3 Fractionation Effects in Clinical Practice
Radiation dose fractionation—the temporal programming of radiation delivery—has been a fruitful field of clinical research throughout the history of radiation therapy and has been one of the main arenas for attempting to improve the therapeutic ratio.* With the further development and wider use of the linear-quadratic (LQ) bioeffect formula around 1980 and its success in explaining a flurry of clinical and experimental fractionation data during the next decade or so, many clinical radiation researchers believed that the main effects of dose fractionation on tumors and normal tissues were finally well understood; everything that could be discovered had been discovered, and it was only a question of time before the last outstanding problems were cleared away. Interestingly, the whole field of clinical fractionation research has undergone a veritable renaissance since then, and there seems to be even more research challenges and opportunities today than there were back in the 1980s and 1990s. This revived interest has been stimulated by the introduction of new treatment planning and delivery technologies during this period. In addition, the wider use of combined treatment modalities has reopened many questions relating to optimal dose fractionation. But more than anything, a large body of clinical and biologic research has challenged many of the dogmas taught just 10 or 15 years ago. Although much of the teaching of dose fractionation biology has traditionally relied on assumed parallelisms between humans and a variety of biologic model systems, the discussion can now largely be based on clinical data from case series or even from randomized controlled trials. In this chapter, results of randomized trials are cited whenever relevant, but, rather than tabulating a long string of individual trials, the focus of this chapter is on using and understanding the biologic insights derived from clinical studies.
Conventional and Altered Fractionation
The Origins of Conventional Fractionation
Within a few years after Röntgen’s discovery of x-rays in 1895, the first attempts were made at using these new rays in the treatment of benign and malignant disease and the first cures of human skin cancer with radiation therapy were reported independently by two Swedish physicians, Tage Sjögren2 and Thor Stenbeck,3 in 1899 and 1900. It became almost immediately clear that the biologic effects of a given physical absorbed dose depend strongly on the details of how this dose is delivered over time. As early as in 1902, Ludwig Teleky recommended dose fractionation as a means of reducing the level of late side effects in a discussion in the Royal Society of Physicians in Vienna.4
With advances in x-ray technology, it became technically possible to deliver a large enough dose to produce a visible skin reaction in a single sitting. Biologic reasons for preferring short, intensive schedules were mainly speculative rather than based on empirical data, but these schedules won many supporters especially in Germany and Sweden. A few systematic experimental studies tried to compare single dose versus fractionated radiotherapy for various tissues. In 1927, Regaud and Ferroux showed that it was possible to sterilize a ram’s testis without excessive skin reactions using fractionated radiation but not using single doses,5 experiments that have become part of radiotherapy folklore. Regaud proposed, controversially at the time of publication, that spermatogenesis could serve as a model of human cancers. We now know that the “inverse fractionation effect” he observed (i.e., a larger effect with decreasing dose per fraction [F]) is unique to the spermatogenic system. What Regaud did realize was that it is not the biologic effect of higher dose per fraction in itself than matters, but rather the differential effect of changes in dose per fraction for tumors and the relevant normal tissues. Jens Juul in Denmark experimented on transplantable murine tumors in the late 1920s and used skin as the relevant normal tissue in a series of studies based on the idea of treating to isotoxicity6 and concluded that fractionated radiotherapy yielded a superior therapeutic index. For a more detailed account of these studies, see Bentzen and Thames.7 The fractionation debate was finally settled by clinical observations and by the early 1930s there was a widespread consensus that curative radiation therapy should be delivered in multiple fractions (so-called fractionated-protracted radiotherapy) rather than as a large single dose. What is even today referred to as conventional fractionation originated from a series of systematic clinical studies conducted by Baclesse and Coutard between approximately 1910 and the mid-1920s in Paris. These studies aimed to mimic Regaud’s fractionated daily external radium treatments and succeeded in reproducing the good clinical outcome achieved by Regaud. It was this superior clinical outcome of the “Paris schedule” that convinced the doubters and at least temporarily created a consensus on dose fractionation. It is worth stipulating that conventional fractionation originated from a series of clinical experiments aiming at optimizing the local control of head and neck squamous cell carcinoma (HNSCC) while maintaining a tolerable level of skin reactions. This schedule influenced what became conventional fractionation in many other tumor sites. It is only relatively recently that clinical researchers have fully embraced the idea that fractionation should be optimized separately for each tumor type and for the most clinically relevant toxicities associated with treating that tumor.
As a result of strained resources in the wake of World War II, Ralston Patterson at the Christie Hospital in Manchester introduced a 3-week schedule delivering 52.5 or 55 Gy in 15 or 16 F. In the early 1950s, Patterson looked at the clinical outcome of these hypofractionated schedules and concluded that they warranted continued use even as the shortage of resources were partly relieved. A variant of the Manchester schedule was developed in Edinburgh in Scotland delivering therapy over 4 instead of 3 weeks.7a The Manchester and Edinburgh schedules were adopted in many institutions, mainly in the British Commonwealth. These schedules coexisted—not always peacefully—with the standard fractionation schedule for decades, and it was only much later, with an improved quantitative understanding of dose-time-fractionation effects, that it became clear why these schedules produce near-equivalent outcomes to that of standard fractionation.
The Four “Rs” of Fractionated Radiotherapy in a Clinical Perspective
Withers8 reviewed the radiobiologic basis of fractionated radiotherapy in 1975 and summarized the main rationale for dose fractionation in his now famous mnemonic referring to the four “Rs” of radiotherapy: repair, repopulation, redistribution, reoxygenation. All of these are biologic effects that occur in the time interval between dose fractions. They are characterized by the magnitude of the effects as well as by their kinetics, and it is the differential action of these in tumors and normal tissues that determine whether a change in dose fractionation will generate an improved therapeutic ratio. A fifth R, radiosensitivity, has been proposed9 as a major factor determining radiotherapy outcome. However, this R is clearly of a different nature than the four Rs proposed by Withers as it does not take place in the interfraction interval. The most elegant experiments illustrating the four Rs of radiotherapy are in vitro or small animal studies, but these are not reviewed in this chapter. Instead, a brief summary is given of the status of the four Rs as deduced from clinical observations. A distinction is made between the observable clinical effect and the underlying radiobiologic interpretation.
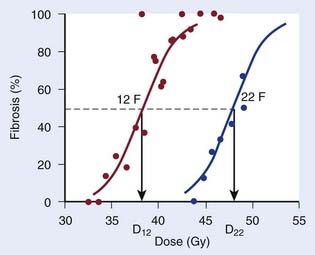
Repair at the cellular level has traditionally been seen as a manifestation of sublethal damage repair. At the tissue (or clinical) level, the term recovery is preferred by some authors as a more phenomenologic description of the recovery from damage observed when splitting the same physical dose into fractions, without assuming a specific cellular or molecular mechanism behind this effect. The clinical manifestation of recovery is the decreased biologic effect resulting from delivering a constant physical absorbed dose with decreasing dose per fraction. Fig. 3-1 shows dose-response curves for the incidence of moderate and severe (G2+) subcutaneous fibrosis. Note that the doses are estimated at the relevant reference depth for subcutaneous fibrosis10; in other words, they are not identical to the prescribed dose to the target delivered in 12 or 22 F. The dose, D12, corresponding to a 50% incidence of G2+ subcutaneous fibrosis is lower than the dose, D22, that would produce the same incidence in the 22 F group. The two doses are related by a mathematic relationship—the LQ model. This fractionation effect has been observed in countless studies, perhaps most clearly in clinical studies in which a larger dose per fraction has been introduced without any reduction in total dose. The classical example is the breast radiotherapy study by Montague11 in which 35 to 40 Gy was delivered across 4 weeks, giving 5 F per week in 88 patients and 3 F per week without a total dose reduction in 30 patients. The incidence of late complication was much higher in the large dose-per-fraction group. More support comes from studies in which the dose per fraction has been increased, but although a reduction of total dose was implemented, this later turned out to be insufficient. This occurred, for example, in studies12,13 using the nominal standard dose formula created by Ellis14 as a guide for reducing the dose, which effectively led to an over-dosage of late normal-tissue endpoints.
In addition to the magnitude of recovery, the kinetics of recovery is important for biologic effect. A dramatic illustration of this is the study by Nguyen et al.15 in which 39 patients received rapid hyperfractionated radiotherapy: 6 to 8 F of 0.9 Gy per day with a 2-hour interval between fractions were delivered five days a week for a total dose of 66 to 72 Gy. Radiotherapy was delivered in two series of 33 to 36 Gy separated by a 2- to 4-week rest interval. If the 2-hour interval had been sufficient for complete or near-complete recovery, this dose should have been relatively well-tolerated. Instead, after a minimum follow-up of 2 years, 70% of patients experienced late complications, and in 54% of cases these reactions were considered severe, causing death in 13% of patients.
Repopulation is clinically reflected in the influence of overall treatment time on local control of at least some tumor types (in which it is often called accelerated proliferation) and on the incidence of some early normal tissue effects. This phenomenon is often referred to as the time factor, a term that has the advantage of referring to a clinically observable effect rather than to an underlying hypothetical cellular mechanism. Several studies tried to estimate the tumor time factor by comparing patients who completed therapy in the planned overall time versus patients who had protracted overall time resulting from unplanned treatment interruptions.16 A strong case can be made, however, that patients with and without unplanned interruptions are not likely to present with comparable tumor and patient characteristics.17 Much stronger evidence has subsequently been obtained from randomized controlled trials, at least for some tumor types.
Redistribution of the number of cells in various phases of the cell cycle is observed after irradiation of an asynchronous cell population caused by varying radiosensitivity in these phases. There is no directly observable clinical parallel to redistribution. It is interesting to note that the European Organisation for Research and Treatment of Cancer (EORTC) 22791 trial by Horiot et al.18 used two fractions per day in an attempt to “catch” the more rapidly proliferating tumor cells that were hypothesized to have progressed into a more sensitive phase of the cell cycle at the time of the second daily fraction.19 When this trial was completed, the data were widely interpreted in terms of a differential sensitivity to dose per fraction (i.e., as a manifestation of differences in repair or recovery capacity) as predicted by the LQ model. Redistribution is also of great interest in combining drugs with radiation and many cytotoxic drugs such as cisplatin, 5-fluorouracil (5-FU), and gemcitabine require cell-cycle progression to act as radiosensitizers.20 Carefully timed administration of drugs and radiation has been proposed in an attempt to synchronize surviving tumor cell populations and then irradiate them in a sensitive phase of the cycle—an idea that so far has not worked convincingly in clinical trials, most likely because human cell populations in vivo are so heterogeneous in their cell kinetics parameters that synchrony is quickly lost in a population of patients.
Reoxygenation refers to the empirical observation that hypoxic regions in tumors may improve their oxygenation during fractionated radiotherapy. Traditionally, this has been viewed as a consequence of preferential killing by radiation of well-oxygenated cells that in turn leads to a lower metabolic consumption of oxygen, which again leads to improved oxygenation of the predominantly hypoxic cells surviving a dose fraction. The kinetics of reoxygenation is not well-studied in humans. However, a wide tumor-to-tumor variability in reoxygenation has been detected in human tumors by means of positron emission tomography scan using hypoxia-sensitive tracers.21 Because many human tumors, in contrast to most normal tissues, contain hypoxic cell populations, reoxygenation provides further rationale for fractionated radiation therapy. Advances in hypoxia-related cancer research suggest that this rather simple model of hypoxia and its modification during fractionated radiotherapy is an over-simplification. Hypoxia is now seen as an integral aspect of malignant progression and as an active biologic process rather than the passive push of cells into oxygen deprivation.22 Hypoxia-mediated radioresistance is also emerging as an actively controlled phenomenon, with the hypoxia-inducible factor–1α as a key player, and not just a direct consequence of the physicochemical absence of molecular oxygen.23–25 From a clinical radiation research perspective, many strategies have been devised to modify tumor hypoxia, including treatment in hyperbaric oxygen, administration of electron-affinic oxygen mimetic drugs such as nitroimidazoles,26 or hypoxic cytotoxins such as tirapazamine.27 These have been plagued by logistic difficulties and unexpected toxicities, but more discouragingly have yielded modest improvements in tumor control.28
The Linear-Quadratic Model
Mechanistic Background
The LQ model originated from work by Lea and Catcheside in Cambridge around the time of World War II.28a In a series of published studies, they investigated the influence of dose and dose rate on chromosome damage in cells. Lea and colleagues developed a framework for interpreting their experimental data in terms of inactivation of discrete targets.28b They hypothesized that inactivation could result from single or double “hits” and fitted an expression of the form α · D + β · D2 to experimental data on the number of chromatid exchanges per cell as a function of dose. Curiously, they used the notations α and β for the two coefficients in their “linear-quadratic” equation.28c Numerous investigators derived LQ bioeffect relationship under various assumptions; for example, Kellerer and Rossi29 published an elaborate theory of dual radiation action in 1972. However, it was the 1976 paper by Douglas and Fowler,30 in which they used the LQ model to fit data on skin reactions after fractionated radiotherapy, that stimulated the modern interest in the LQ model. The full effect plot devised by Douglas and Fowler facilitated the analysis of isoeffect data for any sign or symptom of radiation therapy without the need to know the underlying target cell survival curve. At the same time, the Fe plot provided a simple graphic test of the fit of the LQ model to the data. A few years later, Thames and colleagues31 realized that the numeric value of the ratio of the two model parameters, α/β, differed between early and late effects of radiation therapy, with values for early effects typically ranging from 8 to 12 Gy and for late effects between 1 and 5 Gy. Thames and colleagues hypothesized that the difference in α/β reflected differences in the curvature of the dose-survival curve of the putative target cells. The compelling link between the LQ model and the target cell hypothesis remained strong for many years, but this has come under increasing pressure recently.
Beyond the Target Cell Hypothesis
Advances in molecular pathology have considerably improved our understanding of radiation-induced pathologic conditions of late normal tissue effects in particular.32 It has become clear that many late effects result from a powerful, sustained wound healing response at the tissue level rather than simple parenchymal cell loss. The biology of this “fibrogenic-atrophic” radiation response pathway has been unraveled in quite some detail during the preceding decade or so. A key switch in this response is the activation of the multifunctional, strongly profibrotic cytokine transforming growth factor (TGF)-β. Ionizing radiation is one of the few exogenous factors that can activate TGF-β directly. Thus the target cell hypothesis may not explain the main pathogenic pathways for late effects. This is in contrast to the radiation-induced pathologic conditions of early effects that typically occur in tissues with a hierarchical proliferative organization and depend on the relative depletion of the stem cell pool.33 In the present context, this puts a question mark over the traditional explanation of the difference in α/β ratio between early and late effects, namely that this reflects differences in the shape of the (in vivo) cellular dose-survival curve for the putative target cells giving rise to the two types of radiation effects. At the time of writing, it is not clear what part of the TGF-β pathways determines the relatively high fractionation sensitivity of late effects. Suffice it to say that an overwhelming body of clinical and experimental data show that the dose adjustment required for maintaining a constant level of side effects when dose per fraction is changed is much larger for late than for early endpoints. This “postmodern” view of the LQ model simply acknowledges the empirical success of the model in estimating biologically equivalent doses when changing dose-fractionation—at least within a limited dose range—but does not presume any deeper mechanistic validity of the mathematic form of this model.
Pragmatic Derivation—Correction for Dose per Fraction
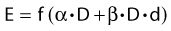
Other functional forms could work as well, but this expression is as simple as it gets. Now we are ready to derive the isoeffect formula, referred to as the Withers formula. Two dose fractionation schedules, delivering a dose D1 in dose per fraction d1, produces the same biologic effect as a dose D2 delivered in dose per fraction d2, if and only if:
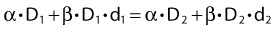
This follows from the assumption that f is a strictly increasing function. We assume that the overall treatment time is identical in the two schedules and that redistribution and reoxygenation can be ignored. Equation 2 can easily be rearranged to yield:
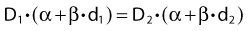
Dividing by β on both sides of the equation yields:
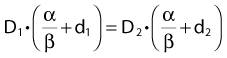
Finally, we isolate D1 on the left-hand side:
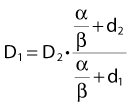
which is the so-called Withers formula.34 Note that there is only one parameter in this formula that needs to be estimated from clinical or experimental data: the α/β ratio. This formula describes the relationship between the two isoeffective dose in Fig. 5-1; or, alternatively, if we observe the shift in the dose-response curves, we can calculate the α/β ratio for the endpoint in question, subcutaneous fibrosis (in turns out to be α/β = 1.8 Gy10). More precise estimates can be obtained using statistical techniques and these also allow estimation of the uncertainty of the α/β (as well as other model parameters) and allow correction for the latent period and censoring in patients who were alive and well at the last follow-up.35,36 In other words, it is not necessary to know the separate values of α and β to estimate the biologically equivalent dose when changing from one fraction size to another. Up-to-date compilations of α/β values for experimental37 and clinical38 normal-tissue and tumor endpoints have been published recently.
For an endpoint with a known α/β ratio, Equation 3 can be applied to convert an arbitrary dose, D, delivered with dose per fraction, d, into an equivalent dose in 2-Gy fractions, (EQD2).38 This quantity is equivalent to the normalized total dose introduced by Withers et al.34:
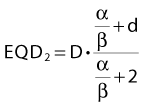
A simple numeric example follows: A simultaneous integrated boost plan is being considered for a patient with HNSCC. The plan will deliver 66 Gy in 22 F to the gross tumor volume. Assuming that α/β = 10 Gy for HNSCC, what is the equivalent dose in 2-Gy fractions, EQD2? Inserting the known values in Equation 4, we get:
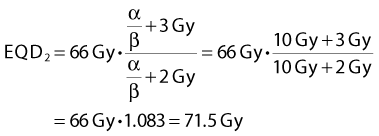
Recovery Kinetics
Split-dose recovery was discovered in cell lines in vitro by Elkind and Sutton39 in 1960. By varying the interval between two fractions (“splitting” the same physical dose in two), Elkind and Sutton were able to show how the two fractions had the same biologic effect as a single dose of the same size if the interval between fractions was very short, and how this effect asymptotically approached a certain minimum effect as the interval became very long. The same effect can be seen in clinical studies when the interval between fractions is varied, and this is also an important component in explaining the dose-rate effect in continuous irradiations. Mathematically, the effect of protracting therapy (i.e., delivering each dose fraction over longer and longer time) can be described by introducing the Lea-Catcheside factor (g), which can be seen as a factor modifying the effective fraction size:
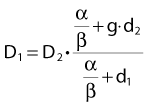
Under fairly simple assumptions, the g-function can be expressed in analytic form for many of the situations arising in clinical radiation oncology such as continuous irradiation or multiple fractions per day (MFD) with incomplete repair between fractions (see, for example, Joiner and Bentzen37).
Correction for Overall Treatment Time
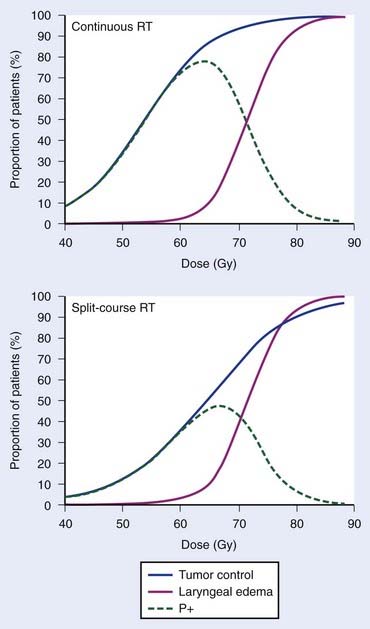
Empirically, the biologic effect of a given total dose delivered in a fixed number of fractions decreases with increasing overall treatment time (i.e., with increasing time between the first and last dose fraction). In case of tumor control, this effect is often referred to as the tumor time factor, and it is often interpreted as the result of tumor clonogen (cancer stem cell) proliferation in this time interval. Fig. 3-2 summarizes data from the Danish Head and Neck Cancer collaborative group (DAHANCA) comparing the outcome of fractionated radiotherapy delivered with 2-Gy fractions over 6 weeks and 10 weeks.40 With longer overall treatment time, the tumor dose-control curve shifted to the right; in other words, an increased dose was required to maintain the same level of tumor control. In contrast, the dose-response curve for laryngeal edema did not change. This caused a narrowing of the therapeutic window. Assuming that laryngeal edema and local failure are statistically independent,41 the probability of uncomplicated cure (P+) becomes TCPx (1 − normal tissue complication probability [NTCP]). The maximum value of P+ dropped from 78% for continuous course to 47% for split-course radiation therapy.
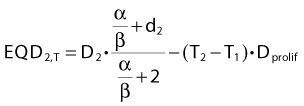
In other words, if total dose D2 is delivered in T2 days and T2 is greater than T1, we will subtract a (positive) dose from the estimated equivalent dose, reflecting the fact that biologic effect is lost because of the prolonged treatment time. It is assumed that both T1 and T2 are sufficiently long for accelerated proliferation to occur at a constant rate. As this formula is likely to be used in the comparison of two fractionation schedules, the arbitrary choice of the length of the reference schedule (T1) will cancel out.
The majority of empirical Dprolif estimates are derived from HNSCC data, in which this parameter consistently across a large number of studies38,42 comes out at approximately 0.65 Gy/day. This means that if a schedule is prolonged by, for example, 5 days, the estimated decrease in the EQD2 is 5 days × 0.65 Gy/day = 3.25 Gy. Mechanistically, the cellular proliferative response to a cytotoxic insult is a complex phenomenon,33,43,44 but this simple linear correction is likely to be a good approximation over a narrow range of treatment times, in the order of 1 to 2 weeks perhaps for a 6- or 7-week schedule. The numeric value of Dprolif cited previously is estimated toward the end of treatment schedules with an overall treatment time of some 5 to 7 weeks. Proliferation before the start of radiotherapy corresponds to a much lower dose per day. This phenomenon is called accelerated proliferation (or accelerated repopulation).
When does accelerated proliferation start? In their classic paper from 1988, Withers et al.45 proposed that isoeffect doses for local control of HNSCC remained largely constant up until approximately 4 weeks of overall treatment time, after which accelerated proliferation would kick in and the dose lost per day because of proliferation would be approximately 0.6 to 0.7 Gy/day. The appearance of this biphasic relationship, dubbed the dog-leg, depended on the exact assumptions of the modeling performed by Withers et al. to pool data from multiple studies on the same graph, and some authors—including the present one—argued that these assumptions were unrealistic46 and that the data could in reality not discriminate between the dog-leg shape and a straight line all the way down to very short schedules. A specific weakness of the available data was an absence of schedules treating in less than 4 weeks.
The continuous, hyperfractionated, accelerated radiotherapy (CHART) trial is very interesting in this context, as the experimental CHART arm finished radiotherapy in only 12 days. This provides a sensitive test for the existence of the “kink” on the dog-leg graph (Fig. 3-3). As CHART employed 1.5-Gy fractions, we use Equation 4 with an assumed α/β of 10 Gy (this parameter should ideally be estimated from the data as well) to calculate the EQD2 of the CHART arm: 51.75 Gy. From the reported hazard ratio (HR) derived from the 918 patients in the trial, we can calculate the apparent dose in 2-Gy fractions that would be isoeffective with 66 Gy in 45 days: the result is 51.3 Gy with 95% confidence limits 49.3 Gy and 53.3 Gy, plotted in Fig. 3-3. This is considerably higher than the 44.6 Gy estimated from back-extrapolating the 66 Gy by 0.65 Gy/day all the way down to 12 days’ overall time. In other words, delivering dose in just 12 days is not nearly as effective as one would estimate if the dose recovered per day was constant all the way down to 12 days.*
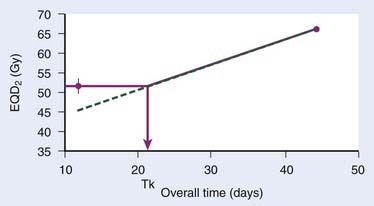
Thus, Withers turned out to be right: our current best estimates of the radiobiologic parameters for accelerated repopulation are almost exactly as derived in the 1988 paper. This has huge implications for how far accelerated fractionation schedules can be pushed. It is difficult from the available data to decide whether dose recovery with extended treatment time is truly a biphasic relationship or whether there are more phases. What is clear, however, is that a constant rate of proliferation corresponding to a recovered dose of Dprolif = 0.6-0.7 Gy/day cannot be back-extrapolated all the way down to schedules of 1 or 2 weeks’ duration. The standard way to incorporate this phenomenon in the modeling of the overall time effect is to assume Dprolif = 0 Gy/day up until a defined kick-off time (Tk) and then assume a constant Dprolif thereafter (see Fig. 3-3). In practice, this means that if T1 or T2 (or both) is shorter than Tk then this (or these) times are set equal to Tk when entered into equation 6.
Alternative Formulations of the LQ Model
A couple of mathematically equivalent formulations of the LQ model have been used as alternatives to the EQD2 formula. Among these, only the biologically effective dose (BED) formula has found any wider use.47 The mathematic equivalence ensures that calculations made in any of these frameworks will produce the same end result and the choice between these formulations typically comes down to how this was first taught to an individual or a more subjective preference for one or the other of these methods. The BED formula and the language associated with its use are solidly anchored in the target cell hypothesis: “logs of cell kill,” “potential doubling times,” “repairable and irreparable damage,” “α- and β-component of damage”—in contrast to the EQD2 formula, which is more naturally linked to clinical dose fractionation and observable clinical outcomes. Apart from this difference in conceptual context, the EQD2 formula has the advantage that all doses calculated for parts or the whole of a fractionated radiotherapy schedules, are immediately recognized as “real” doses in 2-Gy fractions, a circumstance that reduces the risk of making numerical errors.
Range of Applicability of the LQ Model
Limitations to the range of dose per fraction in which the LQ model can be usefully applied spring in part from experimental data suggesting that the mathematic form of the LQ model may not be adequate at very low or high dose per fraction and in part from the statistical uncertainty of available parameter estimates. Fig. 3-4 shows the range (green) from approximately 1 Gy to approximately 5 Gy, in which the applicability of the LQ model has been supported by clinical data from a variety of normal-tissue endpoints and, to a smaller extent, tumor endpoints. This range could obviously depend on the endpoint in question and should be taken as a crude reminder of the fraction sizes in which we possibly could see deviations from the simple LQ formula, as briefly discussed in the following.
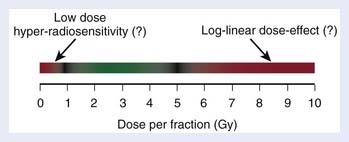
Deviations From the LQ Model at Low Dose Per Fraction
Modern delivery technologies typically irradiate large normal-tissue volumes to relatively low doses. Some in vitro tumor cell survival curves display excess cell killing at low dose per fraction relative to that predicted from the LQ model.48 This phenomenon is called low-dose hyper-radiosensitivity (HRS). Extensive studies have shown that HRS leads to apoptotic death of cells in the G2 phase of the cell cycle after doses of less than about 0.3 Gy. At higher doses a G2 checkpoint is activated that allows deoxyribonucleic acid repair to take place, which leads to increased cell survival, an effect referred to as induced radiation resistance. Dose planning studies have shown that if HRS plays a major role for the dose-limiting late side effects, this could reduce the benefit from intensity-modulated radiation therapy (IMRT) in some cases.49 It remains an open question, however, whether this is actually the case. Under the target cell hypothesis, the expectation is that, because of the very low G2 phase fraction in the cell population involved in late effects, HRS is unlikely to be a main issue for late effects. It is not clear if this expectation can be extended to biologic damage-induction or damage-processing models.32
Deviations From the LQ Model at High Dose Per Fraction
At the other extreme, there are in vitro and small animal data suggesting that the LQ model overestimates the effect of dose per fraction exceeding 8 to 10 Gy; for example, see Guerrero and Li.50 This range of fraction sizes has become increasingly important with the clinical exploration of stereotactic body radiation therapy (SBRT), stereotactic radiation surgery, and intraoperative radiation therapy, as well as some schedules used in high-dose rate (HDR) brachytherapy.
Uncertainty of Radiobiologic Parameter Estimates
Numeric values of radiobiologic parameters for a specific tumor or normal-tissue endpoint should ideally be estimated empirically from clinical observations. Using generic values (e.g., α/β = 3 Gy for late effects) should be the last resort when no empirical estimates are available for the endpoint of interest. These clinical estimates, however, are associated with a statistical uncertainty, typically specified by the standard error of the estimate or the 95% confidence limits around the estimate. Fig. 3-5 shows an example for prostate cancer. Brenner et al.51 estimated α/β at 1.1 Gy with 95% confidence limits 0.03 Gy and 4.1 Gy. Using α/β = 4.1 Gy, we can estimate the dose for a given fraction size that will be isoeffective with 76 Gy in 38 fractions (red curve in Fig. 3-5). For example, for a fraction size of 5 Gy, we see that 5 Gy × 8 is expected to produce approximately the same effect as 76 Gy in 38 F. However, the interval from 0.03 Gy to 4.1 Gy includes with 95% probability the “true” value of α/β. Depending on what this “true” value actually is, the 40 Gy in 8 F produces a tumor effect equivalent to somewhere between 59 Gy and 97 Gy in 2-Gy fractions. A more precise titration of the equivalent dose in 8 F requires an improved α/β estimate, most likely to result from clinical trials using fraction sizes in the relevant range.
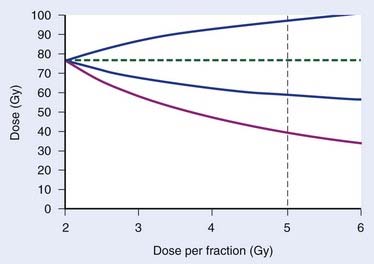
FIGURE 3-5 • Effect of uncertainty in α/β on the estimated equivalent dose in 2-Gy fractions is illustrated for prostate cancer. The red curve is the estimated dose as a function of fraction size for a hypofractionated regimen that is equivalent to 76 Gy in 38 F using Brenner et al.’s estimate,51 α/β = 1.1 Gy with 95% confidence limits 0.03 Gy and 4.1 Gy. The stippled, vertical line indicates a dose per fraction of 5 Gy. The blue lines define the upper and lower 95% confidence limits on the estimated equivalent dose for the “true” tumor effect of the hypofractionated schedule resulting from the uncertainty in the α/β estimate.
Altered Fractionation
In the wake of the wide interest in the LQ model, it was hypothesized that standard fractionation would not be optimal for some tumor types and some (especially late) toxicities.52 There is no general consensus on the terminology used to describe classes of altered fractionation schedules. The definitions proposed by myself53 have the advantage that any schedule can be classified using these terms:



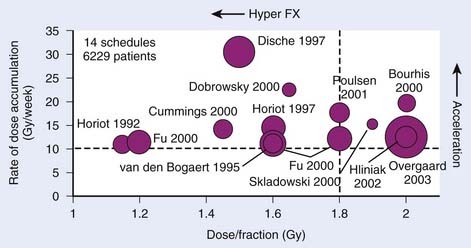
Note that there is no consideration of total dose in any of these definitions, nor is there any requirement in terms of number of fractions per day or per week. Fig. 3-6 shows the dose per fraction and dose accumulation tested in the experimental arms of a large number of altered fractionation trials in HNSCC.
Fractionated Radiotherapy Alone or Combined With Other Modalities
Head and Neck Cancer
Hyperfractionation in Head and Neck Squamous Cell Carcinoma
As an example, the EORTC 22791 trial54 gave 80.5 Gy delivered as 70 F of 1.15 Gy per fraction, 2 F per day, over 7 weeks as definitive therapy in patients with T2-T3 oropharyngeal carcinomas. For a tumor with α/β = 10 Gy, the equivalent dose in 2-Gy fractions would be:
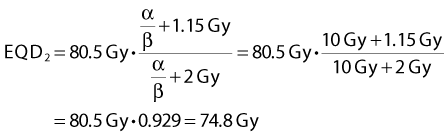
Whereas for late effects with an α/β ratio of, for example, 2 Gy, the equivalent dose in 2 Gy fractions would become:
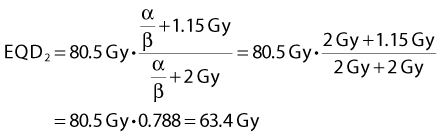
Thus the equivalent dose delivered to the tumor would be 4.8 Gy (6.9%) higher than the 70 Gy in 35 F delivered in the control arm. At the same time, the equivalent dose in 2-Gy fractions for a late endpoint would be reduced by 6.6 Gy (9.4%). Applying estimates from the literature of the steepness of dose-response curves,* these changes in dose can be converted into expected changes in tumor control and incidence of neck fibrosis. For tumor control, the 6.9% higher dose is estimated to yield a 12% increase in control. This is in reasonable agreement with the observed 19% difference in local control in the two arms of the randomized controlled trial when the statistical uncertainty in the observed local control estimates are taken into account.54 However, the expected 28% reduction in the incidence of neck fibrosis was not observed: there was no clear difference between the incidence of this endpoint in the two arms of the trial. This observation has been suggested to reflect incomplete recovery in the 6-hour interval between dose fractions in view of the long recovery halftimes estimated from the UK CHART HNSCC trial.56
Accelerated Fractionation in Head and Neck Squamous Cell Carcinoma
The clinical radiobiologic rationale for accelerated fractionation in HNSCC was the observed loss of tumor control57 when the same total dose was delivered as “split-course” therapy (i.e., extending the overall treatment time by including a planned gap in the schedule, typically of 2 to 3 weeks’ duration). The importance of overall treatment time in HNSCC was further supported by Withers’s “dog-leg” graph in which he plotted the dose required to control 50% of tumors as a function of overall treatment time and found a linear relationship (i.e., a constant dose lost per day of treatment time) at least after a lag time of 3 to 4 weeks.45 Although the details of this analysis were questioned at the time of publication,46 it became hugely influential. In hindsight, the conclusions of the paper by Withers and colleagues have been shown to be correct (see Fig. 3-3). The significant tumor time factor together with the expectation—later supported by clinical data—that the overall time factor would be negligible for late side-effects58 provided a strong rationale for shortening the overall treatment time (i.e., increasing the dose delivered per week; see Fig. 3-2).
The cleanest test of accelerated fractionation in HNSCC was probably the trial conducted by DAHANCA in which the total dose of 66 to 68 Gy and the 2-Gy fraction size was kept identical in the two trial arms, but acceleration was achieved by delivering 6 F per week in the experimental arm versus the standard 5 F per week in the control arm, which shortened the overall treatment time by 7 days, from 46 to 39 days.59 This roughly increased the rate of dose accumulation from 10 Gy per week to 12 Gy per week. Between January 1992 and December 1999, 1476 patients treated with primary radiotherapy alone were randomly allocated to the two trial arms. Using Equation 6 with Dprolif = 0.65 Gy/day, it is estimated that the 7-days-shorter treatment time corresponds to a dose escalation of 7 days × 0.65 Gy/day = 4.6 Gy. This is equivalent to a 6.7 to 6.9% change in the total dose. Using the previously discussed approximation, we estimate that this should result in a 12% improvement in tumor control probability. This is in agreement with the improvement from 64% to 76% (P = 0.0001) observed in the trial.59 There was no statistically significant increase in late toxicity in the 6 F per week arm relative to the 5 F per week arm. Thus the DAHANCA trial provides direct evidence, without the possible confounding effect of differences in total dose or dose per fraction, for the importance of the overall time factor in HNSCC and that a therapeutic gain is achievable by treatment acceleration.
Current Status of Altered Fractionation in Head and Neck Squamous Cell Carcinoma
HNSCC is the most intensively studied tumor type with respect to its response to altered fractionation. As shown in Fig. 3-6, a broad region of time- and dose-per-fraction space was almost systematically sampled by the cumulated research effort of a large number of trials conducted by investigators all over the world: a meta-analysis published in 2006 included data from 15 randomized controlled trials including 6515 patients with locally advanced HNSCC, mainly oropharyngeal or laryngeal carcinomas.60 With a median follow-up of 6 years, altered fractionated radiotherapy provided an absolute 5-year survival benefit of 3.4% (P = 0.003). The absolute benefit was 8% with hyperfractionated radiotherapy versus 2% with accelerated radiotherapy.60 There are two limitations to this analysis. First of all, toxicity data were not analyzed because they could not be retrieved in a format suited for pooled analysis. Second, the use of standard meta-analysis techniques required a rather crude grouping of trials—with varying modeled biologic efficacy within each group—into broad categories, thereby averaging the treatment benefit over more and less effective schedules. In a way, one could argue that the whole is less than the sum of the parts in this case—that the information content in the highest quality trials exceeds that of the meta-analysis.
A head-to-head comparison of accelerated fractionation with hyperfractionation was conducted in the four-arm Radiation Therapy Oncology Group (RTOG) 9003 phase III trial.61 Both altered fractionation schedules gave a significantly improved local control compared with conventional fractionation for a comparable incidence of late effects. The outcomes in the accelerated and hyperfractionated arms were similar and both agreed well with the expectations from radiobiologic modeling.
Lessons from the Head and Neck Squamous Cell Carcinoma Fractionation Trials
Breast Cancer
Data are now available from four large randomized controlled trials63–67 including a total of more than 7000 patients receiving standard 50 Gy in 25 F whole-breast radiotherapy versus hypofractionated radiotherapy. The dose per fraction varied from 2.65 to 3.3 Gy among the tested hypofractionation schedules. These large trials have demonstrated that the α/β ratio for subclinical breast cancer is in the same range as those for the relevant late side effects65: α/β for tumor control was estimated at 4.6 Gy (95% confidence interval [CI], 1.1-8.1 Gy) and for late change in breast appearance at 3.4 Gy (95% CI, 2.3-4.5 Gy). Further attempts at reducing the number of fractions in this indication are in progress in the United Kingdom with John Yarnold as the principal investigator.
Prostate Cancer
The seminal analysis by Brenner and Hall68 published 10 years ago, stimulated a flurry of theoretical studies on the α/β ratio for prostate cancer. Brenner and Hall’s analysis was based on comparing the biochemical failure-free rate after 125I permanent prostate implants (PPIs) and after external-beam radiotherapy (EBRT). In the case of 125I, the Lea-Catcheside factor, g in Equation 5, is essentially zero.68 Now assume that we can estimate two doses, DEBRT and DPPI, that are isoeffective (i.e., that produce the same tumor control after EBRT and brachytherapy, respectively). Then, by rearrangement of Equation 5, we find a simple expression for α/β:
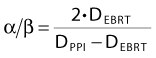
If, for example, 140 Gy from PPI is isoeffective with 60 Gy from EBRT, then α/β becomes 1.5 Gy. This whole argument hinges on the recurrences seen after PPI being dose-limited rather than geographic misses. If the tumor control curve at 140 Gy has maxed out, then it is possible that a lower dose than 140 Gy would produce the same tumor control probability (i.e., that the “true” value of DPPI would be lower than 140 Gy in Equation 7). This would lead to a higher estimate of α/β; in the hypothetical limit in which DPPI approaches DEBRT, the α/β estimate would tend to infinity.
The stark difference in the dose distributions and the possibility that some (or most?) recurrences after PPI are geographical misses (rather than dose-limited) weakened the original estimation of α/β. A subsequent study by Brenner et al.51 got around the first of these concerns by analyzing data from patients treated with EBRT plus two or three HDR brachytherapy implants, still arriving at a low estimate of α/β = 1.2 Gy (95% CI: 0.03, 4.1 Gy). Further support for a low α/β for prostate cancer comes from large randomized studies of conventional versus hypofractionated EBRT.69,70 A definitive analysis of α/β from the EBRT trials has not yet been published; an analysis71 of a preliminary report from the National Cancer Institute of Canada trial72 estimated α/β at 1.1 Gy. The upper bound on the 95% confidence interval for this estimate was 5.6 Gy, but the method of estimation requires knowledge of the steepness of the dose-response curve for prostate cancer, and the confidence interval should be taken with a pinch of salt. There is one further caveat—all analyses published to date have assumed a zero time factor for prostate cancer—an assumption that is not supported by strong clinical data.
Several prostate cancer fractionation trials are in progress and it is beyond the scope of the present chapter to review these. One trial that does deserve mention because of its design is the large multicenter phase I/II trial conducted by Ritter et al.73 The design is a three-level dose-per-fraction escalation phase I/II trial: 2.94 Gy × 22 (= 64.68 Gy); 3.63 Gy × 16 (= 58.08 Gy); 4.3 Gy × 12 (= 51.6 Gy), for level I, II, and III. The dose-fractionations were selected74 to yield near-isoeffective schedules with respect to both rectal toxicity (assuming α/β = 3 Gy) and tumor control (assuming α/β = 1.5 Gy). Levels I and II are closed, having accrued 103 and 109 patients, respectively. The median follow-up for level I patients was 47 months and for level II patients 24 months (analyzed March 2009).
Non–Small Cell Lung Cancer
The largest phase III trial of altered fractionation in patients with non–small cell lung cancer (NSCLC) is the UK Medical Research Council CHART Lung trial.75 The CHART schedule used in NSCLC was identical to that used in HNSCC delivering 54 Gy in 36 F (1.5 Gy per fraction) with 3 F per day in only 12 consecutive days: patients would start radiotherapy on a Monday morning and would finish their treatment course on Friday afternoon of week 2. Between April 1990 and March 1995, 563 patients with disease confined to the thorax were accrued. A 9% absolute improvement in 2-year survival, from 20% to 29% (P = 0.004), was seen in the CHART arm relative to the conventional arm in which patients got 60 Gy in 30 F over 6 weeks.75
Despite the significant advantage of CHART over conventional fractionation—and the proof of principle demonstration of improved survival from intensified locoregional therapy—wider uptake of the CHART schedule has been slow,76 most likely because of the logistical difficulties in treating three times a day as well as on Saturday and Sunday. This led to attempts to use the radiobiologic model parameters derived from the CHART trial to develop a weekend-less, dose-escalated schedule (CHARTWEL)77 and a 2-F-per-day, weekend-less, dose-escalated regimen (Hi-CHART). The latter has been piloted in a large phase I/II trial by the University Medical Center in Maastricht, the Netherlands.78
A CHARTWEL variant, hyperfractionated, accelerated radiotherapy (HART)—dropping “continuous”—was tested in the randomized phase III Eastern Cooperative Oncology Group 2597 trial79 but preceded by induction chemotherapy consisting of two cycles of carboplatin (area under time concentration curve 6 mg/ml/min) on days 1 and 22 plus paclitaxel (225 mg/m2) on day 1. HART delivered 57.6 Gy in 36 F, 3 F per day, 5 days a week. The first and third fraction in each day delivered 1.5 Gy on parallel-opposing anterior-posterior fields, and the mid fraction delivered 1.8 Gy on smaller-sized oblique or lateral fields excluding the spinal cord and striving to reduce the esophageal volume irradiated. The inter-fraction interval was only 4 hours. Conventional fractionation consisted of 64 Gy in 2-Gy fractions, 5 F per week. The trial was stopped prematurely after reaching approximately one-third of the planned accrual: 60 patients were randomized to HART and 59 patients to conventional radiotherapy. The early stopping of the trial reduced its power and the differences between arms were not statistically significant. However, from data in the published report it is possible to derive point estimates of the HR for survival between the two trial arms: HART reduced the HR to 0.73 when estimated from the median survival and to HR = 0.55 when estimated from the 3-year survival figures. These estimates are remarkably similar to the HR = 0.76 estimated from the CHART trial,80 supporting the role of strongly accelerated fractionation in NSCLC.
Although patients in the CHART trial were typically treated using parallel-opposing fields, treating a relatively large volume in the first phase of the treatment course (to 44 Gy in the conventional arm, 37.5 Gy in the CHART arm), the introduction of 3-D CRT and IMRT techniques—combined with advances in anatomic and molecular imaging for better target selection and delineation—may allow intensification of therapy, especially when combined with personalized dose-fractionation schedules. One such strategy was tested in a phase I/II trial at the University of Wisconsin–Madison81 and included acceleration by dose-per-fraction escalation with patients stratified according to the risk of developing radiation toxicity estimated from NTCP modeling.
Stereotactic Body Radiotherapy in Lung Cancer
An unexpected region of dose-per-fraction and time space has opened up for exploration through the use of SBRT, mainly in early stage NSCLC but it is also of interest in an increasing number of other tumor types.82 Intracranial stereotactic radiosurgery was originally developed by Leksell83 in 1951 and later for extracranial sites (SBRT) by Lax et al. at the Karolinska Hospital in Stockholm.84,85 SBRT is often referred to as ablative radiotherapy86 and is typically delivered using fraction sizes larger than 8.0 Gy. Application of the simple LQ model at these large fraction sizes gives rise to ludicrous EQD2s, especially for endpoints with a low α/β ratio. As an example, 22 Gy × 3 has been used in stage I NSCLC87 and for an endpoint with α/β = 3 Gy, the EQD2 is estimated at 330 Gy, a dose that is difficult to relate to experience with larger-volume radiation therapy. Fowler et al.88 have called SBRT “a challenge to traditional radiation oncology”—which, in a way, it is not. Rather, the apparent clinical feasibility of such regimens shows the limitations of our current radiobiologic models when extrapolated far outside the range of data in which they have been validated. The feasibility of SBRT fractionation regimens is mainly driven by the volume effect in parallel tissues with a physiologic reserve capacity. In addition, the simple LQ model overestimates the biologic effect of large-dose fractions, as mentioned previously.
A flurry of modifications of the LQ model have been proposed (e.g., see Park et al.89 and related correspondence) to achieve a mathematic expression that smoothly transitions from the LQ behavior at dose per fraction of less than some 5 Gy to the log-linear behavior seen in vitro at higher doses per fraction. Most of these are purely mathematic manipulations without any underlying mechanistic background. At the time of writing, none of these have come into wider use, and the real question is whether it is possible, or even useful, to extrapolate the clinical outcome after SBRT-type fractionation schedules all the way back to 2-Gy fraction sizes.
Several of the other four Rs of radiotherapy could be influenced by these very short, intensive schedules: reoxygenation, redistribution, and proliferation (for early normal tissue endpoints and for tumors) could affect toxicity as well as efficacy. However, very little data are available on these effects, and the standard position among SBRT proponents seem to be that the very large fraction sizes will dominate efficacy considerations and that the volume effect will take care of any normal tissue concerns. Data are emerging, however, suggesting that toxicity may become an issue, also in the beam transit zone away from the target volume.90,91
Other Tumor Histologies
There is every reason to believe that the biology of fractionation effects is similar in other tumor histologies than those discussed previously—the problem is that radiobiologic parameters allowing a quantitative discussion of fractionation effects are sparse in all of these cases. Estimates of α/β are available from single studies of malignant melanoma92 and liposarcoma93; both turn out to be low: 0.6 Gy and 0.4 Gy, respectively. There are also values for skin cancer, estimated at 8.5 Gy with 95% CI (4.5 Gy, 11.3 Gy) and for esophageal cancer 4.9 Gy with 95% CI (1.5 Gy, 17 Gy), but, as can be seen, the confidence intervals for these estimates are wide. That is all that’s available; for most other tumor types there are no strong empirical human data. Most isoeffect estimates in other histologic grades are derived from assumed, “generic” values, typically α/β = 10 Gy for tumors, but as the breast and prostate examples show, this could turn out to be very misleading.
Palliative Therapy
Palliation is an important indication for radiation therapy in a large number of patients. Hypofractionated schedules are attractive because of their convenience to the patient, especially when the treatment course spans a sizable proportion of the patient’s life expectancy. Decision making in prescription of palliative radiation therapy is complex and involves weighing a number of factors against each other, including the patient’s age, performance status, disease burden, and prognosis. It cannot be assumed that hypofractionation always offers the best balance between symptom relief and patient convenience, although in many cases it probably does.94–96
From the perspective of clinical radiobiology, the largest body of evidence on fractionation in palliative radiotherapy stems from several randomized, controlled trials comparing single-radiation-dose fractions versus multifraction (typically also hypofractionated) schedules. Wu et al.97 performed a meta-analysis of trials published before 2001 and concluded that there was no significant difference in complete and overall pain relief between single and multifraction palliative radiotherapy for bone metastases. This conclusion was further strengthened in 2005 when the results of the RTOG 9714 trial were published.98 This trial randomized 898 patients to 8 Gy × 1 versus 3 Gy × 10. Both regimens were equivalent in terms of pain and narcotic relief at 3 months and were well tolerated. The 8-Gy arm had a higher rate of retreatment, but had less acute toxicity than the 30-Gy arm. Further analyses of the Dutch bone pain trial have shown equal levels of palliation also in patients with a relatively long survival time99 and have shown a favorable cost-utility of the single fraction schedule.100 Nevertheless, single-fraction schedules for metastatic bone pain have obtained wider acceptance in Europe and Canada than in the United States.101 Speculating on the implications for evidence-based radiation oncology or on the possible influence of remuneration in various health care systems on the choice of fractionation102 is not the topic of the present chapter.
With respect to radiobiology, the experience summarized in this section suggests that standard isoeffect calculations probably are of little value in comparing palliative fractionation schedules, pointing to the mechanism of palliation being different from that of curative treatment. Studies on urinary or serum markers of bone resorption suggest that pain relief is more closely related to an effect on normal bone than on the tumor itself.103,104
Where Next in Fractionated Radiotherapy?
Dose Distribution and Fractionation
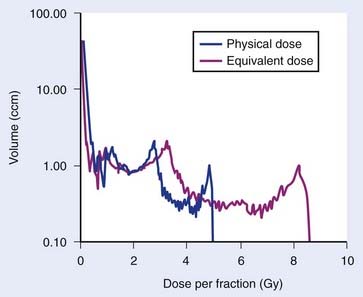
At the same time, the typical dose distribution in surrounding organs at risk (OAR) have changed from partial organ irradiation, in which a fraction of the organ receives a dose equal to the prescription dose and very limited dose (except for a relative narrow penumbra) elsewhere, to a broad spectrum of absorbed doses throughout the OAR. The varying dose to voxels in the OAR also means that these are irradiated with a varying dose per fraction (Fig. 3-7). How this local fractionation effect influences local damage, and how this in turn determines organ-level functional endpoints, is not yet clear in most tissues and organs. What is clear is that fractionation effects and dose distribution cannot be considered separately. An American Society for Therapeutic Radiology and Oncology and American Association of Physicists in Medicine task force, Quantitative Analysis of Normal Tissue Effects in the Clinic, has undertaken a systematic review of our current knowledge of dose-volume effects in approximately 20 OARs. These organ-related reviews together with a number of methodologic reviews and scientific vision papers are planned to be published at the end of 2009. Much has been learned during the preceding 15 years, but there are still large white areas on the map, and further research is urgently needed.
Combined Modality Therapy and Fractionation
Indications for multimodality therapy, with radiation therapy as one component, are widening. Cytotoxic and molecular targeted agents are being tested in combination with fractionated radiotherapy in clinical trials. Among the emerging rationales for combining drugs and radiation are the possibilities of hitting distinct cellular targets as well as modulation of radiation fractionation effects.20,105 The improved locoregional relapse-free survival seen in a large HNSCC trial106 randomizing between radiation therapy with and without cetuximab, an antibody against the epidermal growth factor receptor (EGFR), has created much excitement among radiobiologists. Although several biologic mechanisms may be targeted by cetuximab, it is very likely that partly abrogating the accelerated proliferation response during fractionated radiotherapy is a main contributor. This would open exciting possibilities for reconsidering hyperfractionated regimens if the tumor time factor really is reduced by administering cetuximab.
Few studies have directly addressed the possible interaction between fractionation and chemotherapy. One example is the elegant randomized study with a factorial design, conducted by Anne Lee and her colleagues,107 testing conventional (66 Gy in 33 F, 5 F/week) versus accelerated (same total dose and fraction size but 6 F/week) radiation therapy with or without chemotherapy in patients with T3-4N0-1M0 nasopharyngeal carcinoma. Chemotherapy was given as concurrent cisplatin plus adjuvant cisplatin and 5-FU. Preliminary results suggested a significant interaction between chemotherapy and accelerated fractionation, with the combination of the two being the superior arm of the four. A full analysis of the outcome of this trial awaits final analysis of the mature data from the study.
Optimizing fractionation in the context of combined surgery and radiotherapy is also an under-studied field. In postoperative radiotherapy for HNSCC it is conceivable that the accelerated proliferation is triggered at the time of surgery (i.e., the 3- to 4-week delay before the onset of accelerated proliferation will already have passed at the time of commencement of radiation therapy). This would revive the rationale for very short, intensive schedules like CHART. Support for this hypothesis comes from the randomized phase III trial from the National Cancer Institute in Cairo, Egypt.108 An independent trial addressing this issue was activated by the UK Medical Research Council, but was closed prematurely because of slow accrual.
Personalized Medicine and Fractionation
High-throughput assays and a large number of immunohistochemical biomarkers have opened new research avenues in predictive oncology. Although many prognostic markers are of relevance for patients receiving radiation therapy as well as other therapies, relatively few studies have looked specifically for biomarkers that would select for a specific dose-fractionation. An example of a study in which a molecular biomarker did prove to be a predictive factor for a benefit from a specific dose-fractionation schedule has come from the CHART HNSCC trial.109 Although overall there was no statistically significant benefit from being randomized to CHART rather than to conventional fractionation, patients harboring tumors with an EGFR index assessed by immunohistochemistry above the population median value did have a significant benefit from CHART. The interaction between the randomization arm and high or low EGFR expression was statistically significant with respect to locoregional control. This is the first among a large number of clinic-pathologic and immunohistochemical markers that has shown a significant association with a benefit from strongly accelerated radiotherapy. In the future, it may be possible to use an array of markers for personalized prescription of dose fractionation.
Because radiation therapy is a locoregional modality, predictive competing risks models110,111 that allow stratification of patients according to their relative risks of local, regional, and distant failure would be of great potential value in stratifying patients for differently weighted combinations of locoregional and systemic therapy. Radiation dose fractionation and scheduling of the combined modalities would need to be optimized in various subgroups of patients. Again, more research is needed in this area.
Finally, recent advances in functional and molecular imaging are hugely interesting in the context of dose fractionation. Novel imaging targets may allow noninvasive mapping of inter- and intralesion variations in surrogates for the four Rs of radiotherapy. Imaging surrogates for hypoxia and cellular proliferation are the most intensely investigated at the moment. Ultimately, this may pave the way for theragnostic radiation oncology112: voxel-based dose-fraction prescriptions, so-called dose painting by numbers, according to maps of cellular or microenvironmental variation throughout a tumor volume. Although this research field is still in its infancy, the potential is huge. Theragnostic radiation oncology is at the crossroad between technology and biology and is close to the heart of what makes radiation unique in cancer management.
1 Bentzen SM. Design of clinical trials in radiation oncology: saving lives, not Grays. In: Dale RG, Jones B, editors. Modelling in radiation oncology. British Institute of Radiology, 2007.
2 Sjögren TAU. Fall av epitheliom behandlat med röntgenstrålar. Forh Svenska Läkare Sällskapets Sammankomster. 1899:208.
3 Stenbeck T. Fall av hudkräfta, läkt genom behandling med röntgenstrålar. Hygiea. 1900;68:18.
4 Teleky L. Discussion in the K.K. Gesellschaft der Ärzte in Wien, 1902. Fortschritte auf dem Gebiete der Röntgenstrahlen. 1902;5:362.
5 Regaud C, Ferroux R. Discordance des effects des rayons X, d’une part dans la peau, d’autre part dans le testicule, par le fractionement de la dose: Diminution de l’afficacite dans la peau, maintien de l’efficacite dans le testicule. Comptes Rendus Societé Biologique. 1927;97:431.
6 Juul J, Experimental studies on roentgen treatment of malignant tumors, Acta Radiol; Suppl. VIII; 1929:1.
7 Bentzen SM, Thames HD. A 100-year Nordic perspective on the dose-time problem in radiobiology. Acta Oncol. 1995;34:1031.
7a Paterson R, Tod M, Russell M. The results of radium and X-ray therapy in malignant disease, being the third statistical report from the Christie Hospital and Holt Radium Institute. Manchester, Edinburg: E.S. Livingstone Ltd; 1950.
8 Withers HR. The four R’s of radiotherapy. Adv Radiat Biol. 1975;5:241.
9 Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. Int J Radiat Biol. 1989;56:1045.
10 Bentzen SM, Christensen JJ, Overgaard J, Overgaard M. Some methodological problems in estimating radiobiological parameters from clinical data. Alpha/beta ratios and electron RBE for cutaneous reactions in patients treated with postmastectomy radiotherapy. Acta Oncol. 1988;27:105.
11 Montague ED. Experience with altered fractionation in radiation therapy of breast cancer. Radiology. 1968;90:962.
12 Bates TD, Peters LJ. Dangers of the clinical use of the NSD formula for small fraction numbers. Br J Radiol. 1975;48:773.
13 Overgaard M, Bentzen SM, Christensen JJ, Hjøllund Madsen E. The value of the NSD formula in equation of acute and late radiation complications in normal tissue following 2 and 5 fractions per week in breast cancer patients treated with postmastectomy radiotherapy. Radiother Oncol. 1987;9:1.
14 Ellis F. Dose, time and fractionation: a clinical hypothesis. Clin Radiol. 1969;20:1.
15 Nguyen TD, Panis X, Froissart D, et al. Analysis of late complications after rapid hyperfractionated radiotherapy in advanced head and neck cancers. Int J Radiat Oncol Biol Phys. 1988;14:23.
16 Fowler JF, Lindstrom M. Loss of local control with prolongation in radiotherapy. Int J Radiat Oncol Biol Phys. 1992;23:457.
17 Bentzen SM. Time-dose relationships for human tumors: estimation from non-randomized studies. In: Beck-Bornholt HP, editor. Current topics in clinical radiobiology of tumours. Medical radiology. Berlin: Springer-Verlag; 1993:11.
18 Horiot JC, Le Fur R, N’Guyen T, et al. Hyperfractionated compared with conventional radiotherapy in oropharyngeal carcinoma: an EORTC randomized trial. Eur J Cancer. 1990;26:779.
19 Withers HR. The EORTC hyperfractionation trial. Radiother Oncol. 1992;25:229.
20 Wilson GD, Bentzen SM, Harari PM. Biologic basis for combining drugs with radiation. Semin Radiat Oncol. 2006;16:2.
21 Koh WJ, Bergman KS, Rasey JS, et al. Evaluation of oxygenation status during fractionated radiotherapy in human nonsmall cell lung cancers using [F-18]fluoromisonidazole positron emission tomography. Int J Radiat Oncol Biol Phys. 9-30-1995;33:391.
22 Harris AL. Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38.
23 Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer. 2008;8:425.
24 Chan N, Milosevic M, Bristow RG. Tumor hypoxia, DNA repair and prostate cancer progression: new targets and new therapies. Future Oncol. 2007;3:329.
25 Williams KJ, Telfer BA, Xenaki D, et al. Enhanced response to radiotherapy in tumours deficient in the function of hypoxia-inducible factor-1. Radiother Oncol. 2005;75:89.
26 Adams GE, Dische S, Fowler JF, et al. Hypoxic cell sensitisers in radiotherapy. Lancet. 1-24-1976;1:186.
27 Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297-321. 297
28 Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25:4066.
28a Lea DE, Catcheside DG. The mechanism of the induction by radiation of chromosome aberrations in Tradescantia. J Genet. 1942;44:216-245.
28b Lea DE. A theory of the actions of radiation on biological materials capable of recovery. Part I: The time-intensity factor. Brit J Radiol. 1938;11:489-497.
28c Catcheside DG, Lea DE, Thoday JM. The production of chromosome structural changes in TRADESCANTIA microspores in relation to dosage, intensity and temperature. J Genet. 1945;47:137-149.
29 Kellerer AM, Rossi HH. The theory of dual radiation action. Curr Top Radiat Res. 1972;8:85.
30 Douglas BG, Fowler JF. The effect of multiple small doses of x-rays on skin reactions in the mouse and a basic interpretation. Rad Res. 1976;66:401.
31 Thames HD, Withers HR, Peters LJ, et al. Changes in early and late radiation responses with altered dose fractionation: Implications for dose-survival relationships. Int J Radiat Oncol Biol Phys. 1982;8:219.
32 Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6:702.
33 Dorr W. Three A’s of repopulation during fractionated irradiation of squamous epithelia: asymmetry loss, acceleration of stem-cell divisions and abortive divisions. Int J Radiat Biol. 1997;72:635.
34 Withers HR, Thames HD, Peters LJ. A new isoeffect curve for change in dose per fraction. Radiother Oncol. 1983;1:187.
35 Bentzen SM, Thames HD, Travis EL, et al. Direct estimation of latent time for radiation injury in late-responding normal tissues: gut, lung and spinal cord. Int J Radiat Biol. 1989;55:27.
36 Taylor JMG, Withers HR, Vegesna V, et al. Fitting the linear-quadratic model using time of occurrence as the endpoint for quantal response multifraction experiments. Int J Radiat Biol. 1987;52:459.
37 Joiner MC, Bentzen SM. Fractionation: the linear-quadratic approach. In: Joiner MC, van der Kogel AJ, editors. Basic clinical radiobiology, vol 4. London, UK: Edward Arnold; 2009:102.
38 Bentzen SM, Joiner MC. The linear-quadratic approach in clinical practice. In: Joiner MC, van der Kogel AJ, editors. Basic clinical radiobiology, vol 4. London, UK: Edward Arnold; 2009:120.
39 Elkind MM, Sutton H. Radiation response of mammalian cells grown in culture. 1. Repair of x-ray damage in surviving Chinese hamster cells. Radiat Res. 1960;13:556.
40 Overgaard J, Hjelm-Hansen M, Johansen LV, et al. Comparison of conventional and split-course radiotherapy as primary treatment in carcinoma of the larynx. Acta Oncol. 1988;27:147.
41 Holthusen H. Erfahrungen über die Verträglichkeitsgrenze für Röntgenstrahlen und deren Nutzanwendung zur Verhütung von Schäden. Strahlentherapie. 1936;57:254.
42 Bentzen SM, Dorr W, Anscher MS, et al. Normal tissue effects: reporting and analysis. Semin Radiat Oncol. 2003;13:189.
43 Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Rev Cancer. 2005;5:516.
44 Bentzen SM. Repopulation in radiation oncology: perspectives of clinical research. Int J Radiat Biol. 2003;79:581.
45 Withers HR, Taylor JMG, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131.
46 Bentzen SM, Thames HD. Clinical evidence for tumor clonogen regeneration: interpretations of the data. Radiother Oncol. 1991;22:161.
47 Fowler JF. What next in fractionated radiotherapy? Br J Cancer. 1984;49(Suppl. VI):285.
48 Marples B, Collis SJ. Low-dose hyper-radiosensitivity: past, present, and future. Int J Radiat Oncol Biol Phys. 2008;70:1310.
49 Honore HB, Bentzen SM. A modelling study of the potential influence of low dose hypersensitivity on radiation treatment planning. Radiother Oncol. 2006;79:115.
50 Guerrero M, Li XA. Extending the linear-quadratic model for large fraction doses pertinent to stereotactic radiotherapy. Phys Med Biol. 2004;49:4825.
51 Brenner DJ, Martinez AA, Edmundson GK, et al. Direct evidence that prostate tumors show high sensitivity to fractionation (low alpha/beta ratio), similar to late-responding normal tissue. Int J Radiat Oncol Biol Phys. 2002;52:6.
52 Thames HD, Peters LJ, Withers HR, et al. Accelerated fractionation vs. hyperfractionation: Rationales for several treatments per day. Int J Radiat Oncol Biol Phys. 1983;9:127.
53 Bentzen SM. Quantitative clinical radiobiology. Acta Oncol. 1993;32:259.
54 Horiot JC, Le Fur R, N’Guyen T, et al. Hyperfractionation versus conventional fractionation in oropharyngeal carcinoma: Final analysis of a randomized trial of the EORTC cooperative group of radiotherapy. Radiother Oncol. 1992;25:231.
55 Bentzen SM. Dose-response relationships in radiotherapy. In: Joiner MC, van der Kogel AJ, editors. Basic clinical radiobiology, vol 4. London, UK: Edward Arnold; 2009:56.
56 Bentzen SM, Saunders MI, Dische S. Repair halftimes estimated from observations of treatment-related morbidity after CHART or conventional radiotherapy in head and neck cancer. Radiother Oncol. 1999;53:219.
57 Parsons JT, Bova FJ, Million RR. A re-evaluation of split-course technique for squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys. 1980;6:1645.
58 Bentzen SM, Overgaard J. Clinical normal-tissue radiobiology. In: Tobias JS, Thomas PRM, editors. Current radiation oncology. London: Arnold; 1996:37.
59 Overgaard J, Hansen HS, Specht L, et al. Five compared with six fractions per week of conventional radiotherapy of squamous-cell carcinoma of head and neck: DAHANCA 6 and 7 randomised controlled trial. Lancet. 2003;362:933.
60 Bourhis J, Overgaard J, Audry H, et al. Hyperfractionated or accelerated radiotherapy in head and neck cancer: A meta-analysis. Lancet. 2006;368:843.
61 Fu KK, Pajak TF, Trotti A, et al. a Radiation Therapy Oncology Group (RTOG) phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol Biol Phys. 2000;48:7.
62 Bentzen SM, Saunders MI, Dische S, et al. Radiotherapy-related early morbidity in head and neck cancer: quantitative clinical radiobiology as deduced from the CHART trial. Radiother Oncol. 2001;60:123.
63 Whelan T, MacKenzie R, Julian J, et al. Randomized trial of breast irradiation schedules after lumpectomy for women with lymph node-negative breast cancer. J Natl Cancer Inst. 2002;94:1143.
64 Bentzen SM, Agrawal RK, Aird EG, et al. The UK Standardisation of Breast Radiotherapy (START) Trial A of radiotherapy hypofractionation for treatment of early breast cancer: a randomised trial. Lancet Oncol. 2008;9:331.
65 Bentzen SM, Agrawal RK, Aird EG, et al. The UK Standardisation of Breast Radiotherapy (START) Trial B of radiotherapy hypofractionation for treatment of early breast cancer: a randomised trial. Lancet. 2008;371:1098.
66 Owen J, Ashton A, Regan J, et al. Fractionation sensitivity of breast cancer. Results of a randomised trial. Eur J Cancer Suppl. 2003;1:S9.
67 Yarnold J, Ashton A, Bliss J, et al. Fractionation sensitivity and dose response of late adverse effects in the breast after radiotherapy for early breast cancer: long-term results of a randomised trial. Radiother Oncol. 2005;75:9.
68 Brenner DJ, Hall EJ. Fractionation and protraction for radiotherapy of prostate carcinoma. Int J Radiat Oncol Biol Phys. 1999;43:1095.
69 Lukka H, Hayter C, Julian JA, et al. Randomized trial comparing two fractionation schedules for patients with localized prostate cancer. J Clin Oncol. 2005;23:6132.
70 Yeoh EE, Holloway RH, Fraser RJ, et al. Hypofractionated versus conventionally fractionated radiation therapy for prostate carcinoma: updated results of a phase III randomized trial. Int J Radiat Oncol Biol Phys. 2006;66:1072.
71 Bentzen SM, Ritter MA. The alpha/beta ratio for prostate cancer: what is it, really? Radiother Oncol. 2005;76:1.
72 Lukka H, Hayter C, Warde P, et al. A randomized trial comparing two fractionation schedules for patients with localized prostate cancer. Radiother Oncol. 2003;69(Suppl. 1):S7.
73 Ritter MA, Forman JD, Kupelian PA, et al. A phase I/II trial of dose-per-fraction escalation for prostate cancer. Int J Radiat Oncol Biol Phys. 2007;69:S174.
74 Ritter M. Rationale, conduct, and outcome using hypofractionated radiotherapy in prostate cancer. Semin Radiat Oncol. 2008;18:249.
75 Saunders MI, Dische S, Barrett A, et al. Continuous hyperfractionated accelerated radiotherapy (CHART) versus conventional radiotherapy in non-small-cell lung cancer: a randomised multicentre trial. CHART Steering Committee. Lancet. 1997;350:161.
76 Macbeth F. An uncharted country. Clin Oncol (R Coll Radiol). 1999;11:71.
77 Bentzen SM, Saunders MI, Dische S. From CHART to CHARTWEL in non-small cell lung cancer: clinical radiobiological modelling of the expected change in outcome. Clin Oncol (R Coll Radiol). 2002;14:372.
78 De Ruysscher D, Wanders R, van Haren E, et al. HI-CHART: a phase I/II study on the feasibility of high-dose continuous hyperfractionated accelerated radiotherapy in patients with inoperable non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2008;71:132.
79 Belani CP, Wang W, Johnson DH, et al. Phase III study of the Eastern Cooperative Oncology Group (ECOG 2597): induction chemotherapy followed by either standard thoracic radiotherapy or hyperfractionated accelerated radiotherapy for patients with unresectable stage IIIA and B non-small-cell lung cancer. J Clin Oncol. 6-1-2005;23:3760.
80 Saunders M, Dische S, Barrett A, et al. Continuous hyperfractionated accelerated radiotherapy (CHART) versus conventional radiotherapy in non-small-cell lung cancer: a randomised multicentre trial. CHART Steering Committee. Lancet. 1997;350:161.
81 Adkison JB, Khuntia D, Bentzen SM, et al. Dose escalated, hypofractionated radiotherapy using helical tomotherapy for inoperable non-small cell lung cancer: preliminary results of a risk-stratified phase I dose escalation study. Technol Cancer Res Treat. 2008;7:441.
82 Lo SS, Cardenes HR, Teh BS, et al. Stereotactic body radiation therapy for nonpulmonary primary tumors. Expert Rev Anticancer Ther. 2008;8:1939.
83 Leksell L. The stereotaxic method and radiosurgery of the brain. Acta Chir Scand. 1951;102:316.
84 Lax I, Blomgren H, Naslund I, et al. Stereotactic radiotherapy of malignancies in the abdomen. Methodological aspects. Acta Oncol. 1994;33:677.
85 Blomgren H, Lax I, Naslund I, et al. Stereotactic high dose fraction radiation therapy of extracranial tumors using an accelerator. Clinical experience of the first thirty-one patients. Acta Oncol. 1995;34:861.
86 Timmerman RD. An overview of hypofractionation and introduction to this issue of seminars in radiation oncology. Semin Radiat Oncol. 2008;18:215.
87 Fakiris AJ, McGarry RC, Yiannoutsos CT, et al. Stereotactic body radiation therapy for early-stage non-small-cell lung carcinoma: four-year results of a prospective phase II study. Int J Radiat Oncol Biol Phys. 2009;75:677-682.
88 Fowler JF, Tome WA, Fenwick JD, et al. A challenge to traditional radiation oncology. Int J Radiat Oncol Biol Phys. 2004;60:1241.
89 Park C, Papiez L, Zhang S, et al. Universal survival curve and single fraction equivalent dose: Useful tools in understanding potency of ablative radiotherapy. Int J Radiat Oncol Biol Phys. 2008;70:847.
90 Pettersson N, Nyman J, Johansson KA. Radiation-induced rib fractures after hypofractionated stereotactic body radiation therapy of non-small cell lung cancer: a dose- and volume-response analysis. Radiother Oncol. 2009;91:360.
91 Dunlap NE, Cai J, Biedermann GB, et al. Chest wall volume receiving >30 Gy predicts risk of severe pain and/or rib fracture after lung stereotactic body radiotherapy. Int J Radiat Oncol Biol Phys. 2009;76:796-801.
92 Bentzen SM, Overgaard J, Thames HD, et al. Clinical radiobiology of malignant melanoma. Radiother Oncol. 1989;16:169.
93 Thames HD, Suit HD. Tumor radioresponsiveness versus fractionation sensitivity. Int J Radiat Oncol Biol Phys. 1986;12:687.
94 Timothy AR, Girling DJ, Saunders MI, et al. Radiotherapy for inoperable lung cancer. Clin Oncol (R Coll Radiol). 2001;13:86.
95 Hoskin PJ, Yarnold JR, Roos DR, et al. Radiotherapy for bone metastases. Clin Oncol (R Coll Radiol). 2001;13:88.
96 Hoskin PJ, Brada M. Radiotherapy for brain metastases. Clin Oncol (R Coll Radiol). 2001;13:91.
97 Wu JS, Wong R, Johnston M, et al. Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys. 2003;55:594.
98 Hartsell WF, Scott CB, Bruner DW, et al. Randomized trial of short- versus long-course radiotherapy for palliation of painful bone metastases. J Natl Cancer Inst. 2005;97:798.
99 Van der Linden YM, Steenland E, van Houwelingen HC, et al. Patients with a favourable prognosis are equally palliated with single and multiple fraction radiotherapy: Results on survival in the Dutch Bone Metastasis Study. Radiother Oncol. 2006;78:245.
100 Van den Hout WB, van der Linden YM, Steenland E, et al. Single- versus multiple-fraction radiotherapy in patients with painful bone metastases: cost-utility analysis based on a randomized trial. J Natl Cancer Inst. 2003;95:222.
101 Kachnic L, Berk L. Palliative single-fraction radiation therapy: how much more evidence is needed? J Natl Cancer Inst. 2005;97:786.
102 Lievens Y, van den Bogaert W, Rijnders A, et al. Palliative radiotherapy practice within Western European countries: impact of the radiotherapy financing system? Radiother Oncol. 2000;56:289.
103 Papatheofanis FJ. Serum PICP as a bone formation marker in 89Sr and external beam radiotherapy of prostatic bony metastases. Br J Radiol. 1997;70:594.
104 Hoskin PJ, Stratford MR, Folkes LK, et al. Effect of local radiotherapy for bone pain on urinary markers of osteoclast activity. Lancet. 2000;355:1428.
105 Bentzen SM, Harari PM, Bernier J. Exploitable mechanisms for combining drugs with radiation: concepts, achievements and future directions. Nat Clin Pract Oncol. 2007;4:172.
106 Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567.
107 Lee AW, Tung SY, Chan AT, et al. Preliminary results of a randomized study (NPC-9902 Trial) on therapeutic gain by concurrent chemotherapy and/or accelerated fractionation for locally advanced nasopharyngeal carcinoma. Int J Radiat Oncol Biol Phys. 2006;66:142.
108 Awwad HK, Lotayef M, Shouman T, et al. Accelerated hyperfractionation (AHF) compared to conventional fractionation (CF) in the postoperative radiotherapy of locally advanced head and neck cancer: influence of proliferation. Br J Cancer. 2002;86:517.
109 Bentzen SM, Atasoy BM, Daley FM, et al. Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. J Clin Oncol. 2005;23:5560.
110 Ataman OU, Bentzen SM, Saunders MI, et al. Failure-specific prognostic factors after continuous hyperfractionated accelerated radiotherapy (CHART) or conventional radiotherapy in locally advanced non-small-cell lung cancer: a competing risks analysis. Br J Cancer. 2001;85:1113.
111 Ataman OU, Bentzen SM, Wilson GD, et al. Molecular biomarkers and site of first recurrence after radiotherapy for head and neck cancer. Eur J Cancer. 2004;40:2734.
112 Bentzen SM. Theragnostic imaging for radiation oncology: dose-painting by numbers. Lancet Oncol. 2005;6:112.
* We follow the in many ways unsatisfactory convention in the literature of using therapeutic ratio in a loose sense when referring to efficacy relative to toxicity. For a discussion of attempts to treat this more quantitatively, see Bentzen.1
* A formal analysis of this problem requires that the uncertainty in Dprolif be taken into account, an analysis that is beyond the scope of this chapter.
* Values of γ50 equivalent to 1.8 for tumor control and 3.0 for neck fibrosis are assumed. For details of this method, see Bentzen.55