CHAPTER 252 Fractionated Radiotherapy for Spine Tumors
In the 1920s, after biologic experiments, Coutard and Regaud showed that by dividing the total dose into many smaller treatments (i.e., fractions) delivered over a period of several weeks, tumors could be cured without severe normal tissue damage.1 Since that time, fractionated radiotherapy had been used for the treatment of tumors (benign or malignant) of the spine. Fractionated radiotherapy takes advantage of the differential radiosensitivity of normal tissues and the target lesion. The time between fractions allows some recovery of normal tissues, which reduces the risk associated with radiation injury. The inherent advantages of fractionated radiotherapy are especially appealing when treating near critical dose-limiting structures such as the spinal cord. Before proceeding with a discussion of the treatment of spine tumors, it is essential to have a solid understanding of the tolerance of the spinal cord to fractionated radiotherapy.
Radiation Tolerance of the Spinal Cord
Because spinal cord injury is disabling and untreatable, understanding the radiation tolerance of the spinal cord and risk factors for injury is of paramount importance for radiation oncologists. Two types of radiation injury have been well documented. The first is a transient, self-limited, reversible myelopathy called Lhermitte’s sign that occurs 2 to 6 months after radiotherapy. Characterized by numbness, tingling, and shock-like sensations radiating to the hands and feet when the neck is flexed, Lhermitte’s sign is a classic finding in patients with transient myelopathy. It is believed that this phenomenon is related to transient demyelination mediated by damage to oligodendrocytes. This syndrome is self-limited and resolves within weeks to months. It is not associated with chronic progressive myelitis when standard radiation doses have been delivered.2
Chronic progressive or delayed myelopathy can occur months to years after radiotherapy but fortunately is quite rare with conventional fractionation and adequate time between radiation treatments.3 Permanent myelopathy is characterized by progressive neurological signs and symptoms, including paresthesias, motor weakness, and loss of pain or temperature sensation. Patients ultimately lose bowel and bladder control and exhibit complete sensory and motor function loss. The latency period of chronic myelopathy is bimodal, with peaks of incidence occurring at 13 and 29 months. It is hypothesized that the early peak is due to white matter injury with subsequent demyelination whereas the latter peak results from radiation injury to the cord microvasculature.4 Because of the rarity of radiation myelitis after conventional treatment, spinal cord myelopathy caused by radiation is a diagnosis of exclusion, with recurrence of the tumor being the most common cause. Magnetic resonance imaging (MRI) may assist in the diagnosis, both to confirm that the changes have occurred within the irradiated volume and to substantiate the presence of the classic findings associated with radiation-induced myelopathy. In the early delayed phase, cord edema is seen frequently. Within 8 months of the onset of symptoms, T1-weighted images may show low intensity, whereas T2-weighted images show high intensity. Gadolinium enhancement is also common. Late changes may include spinal cord atrophy.5
The occurrence of chronic progressive myelopathy is dependent on the total dose, fraction size, volume irradiated, and use of chemotherapy.4,6,7 Historically, radiation oncologists have limited the spinal cord dose to 45 to 50 Gy, with conventional fractionation schedules of 1.8 to 2.0 Gy/day. A review of 1112 patients treated with multiple different fractionation schedules and a range of doses but no chemotherapy found only two cases of myelopathy in patients receiving less than 50 Gy. Because there were no identifiable risk factors that set these patients apart from others receiving similar doses, the authors argued that the onset of permanent myelopathy in patients receiving less than 50 Gy was idiosyncratic. The actual incidence of myelopathy with these conventionally fractionated doses is less than 0.2% to 0.5% after 50 Gy and 1% to 5% after 60 Gy.3,4 The dose required to cause a 5% risk for myelopathy at 5 years (TD 5/5) is in the range of 57 to 61 Gy, and the 50% rate of myelopathy at 5 years (TD 50/5) is believed to be 68 to 73 Gy.8 Risk for spinal cord injury increases with fraction sizes above 4 to 6 Gy.4 Time between fractions of radiotherapy is also important. To minimize risk for injury, time between radiation treatments should be greater than 6 hours, and total dose constraints to the spinal cord should be reduced in treatment regimens administering more than one fraction per day.4,9 Although historically the thoracic spinal cord was thought to be more radiosensitive than other regions of the spinal cord, newer data do not support differential sensitivities based on region of the cord. The total volume of spinal cord irradiated has been associated with risk for radiation injury, with larger volumes of cord treated associated with higher risk for myelopathy.4
Increasingly, combined-modality therapy that includes chemotherapy and radiation therapy is used for the optimal treatment of many tumors. Some chemotherapeutic agents are radiosensitizing in addition to their antitumor effects. Unfortunately, this radiosensitization is often not selective and may sensitize the spinal cord as well as the tumor to the effects of radiotherapy. Reports of permanent myelopathy after doses as low as 37 Gy have been noted in patients with Hodgkin’s lymphoma when combined with intensive chemotherapy and bone marrow transplantation.10 Nitrosoureas, cytosine arabinose, 5-fluorouracil, and vincristine have all been associated with myelopathy when used in combination with radiotherapy.6 Although reports are still rare, care should be taken with patients who have been heavily treated with radiosensitizing or neurotoxic chemotherapy until further data are available.
If left untreated, most spinal tumors, especially intramedullary tumors, would be expected to cause severe neurological dysfunction and, eventually, myelopathy. When treating a patient with a spinal cord tumor, the radiation oncologist must weigh the risk of causing myelopathy against the risk of tumor progression and subsequent severe neurological dysfunction. Radiation-induced myelopathy remains rare, in part because of the unwillingness of radiation oncologists to cause spinal cord complications as a result of treatment; this extremely low risk for permanent spinal cord damage after radiotherapy is achieved at the cost of a decrease in the probability of tumor control. Therefore, tumor progression is the most common reason for myelopathy and neurological morbidity, partially because of this caution.4
Intramedullary Tumors
Astrocytoma
Astrocytoma is the most common intramedullary spinal cord tumor and accounts for 40% to 45% of all reported cases. In children, 75% to 90% of intramedullary spinal cord tumors are astrocytomas, and about 85% of them are low-grade fibrillary or juvenile pilocytic astrocytomas.11,12 Long-term survival in patients with spinal cord astrocytomas is most strongly correlated with histology: patients with high-grade astrocytomas have a median survival of 8 to 10 months, whereas those with low-grade histology have a median survival greater than 15 years.13–15 Low-grade tumors often have a low infiltrative nature, and microsurgical resection is the treatment of choice.12 In contrast, radical surgery in patients with high-grade astrocytomas has no impact on outcome and may contribute to poor postoperative neurological function. For malignant, infiltrative tumors, maximal safe resection is advised. Malignant spinal cord tumors appear to have a higher risk for leptomeningeal dissemination, thus confirming the limited role of aggressive surgery.13 Adjuvant radiotherapy is recommended for patients with high-grade or infiltrative tumors associated with neurological dysfunction in whom gross total resection is not possible.12,13,16
The role of adjuvant radiotherapy for subtotally resected low-grade astrocytic tumors of the spinal cord is controversial, with treatment decisions primarily being dependent on tumor grade and age of the patient. In young children, because the late toxicity of radiotherapy is more pronounced, radiotherapy can be reserved until after a second operation for clinical recurrence.12 Young children in whom astrocytic tumors are diagnosed before their pubertal growth spurt are at significant risk for the development of radiation-induced delay in bone growth with kyphoscoliosis or short stature, especially affecting their sitting height. This radiation-induced deformity is most severe in patients who have extensive tumors or holocord involvement of the spine.17,18 In these young children, if their neurological function is good or improved after subtotal resection, close follow-up without adjuvant therapy is a reasonable course of action. Delaying radiotherapy until recurrence or early tumor progression may allow the child to grow at a normal rate for several years before receiving radiotherapy. Careful radiographic and neurological follow-up after surgery is required to ensure that irreversible neurological injury does not develop during these periods of “observation.” Radiotherapy delays the progression of neurological symptoms, but it is unlikely to reverse long-standing neurological deficits. To maximize long-term neurological function, radiotherapy should be initiated after radiologic progression but before clinical neurological deterioration for patients in whom no further surgery is possible or postoperatively for patients with multiply recurrent tumors in whom further surgery is likely to cause significant morbidity. Collaboration between the radiation oncologist and neurosurgeon is critical to determine the most appropriate timing for adjuvant radiotherapy in this group of patients.
In teenagers and adults, observation is reasonable for pilocytic astrocytomas, but adjuvant radiotherapy should be considered for all patients with World Health Organization grade II, III, and IV astrocytomas.19 The largest experience of spinal cord astrocytomas comes from the Mayo Clinic: 69 pilocytic and 67 infiltrative (nonpilocytic) astrocytomas. The vast majority of the patients in this series were adults (mean age, 35 years). Tumor grade was a strong predictor of prognosis (median survival for grade I not reached; grade II, 4.1 years; grade III, 1.6 years; and grade IV, 0.8 years). Greater extent of resection did not predict a better outcome, but adjuvant radiotherapy did for infiltrative astrocytomas, and this held true on multivariate analysis. Therefore, based on these results and the work of others, adjuvant radiotherapy should be strongly considered for all teenagers and adults with infiltrative (nonpilocytic) astrocytomas.20
Radiotherapy Dose and Techniques
Dose-response data from randomized trials in the cerebrum suggest that there is no difference in outcomes for doses between 45 and 69.4 Gy.21,22 Retrospective dose-response data from spinal cord tumors suggest an improvement in time to progression for patients receiving greater than 40 Gy versus those who received lower doses.23 The most commonly used doses for the treatment of both low- and high-grade spinal cord astrocytomas range from 45 to 55 Gy,8 depending on the volume of spinal cord being irradiated and the patient’s neurological function. Unfortunately, even with these doses, local recurrence is the predominant pattern of treatment failure, especially for low-grade tumors.14,24 In rare cases in which the tumor is so advanced that no meaningful recovery of function is expected, dose escalation resulting in anticipated “radiocordectomy” has been shown to control disease, albeit with permanent disability.25
For the treatment of adults with radiotherapy, the chance of damage to surrounding soft tissues is exceedingly small; nonetheless, the benefits gained from multifield treatment in reducing acute toxicity outweigh the remote risk for second malignancy, especially since the prognosis is not as favorable as in children. Although a second malignancy is a greater concern in children, higher doses of radiation are associated with reduced vertebral body bone growth17 and increased risk for muscle and soft tissue hypotrophy, which can result in reduced paraspinal support in an already unstable spine.18 Multifield techniques should help reduce the risk for significant hypotrophy in children, although no long-term data exist as yet to support this hypothesis.24,26
Modern radiotherapy uses imaging information from preoperative and postoperative MRI, as well as treatment-planning computed tomography (CT), to design a conformal radiotherapy plan. With more precise dose administration, it is becoming increasingly important to accurately delineate the radiotherapy target volumes. The gross tumor volume (GTV) is generally the residual tumor visible on imaging. A margin for subclinical spread, generally 1 cm for low-grade tumors and 1 to 2 cm for higher grade tumors, is anatomically confined to the spinal canal to create a clinical target volume (CTV). These tumors appear to spread craniocaudally, so a larger margin superiorly and inferiorly may be advisable for infiltrative, high-grade lesions.27 Careful review of imaging and communication with the neurosurgeon and pathologist to help determine the extent of spread along nerve roots are also important. An additional 0.3- to 0.5-cm margin is added to account for setup error, depending on the treating institution’s immobilization technique, to create the planning target volume (PTV). The dose is typically prescribed so that 95% of the prescription covers the PTV. Three-dimensional conformal techniques use CT guidance to design radiotherapy target volumes, design beams and blocks, and model a radiation plan. Another treatment technique is intensity-modulated radiotherapy (IMRT); it uses sophisticated computer software to control the blocking leaves on the linear accelerator, which then move throughout the treatment to generate a more complex and conformal (around the radiotherapy target) plan.
Craniospinal or spinal axis irradiation is not generally indicated for the treatment of spinal cord tumors unless neuraxis dissemination is present at the time of diagnosis. In adults with neuraxis dissemination, craniospinal axis irradiation is rarely indicated because of the poor tolerance of craniospinal irradiation in adults and the poor prognosis; for adults, treatment is usually limited to bulky or symptomatic sites (or both). For younger patients with neuraxis dissemination, doses of 39.6 to 50.4 Gy in 1.8-Gy fractions have been used in combination with a boost dose given to bulky areas of disease, especially for higher grade tumors.24 If more than half the spinal cord is irradiated, the total dose should probably not exceed 45 Gy, but small segments may safely tolerate 55 Gy. In young children, the dose to the spinal cord should be limited to between 40 and 45 Gy in 1.5- to 1.75-Gy daily fractions because the developing cord may have a lower tolerance for radiotherapy.
Ependymoma
Ependymomas are usually histologically benign, and they may have a long and often indolent course. Poorly differentiated ependymomas in the spine are rare. Rostral tumors are more frequently cellular variants, whereas distal tumors (including those of the cauda equina) are more commonly a myxopapillary variant. Approximately two thirds of ependymomas occur in the region of the cauda equina; they are not truly intramedullary and are reviewed in the section “Extramedullary Tumors.” Spinal ependymomas are more common in adults than in children.28,29
Complete resection is more frequently achieved for ependymomas than for astrocytomas, and intraoperative assessment of tumor resection is more reliable with ependymomas than with astrocytomas. Advances in microsurgical techniques have contributed to improved postoperative outcomes for patients with intramedullary spinal cord tumors. The frequency of complete excision varies from 33% to 94%.30,31 For patients undergoing complete en bloc gross excision, the prognosis is excellent, with cure rates exceeding 90% in some series.32 Adjuvant radiotherapy is not recommended in these cases.
Substantial evidence supports the use of postoperative radiotherapy in patients with ependymoma after incomplete or piecemeal excision.23,33–35 Modern series with long-term follow-up confirm these results. Wahab and colleagues reported an 80% progression-free survival rate at 15 years in patients receiving adjuvant radiotherapy after incomplete or piecemeal resection.36 Dose-response data also indirectly suggest a role for postoperative therapy. Some radiotherapy series have demonstrated that increasing doses of radiation are associated with better tumor control in patients with ependymomas. Shaw and colleagues reported that the local failure rate was 35% in patients receiving 50 Gy or less as opposed to just 20% in patients receiving more than 50 Gy.34 Local control and overall survival in patients who receive adjuvant radiotherapy after incomplete resection for low-grade ependymomas are excellent and similar to rates in patients who undergo gross total resection.35–38
The radiotherapy dose and techniques for intramedullary ependymomas are similar to those for astrocytomas (see earlier). CTV is generally defined as a 1- to 2-cm margin on residual tumor on T1-weighted contrast-enhanced MRI plus any edema seen on T2-weighted or fluid-attenuated inversion recovery (FLAIR) sequences. As with astrocytomas, a more generous CTV is recommended for malignant and infiltrative tumors. If no residual tumor is visible, the postoperative cavity (which preoperative imaging can help delineate) is considered the GTV. A total dose of 45 to 50.4 Gy is typically given in 1.8- to 2-Gy daily fractions to the CTV, along with a boost for a total dose of 54 to 55.8 Gy to areas of bulky residual disease (GTV) (Fig. 252-1).
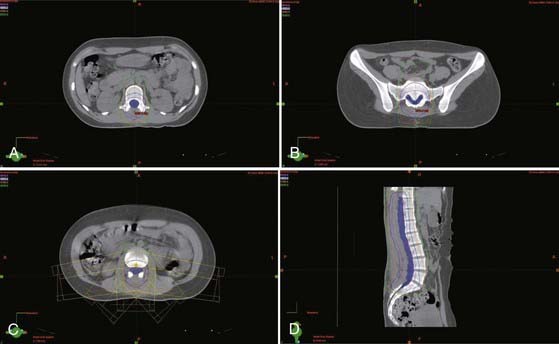
Extramedullary Tumors
Meningioma
Surgery is the mainstay of treatment of spinal meningiomas. Complete resection is achievable in more than 80% of patients and is associated with a low risk for local recurrence.39 Subtotal resection is associated with a much higher risk for local recurrence ranging from 17% to 100%. Because of this high risk for local recurrence, some have advocated adjuvant radiotherapy by extrapolation of data from the brain. Because of the rarity of the tumor and the long latency period before recurrence, the role of radiotherapy is not well understood. For patients with multiply recurrent disease with atypical or malignant histology, doses of 50 to 54 Gy in 1.8- to 2-Gy fractions have been used.39,40
Nerve Sheath Tumor
Most nerve sheath tumors are benign, well-encapsulated lesions that are amenable to total surgical excision. A recent study suggests that multiply recurrent tumors may be candidates for radiosurgery.41 The rare malignant nerve sheath tumors have a natural history similar to that of soft tissue sarcomas, and they should be treated as such. Treatment commonly includes wide excision followed by high-dose (>60 Gy) radiotherapy.42
Ependymoma
About two thirds of all spinal canal ependymomas occur in the lumbosacral region, and 40% arise from within the filum terminale. Most intradural extramedullary ependymomas in the lumbosacral spine are of the myxopapillary type.29,34 Because of their location, they can frequently be excised completely. Care must be taken to remove the tumor whole as opposed to piecemeal to prevent upward seeding. In many circumstances, these tumors tightly envelop the nerve roots of the cauda equina, thus making en bloc excision difficult. Gross total resection of these tumors requires piecemeal removal. It is unclear whether the favorable prognosis of these tumors is related to an inherent biologic tendency or the fact that they are more likely to be completely resected. It does appear that biologically these tumors grow slowly, and long-term follow-up is recommended even with gross total resection because of reports of late recurrences.29
Patients who have undergone gross total resection of an ependymoma have an excellent local control rate without additional therapy.29,43 Patients who have undergone complete excision of cauda equina ependymoma by piecemeal removal have a local failure rate ranging from 20% to 43%.29,34,35 The addition of radiotherapy in such patients produces a local recurrence rate equal to that of patients undergoing gross total resection.34,44,45 A retrospective study from MD Anderson found a significant benefit in terms of local control (10-year local control rate of 86% versus 46%) and progression-free survival (10-year rate of 75% versus 37%) for adjuvant radiotherapy (median dose, 50.4 Gy) when compared with observation, even though significantly more patients had undergone gross total resection in the observation cohort.46
Treatment of myxopapillary ependymomas is similar to that for intramedullary tumors (see the earlier sections on astrocytoma and ependymoma). The treatment field should be extended to encompass the entire thecal sac, with the field widened inferiorly to the sacroiliac joints to ensure adequate coverage of the meningeal sleeves within the intervertebral foramina (CTV). Failure to adequately encompass the thecal sac has been associated with an increased rate of treatment failure.35 The lateral beam technique described earlier adequately covers lateral cerebrospinal fluid extension of the thecal sac within the sacrum and minimizes the dose to the pelvic organs. A total dose of 45 to 50.4 Gy is typically given in 1.8- to 2-Gy daily fractions to the CTV, along with a boost for a total dose of 54 to 55.8 Gy to areas of bulky residual disease (GTV).
Extradural Tumors
Tumors of the Vertebral Column—Metastatic
Bones are a common site of cancer metastasis, and they cause significant morbidity as a result of pain, pathologic fractures, and spinal cord compression. Patients with metastatic cancer are generally incurable. For most patients the goals of therapy are to maintain quality of life and functional independence for as long as possible. Spinal cord compression can be both neurologically and psychologically devastating, and efforts to improve diagnosis and treatment have resulted in improvement in both neurological independence47 and survival.48,49
Metastatic cancer may involve either the epidural space or the bones of the vertebral column itself. The most common primary cancers that involve the spinal column are those arising from the breast, lung, and prostate.50 Spinal cord compression can occur by direct extension from a metastatic lesion in the vertebral body, extension from a paraspinal metastasis through a neural foramen, or enlargement of an epidural mass. Rarely, intramedullary metastases can occur.51
The most common initial symptom of spinal cord compression is pain; it often precedes other symptoms such as weakness, sensory loss, or autonomic dysfunction.52 Back pain can be localized to the metastatic lesion, or radicular pain may radiate along a dermatome caused by spinal nerve compression or invasion. New-onset back pain in a patient with a history of malignancy should be considered cord compression until proved otherwise. Once neurological deficits begin, rapid progression is common. Metastatic epidural spinal cord compression is a medical emergency that requires rapid diagnosis and the cooperation of a multidisciplinary team to facilitate urgent therapy.51 It cannot be overemphasized that prompt initiation of therapy before the onset of neurological signs and symptoms is important to minimize long-term neurological dysfunction.47,48,51
Treatment of spinal cord compression depends on the patient’s prognosis, extent of disease in the spine, and tumor radiosensitivity. Patients who have an expected survival of more than 3 months, have only one site of cord compression, and are medically able to undergo surgery should receive direct decompressive surgery followed by radiotherapy. An exception to this paradigm would be exquisitely radiosensitive tumors such as lymphoma and myeloma; for such radiosensitive tumors, radiotherapy should be the local modality of choice with surgery reserved for salvage. In a phase III randomized trial by Patchell and others, patients who underwent direct decompressive surgery followed by radiotherapy were more likely to be ambulatory (84% versus 57%), regain their ability to walk (62% versus 19%), and maintain their ability to walk (122 versus 13 days) than those treated by radiotherapy alone. Patients paraplegic longer than 48 hours were not eligible for this study. Even with a relatively short duration of neurological symptoms, radiotherapy alone was rarely able to help patients regain function.47 This again underscores the importance of early diagnosis and rapid treatment.
In patients who are not candidates for surgical resection, emergency initiation of radiotherapy is critical. Ambulatory status at the time of initiation of radiotherapy is the strongest prognostic factor for remaining ambulatory after treatment and for overall survival. Sixty percent to 80% of patients who are ambulatory at the time of radiation therapy remain ambulatory, only 45% remain ambulatory if they had significant motor weakness before therapy, and just 5% to 20% will become ambulatory if they are paralyzed at the initiation of radiotherapy.49,53
Histology of the primary tumor may be a prognostic factor for neurological recovery. Radiosensitive tumors such as myeloma, lymphoma, small cell lung cancer, and seminoma have a rapid response to radiation, and recovery of neurological function occurs frequently. Unfavorable histologic findings such as non–small cell lung cancer and melanoma have a less favorable response and should be evaluated for surgical decompression.48,54
Time to development of motor deficits is another prognostic factor in spinal cord compression. Patients with a history of slowly progressive motor deficits have a better neurological prognosis than do those with a rapid decline in motor function. In a prospective study, Rades and colleagues found that patients with more than 14 days of progressive motor symptoms had an 86% chance of neurological recovery, as opposed to only a 10% chance of recovery if the motor deficits progressed over a period of less than 7 days. It has been suggested that patients with a rapid onset of paralysis may actually experience spinal cord ischemia or infarction. The antineoplastic effect of radiation is not likely to reverse such a vascular event.55
Although evidence of efficacy is controversial, it is common practice to initiate corticosteroids in anticipation of radiotherapy-induced edema, which may contribute to an initial worsening in neurological functioning.56,57
The fractionation schedule of radiotherapy has recently been shown to affect local control and progression-free survival, defined as maintenance of motor strength, with longer fractionation schedules being associated with improved durability of treatment.58 Initial recovery of neurological function appears to be similar between different fractionation schedules. Fractionation schedules of 800 cGy for 1 fraction, 200 cGy for 20 fractions, 250 cGy for 15 fractions, 300 cGy for 10 fractions, or 400 cGy for 5 fractions to cumulative doses of 800, 4000, 3000, 3000, and 2000 cGy, respectively, were used in these studies.59 These data suggest that patients with a more favorable prognosis should receive treatment with a longer fractionation regimen, most commonly 3000 cGy in 10 fractions. Shorter overall courses are most appropriate for patients with limited expected survival.54,58,60
The radiation treatment volume should encompass the entirety of the lesion on MRI, with a generous margin above and below the mass. Adding a 1.5-cm margin around the GTV results in a CTV that typically encompasses a vertebral body above and below the involved disease. An attempt is made to limit field borders to vertebral body interspaces to facilitate matching future radiation fields to adjacent vertebral bodies while minimizing the risk of cord injury from treatment overlap (Fig. 252-2). Field width is 8 to 10 cm on average and is typically at the lateral edge of the spinous process or 1 to 2 cm beyond the most lateral extent of gross disease if paraspinal extension is present.
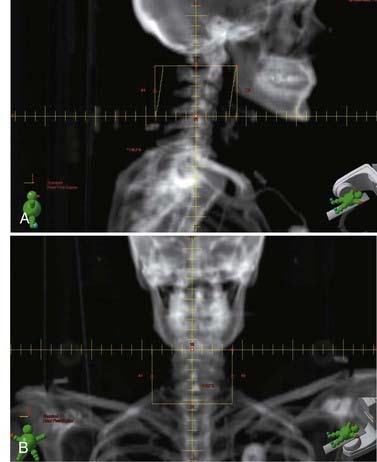
Tumors of the Vertebral Column—Primary
Chordoma
Chordomas are rare tumors that arise from notochordal remnants in the axial skeleton. Nearly 50% of all chordomas arise from the sacrococcygeal region, another 35% arise from the clivus, and the remainder occur within the vertebral column, primarily the cervical spine.61 Chordomas are slow-growing neoplasms that destroy bone and infiltrate soft tissues. Metastases are uncommon at initial evaluation, but metastatic disease will eventually develop in up to 25% of patients. Gross total en bloc resection is necessary to minimize the risk for local recurrence.62 Because of their location, chordomas are rarely completely resectable. Local recurrence rates of 80% to 100% are seen after surgical resection.63
Postoperative radiotherapy can reduce the risk for local recurrence. Chordomas are radioresistant tumors and are often located adjacent to sensitive, critical normal structures such as the brainstem, which limits the tumor dose achievable with conventional three-dimensional radiotherapy. Conventional radiotherapy and doses in the range of 50 to 60 Gy delay tumor progression but do not usually provide durable local control.62,64,65
Because of the poor results with conventional photon treatment, highly conformal radiation techniques have been studied extensively in patients with this disease. Charged particles—protons and carbon ions are examples currently being used in human treatment—have unique physical properties that result in very rapid energy loss at a certain distance in tissue that is dependent on the energy of the particle. Because of this rapid falloff, high doses of radiation can be delivered to tumors immediately adjacent to critical normal structures.63
Results from proton therapy centers demonstrate dramatic improvements in local control relative to historical patients treated with lower doses of conventional photon radiotherapy. An updated report from the Massachusetts General Hospital reported a 42% to 64% 10-year local control rate with tumor doses ranging from 66 to 83 GyE (gray equivalents).66 The proton therapy experience from Loma Linda for skull base chordomas confirmed the effectiveness of high-dose conformal proton therapy. With a median follow-up of 33.2 months, the local control rate for patients with chordomas was 92% (23 of 25).63 Noel and coauthors reported favorable early results with proton beam therapy in a French series, with an 83.1% 3-year local control rate in 34 patients treated for chordoma at a median dose of 67 GyE.67 Recent results from Switzerland in pediatric patients are similar and consistent with other proton results.68 Data are consistent from all proton therapy sites in that local control is improved with minimal residual disease. The authors recommend maximal debulking before radiotherapy to improve treatment outcomes by allowing more physical space between the tumor and critical normal structures.63,66,67
Heavy charged particle radiotherapy has been explored in the management of chordoma. Heavy charged particles are particularly well suited to treat radioresistant tumors. Not only do they have the unique physical properties of protons, which allows rapid dose falloff, but the high linear energy transfer of particle therapy is also believed to offer a radiobiologic advantage in treating chordoma that may help overcome the relative radioresistance of these slow-growing tumors. Carbon ion radiotherapy has been used to treat chordomas in Darmstadt, Germany, since 1997. The local control rate of 70% at 5 years for base of the skull chordomas compares favorably with the proton experience.69 Carbon ion has been used as an alternative to radical surgery in Japan for large sacral chordomas (Fig. 252-3). In one series, the 5-year local control rate was 96% with doses ranging from 52.8 to 73.6 GyE given in 16 fractions over a period of 4 weeks.70
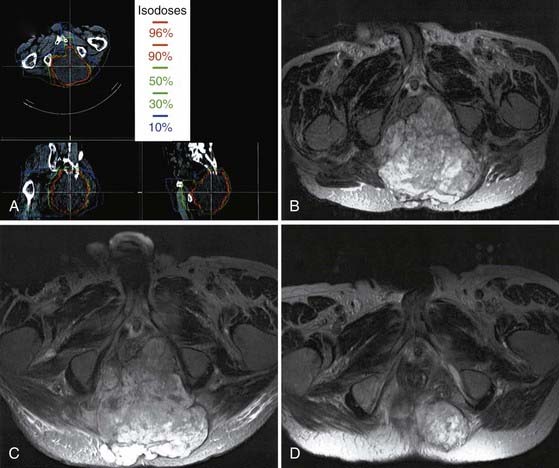
Stereotactic radiotherapy and radiosurgery have been evaluated for the treatment of chordoma. Martin and colleagues from the University of Pittsburgh reported an actuarial tumor control rate of 63% at 5 years.71 The average target volume was small (9.8 cm3) in this and other radiosurgical series, in contrast to patients treated with heavy ion therapy (often 80 to 500 cm3). Fractionated stereotactic radiotherapy is applicable to larger tumor volumes. Debus and coworkers reported a 50% 5-year local control rate for chordomas treated with fractionated stereotactic radiotherapy. The median target volume was 56 cm3 (range, 17 to 215 cm3). Radiotherapy was delivered at a median dose of 66.6 Gy in 1.8-Gy fractions under stereotactic guidance.72 Until carbon ion and proton treatment centers become more readily available, IMRT, radiosurgery, and stereotactic fractionated radiotherapy are promising alternatives for the treatment of chordomas.
Based on treatment guidelines for proton therapy, doses of at least 70 to 80 GyE to the GTV are recommended. The CTV, defined as the GTV with an additional 0.5- to 1-cm margin for subclinical spread, should receive 50 to 60 GyE. CT is valuable for detailing bone invasion or destruction, and MRI is helpful in determining the extent of soft tissue invasion. These two imaging modalities are complementary in planning both surgery and radiotherapy.73
Chondrosarcoma
Chondrosarcomas arise from bone or cartilage and are more common than chordomas. They appear similar to chordomas radiologically and even pathologically. The most common sites of origin are the pelvis and femur. In contrast to chordomas, the thoracic spine is the most common site of origin for axial tumors. Patients with chondrosarcoma tend to be slightly younger than those with chordoma. As with chordomas, surgery is the treatment of choice, but in the axial skeleton, gross total excision is difficult. Also like chordomas, the local recurrence rate approaches 100% for patients who do not undergo gross total resection.74 Chondrosarcomas of the clivus or sacrococcygeal areas are often treated similarly to chordomas. Chondrosarcomas generally have a better prognosis than do chordomas in the same location, and excellent results can be achieved with radiotherapy or radiosurgery.63,66,67 Like chordomas, they have been treated with high-dose conformal proton therapy. Adjuvant or definitive proton radiotherapy at doses ranging from 66 to 83 Gy results in 5-year local control rates higher than 90%.63,66 The results have remained stable for up to 10 years.66
Carbon ions have also been used with success to treat chondrosarcomas. With a median follow-up of 33 months, the actuarial local control rate at 4 years for base of the skull chondrosarcomas was 90%.75 In the absence of heavy ions, highly conformal photon radiotherapy has also been evaluated. Radiosurgery for small base of the skull tumors has resulted in an actuarial local tumor control rate of 80% at 5 years.71 With fractionated stereotactic radiotherapy for larger tumors, preliminary 2-year local control rates of 100% have been reported.72,76
Radiotherapy target delineation is similar to that for chordomas. Because they appear to be slightly more radiosensitive than chordomas, boost doses to the GTV are often slightly lower (66 to 74 GyE).68
Ewing’s Sarcoma
Ewing’s sarcoma is an undifferentiated small, round blue cell tumor of childhood. It is most commonly diagnosed in adolescents and young adults. Nearly a third of children with Ewing’s sarcoma have metastatic disease at the time of diagnosis, commonly in the axial skeleton. As with most childhood tumors, surgery is preferred over radiotherapy for local control to reduce the risk for second malignancies. Patients with close or positive surgical margins are treated with postoperative radiotherapy. Unresectable tumors are treated with primary radiotherapy, with similar success.77,78 Tumors of the axial skeleton, including the vertebral column, are often unresectable and treated with combined chemotherapy and radiotherapy. Current protocols also call for irradiation of all metastatic sites. Radiation doses of up to 55.8 Gy in conventional fraction sizes of 1.8 Gy/day to the primary site and areas of gross disease are recommended. Because of concerns of spinal cord toxicity, doses for vertebral body disease have historically been limited to 45 Gy. Advances in conformal radiotherapy techniques are allowing dose escalation while still respecting the normal tissue constraints (Fig. 252-4). Because of these advances, it is now possible to treat bony disease with doses of 50 to 55 Gy while still keeping doses to the spinal cord below 45 to 50 Gy and minimizing the risk for paraplegia.
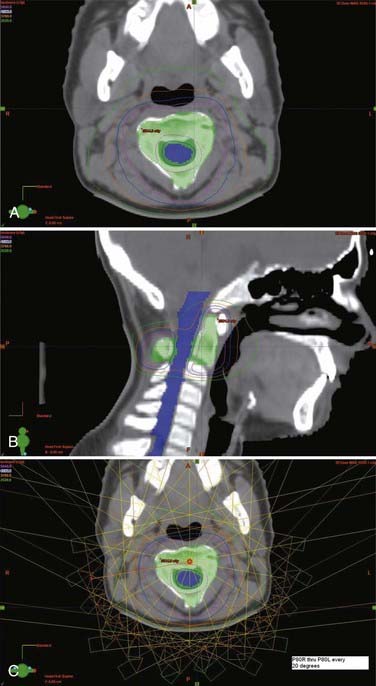
Abdel-Wahab M, Etuk B, Palermo J, et al. Spinal cord gliomas: a multi-institutional retrospective analysis. Int J Radiat Oncol Biol Phys. 2006;64:1060-1071.
Cole JS, Patchell RA. Metastatic epidural spinal cord compression. Lancet Neurol. 2008;7:459-466.
de Jonge T, Slullitel H, Dubousset J, et al. Late-onset spinal deformities in children treated by laminectomy and radiation therapy for malignant tumours. Eur Spine J. 2005;14:765-771.
Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
Hartley KA, Li C, Laningham FH, et al. Vertebral body growth after craniospinal irradiation. Int J Radiat Oncol Biol Phys. 2008;70:1343-1349.
Jallo GI, Freed D, Epstein F. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19:641-649.
Maranzano E, Latini P, Beneventi S, et al. Radiotherapy without steroids in selected metastatic spinal cord compression patients. A phase II trial. Am J Clin Oncol. 1996;19:179-183.
Merchant TE, Kiehna EN, Thompson SJ, et al. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg. 2000;32:30-36.
Merchant TE, Nguyen D, Thompson SJ, et al. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999;30:1-5.
Minehan KJ, Shaw EG, Scheithauer BW, et al. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83:590-595.
Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(suppl 2):57-63.
Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366:643-648.
Rades D, Dunst J, Schild SE. The first score predicting overall survival in patients with metastatic spinal cord compression. Cancer. 2008;112:157-161.
Rades D, Heidenreich F, Karstens JH. Final results of a prospective study of the prognostic value of the time to develop motor deficits before irradiation in metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2002;53:975-979.
Robinson CG, Prayson RA, Hahn JF, et al. Long-term survival and functional status of patients with low-grade astrocytoma of spinal cord. Int J Radiat Oncol Biol Phys. 2005;63:91-100.
Sandalcioglu IE, Gasser T, Asgari S, et al. Functional outcome after surgical treatment of intramedullary spinal cord tumors: experience with 78 patients. Spinal Cord. 2005;43:34-41.
Schild SE, Nisi K, Scheithauer BW, et al. The results of radiotherapy for ependymomas: the Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42:953-958.
Schulz-Ertner D, Karger CP, Feuerhake A, et al. Effectiveness of carbon ion radiotherapy in the treatment of skull-base chordomas. Int J Radiat Oncol Biol Phys. 2007;68:449-457.
Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
Volpp PB, Han K, Kagan AR, et al. Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys. 2007;69:1199-1204.
Wahab SH, Simpson JR, Michalski JM, et al. Long term outcome with post-operative radiation therapy for spinal canal ependymoma. J Neurooncol. 2007;83:85-89.
1 del Regato JA. Fractionation: a panoramic view. Int J Radiat Oncol Biol Phys. 1990;19:1329-1331.
2 Fein DA, Marcus RBJr, Parsons JT, et al. Lhermitte’s sign: incidence and treatment variables influencing risk after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1993;27:1029-1033.
3 Marcus RBJr, Million RR. The incidence of myelitis after irradiation of the cervical spinal cord. Int J Radiat Oncol Biol Phys. 1990;19:3-8.
4 St Clair WH, Arnold SM, Sloan AE, et al. Spinal cord and peripheral nerve injury: current management and investigations. Semin Radiat Oncol. 2003;13:322-332.
5 Rampling R, Symonds P. Radiation myelopathy. Curr Opin Neurol. 1998;11:627-632.
6 Keime-Guibert F, Napolitano M, Delattre JY. Neurological complications of radiotherapy and chemotherapy. J Neurol. 1998;245:695-708.
7 Wara WM, Phillips TL, Sheline GE, et al. Radiation tolerance of the spinal cord. Cancer. 1975;35:1558-1562.
8 Schultheiss TE, Kun LE, Ang KK, et al. Radiation response of the central nervous system. Int J Radiat Oncol Biol Phys. 1995;31:1093-1112.
9 Saunders MI, Dische S, Grosch EJ, et al. Experience with CHART. Int J Radiat Oncol Biol Phys. 1991;21:871-878.
10 Gatcombe H, Lawson J, Phuphanich S, et al. Treatment related myelitis in Hodgkin’s lymphoma following stem cell transplant, chemotherapy and radiation: a case report and review of the literature. J Neurooncol. 2006;79:293-298.
11 Rossitch EJr, Zeidman SM, Burger PC, et al. Clinical and pathological analysis of spinal cord astrocytomas in children. Neurosurgery. 1990;27:193-196.
12 Jallo GI, Freed D, Epstein F. Intramedullary spinal cord tumors in children. Childs Nerv Syst. 2003;19:641-649.
13 Santi M, Mena H, Wong K, et al. Spinal cord malignant astrocytomas. Clinicopathologic features in 36 cases. Cancer. 2003;98:554-561.
14 Minehan KJ, Shaw EG, Scheithauer BW, et al. Spinal cord astrocytoma: pathological and treatment considerations. J Neurosurg. 1995;83:590-595.
15 Kim MS, Chung CK, Choe G, et al. Intramedullary spinal cord astrocytoma in adults: postoperative outcome. J Neurooncol. 2001;52:85-94.
16 Robinson CG, Prayson RA, Hahn JF, et al. Long-term survival and functional status of patients with low-grade astrocytoma of spinal cord. Int J Radiat Oncol Biol Phys. 2005;63:91-100.
17 Hartley KA, Li C, Laningham FH, et al. Vertebral body growth after craniospinal irradiation. Int J Radiat Oncol Biol Phys. 2008;70:1343-1349.
18 de Jonge T, Slullitel H, Dubousset J, et al. Late-onset spinal deformities in children treated by laminectomy and radiation therapy for malignant tumours. Eur Spine J. 2005;14:765-771.
19 Minehan KJ, Brown PD, Scheithauer BW, et al. Spinal cord astrocytomas: treatment considerations. Int J Radiat Oncol Biol Phys. 1997;69(3 suppl S):S104.
20 Abdel-Wahab M, Etuk B, Palermo J, et al. Spinal cord gliomas: a multi-institutional retrospective analysis. Int J Radiat Oncol Biol Phys. 2006;64:1060-1071.
21 Karim AB, Maat B, Hatlevoll R, et al. A randomized trial on dose-response in radiation therapy of low-grade cerebral glioma: European Organization for Research and Treatment of Cancer (EORTC) Study 22844. Int J Radiat Oncol Biol Phys. 1996;36:549-556.
22 Shaw E, Arusell R, Scheithauer B, et al. Prospective randomized trial of low- versus high-dose radiation therapy in adults with supratentorial low-grade glioma: initial report of a North Central Cancer Treatment Group/Radiation Therapy Oncology Group/Eastern Cooperative Oncology Group study. J Clin Oncol. 2002;20:2267-2276.
23 Garcia DM. Primary spinal cord tumors treated with surgery and postoperative irradiation. Int J Radiat Oncol Biol Phys. 1985;11:1933-1939.
24 Merchant TE, Kiehna EN, Thompson SJ, et al. Pediatric low-grade and ependymal spinal cord tumors. Pediatr Neurosurg. 2000;32:30-36.
25 Shirato H, Kamada T, Hida K, et al. The role of radiotherapy in the management of spinal cord glioma. Int J Radiat Oncol Biol Phys. 1995;33:323-328.
26 Hua C, Gray JM, Merchant TE, et al. Treatment planning and delivery of external beam radiotherapy for pediatric sarcoma: the St. Jude Children’s Research Hospital experience. Int J Radiat Oncol Biol Phys. 2008;70:1598-1606.
27 Merchant TE, Nguyen D, Thompson SJ, et al. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999;30:1-5.
28 Shaw EG, Evans RG, Scheithauer BW, et al. Postoperative radiotherapy of intracranial ependymoma in pediatric and adult patients. Int J Radiat Oncol Biol Phys. 1987;13:1457-1462.
29 Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883-893.
30 Hoshimaru M, Koyama T, Hashimoto N, et al. Results of microsurgical treatment for intramedullary spinal cord ependymomas: analysis of 36 cases. Neurosurgery. 1999;44:264-269.
31 Sandalcioglu IE, Gasser T, Asgari S, et al. Functional outcome after surgical treatment of intramedullary spinal cord tumors: experience with 78 patients. Spinal Cord. 2005;43:34-41.
32 Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg. 1993;79:204-209.
33 Guidetti B, Mercuri S, Vagnozzi R. Long-term results of the surgical treatment of 129 intramedullary spinal gliomas. J Neurosurg. 1981;54:323-330.
34 Shaw EG, Evans RG, Scheithauer BW, et al. Radiotherapeutic management of adult intraspinal ependymomas. Int J Radiat Oncol Biol Phys. 1986;12:323-327.
35 Wen BC, Hussey DH, Hitchon PW, et al. The role of radiation therapy in the management of ependymomas of the spinal cord. Int J Radiat Oncol Biol Phys. 1991;20:781-786.
36 Wahab SH, Simpson JR, Michalski JM, et al. Long term outcome with post-operative radiation therapy for spinal canal ependymoma. J Neurooncol. 2007;83:85-89.
37 McLaughlin MP, Marcus RBJr, Buatti JM, et al. Ependymoma: results, prognostic factors and treatment recommendations. Int J Radiat Oncol Biol Phys. 1998;40:845-850.
38 Schild SE, Nisi K, Scheithauer BW, et al. The results of radiotherapy for ependymomas: the Mayo Clinic experience. Int J Radiat Oncol Biol Phys. 1998;42:953-958.
39 Roux FX, Nataf F, Pinaudeau M, et al. Intraspinal meningiomas: review of 54 cases with discussion of poor prognosis factors and modern therapeutic management. Surg Neurol. 1996;46:458-463.
40 Schiebe ME, Hoffmann W, Kortmann RD, et al. Radiotherapy in recurrent malignant meningiomas with multiple spinal manifestations. Acta Oncol. 1997;36:88-90.
41 Gerszten PC, Burton SA, Ozhasoglu C, et al. Radiosurgery for benign intradural spinal tumors. Neurosurgery. 2008;62:887-895.
42 Wong WW, Hirose T, Scheithauer BW, et al. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int J Radiat Oncol Biol Phys. 1998;42:351-360.
43 Volpp PB, Han K, Kagan AR, et al. Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys. 2007;69:1199-1204.
44 Whitaker SJ, Bessell EM, Ashley SE, et al. Postoperative radiotherapy in the management of spinal cord ependymoma. J Neurosurg. 1991;74:720-728.
45 Linstadt DE, Wara WM, Leibel SA, et al. Postoperative radiotherapy of primary spinal cord tumors. Int J Radiat Oncol Biol Phys. 1989;16:1397-1403.
46 Akyurek S, Chang EL, Yu TK, et al. Spinal myxopapillary ependymoma outcomes in patients treated with surgery and radiotherapy at M.D. Anderson Cancer Center. J Neurooncol. 2006;80:177-183.
47 Patchell RA, Tibbs PA, Regine WF, et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: a randomised trial. Lancet. 2005;366:643-648.
48 Rades D, Fehlauer F, Schulte R, et al. Prognostic factors for local control and survival after radiotherapy of metastatic spinal cord compression. J Clin Oncol. 2006;24:3388-3393.
49 Maranzano E, Latini P. Effectiveness of radiation therapy without surgery in metastatic spinal cord compression: final results from a prospective trial. Int J Radiat Oncol Biol Phys. 1995;32:959-967.
50 Clouston PD, DeAngelis LM, Posner JB. The spectrum of neurological disease in patients with systemic cancer. Ann Neurol. 1992;31:268-273.
51 Cole JS, Patchell RA. Metastatic epidural spinal cord compression. Lancet Neurol. 2008;7:459-466.
52 Brown PD, Stafford SL, Schild SE, et al. Metastatic spinal cord compression in patients with colorectal cancer. J Neurooncol. 1999;44:175-180.
53 Gilbert RW, Kim JH, Posner JB. Epidural spinal cord compression from metastatic tumor: diagnosis and treatment. Ann Neurol. 1978;3:40-51.
54 Rades D, Rudat V, Veninga T, et al. A score predicting posttreatment ambulatory status in patients irradiated for metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2008;72:905-908.
55 Rades D, Heidenreich F, Karstens JH. Final results of a prospective study of the prognostic value of the time to develop motor deficits before irradiation in metastatic spinal cord compression. Int J Radiat Oncol Biol Phys. 2002;53:975-979.
56 Vecht CJ, Haaxma-Reiche H, van Putten WL, et al. Initial bolus of conventional versus high-dose dexamethasone in metastatic spinal cord compression. Neurology. 1989;39:1255-1257.
57 Maranzano E, Latini P, Beneventi S, et al. Radiotherapy without steroids in selected metastatic spinal cord compression patients. A phase II trial. Am J Clin Oncol. 1996;19:179-183.
58 Rades D, Lange M, Veninga T, et al. Preliminary results of spinal cord compression recurrence evaluation (score-1) study comparing short-course versus long-course radiotherapy for local control of malignant epidural spinal cord compression. Int J Radiat Oncol Biol Phys. 2009;73:228-234.
59 Rades D, Stalpers LJ, Veninga T, et al. Evaluation of five radiation schedules and prognostic factors for metastatic spinal cord compression. J Clin Oncol. 2005;23:3366-3375.
60 Rades D, Dunst J, Schild SE. The first score predicting overall survival in patients with metastatic spinal cord compression. Cancer. 2008;112:157-161.
61 Rich TA, Schiller A, Suit HD, et al. Clinical and pathologic review of 48 cases of chordoma. Cancer. 1985;56:182-187.
62 Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases. J Neurosurg. 1993;78:741-747.
63 Hug EB, Loredo LN, Slater JD, et al. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. J Neurosurg. 1999;91:432-439.
64 Keisch ME, Garcia DM, Shibuya RB. Retrospective long-term follow-up analysis in 21 patients with chordomas of various sites treated at a single institution. J Neurosurg. 1991;75:374-377.
65 Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
66 Munzenrider JE, Liebsch NJ. Proton therapy for tumors of the skull base. Strahlenther Onkol. 1999;175(suppl 2)):57-63.
67 Noel G, Habrand JL, Mammar H, et al. Combination of photon and proton radiation therapy for chordomas and chondrosarcomas of the skull base: the Centre de Protontherapie D’Orsay experience. Int J Radiat Oncol Biol Phys. 2001;51:392-398.
68 Rutz HP, Weber DC, Goitein G, et al. Postoperative spot-scanning proton radiation therapy for chordoma and chondrosarcoma in children and adolescents: initial experience at Paul Scherrer institute. Int J Radiat Oncol Biol Phys. 2008;71:220-225.
69 Schulz-Ertner D, Karger CP, Feuerhake A, et al. Effectiveness of carbon ion radiotherapy in the treatment of skull-base chordomas. Int J Radiat Oncol Biol Phys. 2007;68:449-457.
70 Imai R, Kamada T, Tsuji H, et al. Carbon ion radiotherapy for unresectable sacral chordomas. Clin Cancer Res. 2004;10:5741-5746.
71 Martin JJ, Niranjan A, Kondziolka D, et al. Radiosurgery for chordomas and chondrosarcomas of the skull base. J Neurosurg. 2007;107:758-764.
72 Debus J, Schulz-Ertner D, Schad L, et al. Stereotactic fractionated radiotherapy for chordomas and chondrosarcomas of the skull base. Int J Radiat Oncol Biol Phys. 2000;47:591-596.
73 Oot RF, Melville GE, New PF, et al. The role of MR and CT in evaluating clival chordomas and chondrosarcomas. AJR Am J Roentgenol. 1988;151:567-575.
74 Shives TC, McLeod RA, Unni KK, et al. Chondrosarcoma of the spine. J Bone Joint Surg Am. 1989;71:1158-1165.
75 Schulz-Ertner D, Nikoghosyan A, Hof H, et al. Carbon ion radiotherapy of skull base chondrosarcomas. Int J Radiat Oncol Biol Phys. 2007;67:171-177.
76 Gwak HS, Yoo HJ, Youn SM, et al. Hypofractionated stereotactic radiation therapy for skull base and upper cervical chordoma and chondrosarcoma: preliminary results. Stereotact Funct Neurosurg. 2006;83:233-243.
77 Dunst J, Jurgens H, Sauer R, et al. Radiation therapy in Ewing’s sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys. 1995;32:919-930.
78 Yock TI, Krailo M, Fryer CJ, et al. Local control in pelvic Ewing sarcoma: analysis from INT-0091—a report from the Children’s Oncology Group. J Clin Oncol. 2006;24:3838-3843.






