasis revisited. Ann Surg. 2004;239:28-33.
251. Fernandez M, Csendes A, Yarmuch J, et al. Management of common bile duct stones: The state of the art in 2000. Int Surg. 2003;88:159-63.
252. Deacu A, Alecu L, Costan I, et al. Intraoperative diagnosis of common biliary duct using laparoscopic ultrasonography. Chirurgia (Bucur). 2003;98:547-52.
253. Yusuf TE, Baron TH. AIDS cholangiopathy. Curr Treat Options Gastroenterol. 2004;7:111-17.
254. Cotton PB. Endoscopic retrograde cholangiopancreatography and laparoscopic cholecystectomy. Am J Surg. 1993;165:474-8.
255. Hill J, Martin DF, Tweedle DE. Risks of leaving the gallbladder in situ after endoscopic sphincterotomy for bile duct stones. Br J Surg. 1991;78:554-7.
256. Bornman PC, van Beljon JI, Krige JE. Management of cholangitis. J Hepatobiliary Pancreat Surg. 2003;10:406-14.
257. Lillemoe KD. Surgical treatment of biliary tract infections. Am Surg. 2000;66:138-44.
258. Pitt HA, Cameron JL. Acute cholangitis. In: Way LW, Pellegrini CA, editors. Surgery of the Gallbladder and Bile Ducts. Philadelphia: WB Saunders; 1987:295.
259. Hanau LH, Steigbigel NH. Acute (ascending) cholangitis. Infect Dis Clin North Am. 2000;14:521-46.
260. Bennett GL, Balthazar EJ. Ultrasound and CT evaluation of emergent gallbladder pathology. Radiol Clin North Am. 2003;41:1203-16.
261. Glenn F, Reed C, Grafe WR. Biliary enteric fistula. Surg Gynecol Obstet. 1981;153:527-31.
262. Lassandro F, Gagliardi N, Scuderi M, et al. Gallstone ileus analysis of radiological findings in 27 patients. Eur J Radiol. 2004;50:23-9.
263. Gencosmanoglu R, Inceoglu R, Baysal C, et al. Bouveret’s syndrome complicated by a distal gallstone ileus. World J Gastroenterol. 2003;9:2873-5.
264. Abou-Saif A, Al-Kawas FH. Complications of gallstone disease: Mirizzi syndrome, cholecystocholedochal fistula, and gallstone ileus. Am J Gastroenterol. 2002;97:249-54.
265. Yeh CN, Jan YY, Chen M. Laparoscopic treatment for Mirizzi syndrome. Surg Endosc. 2003;17:1573.
266. Hazzan D, Golijanin D, Reissman P, et al. Combined endoscopic and surgical management of Mirizzi syndrome. Surg Endosc. 1999;13:618-20.
267. Stephen AE, Berger DL. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery. 2001;129:699-703.
268. Kwon AH, Inui H, Matsui Y, et al. Laparoscopic cholecystectomy in patients with porcelain gallbladder based on the preoperative ultrasound findings. Hepatogastroenterology. 2004;51:950-3.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]<
[/not-level-membership-for-gastroenterology-and-hepatology-category]
