CHAPTER 251 Fractionated Radiation Therapy for Benign Brain Tumors
Meningioma
Meningiomas are the most common nonglial intracranial neoplasms in adults,1,2 with the majority of meningiomas being benign. Collectively, they form the most common group of benign, intracranial neoplasms in adults and account for about 30% of primary central nervous system (CNS) tumors.2–4 Many are identified solely from findings on imaging; in recent studies, 35% to 62% were diagnosed on the basis of imaging alone.5,6 The risk of development of meningioma increases with age.7 They are infrequent in the pediatric population, except in the setting of neurofibromatosis type 2 (NF2).8
Overall, meningiomas are more commonly diagnosed in females, in whom they occur at a ratio of approximately 2 : 1.2,9 Nonetheless, atypical and anaplastic meningiomas are reported to occur more frequently in men.10 The reason for this gender distribution is unclear. Progesterone and estrogen receptors have been identified in greater than 70% and 30% of meningiomas, respectively.10 Increased estrogenicity is believed to be associated with an increase in tumor-related symptoms during pregnancy or the luteal phase of the menstrual cycle.11
Meningiomas may be accompanied by focal neurological symptoms, depending on their intracranial location. Seizures are a common initial sign and occur in 26% of patients with meningioma.12 Many meningiomas of the skull base cause lateralized cranial nerve deficits.13 Raised intracranial pressure is uncommon at initial evaluation but can occur secondary to obstructive hydrocephalus. Therapy is therefore aimed at relieving neurological symptoms and providing long-term tumor control.
Meningiomas are neoplasms of the meningothelial cells of the arachnoid layer and occur most frequently at sites where arachnoid cells are most numerous, namely, within the arachnoid villi or granulations that lie along the dural venous sinuses.14 Thus, the most common locations include the convexity (parasagittal dura or attached to the sagittal sinus), sphenoid ridge, and planum sphenoidale. Other locations include the sylvian fissure, parasellar region, and olfactory grooves. Meningiomas can also arise intraventricularly from pia-arachnoid rests, and about 10% occur infratentorially along the free edge of the tentorium, clivus, foramen magnum, petroclival ligament, and petrous ridge.15 Infrequently, optic nerve sheaths and the spinal canal may be involved.
Benign Meningiomas
Not all tumors can be resected completely without considerable morbidity. Safe and complete resection is frequently precluded by tumor involvement of the cranial nerves or arteries or invasion of the venous sinuses. Resectability also depends on the site and thus the surgical accessibility of the tumor. Gross total resection (GTR) is often not possible in the cavernous sinus, clivus, cerebellopontine angle, and sellar regions. It is estimated that a third of meningiomas are not fully resectable.16 In these situations, when complete and safe tumor removal is not possible, an effective strategy is subtotal resection (STR) followed by postoperative RT or RT alone. Clinical follow-up and serial imaging are recommended at annual or semiannual intervals for all patients who have undergone incomplete resection.
For benign meningiomas, the extent of resection is a major clinical predictor of recurrence in the absence of postoperative RT.17–19 GTR of benign meningiomas is considered definitive treatment (Table 251-1 and Fig. 251-1). Mirimanoff and coauthors reported 5-, 10-, and 15-year recurrence rates of 7%, 20%, and 32% and second operation rates of 6%, 15%, and 20%, respectively, among 145 patients in whom GTR was achieved.16 These rates were later confirmed in a large series from the Mayo Clinic.20 Additionally, Condra and coworkers confirmed the importance of complete excision but found no association between Simpson grade and local control or cause-specific survival, as long as GTR (Simpson grades 1, 2, and 3) was achieved.21 For patients treated by surgery alone, GTR resulted in 5-, 10-, and 15-year actuarial recurrence rates of 7%, 20%, and 24%, respectively.21
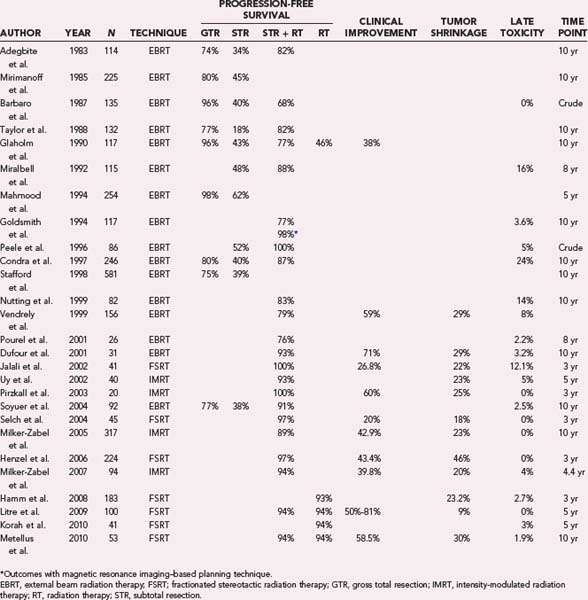
Recurrence rates after STR alone are substantially higher (see Table 251-1 and Fig. 251-1). For example, among 116 patients with STR, Stafford and associates found recurrence in 39% at 5 years and in 61% at 10 years.20 Condra and colleagues documented local recurrence at 5, 10, and 15 years in 47%, 60%, and 70% of patients who underwent STR, respectively.21 Overall, it is estimated that local progression develops in 40% to 50% of patients after STR within 5 years, in 60% within 10 years, and in at least 70% within 15 years.21,22
Because GTR is frequently not possible, RT, whether fractionated external beam RT or stereotactic radiosurgery (SRS), is currently the only accepted nonsurgical treatment option. Historically, meningiomas were considered to be resistant to RT, with authors of many older retrospective studies reporting infrequent radiographic regression.23 Additionally, concerns about malignant degeneration were cited as a reason to refrain from either definitive or postoperative RT.24,25 Consequently, many patients with inoperable or subtotally resected meningiomas underwent observation. At present, malignant degeneration has not been explicitly linked to RT; instead, it is believed to be related to the natural history of a subset of meningiomas. Strojan and coauthors reviewed the case files of 170,000 meningioma patients treated in Slovenia over a 31-year period and reported the actuarial risk of a secondary meningioma developing after cranial irradiation to be 0.53% at 5 years and 8.18% at 25 years.26
Most retrospective studies since the 1980s have demonstrated that postoperative RT after STR improves local control (see Table 251-1 and Fig. 251-1) and perhaps even survival.21,27 Improvement in local control, as well as frequent improvement in tumor-related neurological symptoms, was observed despite the fact that only about a fourth to a third of treated meningiomas exhibit tumor shrinkage (Fig. 251-2; also see Table 251-1).
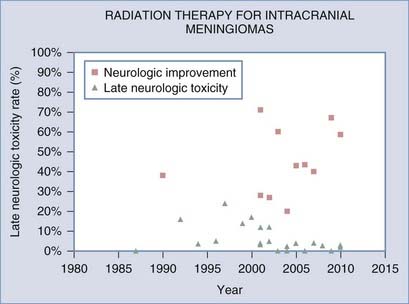
FIGURE 251-2 Rates of neurological improvement and late post–radiation therapy toxicity as a function of treatment period.
RT can also be used as primary treatment of meningiomas diagnosed radiographically or by biopsy. In one of the earliest reports, Glaholm and associates reported on the outcome of 32 patients treated by primary RT without resection at the Royal Marsden Hospital; a 47% disease-free survival rate was observed at 15 years.24,25 This was only modestly lower than a corresponding 56% disease-free survival rate in patients treated with STR plus RT. More recent series, however, demonstrate markedly improved local control rates and progression-free survival rates greater than 93% after 5 to 10 years of follow-up (see Table 251-1).28–31 These outcomes compare favorably with those observed after GTR of benign meningiomas (see Fig. 251-1).
The discrepant outcomes of older studies (before 2000) and more recent RT studies, whether in the postoperative or primary setting, may be attributed to technical advancements in RT treatment planning and delivery techniques. This trend is clearly illustrated in Figure 251-1 for progression-free survival and in Figure 251-2 for RT-related late neurological toxicity, as well as neurological improvement. These observations are more directly supported by Goldsmith and colleagues’ analysis of 140 patients treated at the University of California, San Francisco, with a median dose of 54 Gy in the postoperative setting; a progression-free survival rate of 98% was noted in those treated after 1980 (when computed tomography [CT] and magnetic resonance imaging [MRI] was used for planning therapy) as compared with a rate of 77% for patients treated before 1980, when advanced planning techniques were not available.32
Newer treatment series also report on the use of intensity-modulated RT (IMRT), as well as fractionated stereotactic RT (FSRT) (see Table 251-1). IMRT techniques allow the delivery of doses via multiple convergent beams (Fig. 251-3), each of which can be divided into zones of different intensities so that the integral dose can tightly conform to the shape of the target. This technique can potentially minimize unnecessary irradiation of surrounding normal tissues, including the critical anterior optic structures. FSRT is a variant of conventional RT characterized by a highly accurate patient immobilization setup, use of a stereotactic coordinate system, and submillimeter mechanical and beam tolerances of the treatment equipment. All modern RT techniques rely on the use of sophisticated imaging for planning treatment and verifying delivery. It is believed, but not yet definitively proved, that these newer treatment methods will continue to improve treatment outcomes and reduce RT-related toxicity.
It is worth noting, however, that serious side effects of modern methods of treatment planning and delivery are already quite uncommon. For example, Debus and coauthors reported on 189 patients treated by FSRT for large skull base meningiomas with median daily fractions of 1.8 Gy to a total mean dose of 56.8 Gy.33 With a nearly 3-year median follow-up, the rate of clinically significant toxicity (grade 3) was 2.2% (4 patients), and 1.6% (3 patients) in the absence of a preexisting neurological deficit. The neurological deficits reported in the study included reduced vision, a new visual field loss, and trigeminal neuropathy. Goldsmith and coworkers noted complications in 3.6% of 140 patients treated with RT after STR from 1967 to 1990.32 The RT-related toxicity observed in this study included retinopathy, optic neuropathy, and cerebral necrosis. Further analysis of the data by Goldsmith and associates permitted construction of a model to predict that optic nerve tolerance is 890 optic ret, equivalent to 54 Gy in 30 fractions.34 This threshold dose is supported by only rare observations of optic nerve injury with doses lower than 54 Gy, particularly with fractional doses of 2.0 Gy or less.24,35,36 Additionally, Uy and colleagues reported no anterior optic pathway injury with a median dose of 50.4 Gy in fractions of 1.7 to 2.0 Gy.37 Shrieve and associates further confirmed these findings, as well as the relative benefit of a fraction size of 2.0 Gy or less.38
Other cranial neuropathies (nonoptic) are uncommon with modern treatment techniques but have been reported in older series. For example, Selch and coworkers found no RT-related neuropathy in 45 patients treated for cavernous sinus meningiomas with a median dose of 50.4 Gy (1.8 Gy/fraction).39 Similar findings were noted earlier by Urie and colleagues.40 Brain or brainstem injury, including necrosis, is quite uncommon in the modern era with conventional doses and techniques. Similarly, edema is not generally observed after fractionated RT, unlike single-session SRS. For this reason, fractionated RT is preferred over SRS for tumors in locations where edema is likely to lead to significant morbidity (e.g., larger cerebellopontine angle tumors).
Optic Nerve Sheath Meningiomas
Management of optic nerve sheath meningiomas (ONSMs) is almost exclusively nonsurgical. These rare tumors represent less than 2% of all meningiomas and arise from the meningeal lining of the optic nerves.41,42 They generally grow very slowly within the subarachnoid space and the confines of the optic canal but eventually compress the optic nerve itself, its vasculature, or both. Patients usually have progressive visual loss, although some may exhibit rapid loss of vision. In a small minority of patients, ONSMs are found incidentally on imaging. Less common complaints include visual field defects, loss of or alterations in color vision, and orbital pain.
Resection of ONSMs while keeping the optic nerve in place has been tried43–47 but carries an unacceptably high risk for visual complications and local recurrence. Resection commonly impairs the blood supply and results in blindness, even though the nerve is left grossly intact. For these reasons, fractionated RT has become the centerpiece of management of ONSM. In one of the largest series published, Turbin and coauthors reported on 64 patients with ONSM and found that fractionated RT alone provided more favorable outcomes than did observation, surgery alone, or even a combination of surgery and RT.47 Patients in the RT-only group were the only ones who did not demonstrate a statistically significant decline in visual acuity after a mean follow-up period of 11.5 years (range, 4.75 to 23 years); all other groups, including patients who underwent surgery plus RT, demonstrated a significant decrement in visual acuity, including complete visual loss. Irradiated patients received 40 to 55 Gy of fractionated RT (1.8 to 2.0 Gy/fraction) over an approximately 5-week period. In another study from Japan, Narayan and colleagues evaluated 14 patients with ONSM who were treated with conformal, fractionated RT to a total dose of 50.4 to 56 Gy.48 After a median follow-up of 4.1 years, 86% of the treated patients had either improved or stable visual acuity. These studies and six additional ones with similar results established highly conformal, fractionated RT as the treatment of first choice for patients with ONSM. In aggregate, local control is excellent (95%), early visual improvement (<3 months after completion of RT) is attained in about half of the patients (54.7%), and complications of therapy are relatively uncommon.41 It is our institutional policy to treat all symptomatic ONSM patients with highly conformal, fractionated RT to total doses of 50.4 to 54 Gy in daily fractions of 1.8 Gy; however, there is no uniform agreement on whether patients with stable ONSM and stable vision should be managed by observation or early RT. Patients with bilateral disease are generally treated, whereas those with unilateral tumors are observed, provided that that they can be expected to comply with serial visual field testing and regular clinical and imaging follow-up.
Atypical Meningiomas
Atypical, WHO grade II meningiomas may account for up to 20% of all diagnosed meningiomas.10 Atypical meningiomas are a heterogeneous group, largely because of the imprecise pathologic classification criteria and varying classification schemes over time. Most investigators have recommended irradiation, irrespective of the extent of resection.21,49 However, Goyal and coauthors argued that fractionated RT has no significant impact on local control and overall survival.50 Their retrospective analysis was based on a cohort of 22 patients, 15 of whom underwent GTR (Simpson grades 1 to 3), 4 underwent STR, and 3 had resection of unknown extent; 8 patients received RT (2 after initial resection and 6 at the time of recurrence). The median radiation dose was 54 Gy (range, 35 to 59.4 Gy). After a median follow-up of 5.5 years, the local control rate was 87% at 5 and 10 years after GTR, and RT had no significant impact on local control or overall survival. This study highlighted some of the difficulties relevant to atypical tumors, including inconsistent use, dosing, and timing of RT, as well as wide variations in the extent of resection.49 Hug and coworkers found that that local control of atypical meningiomas was significantly enhanced by cumulative doses of at least 60 cobalt gray equivalents (CGE).49 Perry and coauthors reported on 108 patients with atypical meningiomas treated with modern surgical techniques, grading, and postoperative imaging and reported a 5-year recurrence rate of 40% even after GTR.51 In another study, recurrence rates of atypical meningiomas after either STR or “radical subtotal” resection were 39% and 61% at 5 and 10 years, respectively.20 Because of these unacceptably high recurrence rates, there is a relative consensus that irradiation to a total dose of 59.4 Gy (1.8 Gy/fraction) or 60 Gy (2 Gy/fraction) is appropriate after STR of an atypical meningioma.
Adjuvant treatment recommendations for atypical meningiomas after GTR are more controversial. Many neurosurgeons argue in favor of close radiographic and clinical follow-up after Simpson grade 1 and 2 resection, but not necessarily Simpson grade 3.52 In a recent study, Aghi and colleagues retrospectively reviewed the outcomes of 108 patients with atypical meningiomas who underwent Simpson grade 1 GTR at the Massachusetts General Hospital between 1993 and 2004.53 With a mean follow-up period of 3.25 years, the actuarial tumor recurrence rates were 7% at 1 year, 41% at 5 years, and 48% at 10 years. Most recurrences occurred within the first 5 years after surgical resection. Of the 108 patients, 8 received immediate adjuvant RT to a total dose of 59.4 to 61.2 Gy (1.5 to 1.8 Gy/fraction) and experienced no recurrences during the follow-up period. This study could be interpreted as evidence in favor of either immediate postoperative fractionated RT or close clinical and radiographic follow-up (every 3 to 6 months) for the first 5 years, with salvage RT being performed as needed. The key question in this patient subset is whether early adjuvant or late salvage RT is more appropriate; the existing publications do not answer this question.
Recurrent Meningiomas
Recurrent meningiomas, after surgery alone, exhibit markedly increased progression rates over newly diagnosed tumors,16,21,35,54 with a mean interval to clinical recurrence of 4 years.16,23 Miralbell and coauthors reported a 78% progression-free survival rate at 8 years in patients treated with surgery plus RT for recurrent tumors as compared with just 11% with surgery alone.35 Similarly, Taylor and colleagues reported on 30 patients in whom recurrent disease developed after their initial treatment; 15 were treated by surgery alone, 10 underwent surgery and RT, and 5 patients either refused treatment or were deemed inoperable.54 The reported local control rate was 30% at 10 years in patients who underwent surgery alone for recurrence, whereas the 10 patients treated by salvage RT (with or without resection) had an actuarial local control rate of 89%. The study of Taylor and associates54 provides compelling support for the use of salvage RT to cumulative doses of 54 to 59.4 Gy (1.8 Gy/fraction or similar regimens at 2 Gy/fraction), not only because of the superior local control rates but also because of the improved actuarial survival in the salvaged patients (89% for surgery plus RT at 10 years versus just 43% for surgery alone). These data support aggressive treatment of recurrent meningiomas.
Pituitary Adenoma
The pituitary gland consists of the anterior and posterior lobes, also referred to as the adenohypophysis and neurohypophysis, respectively. The majority of pituitary adenomas arise from adenohypophysial cells in the anterior pituitary.55 Other anterior pituitary tumors (sarcomas, melanomas, and carcinomas) account for less than 1% of all pituitary neoplasms. Posterior pituitary tumors (gangliomas, choristomas, and astrocytomas) also represent less than 1% of all pituitary neoplasms. In aggregate, pituitary tumors account for approximately 10% to 20% of all CNS tumors.55 Autopsy studies suggest that the incidence of pituitary adenoma in the general population may be as high as 25%.56,57 Both the anterior and posterior pituitary lobes lack a blood-brain barrier.58 Metastatic tumor deposits can also be found in the posterior pituitary but are rarely, if ever found in the anterior lobe at autopsy.59,60 In addition to pituitary adenoma, the differential diagnosis of a sellar or parasellar lesion must include such entities as Rathke’s pouch cysts, craniopharyngiomas, germ cell tumors, and autoimmune conditions such as lymphocytic hypophysitis, among others.61
Pituitary adenomas are classified by several properties descriptive of the tumor, including size, secretory products, and the characteristics of tumor growth or extension.62 Pituitary adenomas smaller than 1.0 cm in diameter are referred to as microadenomas, whereas adenomas 1.0 cm or greater in size are classified as macroadenomas. Macroadenomas are characteristically more nodular and infiltrative and often result in compression of surrounding neurological structures.63 Functional pituitary adenomas are also classified by their secretory products. Prolactinomas are the most common type of pituitary adenoma and account for more than a third of all pituitary tumors.56 This is followed closely by nonfunctioning adenomas (NFAs), which represent a fourth to a third of pituitary tumors. Of the NFAs, null cell tumors (those characterized by the absence of markers that disclose the cell of origin) account for about two thirds of the total, whereas the remaining third are mainly oncocytic. Somatotropic and corticotropic adenomas each constitute about 15% of pituitary tumors. It is also possible, but less common, for tumors to consist of two or more populations of cells that secrete several hormonal products. In vast majority of cases, these multihormonal tumors are macroadenomas and usually synthesize both prolactin and growth hormone (GH).64
Prolactinoma
Prolactinomas are the most common functional pituitary tumors. Nearly 90% of these tumors are entirely intrasellar at diagnosis and rarely increase in size during routine follow-up.65 Elevated levels of prolactin can interfere with the normal pulsatile release of gonadotropin-releasing hormone, thereby impeding the secretion of luteinizing hormone and follicle-stimulating hormone. Consequently, the symptoms of prolactinomas are dependent on the sex of the patient. Hyperprolactinemia in women can result in amenorrhea, galactorrhea, and infertility. The majority of prolactinomas in women are microadenomas.66 Prolactinomas in men tend to be more aggressive and most are macroadenomas.67 Men often report symptoms related to local mass effect such as visual disturbance, neurological dysfunction, and headache with associated hypogonadism, decreased libido, and infertility.
A diagnosis is established by documenting a sustained elevation in serum prolactin above the normal range. Additionally, contrast-enhanced fat-suppressed MRI should be used to confirm a diagnosis. However, a negative MRI scan does not exclude the diagnosis of a prolactinoma in the setting of clinical symptoms, especially in female patients, who are likely to harbor microadenomas.68
The goal of treatment of prolactinoma is restoration of sexual/reproductive function, control of galactorrhea, stabilization of serum prolactin levels, and in cases of macroadenoma, relief of neurological symptoms. The standard initial management of prolactinomas is surgery followed by medical therapy with a dopamine agonist if needed. Bromocriptine, pergolide, and cabergoline are ergot alkaloids that bind the dopamine receptor and relieve symptoms of hyperprolactinemia by causing the cessation of prolactin release.69 Treatment with bromocriptine has been shown to normalize prolactin levels, relieve symptoms, and cause tumor shrinkage in 80% to 90% of patients with microadenomas and in 70% to 80% of patients with macroadenomas.69,70
A significant shortcoming of conservative medical management with dopamine agonists is recurrence of disease in almost 90% of patients after cessation of dopamine agonist therapy.71,72 In a study of 131 patients monitored for an average of 44 months after cessation of bromocriptine therapy, Passos and coworkers reported normalization of prolactin levels in only 20.6% of participants.71 Many of the reported recurrences were primarily biochemical in nature, but tumor regrowth resulting in compression of the optic nerve and associated visual deterioration was reported in a subset of patients.70 These issues argue for an effective adjuvant therapy capable of inducing an extended remission period.
Both surgery and RT have been investigated in this setting. Transsphenoidal pituitary resection is the current second-line therapy and results in 75% to 90% long-term biochemical control in patients with microadenomas.73–75 Pretreatment prolactin levels are a strong risk factor for recurrence, independent of tumor size. The immediate postoperative prolactin level is also prognostic, with levels of less than 5 ng/mL being associated with an 80.5% remission rate at a mean follow-up of 9.2 years.76,77 Gillam and coworkers performed an extensive review and meta-analysis of surgical therapy for prolactinoma and demonstrated average remission rates of 74.7% and 33.9% for microadenomas and macroadenomas, respectively.70 Remission was defined as normalization of prolactin levels within 12 weeks after surgery. The majority of the recurrences were biochemical without any abnormalities noted on imaging. This analysis confirmed the presence of a macroadenoma as a risk factor for recurrence or persistence of disease.
RT has also been investigated for the treatment of prolactinomas. In the current treatment paradigm, RT is typically a second- or third-line treatment, after medical or surgical interventions (or both) have failed or if the patient is not a surgical candidate. Several studies have addressed the efficacy of conventional RT as postoperative treatment and in the primary setting (Table 251-2). Interpretation of these studies is complicated by the use of different end points and definitions of cure.
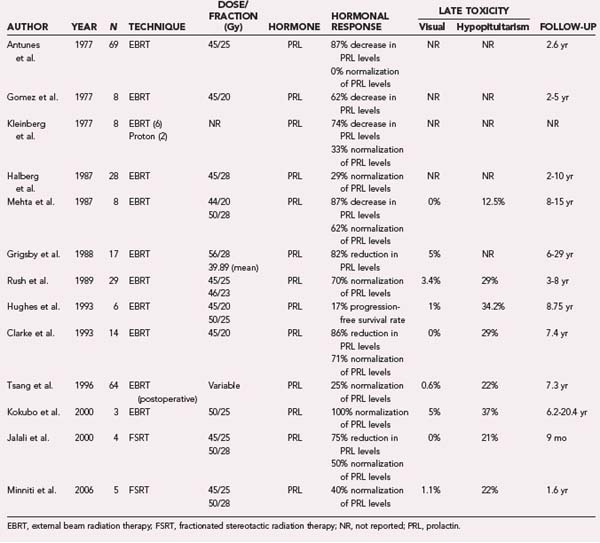
Most of the studies investigating the use of primary conventional RT for the treatment of prolactinoma were conducted between 1977 and 2006 (see Table 251-2). Halberg and Sheline observed 28 patients for 2 to 10 years after treatment and demonstrated normalization of prolactin levels in 29%.76 They used bilateral coronal arc fields with reversing wedge filters to deliver 45 Gy in 25 fractions to the sella turcica while sparing the eyes altogether. Another study by Grigsby and associates analyzed 17 patients treated with a mean dose of 39.89 Gy (range, 3.94 to 56 Gy) in fractional doses of 0.5 to 2 Gy and reported a 10-year disease-free survival rate of 82.3%.78 Patients treated with doses of 45 Gy or greater achieved superior tumor control. Within the treated cohort, 5 of 9 female patients had a return to normal menstrual patterns, and galactorrhea ceased in 4 of 8 patients. Similarly, Rush and Newall reported on 29 patients with a follow-up of 3 to 8 years and reported a 70% rate of prolactin normalization.79 Tsang and colleagues conducted the largest postoperative study consisting of 64 patients treated at the Princess Margaret Hospital in Toronto and demonstrated normalization of prolactin levels in 25% at a median follow-up of 7.3 years.80 Most patients in this study were treated to a total dose of 50 to 52 Gy via right and left parallel-opposed fields targeting the preoperative extent of adenoma with a 1.0- to 1.5-cm margin. More recently, Jalali and coworkers81 and Minniti and colleagues82 reported on the use of FSRT for the treatment of adenomas. However, these preliminary studies have follow-up that is too limited to permit meaningful interpretation. Table 251-2 lists most major published studies that have examined the role of conventional RT for the treatment of prolactinomas.
The aforementioned studies demonstrate less than satisfactory control rates, especially when compared with the efficacy of primary medical therapy with dopamine agonists, thus suggesting that primary fractionated RT may not offer the best chance for long-term disease remission. It has been hypothesized that these suboptimal outcomes are due to RT-induced damage to the hypothalamus83 and subsequent loss of dopamine release and thus loss of the inhibitory signal for release of prolactin. This leads to a paradoxical increase in prolactin secretion from the anterior pituitary. This hypothesis is supported by observations from studies investigating the treatment of nonsecretory adenomas with RT.83
In a study investigating the use of intermittent dopamine agonist therapy and RT, Tsagarakis and associates observed 36 female patients for a mean period of 8.5 years. About half of the patients experienced normalization of prolactin levels, and an additional 28% had prolactin levels just above the normal range (378 to 780 mU/L).84 Only 1 patient exhibited documented disease recurrence by imaging.
Similarly, Grossman and coworkers investigated the impact of 45 Gy in 25 fractions plus interim dopamine agonist therapy in 36 women with prolactinomas.85 Treatment with the dopamine agonist was stopped at a mean of 4.2 years after RT to assess the response in 27 patients, with 26 demonstrating a progressive decline in serum levels of prolactin. Moreover, in patients who were interested in conceiving after treatment, a successful conception rate of 86% was observed. Grigsby and colleagues also analyzed the efficacy of postoperative conventional RT for the treatment of prolactinoma.86 Twenty-six patients with prolactinoma had a 10-year disease-free survival rate of 93.3%, with resolution of galactorrhea in 7 of 13 patients and resumption of menses in 8 of 15 patients. Other studies have reported similar and less satisfactory results, thus prompting investigations of the use of SRS for this disease when large, single-fraction doses can be delivered safely.87,88
Cushing’s Disease
Cushing’s disease is caused by an adrenocorticotropic hormone (ACTH)–secreting pituitary adenoma and, when associated with signs and symptoms of excess ACTH, is referred to as Cushing’s syndrome. Cushing’s disease causes approximately 70% of cases of Cushing’s syndrome and is characterized by the presence of several of the following symptoms: central obesity, glucose intolerance, hypertension, hirsutism, abdominal striae, moon facies, and a buffalo hump.89 Cushing’s disease typically affects men more than women (3 : 1 ratio).90 It is diagnosed with a 24-hour urine collection to quantify the amount of urinary free cortisol, a dexamethasone suppression test, or both. Elevated levels of urinary free cortisol, clinical symptoms, and positive findings on imaging can all, in conjunction, lead to a diagnosis of Cushing’s disease.91 Random measurements of serum cortisol or ACTH (or both) are not diagnostic because of the natural diurnal variations in secretion of ACTH.
Most ACTH-secreting tumors are microadenomas. In up to 25% of patients with this disease, there are no abnormal findings on MRI, and of these patients, about 50% have no identifiable tumor at surgery.92 Transsphenoidal resection is currently the first-line therapy and has yielded control rates of 70% to 85% in several retrospective series.93,94 More recent surgical studies, conducted after 2000, are in line with the historical data and have reported biochemical remission rates ranging from 68.5% to 98%.95–98 For example, Cavagnini and colleagues observed 300 patients for a median follow-up time of 10 years and found a biochemical remission rate of 70% and a recurrence rate of 15% after surgery alone.98 Chen and associates investigated 174 patients treated surgically with a median follow-up of 5 years and found a similar biochemical remission rate of 74%; however, the recurrence rate was just 7% (probably because of a shorter follow-up period).95 This study also revealed that a serum cortisol level of 3 µg/dL or less on the morning of postoperative day 3, after an overnight dexamethasone suppression test, was prognostic of up to a 93% biochemical remission rate at 5 years. Flitsch and coworkers evaluated 147 patients and made similar observations regarding the prognostic significance of low postoperative ACTH levels.96
RT has also been explored as treatment for patients with Cushing’s disease. In a manner analogous to prolactinomas, most studies evaluating the efficacy of primary, conventional RT for the treatment of ACTH-secreting adenomas were conducted before the year 2000 (Table 251-3). Most recent studies explore the efficacy of SRS instead. Orth and Liddle conducted the largest study of RT involving 44 patients treated before 1971 and demonstrated a control rate of 44% and normalization of cortisol levels in 20% of patients observed for a median period of 9 years.99 An analysis by Grigsby and coworkers of 6 patients treated with definitive RT before 1982 showed a 100% tumor control rate with a follow-up ranging from 6 to 29 years.78 A dose of less than 40 Gy to the tumor correlated with lower biochemical control rates and a greater risk for recurrence. A more recent study by Hughes and associates analyzed 40 patients and demonstrated a 10-year progression-free survival rate of 59% (see Table 251-3).100 Large tumor volumes and consequently large treatment fields were found to have a negative prognostic implication.
TABLE 251-3 Summary of Studies on Conventional (Fractionated) Radiotherapy for ACTH-Secreting Pituitary Adenoma
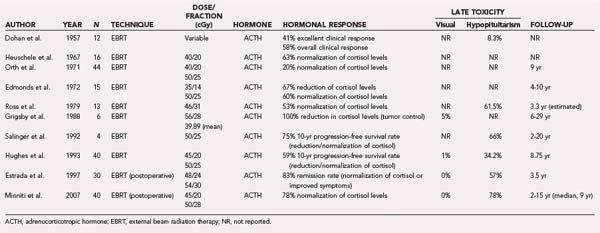
In a 1997 study, Estrada and coworkers investigated the postoperative use of RT consisting of 50 Gy in 25 fractions and demonstrated an 83% biochemical control rate (normalization or improved symptoms) in 30 patients monitored for a median period of 3.5 years.101 Similarly, Minniti and associates evaluated 40 patients treated postoperatively with RT (45 Gy in 20 fractions or 50 Gy in 28 fractions) and reported a 78% biochemical remission rate at 5 years and an 84% remission rate at 10 years.102 Not all retrospective studies were similarly successful, however. For example, an older study by Littley and coauthors analyzed 24 patients and found a biochemical remission rate of only 46%,103 probably because the postoperative dose of RT was just 20 Gy in 10 fractions. In another study, Tsang and colleagues examined 29 patients who received 50 Gy of postoperative RT and found only a fair biochemical remission rate of 53% at 10 years.80 The reasons for these discrepancies among studies are not clear, but it does appear that more modern treatment techniques tend to achieve superior biochemical outcomes.
Nelson’s Syndrome
Nelson’s syndrome is characterized by hyperpigmentation occurring after bilateral adrenalectomy in patients with ACTH-secreting pituitary adenomas.104 Removal of both adrenal glands eliminates the production of cortisol, and the resultant loss of negative feedback allows preexisting pituitary adenomas to grow unchecked. It is estimated that Nelson’s syndrome will eventually develop in 30% to 50% of patients with Cushing’s syndrome who undergo bilateral adrenalectomy.105 These adenomas tend to be more aggressive, frequently extend beyond the sella, and thus are more difficult to cure; surgery in this setting is often complex and morbid.
The role of RT for the treatment of Nelson’s syndrome was investigated in a few studies. Howlett and associates conducted a study of 15 patients with Nelson’s syndrome who were treated with definitive, primary RT to a total dose of 45 Gy in 25 fractions and observed post-treatment biochemical improvement in all but 1 patient after a median follow-up of 9.5 years.106 In 1995, Jenkins and coworkers analyzed the outcomes of 20 patients who underwent “prophylactic” pituitary RT (45 Gy in 25 fractions) at some time after bilateral adrenalectomy and demonstrated that RT reduces the incidence of subsequent Nelson’s syndrome by 50% (75% control rate at a median follow-up of 9.1 years).107
Acromegaly
Acromegaly is caused by excessive secretion of GH after closure of the epiphyseal plates.108 The characteristic bone enlargement found in patients with acromegaly usually involves the frontal bones (frontal bossing), nose, mandible (prognathism), hands, feet, and vertebrae (kyphosis). Adults do not generally become taller with excess GH in serum because the epiphyseal plates of the long bones have already closed. The excessive GH leads to an increase in insulin-like growth factor type I (IGF-I), and this results in several other medical comorbidities in addition to simple bone enlargement. Higher rates of cardiovascular complications, hypertension, diabetes mellitus, and colon cancer have all been described in this patient population and lead to significant decreases in life expectancy.109–111
Because GH secretion varies throughout the day, the diagnosis of acromegaly is established by measuring the GH level after a glucose challenge. A glucose load of 100 g should normally cause marked suppression of GH release, usually below 2 ng/mL.112 Failure to achieve such suppression after a glucose challenge may suggest a presence of a GH-secreting pituitary adenoma. It is also advisable to check serum IGF-I levels because they are less variable throughout the day and may be a more reliable marker for diagnosis.112
The first-line therapy for GH-secreting pituitary tumors is again surgical resection.113 Although somatostatin analogues may also be used to control GH and IGF-I levels, this treatment is successful in only 50% to 79% of patients; there is an associated decrease in tumor size by as much as 50% in about half of the patients.114,115 The concurrent use of dopamine agonists may help reduce GH levels by an additional 10%.116 Both GH and IGF-I levels can be used to monitor the endocrine response (biochemical control) in patients with acromegaly. The goal of therapy is to achieve normalization of GH and IGF-I markers to reduce, stop, or prevent the metabolic complications associated with their elevation.
Transsphenoidal microsurgery is highly successful in the treatment of acromegaly, more successful in the treatment of microadenomas than macroadenomas, just as in other secreting pituitary adenomas. Laws and colleagues analyzed 86 patients treated by microsurgery alone and reported biochemical remission rates of 87% and 51% for microadenomas and macroadenomas, respectively.117 An overall remission rate (normalization of serum IGF-I) of 67% was achieved after 1 year of follow-up. The largest surgical series, by Nomikos and associates in 2005, reported on 688 patients, 506 of whom underwent primary transsphenoidal surgery for their acromegaly.118 An overall biochemical response rate (normalization of serum IGF-I) of 57.3% was achieved in patients treated primarily by surgery during a follow-up period of 10.7 years, with a biochemical recurrence rate of just 0.4%. Again, patients with microadenomas had higher biochemical remission rates than did those with macroadenomas (75% versus 50%, respectively).118
These two large surgical series are representative of outcomes reported in the published literature after transsphenoidal resection of pituitary adenomas for acromegaly. It should be cautioned that a vast range of success rates have been reported in the published literature, principally because of varied definitions of biochemical control or cure. The surgical morbidity in experienced hands is quite low: reported mortality rates are less than 0.5%, and serious complications occur in less than 1.5% of patients.119 Pregnancy after transsphenoidal surgery for microadenomas has been achieved in 86% of women desiring to become pregnant, and the incidence of iatrogenic hypopituitarism in patients with microadenomas is less than 3%.119
Several studies have evaluated the use of conventional RT for the treatment of acromegaly. Table 251-4 lists select studies from 2000 and later that analyzed the outcomes of more than 30 patients and similarly defined biochemical control criteria (GH and IGF-I levels <2 ng/mL). The largest such study, conducted by Jenkins and coworkers, retrospectively analyzed the outcomes of 884 patients treated for acromegaly in 14 centers in the United Kingdom with fractionated RT to a mean total dose of 45 Gy in 25 fractions.120 At a mean follow-up of 2, 10, and 20 years, the biochemical remission rates (serum GH level <2.5 ng/mL) were 22%, 60%, and 77%, respectively. These results demonstrate that biochemical control increases with time after treatment.
TABLE 251-4 Summary of Studies on Conventional (Fractionated) Radiotherapy for GH-Secreting Pituitary Adenoma
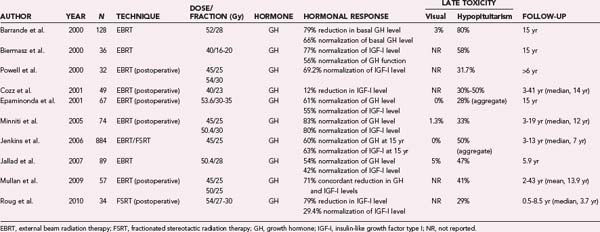
In another large study, Barrande and coworkers monitored 128 patients for a mean period of 11.5 years after definitive RT (52 Gy in 28 fractions) and demonstrated biochemical remission in 35%, 53%, and 66% of patients at 5, 10, and 15 years, respectively.121 This study confirms the temporal evolution of response to therapy after treatment with conventional RT, with the best results occurring 15 years after treatment.
These two large studies, as well as other similar studies (see Table 251-4), also demonstrate that RT effectively reduces tumor size over time. Conventional RT is thus effective not only in achieving endocrinologic remission in a large number of patients, albeit slowly, but also in controlling the growth of these tumors. The risk for injury to the anterior optic structures with conventional RT is less than 5% in the published literature, and the reported rates of iatrogenic hypopituitarism range from 28% to 80%; better toxicity profiles are seen with the use of FSRT (see Table 251-4). The use of single-session SRS techniques is increasingly being explored for the treatment of acromegaly as the technology becomes more available.122
Nonfunctioning Adenomas
NFAs are the most common form of pituitary macroadenoma. Because of their nonsecretory nature, these tumors commonly affect anterior visual structures such as the chiasm and cause visual field defects or have an impact on visual acuity (or both).123,124 Endocrinologic symptoms may also be present as a result of compression of the remaining functional pituitary tissue within the sella.123 One study reported that visual defects occurred in more than two thirds of patients and headaches in an additional 41% of patients.123 This same study found hypogonadism to be the most common clinical manifestation of hypopituitarism.
Surgery is the primary treatment of NFAs and results in immediate relief of the compressive symptoms. Although surgery can acutely address the visual symptoms, several published studies suggest that surgery alone does not result in acceptable long-term control.123,125,126 RT is generally reserved for the postoperative setting to prevent tumor regrowth.127,128
In a 2003 analysis, Greenman and colleagues evaluated 108 patients treated by transsphenoidal surgery alone and found that 72% had STR with identifiable tumor on immediate postoperative imaging.129 The recurrence rate in the STR cohort was 52.6% at a mean follow-up of 4.25 years, whereas the rate was just 20% in the remaining 30 patients who underwent complete resection. In another study, Turner and associates analyzed the outcome data of 73 patients treated by surgery only and observed for a mean of 6.3 years.126 In this study, 5- and 10-year progression-free survival rates were found to be 82% and 56%, respectively. The authors thereby concluded that the regrowth rate of NFAs approaches 50% at 10 years when treated by surgery alone. However, not all published data support this conclusion. In a 2006 study, Dekkers and coworkers found only a 10% recurrence rate in 97 patients treated surgically at a mean follow-up time of 6 years.130 It should be noted that 10 patients were lost to follow-up in this study and could not be included in the analysis.
The majority of studies investigating the role of RT for the management of NFAs were completed before the year 2000; more recent emphasis is on radiosurgical management. In 1993, Jaffrain-Rea and coauthors reported on 24 patients treated with definitive RT between 1970 and 1988 and monitored for a median period of 7.1 years; they achieved a control rate of 92%, but the dose fractionation schemes varied widely.131 Similarly, Tsang and colleagues evaluated a large cohort of 160 patients treated with a median total dose of 45 Gy and found a control rate of 87% in patients who were observed for at least 10 years after treatment.132 Gittoes and coworkers monitored 126 patients for a median of 10 years after conventional RT (45 Gy in 30 to 33 fractions) and demonstrated a 10- and 15-year progression-free survival rate of 93%, thus confirming the durability of response after RT.127
The largest study of conventional RT for the treatment of NFAs was reported by Brada and coauthors in 1993.133 In this study, 252 patients with NFAs underwent postoperative RT to a total dose of 45 to 50 Gy in 25 to 30 fractions via a conventional three-field technique (anterior superior oblique plus right and left opposing portals). After a median follow-up of 10.5 years, 10- and 20-year progression-free survival rates were 97% and 92%, respectively. Treatment-related optic nerve or chiasm injury was seen in 1.5% of the treated patients. More recently, Sasaki and coauthors reported a 98% control rate 10 years after treatment in 81 patients treated with postoperative RT and in 5 treated with primary RT.87
Intracranial Schwannomas
Vestibular schwannomas (acoustic neuromas) account for 6% to 8% of all intracranial tumors and are the most common tumors of the cerebellopontine angle.134 Schwannomas arise from the Schwann cells of various cranial nerves, most commonly cranial nerve VIII. There are currently four management options available, with combinations of two or more being used as required: observation, microsurgery (with neurophysiologic monitoring), SRS, and fractionated RT. Surgical management of vestibular schwannoma and radiosurgery of benign tumors are considered separately in Chapter 130 and Chapter 256, respectively.
Tumor control is the principal aim of the management of these tumors, but because of the benign nature of most of these tumors, long follow-up periods are required to determine this outcome. Conservative management of these tumors relies entirely on their protracted natural history. Studies of patients managed conservatively demonstrate wide-ranging growth rates (between 0 and 30 mm/yr), but with a median growth rate ranging from 0.35 to 2.2 mm/yr (mean, 1.42 mm/yr).135–140 There is also a subset of tumors that do not demonstrate growth on follow-up (18% to 86%; mean, 43%), and there are also some instances of spontaneous tumor regression (<5% of cases).136–138,140–143 Traditionally, conservatively managed patients are elderly and those with medical comorbid conditions that preclude surgery, and therefore the natural history data may not be reflective of the tumor population as a whole. Conservatively managed patients—and probably even those who undergo nonsurgical management—require lifelong follow-up.
Fractionated RT has traditionally been used as an adjunct to surgery after incomplete excision of tumors, but recently, several papers have reported on its virtues as primary treatment.144–146 In one such series, all tumors were treated with 6-MV photons to a total dose of 54 Gy in 30 fractions prescribed to the 90% isodose line; the authors reported a small, but progressive increase in size (<2 mm in any dimension) in 13% of tumors treated after a median follow-up period of 36 months (range, 6 to 74 months).146 They defined local relapse as a sustained increase of greater than 2 mm in any tumor dimension and reported a local tumor control rate of 100% in 48 patients during the follow-up period. Other papers have claimed tumor control rates between 85% and 97%.144,145,147,148 The variance in results seems to largely be related to the length of follow-up and varied definitions of tumor control. Instances of tumor progression can usually be detected within 3 years of treatment, but there are reports of progression at later time points after RT.149 Table 251-5 summarizes the outcomes of selected, modern fractionated RT series.
TABLE 251-5 Summary of Studies on Conventional (Fractionated) Radiotherapy for Vestibular Schwannoma
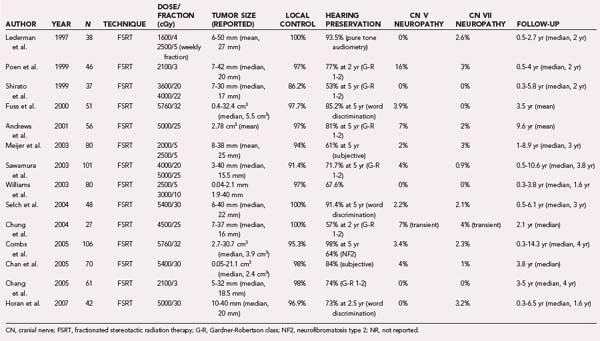
Facial nerve preservation is an important goal in the management of vestibular schwannomas. Facial paresis is disfiguring and associated with significant social and psychological trauma. It is unusual for vestibular schwannoma to primarily be manifested as weakness; this is more common in the setting of previous surgery or NF2, where the likelihood of facial nerve schwannoma is higher. Facial function is also related to the size of the tumor; as the brainstem distortion increases, there is concomitant stretching of the facial nerve, which gives rise to potentially impaired function. Hence, control of tumor growth, by whatever treatment method, has the potential to preserve facial function. A good outcome is widely accepted to be House-Brackmann grade I or II facial function.150 The largest surgical series with the best reported facial nerve outcomes consistently report more than 95% of patients with small tumors retaining grade I or II function.151–154 However, it is also known (and accepted) that such figures are not reproducible in patients with larger tumors or in low-volume centers where surgeon experience is limited. The risk for permanent injury with fractionated RT (and low-dose radiosurgery) varies between 0% and 4% (see Table 251-5),145,146,148 although transient facial nerve injury has a much higher reported rate.149,155 It should be noted that when it occurs, the onset of facial weakness is usually delayed for several months after treatment with either fractionated RT or radiosurgery.156,157
Given that similar tumor control rates and facial nerve function may be achieved with primary surgery or RT, new discriminators for choosing treatment options have been sought. Hearing preservation became such a discriminator and is an additional metric that defines successful treatment. The early experience in the management of vestibular schwannomas did not consider hearing preservation as a treatment objective largely because of preexisting poor hearing in most patients. With earlier access to MRI for the diagnosis of unilateral sensorineural hearing loss, early diagnosis of small tumors became more commonplace. However, analysis of which technique is the best in preserving hearing has been mired in controversy regarding what constitutes good or acceptable hearing.158 Currently, the most widely used scale for assessing useful hearing is the Gardner-Robertson scale,159 which grades hearing into five classes according to pure tone averages and speech discrimination scores; useful or serviceable hearing is considered to be Gardner-Robertson class I or II.
Frequently, patients in whom small vestibular schwannomas are diagnosed but who retain useful hearing have been managed conservatively on the assumption that hearing will be preserved if the tumor remains static. However, there is no direct correlation between the rate of tumor growth and risk for hearing loss even though such a correlation has been suggested,160 and preserved hearing can occur even with large tumors. The converse is also true, with well-described hearing loss in patients with radiographically stable tumors.161,162 After hearing preservation surgery, rates of preserved function vary between 24% and 83%,152,153,163–170 but different series are not directly comparable because of differences in tumor size and the definition of preserved functional hearing. These rates fall dramatically with increasing tumor size,164,171 but overall, a hearing preservation rate of 50% or greater can be expected with small tumors (<2 cm). It is also worth noting that hearing preservation is frequently excellent in the early postoperative period but that delayed hearing loss unrelated to tumor recurrence has been reported increasingly with prolonged follow-up.163,170,172 Subject to these same limitations of nonstandardized data, serviceable hearing after radiosurgical treatment has been reported in 33% to 79% of patients (overall hearing preservation rate of 57%).173 However, in sum, the available data suggest that fractionated RT poses an even smaller risk to hearing. Most studies have reported very high rates of hearing preservation, 53% to 98% (see Table 251-5).144–146
Several other cranial nerves are also at risk as a result of the natural history of the tumor or therapeutic intervention. The most common of these neuropathies involves the trigeminal nerve and, less frequently, the glossopharyngeal/vagal nerve complex. The trigeminal complex gives rise to symptoms in about 10% of patients as part of the natural history of the tumor, and the extent as well as the severity of symptoms is largely a function of tumor size.174 Lower cranial nerve involvement is described in only 3% of patients.174 Of the cranial nerves not entering the internal auditory meatus, radiosurgery and fractionated RT place the trigeminal nerve at the greatest risk for injury. Recent radiosurgical results suggest an injury rate of 2% to 3%.175,176 Similar risk is observed with fractionated RT, with a reported incidence of 2% to 7%.144–146148 Lower cranial nerve dysfunction is very unusual with modern treatment techniques.
Other non–cranial nerve complications that accompany surgery, radiosurgery, or fractionated RT are largely unique to each technique and therefore not easily comparable. The principal complication of fractionated RT is edema within the adjacent brain (or brainstem) tissue, tinnitus, disturbance in balance, and hydrocephalus. When compared with radiosurgery, fractionated RT results in minimal brain edema, and in fact, it is the treatment modality of choice for inoperable patients with large tumors and a significant amount of pretreatment, tumor-related edema. Hydrocephalus was entirely absent in one reported series,146 whereas it was reported in 11% of patients in another.149 Transient or exacerbated tinnitus is not uncommon, but permanent deterioration was seen in less than 4% of patients in a recent series.146
Craniopharyngioma
Craniopharyngioma is a rare benign neoplasm that can arise from vestiges of the stomodeal diverticulum, but it most commonly originates in the region of the infundibulum (remnants of Rathke’s pouch), where squamous epithelial rests are known to occur.177–180 Overall, craniopharyngiomas account for approximately 3% of all intracranial tumors, but the proportion is higher in the pediatric population, where they make up 10% of all pediatric brain tumors.177,181 The estimated incidence is 0.13 per 100,000 cases per year.182 Historically, the lesions were thought to have a bimodal distribution, with peak ages of 5 to 14 years and then 50 to 74 years; however, these lesions are known to occur in patients of all ages.182 Clinically, such patients frequently complain of headache, visual loss, or problems related to hypopituitarism.178 In children, delayed growth and sexual maturation, obesity, and hydrocephalus are also commonly observed.183 The chronic obesity in these patients is probably related to hypothalamic dysfunction.183 Memory loss and other neurocognitive findings are more common in older patients.184 Diabetes insipidus is seen on initial evaluation in 6% to 38% of new patients.185 Radiographically, these tumors are frequently described as calcified, solid or cystic lesions that variably involve the suprasellar space, with 40% to 53% of patients exhibiting intrasellar involvement.181,186 Extension into the anterior, middle, or posterior cranial fossa has been reported, along with invasion of the floor or walls of the third ventricle.187,188 Hydrocephalus occurs in up to 38% of patients and is a more common finding in children.181
The aim of therapy for craniopharyngioma is local control and amelioration of tumor-related sequelae. Surgery is the best initial treatment option but may be limited in the region of the skull base or hypothalamus, where its proximity to critical structures precludes complete resection with acceptable morbidity. Aggressive surgery is more likely to achieve complete tumor removal but frequently at the cost of high treatment mortality and morbidity.189 In contrast, conservative surgery with incomplete tumor removal is associated with a high recurrence rate,188,190–192 which itself is associated with high mortality and morbidity.187 Complete surgical resection (GTR) is achieved in 45% to 90% of patients,193 with a reported 5-year tumor control rate of 70% to 90% in recent adult and pediatric series.187,188,194 The extent of resection correlates with local tumor control and subsequent mortality and morbidity. For example, a recurrence rate of 10% has been reported after GTR (via a transsphenoidal technique),187,195–197 but the recurrence rate increases dramatically (up to 85%) if the craniopharyngioma is incompletely removed.187,188,191,198 The reported surgical mortality rate is 0% to 4%,187,188,191,199 but it increases dramatically to 10% to 17% when aggressive surgery is performed in an attempt at total tumor removal or in patients with tumor recurrence.189 The reported surgical morbidity rate is 20% to 80% and appears to be higher in pediatric patients.181,185,187,188,200 Pituitary or hypothalamic dysfunction that affects one or more hormones, as well as visual and neurocognitive deterioration, has been reported in association with craniopharyngiomas and their management, whether it be surgical or radiotherapeutic.184,185,196,201,202
Several retrospective studies have evaluated the local control and toxicity profiles of surgery alone and surgery plus RT; selected studies are summarized in Table 251-6. For example, Stripp and coauthors reported on 57 patients treated by surgery alone and 18 treated with the combination of surgery and RT and found a 10-year tumor control rate of 42% after surgery alone and 84% after combined therapy.203 Because RT was such an effective adjuvant therapy, 22 patients from the surgery-only arm underwent treatment with salvage RT at the time of failure; the long-term local control rate in the cohort that received immediate postoperative RT was indistinguishable from that of the patients who underwent RT at the time of recurrence.203 The median RT dose used was 54 Gy (range, 44 to 55.8 Gy), and CT or MRI was used for target definition. Similarly, Hetelekidis and coauthors reported a 31% 10-year progression-free survival rate in children treated by surgery versus 86% in those treated with surgery plus RT.204 Others have reported results similar to those shown in Table 251-6, thus suggesting that tumor control and survival outcomes after limited or conservative surgery and RT are better than those after STR alone and that they are comparable to those achieved after GTR. The durability of RT outcomes has been confirmed in multiple large series with reported 10- and 20-year local control rates of 77% to 89% and 54% to 79%, respectively (see Table 251-6).201,203–210
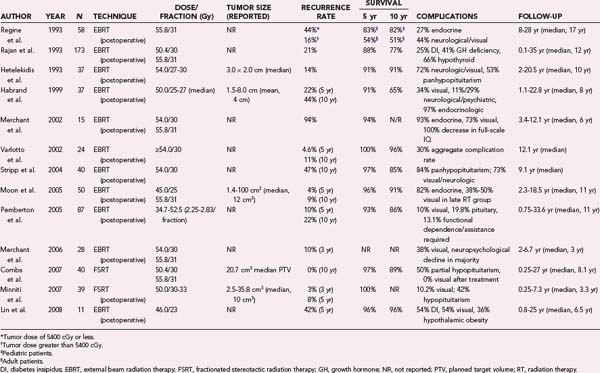
Whether RT should be used in the immediate postoperative period or be reserved for treatment at the time of recurrence is still debated and often depends on the institutional practice pattern. Selected pediatric studies suggest that immediate postoperative RT is preferable because of the reduced morbidity and improved local control in comparison to salvage RT.201,206,209 In adult patients, there does not appear to be a statistically significant difference in either tumor control or survival.207,208,210–212 Because of these studies, immediate RT is generally recommended postoperatively in children after STR, but in adults, it can be given as either adjuvant or salvage therapy.
The optimal RT dose and fractionation for craniopharyngioma have not been established, but retrospective series have used variable doses between 50 and 60 Gy in both adult and pediatric patients (see Table 251-6). In one of the older studies, Regine and associates demonstrated a 44% risk for relapse with doses of less than 54 Gy as compared with just 16% at higher doses,209 but only a small number of patients were used to derive this dosimetric threshold. Similarly, Habrand and coauthors reported that better tumor control was achieved with a dose of 55 Gy or greater than with lesser doses.201 In a series of 24 patients, Varlotto and colleagues noted no failure with a total dose of at least 60 Gy.210 However, several studies have reported 5-year tumor control rates in excess of 90% with conventional fractionated doses of 50 to 54 Gy, thus suggesting that craniopharyngioma may not have a dose-response relationship over the range from 50 to 60 Gy and that the incidence of treatment-related toxicity may be decreased by using doses at the low end of this range.203,205,211–213
Just as with surgery, treatment of craniopharyngioma with fractionated RT is associated with significant acute and late toxicity. During the course of RT or shortly after completion, visual deterioration or hydrocephalus (or both) has been reported in 10% to 15% of patients.208 This is generally due to enlargement of the cystic component of the tumor, although the exact mechanism is unclear. For these reasons, weekly or biweekly imaging during treatment is advisable to ensure that the enlarged cyst remains within the treated volume. Not infrequently, the cystic enlargement resolves with appropriate surgical intervention and does not represent tumor progression. On completion of therapy, the prognosis of patients who experience cystic enlargement during the course of RT remains similar to those without cystic degeneration. It may be necessary to place an Ommaya reservoir or a similar device before RT in patients with large cystic tumors to reduce the treatment volume and to facilitate multiple cyst aspirations; however, such measures may have limited utility with septated, complex cysts.
New pituitary deficits or a worsening of partial hypopituitarism is the most common late complication of fractionated RT and occurs in 20% to 60% of irradiated patients after 5 to 10 years.181,201,203–205,208,209 Pituitary dysfunction is more prevalent in pediatric series and tends to increase with time after treatment.201,204,205 The overall incidence of RT-related optic neuropathy resulting in visual deficits is 2% to 8% (see Table 251-6), but it tends to be lower in FSRT series when highly accurate treatment setup, verification, and delivery techniques are used. The risk for optic nerve injury is related to the total dose and fraction size as described earlier in the section “Meningioma”; optic nerve injury is quite rare with a total dose of up to 54 Gy delivered in 30 fractions.34
The contribution of RT to cognitive decline in this patient population is controversial given the significant baseline deficits observed in these patients.214 Although large-volume RT is recognized to contribute to the developmental problems leading to cognitive impairment,190,215–217 the effect of small-field RT on the brain in adults is not clearly associated with such problems. Most of the published series did not observe significant cognitive decline after RT in adult or pediatric populations, but definitive conclusions should be deferred because comprehensive and formalized neurocognitive testing was performed in only a handful of series.
With the aim of reducing treatment-related morbidity further, new FSRT techniques are increasingly being used for the treatment of these patients.211,218,219 FSRT allows daily delivery of RT with submillimeter precision through the use of an external, stereotactic coordinate system, image-guided verification of patient position, and robotic adjustments. These treatments permit the use of smaller treatment margins and consequently reduce the dose to normal brain tissue.
Chordoma
Chordoma is a rare malignant tumor thought to arise from ectopic rests of embryonal notochord.220 Chordomas may develop in the axial skeleton at any site from the clivus to the sacrum, and they account for 1% to 4% of primary bone tumors.221,222 McMaster and coauthors reported on 400 patients with biopsy-proven chordoma from the Survival, Epidemiology, and End Results (SEER) database and noted an incidence of 0.08 per 100,000, which was higher in men (0.10) than in women (0.06) and very uncommon in African Americans (0.02).220 The age at diagnosis was variable, ranging from 3 to 95 years (median, 58.5 years), but it was rarely less than 40 years.220
Most chordomas (45% to 55%) occur in the sacrococcygeal region, whereas a smaller subset occur at the base of the skull (35%) or the vertebrae (10% to 15%).222 Cranial involvement was more common in young patients (P = .001) and women (P = .037)220; most skull base chordomas arise in the clivus.223 Several investigators have analyzed chordomas in an attempt to identify genetic alterations that may be involved in the pathogenesis of these tumors; the results suggested that several tumor suppressor gene or mismatch repair gene defects may be involved.224–226
Deficits of cranial nerves III and VI, hydrocephalus, and sensorimotor problems are the most common initial symptoms in patients with chordoma.223 Volpe and coauthors reported on 48 patients with skull base chordomas, 52% of whom were initially seen with ocular symptoms such as diplopia, visual impairment as a result of visual field defects, or deterioration of visual acuity.227,228 The diplopia was initially intermittent and associated with cranial nerve VI palsy, but it continued to be progressive in the majority of patients.227 Chordomas tend to be slow growing but locally destructive. Recurrences may be observed several years after treatment, and patients frequently survive for several years after recurrence has been detected. For example, Fagundes and associates reported on 204 patients with chordomas arising in the base of the skull or the cervical spine who were treated between 1975 and 1993; 63 patients (31%) experienced recurrence.229 The reported 3- and 5-year actuarial survival rates after relapse were 43% and 7%, respectively. The majority (73%) of the recurrences were local; 2 patients (3%) experienced nodal failures, 3 patients (5%) had seeding of the surgical track, and distant metastases developed in 13 patients (21%). The most common distant metastatic sites were the lungs and bone. Because the dominant mode of failure is local, complete surgical resection is always the goal; however, the significant surgical morbidity associated with aggressive resection has led to the development of multimodality treatments—conservative surgery to minimize morbidity is usually followed by high-dose, postoperative RT.
Surgical Outcomes
Al-Mefty and Borba reported on 25 patients with skull base chordomas treated between 1990 and 1996; 7 patients had a history of surgical resection and 2 patients had previously reported undergoing RT.230 GTR was achieved in 43% of patients, STR (defined as >90% resection of gross tumor) in 48%, and partial resection (<90% removal of gross tumor) in 9%. Seventeen patients (68%) received immediate, adjuvant photon/proton RT, and 2 patients underwent conventional postoperative photon-only RT. The doses used ranged from 60 to 72 CGE, with a mean dose of 68.8 CGE. At a mean follow-up of 25.4 months, 71% were disease free, 5% (1 patient) died of intercurrent disease, 10% (2 patients) died with disease, and 14% (3 patients) were alive with disease. Gay and coauthors reported on 46 patients with skull base chordoma and 14 patients with chondrosarcoma who underwent surgery between 1984 and 1993 at the University of Pittsburgh, 50% of whom had a history of previous treatment.231 The investigators reported that GTR was achieved in 47%, near-total resection in 20%, STR (≥90% of the tumor removed) in 23%, and partial resection (<90% removal) in 10%. Twelve patients (20%) received postoperative RT. After a median follow-up of 3.9 years (range, 1 to 11 years), 11% of the chordoma patients (5/46) died of the tumor, for a recurrence-free survival rate of 65%. Maira and colleagues reported on 12 patients with clival chordomas who were treated surgically and monitored for a mean period of 40 months (range, 14 to 86 months). Two patients (17%) received postoperative RT, and 8 (67%) had GTR and remained disease free after a mean follow-up of 38 months; local recurrence was reported in 2 of 4 (50%) patients who underwent subtotal or partial resection without RT. Similarly, Thieblemont and associates reported on 8 patients with skull base chordomas treated surgically.232 Two patients in this cohort had GTR; 4 of 8 (50%) received immediate postoperative RT. Four patients were alive with disease at 40 to 59 months of follow-up, 2 patients died with tumor at 13 and 53 months, 1 patient was alive and disease free at 2 months, and 1 patient was alive at 130 months (disease status not reported/unknown).
It is also worth noting that the morbidity associated with skull base surgery, although reduced over time, is not insignificant. For example, Al-Mefty and Borba reported one postoperative death and permanent neurological deficits in two patients, including visual field defects and oculomotor nerve palsy, among those who underwent surgery for skull base chordoma.230 The reported transient postsurgical complications included leakage of cerebrospinal fluid (CSF) with associated meningitis, cranial nerve VII palsy, oronasal fistula, significant epistaxis, and cranial nerve V and VI paresis. In the series by Gay and colleagues, 3 of 46 patients (7%) died within 3 months of the surgical procedure, 80% reported new cranial nerve deficits, 30% experienced CSF leaks, meningitis developed in 10%, and 3% suffered brain infarcts.231 Most new cranial nerve deficits were transient and either improved or resolved, but 40% of patients had permanent deterioration of neurological function after surgery, whereas 20% had improvement.
Radiotherapy Outcomes
The published data after conventional RT are limited, with most data coming from facilities in which patients were treated with protons or charged particles (or both); selected series are summarized in Table 251-7. Historically, proton RT was favored for the treatment of skull base lesions because of improved dose distributions and more advanced treatment planning techniques than were available for conventional (photon) RT, and most published outcomes are from the pre-IMRT era. Fuller and Bloom reported on 13 patients with skull base chordomas treated by conventional RT after STR or biopsy at the Royal Marsden Hospital in London between 1952 and 1981; all patients had follow-up in excess of 5 years.233 The postoperative RT dose ranged from 45 to 65 Gy (median, 55 Gy) delivered in 1.5- to 1.7-Gy daily fractions. Altogether, 9 of 13 patients (69%) died with locally recurrent tumor, 1 patient was alive with recurrent tumor at 49 months, 2 patients (15%) were alive and disease free at 10 and 144 months, and 1 patient was lost to follow-up. The authors found that STR before RT did not significantly affect patient survival.
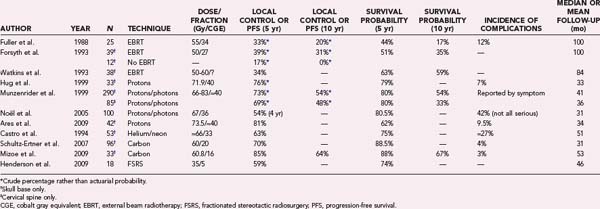
In one of the largest published chordoma series, Terahara and associates reported on 132 patients with skull base chordomas treated by proton/photon RT at the Harvard Cyclotron Laboratory between 1978 and 1993, with a median follow-up of 41 months (range, 5 to 174 months).234 The RT doses ranged from 66.6 to 79.2 CGE (median dose, 68.9 CGE). The reported local control rates at 5 and 10 years were 59% and 44%, respectively. Interestingly, the authors observed that lower local control rates were associated with female gender, lower minimum tumor dose, and greater inhomogeneity. Several authors have confirmed the association between reduced local control and female sex and posited hypotheses for such observations.235–237
Although fractionated RT is an effective adjuvant or definitive treatment of chordomas, it is not without significant treatment-related morbidity. Noel and coauthors reported the following complications after treatment of 65 patients with skull base chordomas or chondrosarcomas: hypopituitarism (25%), memory impairment (2%), oculomotor impairment (3%), profound hearing loss (2%), and bilateral visual loss (2%).223
Chondrosarcoma
Chondrosarcomas are believed to arise from the cartilaginous matrix of the skull base (or from primitive mesenchymal cells) near the petroclival junction or other fused junctions.238 Chondrosarcomas occur less commonly than chordomas and account for 0.15% of all primary intracranial tumors; two studies reported 27 and 37 cartilaginous chondrosarcomas among 16,557 and 24,197 intracranial tumors arising from the skull base, respectively.238,239 Intracranial chondrosarcomas develop most commonly during the fourth and fifth decades240 and do not have a gender predilection.241
Unfortunately, skull base chondrosarcomas (like chordomas) have a propensity for local recurrence, probably because of an inability to achieve complete resection. For example, Korten and coauthors reported a local recurrence rate of 53% in patients treated by surgery alone.241 This was attributed to an inability to remove the entire tumor because of proximity of the lesions to critical neuronal or vascular structures. In another study by Gay and colleagues reporting on the treatment of chordomas (46 patients) and chondrosarcomas (14 patients) of the skull base, the authors found a recurrence-free survival rate of 90% at 5 years in patients with chondrosarcoma (20% of patients received postoperative RT after total or nearly total resection).231 This recurrence-free survival rate compared favorably with that observed in patients with chordoma in the same study (65% at 5 years).
, Andrews DW, Suarez O, Goldman HW, et al. Stereotactic radiosurgery and fractionated stereotactic radiotherapy for the treatment of acoustic schwannomas: comparative observations of 125 patients treated at one institution. Int J Radiat Oncol Biol Phys. 2001;50:1265-1278.
, Boelaert K, Gittoes NJ. Radiotherapy for non-functioning pituitary adenomas. Eur J Endocrinol. 2001;144:569-575.
, Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205.
, Buetow MP, Buetow PC, Smirniotopoulos JG. Typical, atypical, and misleading features in meningioma. Radiographics. 1991;11:1087-1106.
, Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
, Donahue B. Short- and long-term complications of radiation therapy for pediatric brain tumors. Pediatr Neurosurg. 1992;18:207-217.
, Fagundes MA, Hug EB, Liebsch NJ, et al. Radiation therapy for chordomas of the base of skull and cervical spine: patterns of failure and outcome after relapse. Int J Radiat Oncol Biol Phys. 1995;33:579-584.
, Goldsmith BJ, Rosenthal SA, Wara WM, et al. Optic neuropathy after irradiation of meningioma. Radiology. 1992;185:71-76.
, Goldsmith BJ, Wara WM, Wilson CB, et al. Postoperative irradiation for subtotally resected meningiomas. A retrospective analysis of 140 patients treated from 1967 to 1990. J Neurosurg. 1994;80:195-201.
, Grigsby PW, Stokes S, Marks JE, et al. Prognostic factors and results of radiotherapy alone in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 1988;15:1103-1110.
, Halberg FE, Sheline GE. Radiotherapy of pituitary tumors. Endocrinol Metab Clin North Am. 1987;16:667-684.
, Honegger J, Barocka A, Sadri B, et al. Neuropsychological results of craniopharyngioma surgery in adults: a prospective study. Surg Neurol. 1998;50:19-28.
, Howlett TA, Plowman PN, Wass JA, et al. Megavoltage pituitary irradiation in the management of Cushing’s disease and Nelson’s syndrome: long-term follow-up. Clin Endocrinol (Oxf). 1989;31:309-323.
, Jagannathan J, Yen CP, Pouratian N, et al. Stereotactic radiosurgery for pituitary adenomas: a comprehensive review of indications, techniques and long-term results using the Gamma Knife. J Neurooncol. 2009;92:345-356.
, Merchant TE, Kiehna EN, Kun LE, et al. Phase II trial of conformal radiation therapy for pediatric patients with craniopharyngioma and correlation of surgical factors and radiation dosimetry with change in cognitive function. J Neurosurg. 2006;104:94-102.
, Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys. 2002;53:533-542.
, Orth DN, Liddle GW. Results of treatment in 108 patients with Cushing’s syndrome. N Engl J Med. 1971;285:243-247.
, Post KD, Eisenberg MB, Catalano PJ. Hearing preservation in vestibular schwannoma surgery: what factors influence outcome? J Neurosurg. 1995;83:191-196.
, Regis J, Delsanti C, Roche PH, et al. Functional outcomes of radiosurgical treatment of vestibular schwannomas: 1000 successive cases and review of the literature. Neurochirurgie. 2004;50:301-311.
, Rosenberg SI. Natural history of acoustic neuromas. Laryngoscope. 2000;110:497-508.
, Shrieve DC, Hazard L, Boucher K, et al. Dose fractionation in stereotactic radiotherapy for parasellar meningiomas: radiobiological considerations of efficacy and optic nerve tolerance. J Neurosurg. 2004;101(suppl 3):390-395.
, Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
, Stafford SL, Perry A, Suman VJ, et al. Primarily resected meningiomas: outcome and prognostic factors in 581 Mayo Clinic patients, 1978 through 1988. Mayo Clin Proc. 1998;73:936-942.
, Szumacher E, Schwartz ML, Tsao M, et al. Fractionated stereotactic radiotherapy for the treatment of vestibular schwannomas: combined experience of the Toronto-Sunnybrook Regional Cancer Centre and the Princess Margaret Hospital. Int J Radiat Oncol Biol Phys. 2002;53:987-991.
, Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-899.
1 Hoffman S, Propp JM, McCarthy BJ. Temporal trends in incidence of primary brain tumors in the United States, 1985-1999. Neuro Oncol. 2006;8:27-37.
2 Claus EB, Bondy ML, Schildkraut JM, et al. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57:1088-1095.
3 Kuratsu J, Kochi M, Ushio Y. Incidence and clinical features of asymptomatic meningiomas. J Neurosurg. 2000;92:766-770.
4 Kuratsu J, Ushio Y. Epidemiological study of primary intracranial tumors: a regional survey in Kumamoto Prefecture in the southern part of Japan. J Neurosurg. 1996;84:946-950.
5 DiBiase SJ, Kwok Y, Yovino S, et al. Factors predicting local tumor control after gamma knife stereotactic radiosurgery for benign intracranial meningiomas. Int J Radiat Oncol Biol Phys. 2004;60:1515-1519.
6 Flickinger JC, Kondziolka D, Maitz AH, et al. Gamma knife radiosurgery of imaging-diagnosed intracranial meningioma. Int J Radiat Oncol Biol Phys. 2003;56:801-806.
7 Longstreth WTJ, Dennis LK, McGuire VM, et al. Epidemiology of intracranial meningioma. Cancer. 1993;72:639-648.
8 Perry A, Giannini C, Raghavan R, et al. Aggressive phenotypic and genotypic features in pediatric and NF2-associated meningiomas: a clinicopathologic study of 53 cases. J Neuropathol Exp Neurol. 2001;60:994-1003.
9 Bondy M, Ligon BL. Epidemiology and etiology of intracranial meningiomas: a review. J Neurooncol. 1996;29:197-205.
10 Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97-109.
11 Wahab M, Al-Azzawi F. Meningioma and hormonal influences. Climacteric. 2003;6:285-292.
12 Vernooij MW, Ikram MA, Tanghe HL, et al. Incidental findings on brain MRI in the general population. N Engl J Med. 2007;357:1821-1828.
13 Heth JA, Al-Mefty O. Cavernous sinus meningiomas. Neurosurg Focus. 2003;14(6):e3.
14 Commins DL, Atkinson RD, Burnett ME. Review of meningioma histopathology. Neurosurg Focus. 2007;23(4):E3.
15 Buetow MP, Buetow PC, Smirniotopoulos JG. Typical, atypical, and misleading features in meningioma. Radiographics. 1991;11:1087-1106.
16 Mirimanoff RO, Dosoretz DE, Linggood RM, et al. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18-24.
17 De Monte F. Current management of meningiomas. Oncology (Williston Park). 1995;9:83-91. 96
18 Simpson D. The recurrence of intracranial meningiomas after surgical treatment. J Neurol Neurosurg Psychiatry. 1957;20:22-39.
19 Soyuer S, Chang EL, Selek U, et al. Radiotherapy after surgery for benign cerebral meningioma. Radiother Oncol. 2004;71:85-90.
20 Stafford SL, Perry A, Suman VJ, et al. Primarily resected meningiomas: outcome and prognostic factors in 581 Mayo Clinic patients, 1978 through 1988. Mayo Clin Proc. 1998;73:936-942.
21 Condra KS, Buatti JM, Mendenhall WM, et al. Benign meningiomas: primary treatment selection affects survival. Int J Radiat Oncol Biol Phys. 1997;39:427-436.
22 Pollock BE, Stafford SL, Link MJ. Gamma knife radiosurgery for skull base meningiomas. Neurosurg Clin N Am. 2000;11:659-666.
23 King DL, Chang CH, Pool JL. Radiotherapy in the management of meningiomas. Acta Radiol Ther Phys Biol. 1966;3:26-33.
24 Glaholm J, Bloom HJ, Crow JH. The role of radiotherapy in the management of intracranial meningiomas: the Royal Marsden Hospital experience with 186 patients. Int J Radiat Oncol Biol Phys. 1990;18:755-761.
25 Ron E, Modan B, Boice JDJ, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988;319:1033-1039.
26 Strojan P, Popovic M, Jereb B. Secondary intracranial meningiomas after high-dose cranial irradiation: report of five cases and review of the literature. Int J Radiat Oncol Biol Phys. 2000;48:65-73.
27 McCarthy BJ, Davis FG, Freels S, et al. Factors associated with survival in patients with meningioma. J Neurosurg. 1998;88:831-839.
28 Hamm K, Henzel M, Gross MW, et al. Radiosurgery/stereotactic radiotherapy in the therapeutical concept for skull base meningiomas. Zentralbl Neurochir. 2008;69:14-21.
29 Korah MP, Nowlan AW, Johnstone PA, et al. Radiation therapy alone for imaging-defined meningiomas. Int J Radiat Oncol Biol Phys. 2010;76:181-186.
30 Litre CF, Colin P, Noudel R, et al. Fractionated stereotactic radiotherapy treatment of cavernous sinus meningiomas: a study of 100 cases. Int J Radiat Oncol Biol Phys. 2009;74:1012-1017.
31 Metellus P, Batra S, Karkar S, et al. Fractionated conformal radiotherapy in the management of cavernous sinus meningiomas: long-term functional outcome and tumor control at a single institution. Int J Radiat Oncol Biol Phys. 2010:836-843.
32 Goldsmith BJ, Wara WM, Wilson CB, et al. Postoperative irradiation for subtotally resected meningiomas. A retrospective analysis of 140 patients treated from 1967 to 1990. J Neurosurg. 1994;80:195-201.
33 Debus J, Wuendrich M, Pirzkall A, et al. High efficacy of fractionated stereotactic radiotherapy of large base-of-skull meningiomas: long-term results. J Clin Oncol. 2001;19:3547-3553.
34 Goldsmith BJ, Rosenthal SA, Wara WM, et al. Optic neuropathy after irradiation of meningioma. Radiology. 1992;185:71-76.
35 Miralbell R, Linggood RM, de la Monte S, et al. The role of radiotherapy in the treatment of subtotally resected benign meningiomas. J Neurooncol. 1992;13:157-164.
36 Pirzkall A, Debus J, Haering P, et al. Intensity modulated radiotherapy (IMRT) for recurrent, residual, or untreated skull-base meningiomas: preliminary clinical experience. Int J Radiat Oncol Biol Phys. 2003;55:362-372.
37 Uy NW, Woo SY, Teh BS, et al. Intensity-modulated radiation therapy (IMRT) for meningioma. Int J Radiat Oncol Biol Phys. 2002;53:1265-1270.
38 Shrieve DC, Hazard L, Boucher K, et al. Dose fractionation in stereotactic radiotherapy for parasellar meningiomas: radiobiological considerations of efficacy and optic nerve tolerance. J Neurosurg. 2004;101(suppl 3):390-395.
39 Selch MT, Ahn E, Laskari A, et al. Stereotactic radiotherapy for treatment of cavernous sinus meningiomas. Int J Radiat Oncol Biol Phys. 2004;59:101-111.
40 Urie MM, Fullerton B, Tatsuzaki H, et al. A dose response analysis of injury to cranial nerves and/or nuclei following proton beam radiation therapy. Int J Radiat Oncol Biol Phys. 1992;23:27-39.
41 Berman D, Miller NR. New concepts in the management of optic nerve sheath meningiomas. Ann Acad Med Singapore. 2006;35:168-174.
42 Liu JK, Forman S, Moorthy CR, et al. Update on treatment modalities for optic nerve sheath meningiomas. Neurosurg Focus. 14(5), 2003.
43 Andrews BT, Wilson CB. Suprasellar meningiomas: the effect of tumor location on postoperative visual outcome. J Neurosurg. 1988;69:523-528.
44 Cristante L. Surgical treatment of meningiomas of the orbit and optic canal: a retrospective study with particular attention to the visual outcome. Acta Neurochir (Wien). 1994;126:27-32.
45 Ito M, Ishizawa A, Miyaoka M, et al. Intraorbital meningiomas. Surgical management and role of radiation therapy. Surg Neurol. 1988;29:448-453.
46 Kennerdell JS, Maroon JC, Malton M, et al. The management of optic nerve sheath meningiomas. Am J Ophthalmol. 1988;106:450-457.
47 Turbin RE, Thompson CR, Kennerdell JS, et al. A long-term visual outcome comparison in patients with optic nerve sheath meningioma managed with observation, surgery, radiotherapy, or surgery and radiotherapy. Ophthalmology. 2002;109:890-899.
48 Narayan S, Cornblath WT, Sandler HM, et al. Preliminary visual outcomes after three-dimensional conformal radiation therapy for optic nerve sheath meningioma. Int J Radiat Oncol Biol Phys. 2003;56:537-543.
49 Hug EB, Devries A, Thornton AF, et al. Management of atypical and malignant meningiomas: role of high-dose, 3D-conformal radiation therapy. J Neurooncol. 2000;48:151-160.
50 Goyal LK, Suh JH, Mohan DS, et al. Local control and overall survival in atypical meningioma: a retrospective study. Int J Radiat Oncol Biol Phys. 2000;46:57-61.
51 Perry A, Stafford SL, Scheithauer BW, et al. Meningioma grading: an analysis of histologic parameters. Am J Surg Pathol. 1997;21:1455-1465.
52 Palma L, Celli P, Franco C, et al. Long-term prognosis for atypical and malignant meningiomas: a study of 71 surgical cases. J Neurosurg. 1997;86:793-800.
53 Aghi MK, Carter BS, Cosgrove GR, et al. Long-term recurrence rates of atypical meningiomas after gross total resection with or without postoperative adjuvant radiation. Neurosurgery. 2009;64:56-60.
54 Taylor BWJ, Marcus RBJ, Friedman WA, et al. The meningioma controversy: postoperative radiation therapy. Int J Radiat Oncol Biol Phys. 1988;15:299-304.
55 Ghostine S, Ghostine MS, Johnson WD. Radiation therapy in the treatment of pituitary tumors. Neurosurg Focus. 2008;24(5):E8.
56 Burrow GN, Wortzman G, Rewcastle NB, et al. Microadenomas of the pituitary and abnormal sellar tomograms in an unselected autopsy series. N Engl J Med. 1981;304:156-158.
57 Kovacs K, Horvath E. Pathology of pituitary tumors. Endocrinol Metab Clin North Am. 1987;16:529-551.
58 Nussey S, Whitehead S. Endocrinology: An Integrated Approach. Informa Healthcare; Oxford 2001.
59 Aaberg TMJ, Kay M, Sternau L. Metastatic tumors to the pituitary. Am J Ophthalmol. 1995;119:779-785.
60 Max MB, Deck MD, Rottenberg DA. Pituitary metastasis: incidence in cancer patients and clinical differentiation from pituitary adenoma. Neurology. 1981;31:998-1002.
61 Rumana M, Kirmani A, Khursheed N, et al. Lymphocytic hypophysitis with normal pituitary function mimicking a pituitary adenoma: a case report and review of literature. Clin Neuropathol. 2010;29:26-31.
62 Hardy J, Vezina JL. Transsphenoidal neurosurgery of intracranial neoplasm. Adv Neurol. 1976;15:261-273.
63 Grigsby PW. Pituitary adenoma. Front Radiat Ther Oncol. 2001;35:48-56.
64 Scheithauer BW, Horvath E, Kovacs K, et al. Plurihormonal pituitary adenomas. Semin Diagn Pathol. 1986;3:69-82.
65 Schlechte JA, Sherman BM, Chapler FK, et al. Long term follow-up of women with surgically treated prolactin-secreting pituitary tumors. J Clin Endocrinol Metab. 1986;62:1296-1301.
66 Colao A, Sarno AD, Cappabianca P, et al. Gender differences in the prevalence, clinical features and response to cabergoline in hyperprolactinemia. Eur J Endocrinol. 2003;148:325-331.
67 Delgrange E, Trouillas J, Maiter D, et al. Sex-related difference in the growth of prolactinomas: a clinical and proliferation marker study. J Clin Endocrinol Metab. 1997;82:2102-2107.
68 Naidich MJ, Russell EJ. Current approaches to imaging of the sellar region and pituitary. Endocrinol Metab Clin North Am. 1999;28:45-79.
69 Colao A, di Sarno A, Pivonello R, et al. Dopamine receptor agonists for treating prolactinomas. Expert Opin Investig Drugs. 2002;11:787-800.
70 Gillam MP, Molitch ME, Lombardi G, et al. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27:485-534.
71 Passos VQ, Souza JJ, Musolino NR, et al. Long-term follow-up of prolactinomas: normoprolactinemia after bromocriptine withdrawal. J Clin Endocrinol Metab. 2002;87:3578-3582.
72 Zarate A, Canales ES, Cano C, et al. Follow-up of patients with prolactinomas after discontinuation of long-term therapy with bromocriptine. Acta Endocrinol (Copenh). 1983;104:139-142.
73 Charpentier G, de Plunkett T, Jedynak P, et al. Surgical treatment of prolactinomas. Short- and long-term results, prognostic factors. Horm Res. 1985;22:222-227.
74 Fahlbusch R, Buchfelder M. Present status of neurosurgery in the treatment of prolactinomas. Neurosurg Rev. 1985;8:195-205.
75 Randall RV, Laws ERJ, Abboud CF, et al. Transsphenoidal microsurgical treatment of prolactin-producing pituitary adenomas. Results in 100 patients. Mayo Clin Proc. 1983;58:108-121.
76 Halberg FE, Sheline GE. Radiotherapy of pituitary tumors. Endocrinol Metab Clin North Am. 1987;16:667-684.
77 Rodman EF, Molitch ME, Post KD, et al. Long-term follow-up of transsphenoidal selective adenomectomy for prolactinoma. JAMA. 1984;252:921-924.
78 Grigsby PW, Stokes S, Marks JE, et al. Prognostic factors and results of radiotherapy alone in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 1988;15:1103-1110.
79 Rush SC, Newall J. Pituitary adenoma: the efficacy of radiotherapy as the sole treatment. Int J Radiat Oncol Biol Phys. 1989;17:165-169.
80 Tsang RW, Brierley JD, Panzarella T, et al. Role of radiation therapy in clinical hormonally-active pituitary adenomas. Radiother Oncol. 1996;41:45-53.
81 Jalali R, Brada M, Perks JR, et al. Stereotactic conformal radiotherapy for pituitary adenomas: technique and preliminary experience. Clin Endocrinol (Oxf). 2000;52:695-702.
82 Minniti G, Traish D, Ashley S, et al. Fractionated stereotactic conformal radiotherapy for secreting and nonsecreting pituitary adenomas. Clin Endocrinol (Oxf). 2006;64:542-548.
83 Littley MD, Shalet SM, Beardwell CG, et al. Hypopituitarism following external radiotherapy for pituitary tumours in adults. QJM. 1989;70:145-160.
84 Tsagarakis S, Grossman A, Plowman PN, et al. Megavoltage pituitary irradiation in the management of prolactinomas: long-term follow-up. Clin Endocrinol (Oxf). 1991;34:399-406.
85 Grossman A, Cohen BL, Charlesworth M, et al. Treatment of prolactinomas with megavoltage radiotherapy. Br Med J (Clin Res Ed). 1984;288:1105-1109.
86 Grigsby PW, Simpson JR, Emami BN, et al. Prognostic factors and results of surgery and postoperative irradiation in the management of pituitary adenomas. Int J Radiat Oncol Biol Phys. 1989;16:1411-1417.
87 Sasaki R, Murakami M, Okamoto Y, et al. The efficacy of conventional radiation therapy in the management of pituitary adenoma. Int J Radiat Oncol Biol Phys. 2000;47:1337-1345.
88 Sheline GE. Radiation therapy of pituitary tumors. In: Hormone-Secreting Pituitary Tumors: Based on the Proceedings of the Sixth Annual Symposium on Gynecologic Endocrinology, Held March 5-7, 1981 at the University of Tennessee, Memphis, 1982;121.
89 Carpenter PC. Diagnostic evaluation of Cushing’s syndrome. Endocrinol Metab Clin North Am. 1988;17:445-472.
90 Welsh JS, Wharam MD. Pituitary adenoma: a review with an emphasis on radiotherapeutic management. Neurosurgery Q. 1999;9:163-196.
91 Gross BA, Mindea SA, Pick AJ, et al. Diagnostic approach to Cushing disease. Neurosurg Focus. 2007;23(3):E1.
92 Weiss MH, Couldwell WT. Gamma Knife surgery for Cushing disease. J Neurosurg. 2007;106:976-977.
93 Mampalam TJ, Tyrrell JB, Wilson CB. Transsphenoidal microsurgery for Cushing disease. A report of 216 cases. Ann Intern Med. 1988;109:487-493.
94 Tindall GT, Herring CJ, Clark RV, et al. Cushing’s disease: results of transsphenoidal microsurgery with emphasis on surgical failures. J Neurosurg. 1990;72:363-369.
95 Chen JC, Amar AP, Choi S, et al. Transsphenoidal microsurgical treatment of Cushing disease: postoperative assessment of surgical efficacy by application of an overnight low-dose dexamethasone suppression test. J Neurosurg. 2003;98:967-973.
96 Flitsch J, Knappe UJ, Ludecke DK. The use of postoperative ACTH levels as a marker for successful transsphenoidal microsurgery in Cushing’s disease. Zentralbl Neurochir. 2003;64:6-11.
97 Yap LB, Turner HE, Adams CB, et al. Undetectable postoperative cortisol does not always predict long-term remission in Cushing’s disease: a single centre audit. Clin Endocrinol (Oxf). 2002;56:25-31.
98 Cavagnini F, Pecori Giraldi F. Epidemiology and follow-up of Cushing’s disease. Ann Endocrinol (Paris). 2001;62:168-172.
99 Orth DN, Liddle GW. Results of treatment in 108 patients with Cushing’s syndrome. N Engl J Med. 1971;285:243-247.
100 Hughes MN, Llamas KJ, Yelland ME, et al. Pituitary adenomas: long-term results for radiotherapy alone and post-operative radiotherapy. Int J Radiat Oncol Biol Phys. 1993;27:1035-1043.
101 Estrada J, Boronat M, Mielgo M, et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:172-177.
102 Minniti G, Osti M, Jaffrain-Rea ML, et al. Long-term follow-up results of postoperative radiation therapy for Cushing’s disease. J Neurooncol. 2007;84:79-84.
103 Littley MD, Shalet SM, Beardwell CG, et al. Long-term follow-up of low-dose external pituitary irradiation for Cushing’s disease. Clin Endocrinol (Oxf). 1990;33:445-455.
104 Nelson DH, Meakin JW, Thorn GW. ACTH-producing pituitary tumors following adrenalectomy for Cushing’s syndrome. Ann Intern Med. 1960;52:560-569.
105 Minniti G, Brada M. Radiotherapy and radiosurgery for Cushing’s disease. Arq Bras Endocrinol Metabol. 2007;51:1373-1380.
106 Howlett TA, Plowman PN, Wass JA, et al. Megavoltage pituitary irradiation in the management of Cushing’s disease and Nelson’s syndrome: long-term follow-up. Clin Endocrinol (Oxf). 1989;31:309-323.
107 Jenkins PJ, Trainer PJ, Plowman PN, et al. The long-term outcome after adrenalectomy and prophylactic pituitary radiotherapy in adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1995;80:165-171.
108 Lely AJvd. Acromegaly: Pathology, Diagnosis, and Treatment. Boca Raton, FL: Taylor & Francis; 2005.
109 Orme SM, McNally RJ, Cartwright RA, et al. Mortality and cancer incidence in acromegaly: a retrospective cohort study. United Kingdom Acromegaly Study Group. J Clin Endocrinol Metab. 1998;83:2730-2734.
110 Rokkas T, Pistiolas D, Sechopoulos P, et al. Risk of colorectal neoplasm in patients with acromegaly: a meta-analysis. World J Gastroenterol. 2008;14:3484-3489.
111 Wright AD, Hill DM, Lowy C, et al. Mortality in acromegaly. QJM. 1970;39:1-16.
112 Chanson P, Salenave S, Kamenicky P, et al. Pituitary tumours: acromegaly. Best Pract Res Clin Endocrinol Metab. 2009;23:555-574.
113 Cozzi R, Baldelli R, Colao A, et al. AME position statement on clinical management of acromegaly. J Endocrinol Invest. 2009;32:2-25.
114 Melmed S, Casanueva FF, Cavagnini F, et al. Guidelines for acromegaly management. J Clin Endocrinol Metab. 2002;87:4054-4058.
115 Newman CB, Melmed S, Snyder PJ, et al. Safety and efficacy of long-term octreotide therapy of acromegaly: results of a multicenter trial in 103 patients—a clinical research center study. J Clin Endocrinol Metab. 1995;80:2768-2775.
116 Jaffe CA, Barkan AL. Treatment of acromegaly with dopamine agonists. Endocrinol Metab Clin North Am. 1992;21:713-735.
117 Laws ER, Vance ML, Thapar K. Pituitary surgery for the management of acromegaly. Horm Res. 2000;53(suppl 3):71-75.
118 Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical “cure.”. Eur J Endocrinol. 2005;152:379-387.
119 Ciric I, Ragin A, Baumgartner C, et al. Complications of transsphenoidal surgery: results of a national survey, review of the literature, and personal experience. Neurosurgery. 1997;40:225-236.
120 Jenkins PJ, Bates P, Carson MN, et al. Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J Clin Endocrinol Metab. 2006;91:1239-1245.
121 Barrande G, Pittino-Lungo M, Coste J, et al. Hormonal and metabolic effects of radiotherapy in acromegaly: long-term results in 128 patients followed in a single center. J Clin Endocrinol Metab. 2000;85:3779-3785.
122 Jagannathan J, Yen CP, Pouratian N, et al. Stereotactic radiosurgery for pituitary adenomas: a comprehensive review of indications, techniques and long-term results using the Gamma Knife. J Neurooncol. 2009;92:345-356.
123 Ferrante E, Ferraroni M, Castrignano T, et al. Non-functioning pituitary adenoma database: a useful resource to improve the clinical management of pituitary tumors. Eur J Endocrinol. 2006;155:823-829.
124 Snyder PJ. Clinically nonfunctioning pituitary adenomas. Endocrinol Metab Clin North Am. 1993;22:163-175.
125 Boelaert K, Gittoes NJ. Radiotherapy for non-functioning pituitary adenomas. Eur J Endocrinol. 2001;144:569-575.
126 Turner HE, Stratton IM, Byrne JV, et al. Audit of selected patients with nonfunctioning pituitary adenomas treated without irradiation—a follow-up study. Clin Endocrinol (Oxf). 1999;51:281-284.
127 Gittoes NJ, Bates AS, Tse W, et al. Radiotherapy for non-functioning pituitary tumours. Clin Endocrinol (Oxf). 1998;48:331-337.
128 Woollons AC, Hunn MK, Rajapakse YR, et al. Non-functioning pituitary adenomas: indications for postoperative radiotherapy. Clin Endocrinol (Oxf). 2000;53:713-717.
129 Greenman Y, Ouaknine G, Veshchev I, et al. Postoperative surveillance of clinically nonfunctioning pituitary macroadenomas: markers of tumour quiescence and regrowth. Clin Endocrinol (Oxf). 2003;58:763-769.
130 Dekkers OM, Pereira AM, Roelfsema F, et al. Observation alone after transsphenoidal surgery for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab. 2006;91:1796-1801.
131 Jaffrain-Rea ML, Derome P, Bataini JP, et al. Influence of radiotherapy on long-term relapse in clinically non-secreting pituitary adenomas. A retrospective study (1970-1988). Eur J Med. 1993;2:398-403.
132 Tsang RW, Brierley JD, Panzarella T, et al. Radiation therapy for pituitary adenoma: treatment outcome and prognostic factors. Int J Radiat Oncol Biol Phys. 1994;30:557-565.
133 Brada M, Rajan B, Traish D, et al. The long-term efficacy of conservative surgery and radiotherapy in the control of pituitary adenomas. Clin Endocrinol (Oxf). 1993;38:571-578.
134 Lanser MJ, Sussman SA, Frazer K. Epidemiology, pathogenesis, and genetics of acoustic tumors. Otolaryngol Clin North Am. 1992;25:499-520.
135 Bederson JB, von Ammon K, Wichmann WW, et al. Conservative treatment of patients with acoustic tumors. Neurosurgery. 1991;28:646-650.
136 Charabi S, Thomsen J, Mantoni M, et al. Acoustic neuroma (vestibular schwannoma): growth and surgical and nonsurgical consequences of the wait-and-see policy. Otolaryngol Head Neck Surg. 1995;113:5-14.
137 Deen HG, Ebersold MJ, Harner SG, et al. Conservative management of acoustic neuroma: an outcome study. Neurosurgery. 1996;39:260-264.
138 Rosenberg SI. Natural history of acoustic neuromas. Laryngoscope. 2000;110:497-508.
139 Wara WM, Sheline GE, Newman H, et al. Radiation therapy of meningiomas. Am J Roentgenol Radium Ther Nucl Med. 1975;123:453-458.
140 Wiet RJ, Zappia JJ, Hecht CS, et al. Conservative management of patients with small acoustic tumors. Laryngoscope. 1995;105:795-800.
141 Mendenhall WM, Friedman WA, Amdur RJ, et al. Management of acoustic schwannoma. Am J Otolaryngol. 2004;25:38-47.
142 Selesnick SH, Johnson G. Radiologic surveillance of acoustic neuromas. Am J Otol. 1998;19:846-849.
143 Walsh RM, Bath AP, Bance ML, et al. The role of conservative management of vestibular schwannomas. Clin Otolaryngol Allied Sci. 2000;25:28-39.
144 Andrews DW, Suarez O, Goldman HW, et al. Stereotactic radiosurgery and fractionated stereotactic radiotherapy for the treatment of acoustic schwannomas: comparative observations of 125 patients treated at one institution. Int J Radiat Oncol Biol Phys. 2001;50:1265-1278.
145 Fuss M, Debus J, Lohr F, et al. Conventionally fractionated stereotactic radiotherapy (FSRT) for acoustic neuromas. Int J Radiat Oncol Biol Phys. 2000;48:1381-1387.
146 Selch MT, Pedroso A, Lee SP, et al. Stereotactic radiotherapy for the treatment of acoustic neuromas. J Neurosurg. 2004;101(suppl 3):362-372.
147 Maire JP, Caudry M, Guerin J, et al. Fractionated radiation therapy in the treatment of intracranial meningiomas: local control, functional efficacy, and tolerance in 91 patients. Int J Radiat Oncol Biol Phys. 1995;33:315-321.
148 Meijer OW, Vandertop WP, Baayen JC, et al. Single-fraction vs. fractionated LINAC-based stereotactic radiosurgery for vestibular schwannoma: a single-institution study. Int J Radiat Oncol Biol Phys. 2003;56:1390-1396.
149 Sawamura Y, Shirato H, Sakamoto T, et al. Management of vestibular schwannoma by fractionated stereotactic radiotherapy and associated cerebrospinal fluid malabsorption. J Neurosurg. 2003;99:685-692.
150 House JW, Brackmann DE. Facial nerve grading system. Otolaryngol Head Neck Surg. 1985;93:146-147.
151 Briggs RJ, Fabinyi G, Kaye AH. Current management of acoustic neuromas: review of surgical approaches and outcomes. J Clin Neurosci. 2000;7:521-526.
152 Nadol JBJ, Chiong CM, Ojemann RG, et al. Preservation of hearing and facial nerve function in resection of acoustic neuroma. Laryngoscope. 1992;102:1153-1158.
153 Post KD, Eisenberg MB, Catalano PJ. Hearing preservation in vestibular schwannoma surgery: what factors influence outcome? J Neurosurg. 1995;83:191-196.
154 Torres RC, Frighetto L, De Salles AA, et al. Radiosurgery and stereotactic radiotherapy for intracranial meningiomas. Neurosurg Focus. 2003;14(5):e5.
155 Szumacher E, Schwartz ML, Tsao M, et al. Fractionated stereotactic radiotherapy for the treatment of vestibular schwannomas: combined experience of the Toronto-Sunnybrook Regional Cancer Centre and the Princess Margaret Hospital. Int J Radiat Oncol Biol Phys. 2002;53:987-991.
156 Pollock BE, Lunsford LD, Flickinger JC, et al. Vestibular schwannoma management. Part I. Failed microsurgery and the role of delayed stereotactic radiosurgery. J Neurosurg. 1998;89:944-948.
157 Pollock BE, Lunsford LD, Kondziolka D, et al. Vestibular schwannoma management. Part II. Failed radiosurgery and the role of delayed microsurgery. J Neurosurg. 1998;89:949-955.
158 Hardy DG. Acoustic neuroma surgery as an interdisciplinary approach. J Neurol Neurosurg Psychiatry. 2000;69:147-148.
159 Gardner G, Robertson JH. Hearing preservation in unilateral acoustic neuroma surgery. Ann Otol Rhinol Laryngol. 1988;97:55-66.
160 Sughrue ME, Yang I, Aranda D, et al. The natural history of untreated sporadic vestibular schwannomas: a comprehensive review of hearing outcomes. J Neurosurg. 2010;112:163-167.
161 Graamans K, Van Dijk JE, Janssen LW. Hearing deterioration in patients with a non-growing vestibular schwannoma. Acta Otolaryngol. 2003;123:51-54.
162 Walsh RM, Bath AP, Bance ML, et al. Consequences to hearing during the conservative management of vestibular schwannomas. Laryngoscope. 2000;110:250-255.
163 Betchen SA, Walsh J, Post KD. Long-term hearing preservation after surgery for vestibular schwannoma. J Neurosurg. 2005;102:6-9.
164 Gormley WB, Sekhar LN, Wright DC, et al. Acoustic neuromas: results of current surgical management. Neurosurgery. 1997;41:50-58.
165 Mohr G, Sade B, Dufour JJ, et al. Preservation of hearing in patients undergoing microsurgery for vestibular schwannoma: degree of meatal filling. J Neurosurg. 2005;102:1-5.
166 Moriyama T, Fukushima T, Asaoka K, et al. Hearing preservation in acoustic neuroma surgery: importance of adhesion between the cochlear nerve and the tumor. J Neurosurg. 2002;97:337-340.
167 Samii M, Matthies C. Management of 1000 vestibular schwannomas (acoustic neuromas): the facial nerve—preservation and restitution of function. Neurosurgery. 1997;40:684-694.
168 Shelton C, Brackmann DE, House WF, et al. Middle fossa acoustic tumor surgery: results in 106 cases. Laryngoscope. 1989;99:405-408.
169 Tatagiba M, Samii M, Matthies C, et al. The significance for postoperative hearing of preserving the labyrinth in acoustic neurinoma surgery. J Neurosurg. 1992;77:677-684.
170 Umezu H, Aiba T, Tsuchida S, et al. Early and late postoperative hearing preservation in patients with acoustic neuromas. Neurosurgery. 1996;39:267-271.
171 Hecht CS, Honrubia VF, Wiet RJ, et al. Hearing preservation after acoustic neuroma resection with tumor size used as a clinical prognosticator. Laryngoscope. 1997;107:1122-1126.
172 Shelton C, Hitselberger WE, House WF, et al. Hearing preservation after acoustic tumor removal: long-term results. Laryngoscope. 1990;100:115-119.
173 Yang I, Aranda D, Han SJ, et al. Hearing preservation after stereotactic radiosurgery for vestibular schwannoma: a systematic review. J Clin Neurosci. 2009;16:742-747.
174 Matthies C, Samii M. Management of 1000 vestibular schwannomas (acoustic neuromas): clinical presentation. Neurosurgery. 1997;40:1-9.
175 Chopra R, Kondziolka D, Niranjan A, et al. Long-term follow-up of acoustic schwannoma radiosurgery with marginal tumor doses of 12 to 13 Gy. Int J Radiat Oncol Biol Phys. 2007;68:845-851.
176 Regis J, Delsanti C, Roche PH, et al. Functional outcomes of radiosurgical treatment of vestibular schwannomas: 1000 successive cases and review of the literature. Neurochirurgie. 2004;50:301-311.
177 Jane JAJ, Laws ER. Craniopharyngioma. Pituitary. 2006;9:323-326.
178 Karavitaki N, Cudlip S, Adams CB, et al. Craniopharyngiomas. Endocr Rev. 2006;27:371-397.
179 Karavitaki N, Wass JA. Craniopharyngiomas. Endocrinol Metab Clin North Am. 2008;37:173-193. ix-x
180 Prabhu VC, Brown HG. The pathogenesis of craniopharyngiomas. Childs Nerv Syst. 2005;21:622-627.
181 Karavitaki N, Brufani C, Warner JT, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005;62:397-409.
182 Bunin GR, Surawicz TS, Witman PA, et al. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89:547-551.
183 Laws ER. Craniopharyngiomas in children and young adults. Prog Exp Tumor Res. 1987;30:335-340.
184 Honegger J, Barocka A, Sadri B, et al. Neuropsychological results of craniopharyngioma surgery in adults: a prospective study. Surg Neurol. 1998;50:19-28.
185 Honegger J, Buchfelder M, Fahlbusch R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251-257.
186 Sani S, Smith A, Leppla DC, et al. Epidermoid cyst of the sphenoid sinus with extension into the sella turcica presenting as pituitary apoplexy: case report. Surg Neurol. 2005;63:394-397.
187 Fahlbusch R, Honegger J, Paulus W, et al. Surgical treatment of craniopharyngiomas: experience with 168 patients. J Neurosurg. 1999;90:237-250.
188 Van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg. 2002;97:3-11.
189 Yasargil MG, Curcic M, Kis M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73:3-11.
190 Donahue B. Short- and long-term complications of radiation therapy for pediatric brain tumors. Pediatr Neurosurg. 1992;18:207-217.
191 Shi XE, Wu B, Fan T, et al. Craniopharyngioma: surgical experience of 309 cases in China. Clin Neurol Neurosurg. 2008;110:151-159.
192 Symon L, Pell MF, Habib AH. Radical excision of craniopharyngioma by the temporal route: a review of 50 patients. Br J Neurosurg. 1991;5:539-549.
193 Komotar RJ, Roguski M, Bruce JN. Surgical management of craniopharyngiomas. J Neurooncol. 2009;92:283-296.
194 Hoffman HJ. Craniopharyngiomas. The role for resection. Neurosurg Clin N Am. 1990;1:173-180.
195 Abe T, Ludecke DK. Recent results of primary transnasal surgery for infradiaphragmatic craniopharyngioma. Neurosurg Focus. 3(6), 1997.
196 Chakrabarti I, Amar AP, Couldwell W, et al. Long-term neurological, visual, and endocrine outcomes following transnasal resection of craniopharyngioma. J Neurosurg. 2005;102:650-657.
197 Maira G, Anile C, Albanese A, et al. The role of transsphenoidal surgery in the treatment of craniopharyngiomas. J Neurosurg. 2004;100:445-451.
198 Shirane R, Ching-Chan S, Kusaka Y, et al. Surgical outcomes in 31 patients with craniopharyngiomas extending outside the suprasellar cistern: an evaluation of the frontobasal interhemispheric approach. J Neurosurg. 2002;96:704-712.
199 Baskin DS, Wilson CB. Surgical management of craniopharyngiomas. A review of 74 cases. J Neurosurg. 1986;65:22-27.
200 Hoffman HJ, De Silva M, Humphreys RP, et al. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg. 1992;76:47-52.
201 Habrand JL, Ganry O, Couanet D, et al. The role of radiation therapy in the management of craniopharyngioma: a 25-year experience and review of the literature. Int J Radiat Oncol Biol Phys. 1999;44:255-263.
202 Habrand JL, Saran F, Alapetite C, et al. Radiation therapy in the management of craniopharyngioma: current concepts and future developments. J Pediatr Endocrinol Metab. 2006;19(suppl 1):389-394.
203 Stripp DC, Maity A, Janss AJ, et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radiat Oncol Biol Phys. 2004;58:714-720.
204 Hetelekidis S, Barnes PD, Tao ML, et al. 20-year experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys. 1993;27:189-195.
205 Merchant TE, Kiehna EN, Sanford RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys. 2002;53:533-542.
206 Moon SH, Kim IH, Park SW, et al. Early adjuvant radiotherapy toward long-term survival and better quality of life for craniopharyngiomas—a study in single institute. Childs Nerv Syst. 2005;21:799-807.
207 Pemberton LS, Dougal M, Magee B, et al. Experience of external beam radiotherapy given adjuvantly or at relapse following surgery for craniopharyngioma. Radiother Oncol. 2005;77:99-104.
208 Rajan B, Ashley S, Gorman C, et al. Craniopharyngioma—long-term results following limited surgery and radiotherapy. Radiother Oncol. 1993;26:1-10.
209 Regine WF, Mohiuddin M, Kramer S. Long-term results of pediatric and adult craniopharyngiomas treated with combined surgery and radiation. Radiother Oncol. 1993;27:13-21.
210 Varlotto JM, Flickinger JC, Kondziolka D, et al. External beam irradiation of craniopharyngiomas: long-term analysis of tumor control and morbidity. Int J Radiat Oncol Biol Phys. 2002;54:492-499.
211 Combs SE, Thilmann C, Huber PE, et al. Achievement of long-term local control in patients with craniopharyngiomas using high precision stereotactic radiotherapy. Cancer. 2007;109:2308-2314.
212 Minniti G, Saran F, Traish D, et al. Fractionated stereotactic conformal radiotherapy following conservative surgery in the control of craniopharyngiomas. Radiother Oncol. 2007;82:90-95.
213 Merchant TE, Kiehna EN, Kun LE, et al. Phase II trial of conformal radiation therapy for pediatric patients with craniopharyngioma and correlation of surgical factors and radiation dosimetry with change in cognitive function. J Neurosurg. 2006;104:94-102.
214 Waber DP, Pomeroy SL, Chiverton AM, et al. Everyday cognitive function after craniopharyngioma in childhood. Pediatr Neurol. 2006;34:13-19.
215 Crossen JR, Garwood D, Glatstein E, et al. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627-642.
216 Danoff BF, Cowchock FS, Marquette C, et al. Assessment of the long-term effects of primary radiation therapy for brain tumors in children. Cancer. 1982;49:1580-1586.
217 Laack NN, Brown PD. Cognitive sequelae of brain radiation in adults. Semin Oncol. 2004;31:702-713.
218 Kumar S, Burke K, Nalder C, et al. Treatment accuracy of fractionated stereotactic radiotherapy. Radiother Oncol. 2005;74:53-59.
219 Schulz-Ertner D, Frank C, Herfarth KK, et al. Fractionated stereotactic radiotherapy for craniopharyngiomas. Int J Radiat Oncol Biol Phys. 2002;54:1114-1120.
220 McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control. 2001;12:1-11.
221 Sundaresan N, Rosen G, Boriani S. Primary malignant tumors of the spine. Orthop Clin North Am. 2009;40:21-36. v
222 Okazaki H, Scheithauer BW. Atlas of Neuropathology. New York: Gower Medical; 1988.
223 Noel G, Habrand JL, Jauffret E, et al. Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlenther Onkol. 2003;179:241-248.
224 Kelley MJ, Korczak JF, Sheridan E, et al. Familial chordoma, a tumor of notochordal remnants, is linked to chromosome 7q33. Am J Hum Genet. 2001;69:454-460.
225 Riva P, Crosti F, Orzan F, et al. Mapping of candidate region for chordoma development to 1p36.13 by LOH analysis. Int J Cancer. 2003;107:493-497.
226 Scheil S, Bruderlein S, Liehr T, et al. Genome-wide analysis of sixteen chordomas by comparative genomic hybridization and cytogenetics of the first human chordoma cell line, U-CH1. Genes Chromosomes Cancer. 2001;32:203-211.
227 Volpe NJ, Lessell S. Remitting sixth nerve palsy in skull base tumors. Arch Ophthalmol. 1993;111:1391-1395.
228 Volpe NJ, Liebsch NJ, Munzenrider JE, et al. Neuro-ophthalmologic findings in chordoma and chondrosarcoma of the skull base. Am J Ophthalmol. 1993;115:97-104.
229 Fagundes MA, Hug EB, Liebsch NJ, et al. Radiation therapy for chordomas of the base of skull and cervical spine: patterns of failure and outcome after relapse. Int J Radiat Oncol Biol Phys. 1995;33:579-584.
230 al-Mefty O, Borba LA. Skull base chordomas: a management challenge. J Neurosurg. 1997;86:182-189.
231 Gay E, Sekhar LN, Rubinstein E, et al. Chordomas and chondrosarcomas of the cranial base: results and follow-up of 60 patients. Neurosurgery. 1995;36:887-896.
232 Thieblemont C, Biron P, Rocher F, et al. Prognostic factors in chordoma: role of postoperative radiotherapy. Eur J Cancer. 1995;31A:2255-2259.
233 Fuller DB, Bloom JG. Radiotherapy for chordoma. Int J Radiat Oncol Biol Phys. 1988;15:331-339.
234 Terahara A, Niemierko A, Goitein M, et al. Analysis of the relationship between tumor dose inhomogeneity and local control in patients with skull base chordoma. Int J Radiat Oncol Biol Phys. 1999;45:351-358.
235 Halperin EC. Why is female sex an independent predictor of shortened overall survival after proton/photon radiation therapy for skull base chordomas? Int J Radiat Oncol Biol Phys. 1997;38:225-230.
236 O’Connell JX, Renard LG, Liebsch NJ, et al. Base of skull chordoma. A correlative study of histologic and clinical features of 62 cases. Cancer. 1994;74:2261-2267.
237 Catton C, O’Sullivan B, Bell R, et al. Chordoma: long-term follow-up after radical photon irradiation. Radiother Oncol. 1996;41:67-72.
238 Bourgouin PM, Tampieri D, Robitaille Y, et al. Low-grade myxoid chondrosarcoma of the base of the skull: CT, MR, and histopathology. J Comput Assist Tomogr. 1992;16:268-273.
239 Cianfriglia F, Pompili A, Occhipinti E. Intracranial malignant cartilaginous tumours. Report of two cases and review of literature. Acta Neurochir (Wien). 1978;45:163-175.
240 Parker JR, Zarabi MC, Parker JCJ. Intracerebral mesenchymal chondrosarcoma. Ann Clin Lab Sci. 1989;19:401-407.
241 Korten AG, ter Berg HJ, Spincemaille GH, et al. Intracranial chondrosarcoma: review of the literature and report of 15 cases. J Neurol Neurosurg Psychiatry. 1998;65:88-92.






