Chapter 85 Fluid and electrolyte therapy
The management of patients’ fluid and electrolyte status requires an understanding of body fluid compartments as well as an understanding of water and electrolyte metabolism. These principles will be considered along with the commonly encountered fluid and electrolyte disturbances. Recent evidence for the use of fluid therapy in a number of common clinical scenarios will also be presented.
FLUID COMPARTMENTS (Table 85.1, Figure 85.1)
TOTAL BODY WATER
In humans, water contributes approximately 60% of body weight, with organs varying in water content (Table 85.2). The variation of the percentage of total body weight as water, between individuals, is largely governed by the amount of adipose tissue. The average water content as a percentage of total body weight is 60% for males and 50% for females. Total body water as a percentage of total body weight decreases with age, due to a progressive loss of muscle mass, causing bone and connective tissue to assume a greater percentage of total body weight1–3 (Table 85.3).
| Fluid compartment | Volume (ml/kg) | % Total body weight |
|---|---|---|
| Plasma volume | 45 | 4.5 |
| Blood volume | 75 | 7.5 |
| Interstitial volume | 200 | 20 |
| Extracellular fluid volume | 250 | 25 |
| Intracellular fluid volume | 350 | 35 |
| Total body fluid volume | 600 | 60 |
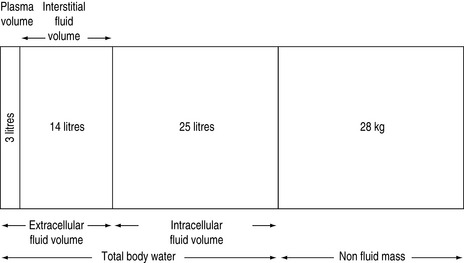
Table 85.2 Water content of various tissues
| Tissue | % Water content |
|---|---|
| Brain | 84 |
| Kidney | 83 |
| Skeletal muscle | 76 |
| Skin | 72 |
| Liver | 68 |
| Bone | 22 |
| Adipose tissue | 10 |
Table 85.3 Water content as a percentage of total body weight
| Age (years) | Males (%) | Females (%) |
|---|---|---|
| 10–15 | 60 | 57 |
| 15–40 | 60 | 50 |
| 40–60 | 55 | 47 |
| > 60 | 50 | 45 |
Total body water is commonly divided into two volumes, the extracellular fluid (ECF) volume and the intracellular fluid (ICF) volume.2 Sodium balance regulates the ECF volume, whereas water balance regulates the ICF volume. Sodium excretion is normally regulated by various hormonal and physical ECF volume sensors, whereas water balance is normally regulated by hypothalamic osmolar sensors.4
WATER METABOLISM
Water balance is maintained by altering the intake and excretion of water. Intake is controlled by thirst, whereas excretion is controlled by the renal action of antidiuretic hormone (ADH). In health, plasma osmolalities of about 280 mOsm/kg suppress plasma ADH to concentrations low enough to permit maximum urinary dilution.5 Above this value, an increase in ECF tonicity of about 1–2% or a decrease in total body water of 1–2 l, causes the posterior pituitary to release ADH, which acts upon the distal nephron to increase water reabsorption. Maximum plasma ADH concentrations are reached at an osmolality of 295 mOsm/kg.5 The osmotic stimulation also changes thirst sensation and, in the conscious ambulant individual, initiates water repletion (drinking), which is more important in preventing dehydration than ADH secretion and action. Thus, in health, the upper limit of the body osmolality (and therefore serum sodium) is determined by the osmotic threshold for thirst, whereas the lower limit is determined by the osmotic threshold for ADH release.6
Increase in osmolality caused by permeant solutes (e.g. urea) does not stimulate ADH release. ADH may also be released in response to hypovolaemia and hypotension, via stimulation of low and high pressure baroreceptors. ADH release is extremely marked when more than 30% of the intravascular volume is lost. ADH may also be stimulated by pain and nausea, which are thought to act through the baroreceptor pathways.4 ADH release may also be stimulated by a variety of pharmacological agents (Table 85.4). Renal response to ADH depends upon an intact distal nephron and collecting duct, and a hypertonic medullary interstitium. The capacity to conserve or excrete water also depends upon the osmolar load presented to the distal nephron.4
| Stimulate | Inhibit |
|---|---|
| Nicotine | Ethanol |
| Narcotics | Narcotic antagonists |
| Vincristine | Phenytoin |
| Barbiturates | |
| Cyclophosphamide | |
| Chlorpropamide | |
| Clofibrate | |
| Carbamazepine | |
| Amitriptylline |
WATER REQUIREMENTS
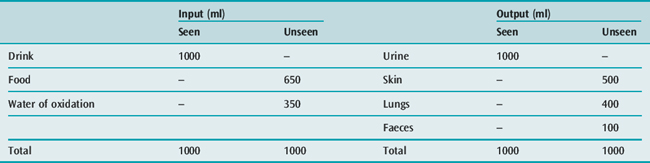
Water is needed to eliminate the daily solute load, and to replace daily insensible fluid loss (Table 85.5). With a normal daily excretion of 600 mOsm solute, maximal and minimal secretions of ADH will cause urine osmolality to vary from 1200 to 30 mOsm/kg respectively, and the urine output to vary from 500 ml to 20 l/day respectively. Skin and lung water losses vary, and may range from 500 ml to 8 l/day depending on physical activity, ambient temperature and humidity.
DISORDERS OF OSMOLALITY
TONICITY
Osmolality is a measure of the number of osmol/kg water. The osmolality of the ECF is due largely to sodium salts. Clinical effects of hyperosmolality, due to excess solute, depend upon whether the solute distributes evenly throughout the total body water (e.g. permeant solutes of alcohol or urea) or distributes in the ECF only (e.g. impermeant solutes of mannitol or glucose). With impermeant solutes, hyperosmolality is associated with a shift of fluid from the ICF to the ECF compartment. Hyperosmolality due to increased impermeant solutes is knownas hypertonicity. This condition may also be associated with a reduction in the serum sodium concentration (see below).
ELECTROLYTES
Chemical compounds in solution may either:
SODIUM
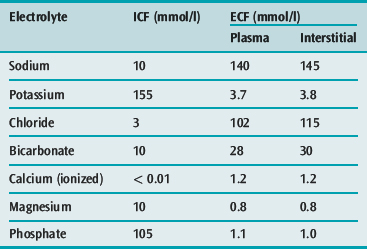
Sodium is the principal cation of the ECF and accounts for 86% of the ECF osmolality. In a 70-kg man, total body sodium content is 4000 mmol (58 mmol/kg) and is divided into a number of compartments (Table 85.7). ECF concentration of sodium varies between 134 and 146 mmol/l. The intracellular sodium concentration varies between different tissues, and ranges from 3 to 20 mmol/l.
| Total (mmol) | (mmol/kg) | |
|---|---|---|
| Total body sodium | 4000 | 58 |
| Non-exchangeable bone sodium | 1200 | 17 |
| Exchangeable sodium | 2800 | 40 |
| Intracellular sodium | 250 | 3 |
| Extracellular sodium | 2400 | 35 |
| Exchangeable bone sodium | 150 | 2 |
The standard Western society dietary sodium intake is about 150 mmol/day, but the daily intake of sodium varies widely, with urinary losses ranging from < 1 to > 240 mmol/day.7 Sodium balance is influenced by renal hormonal and ECF physical characteristics. The complete renal adjustment to an altered sodium load usually requires 3–4 days before balance is restored.
HYPONATRAEMIA
Hyponatraemia is defined as a serum sodium less than 135 mmol/l and may be classified as isotonic, hypertonic or hypotonic, depending upon the measured serum osmolality (Table 85.8).
Isotonic hyponatraemia
Plasma normally contains 93% water and 7% solids (5.5% proteins, 1% salts and 0.5% lipids). If the solid phase is elevated significantly (e.g. in hyperlipidaemia or hyperproteinaemia), any device which dilutes a specific amount of plasma for analysis will give falsely lower values for all measured compounds. This effect produces ‘factitious hyponatraemia’ and is associated with a normal measured serum osmolality.8 Measurement of plasma sodium by an ion-selective electrode is not affected by the volume of plasma ‘solids’ and therefore ‘pseudohyponatraemia’ will not occur with this method.8
Hypertonic hyponatraemia
In patients who have hypertonicity due to increased amounts of impermeant solutes (e.g. glucose, mannitol, glycerol or sorbitol), a shift of water from the ICF to the ECF occurs to provide osmotic equilibration, thus diluting the ECF sodium. Such resultant hyponatraemia is often associated with an increased measured osmolality. For example, in the presence of hyperglycaemia, for every 3 mmol/l rise in glucose, the serum sodium decreases by 1 mmol/l.9
Hypotonic hyponatraemia
Hyponatraemia is almost always caused by an excess of total body water, due to excessive hypotonic or water-generating i.v. fluids (e.g. 1.5% glycine, 0.45% saline or 5% glucose) or excessive ingestion of water, particularly in the presence of high circulating ADH concentrations. It may rarely be caused by loss of exchangeable sodium or potassium. In the latter circumstances, a loss of approximately 40 mmol of sodium or potassium, without a change in total body water content, is required to lower the serum sodium by 1 mmol/l. As hyponatraemia may be associated with an alteration in both total body water and total body sodium, the ECF may be increased (hypervolaemia), decreased (hypovolaemia) or exhibit no change (isovolaemia).5
Transurethral resection of prostate (TURP) syndrome
Clinical features
The TURP syndrome consists of hyponatraemia, cardiovascular disturbances (hypertension, hypotension, bradycardia), an altered state of consciousness (agitation, confusion, nausea, vomiting, myoclonic and generalised seizures) and, when using glycine solutions, transient visual disturbances of blurred vision, blindness and fixed dilated pupils, following TURP. It has also been described following endometrial ablation.10 It may occurwithin 15 minutes or be delayed for up to 24 hours postoperatively,11 and is usually caused by an excess absorption of the irrigating fluid which contains 1.5% glycine with an osmolality of 200 mosm/kg. Hyponatraemic syndromes have also been described when irrigating solutions containing 3% mannitol or 3% sorbitol have been used. Symptomatology usually occurs when > 1 l of 1.5% glycine or > 2–3 l of 3% mannitol or sorbitol are absorbed.12
The excess absorption of irrigating fluid causes an increase in total body water (which is often associated with only a small decrease in plasma osmolality), hyponatraemia (as glycine, sorbitol or mannitol reduces the sodium component of ECF osmolality) and an increase in the osmolar gap.12,13 When glycine is used, other features include hyperglycinaemia (up to 20 mmol/l; normal plasma glycine concentrations range from 0.15 to 0.3 mmol/l), hyperserinaemia (as serine is a major metabolite of glycine), hyperammonaemia (following deamination of glycine and serine), metabolic acidosis and hypocalcaemia (due to the glycine metabolites glyoxylic acid and oxalate). Because glycine is an inhibitory neurotransmitter, and as it passes freely into the intracellular compartment when glycine solutions are used, hyperglycinaemia may be more important in the pathophysiology of this disorder than a reduction in body fluid osmolality and cerebral oedema.14
Treatment
Treatment is largely supportive with the management of any reduction in plasma osmolality being based on the measured plasma osmolality and not the plasma sodium. If the measured osmolality is > 260 mOsm/kg and mild neurological abnormalities exist, and if the patient is haemodynamically stable with normal renal function, close observation and reassurance (e.g. the visual disturbances are reversible and will last for less than 24 hours) are usually all that is needed. If the patient is hypotensive and bradycardic with severe and unresolving neurological abnormalities, haemodialysis may be warranted. Hypertonic saline is only used if the measured osmolality is < 260 mOsm/kg and severe non-visual neurological abnormalities exist.15
Syndrome of inappropriate ADH secretion (SIADH)
This syndrome is a form of hyponatraemia in which there is an increased concentration of ADH inappropriate to any osmotic or volume stimuli that normally affect ADH secretion.16 Diagnostic criteria and common causes are listed in Tables 85.9 and 85.10, respectively.
Table 85.9 Criteria for the diagnosis of syndrome of inappropriate antidiuretic hormone (SIADH)
Table 85.10 Aetiologies of syndrome of inappropriate antidiuretic hormone (SIADH)
Clinical features
While cerebral manifestations are usually absent when the sodium concentration exceeds 125 mmol/l, progressive symptomatology of headache, nausea, confusion, disorientation, coma and seizures are often observed when plasma sodium is below 120 mmol/l.17
Treatment
Initial treatment should be fluid restriction with close monitoring of serum sodium. If serum sodium concentration is not increasing with fluid restriction, judicious administration of sodium as intravenous normal or hypertonic saline may be required. As the true duration and rapidity of onset of hyponatraemia is often unclear, the presence and severity of symptoms may be used as the trigger for active correction of hyponatraemia.17 The evidence base available to guide therapy is limited and there is consequently no consensus on the optimum rate at which to correct the serum sodium concentration. The major concern is to avoid neurological damage from untreated seizures or cerebral oedema and from myelinolysis.17 In the absence of good evidence, recommended rates at which to increase the sodium concentration vary from 0.5 to 2 mmol/l per hour. Unless the treating clinician feels that more rapid correction is indicated, it seems prudent to correct the serum sodium concentration at a slower rather than a faster rate.
Cerebral salt wasting
Cerebral salt wasting (CSW) is a syndrome occurring in patients with a cerebral lesion and an excess renal loss of sodium and chloride.18 The exact aetiology of the syndrome remains unclear. Although hyponatraemia is not necessary for the diagnosis, the syndrome is commonly associated with hyponatraemia.19 The diagnosis of CSW is suspected in patients with a cerebral lesion, such assubarachnoid haemorrhage, traumatic brain injury or a cerebral tumour, when there is an elevated urine output, with elevated urinary sodium in the absence of a physiological cause for increased sodium excretion.19 The syndrome can be differentiated from the SIADH, as patients with CSW will have evidence of ECF depletion (e.g. negative fluid balance, tachycardia, increased haematocrit, increased urea, low central venous pressure) as opposed to the SIADH where ECF volume will be normal or slightly expanded.20 Treatment of patients involves exclusion of other causes of hyponatraemia and increased urine output, replacement of sodium and fluid losses, and possibly fludrocortisone.21
Hypertonic saline
Hypertonic saline, most commonly as 3% saline, is used as a therapy for patients with symptomatic hyponatraemia. Due to the osmolarity (1000 mosm/l) of 3% saline, it must be given through a central line, and care must be taken to avoid the known complications of its use. Complications reported with hypertonic saline therapy include congestive cardiac failure and central pontine and extrapontine myelinolysis (osmotic demyelination syndrome).22,23 Careful haemodynamic and electrolyte monitoring throughout saline administration is required. There is still no uniform agreement that osmotic demyelination is produced by a rapid correction of hyponatraemia.
While hypertonic saline has been proposed as a therapy for raised intracranial pressure in a number of clinical settings,24 its use remains controversial.25 In a recent methodologically sound (adequate allocation concealment, blinded and using intention to treat analysis) randomised clinical trial, which included 229 patients with severe traumatic brain injury, resuscitation with 250 ml of 7.5% saline was not associated with improved mortality or functional outcomes compared to Hartmann’s solution.26
Vasopressin receptor antagonists
V2 receptor antagonists, such as lixivaptan, tolvaptan and OPC-31260, have recently been developed. These agents come from a novel class of non-peptide agents that bind to V2 receptors in the distal tubule of the kidney and prevent vasopressin-mediated aquaporin mobilisation, and thus promote an aquaresis.27 These agents have been trialled as therapeutic options for the treatment of hyponatraemia in a number of clinical settings including hyponatraemia associated with cardiac failure, cirrhosis and SIADH.27,28 At present the role of V2 receptor antagonists in the management of hyponatraemia in the critically ill remains uncertain.
HYPERNATRAEMIA
Hypernatraemia, defined as a serum sodium greater than 145 mmol/l, is always associated with hyperosmolality and may be caused by excessive administration of sodium salts (bicarbonate or chloride), water depletion or excess sodium and loss of water (Table 85.11).
Clinical features
Hypernatraemia usually produces symptoms if the serum sodium exceeds 155–160 mmol/l (i.e. osmolality > 330 mOsm/kg). The clinical features include increased temperature, restlessness, irritability, drowsiness, lethargy, confusion and coma.29 Convulsions are uncommon. The diminished ECF volume may reduce cardiac output, thereby reducing renal perfusion, leading to pre-renal renal failure.
Treatment
For pure water depletion, this consists of water administration. If i.v. fluid is required, 5% glucose or hypotonic saline solutions (0.45% saline) are often used, as sterile water infusion causes haemolysis. In rare cases, i.v. sterile water may be used, by administering through a central venous catheter.30 Since rapid rehydration may give riseto cerebral oedema, the change in serum sodium should be no greater than 0.5 mmol/l per hour.29
POTASSIUM
FACTORS AFFECTING POTASSIUM METABOLISM
Acidosis promotes a shift of potassium from the ICF to the ECF, whereas alkalosis promotes the reverse shift. Hyperkalaemia stimulates insulin release, which promotes a shift of potassium from the ECF to the ICF, an effect independent of the movement of glucose. β2-adrenergic agonists promote cellular uptake of potassium by a cyclic AMP-dependent activation of the Na+/K+ pump, whereas α-adrenergic agonists cause a shift of potassium from the ICF to the ECF.31 Aldosterone increases the renal excretion of potassium; glucocorticoids are also kaliuretic, an effect which may be independent of the mineralocorticoid receptor.
HYPOKALAEMIA
Hypokalaemia is defined as a serum potassium of less than 3.5 mmol/l (or plasma potassium less than 3.0 mmol/l). It may be due to decreased oral intake, increased renal or gastrointestinal loss, or movement of potassium from the ECF to the ICF (Table 85.12).
Clinical features
These include weakness, hypotonicity, depression, constipation, ileus, ventilatory failure, ventricular tachycardias (characteristically torsades de pointes), atrial tachycardias, and even coma.32 With prolonged and severe potassium deficiency, rhabdomyolysis and thirst and polyuria, due to the development of renal diabetes insipidus, may occur. The ECG changes are relatively non-specific, and include prolongation of the PR interval, T-wave inversion and prominent U-waves.
Treatment
Intravenous or oral potassium chloride will correct hypokalaemia, particularly if it is associated with metabolic alkalosis. If the patient has renal tubular acidosis and hypokalaemia, potassium acetate or citrate may be preferred to potassium chloride. Intravenous administration of potassium should normally not exceed 40 mmol/hour, and plasma potassium should be monitored at 1–4-hourlyintervals.33 In patients with acute myocardial infarction and hypokalaemia, some recommend i.v. potassium to maintain the serum potassium at 4.0 mmol/l.34 The plasma concentration should be monitored closely.
HYPERKALAEMIA
Hyperkalaemia is defined as a serum potassium greater than 5.0 mmol/l or plasma potassium greater than 4.5 mmol/l. It may be artifactual (from sampling errors such as in vitro haemolysis); true hyperkalaemia may be due to excessive intake, severe tissue damage, decreased excretion or body fluid compartment shift (Table 85.13).
CALCIUM
Almost all (99%) of the body calcium (30 mmol or 1000 g or 1.5% body weight) is present in bone. A small but significant quantity of ionised calcium exists in the ECF, and is important for many cellular activities, including secretion, neuromuscular impulse formation, contractile functions and clotting. Normal daily intake of calcium is 15–20 mmol, although only 40% is absorbed. The average daily urinary loss is 2.5–7.5 mmol. The total ECF calcium of 40 mmol (2.20–2.55 mmol/l) exists in three forms: 40% (1.0 mmol/l) is bound to protein (largely albumin), 47% is ionised (1.15 mmol/l) and 13% is complexed (0.3 mmol/l) with citrate, sulphate and phosphate. The ionised form is the physiologically important form, and may be acutely reduced in alkalosis which causes a greater amount of the serum calcium to be bound to protein.36 While the serum ionised calcium can be measured, total serum calcium is usually measured, which can vary with the serum albumin concentrations. A correction factor can be used to offset the effect of serum albumin on serum calcium. This is 0.02 mmol/l for every 1 g/l increase in serum albumin (up to a value of 40 g/l), added to the measured calcium concentration. For example, if measured serum calcium is 1.82 mmol/l, and serum albumin is 25 g/l, corrected serum calcium = 1.82 + [(40–25) × 0.02] mmol/l = 2.12 mmol/l. It has been suggested that ionised calcium, where available, is a better indicator of calcium status in the critically ill.37
HYPOCALCAEMIA
Common causes of hypocalcaemia include hypoparathyroidism and pseudohypoparathyroidism, septic shock, acute pancreatitis and rhabdomyolysis.38 Clinical features of a reduced serum ionised calcium include tetany, cramps, mental changes and decrease in cardiac output. Symptomatic hypocalcaemia should be treated with i.v. calcium, either as calcium chloride or calcium gluconate. It should be remembered that 1 ml of calcium chloride has three times as much elemental calcium as 1 ml of calcium gluconate, and so the former is the preferred formulation in acute situations. Calcium should be administered via a central vein when practical due to the risk of tissue damage if extravasated.38
HYPERCALCAEMIA
The clinical features of hypercalcaemia include nausea, vomiting, pancreatitis, polyuria, polydipsia, muscular weakness, mental disturbance and ectopic calcification. Some of the common causes of hypercalcaemia includeendocrine diseases such as hyperparathyroidism and thyrotoxicosis, renal failure, malignancy, thiazide diuretics and prolonged immobilisation.38
Severe hypercalcaemia (> 3.3 mmol/l) or more moderate symptomatic hypercalcaemia will require specific therapy. A cause for the elevated calcium concentration should be sought, and specific treatment may be warranted. General measures include restoration of intravascular volume with normal saline, followed by the use of a loop diuretic such as furosemide to promote calcium excretion. The use of bisphosphonates, such as pamidronate (30–90 mg by i.v. infusion), is recommended for severe cases.39 Other therapies to consider include steroids, calcitonin and mithramycin.
MAGNESIUM
In humans, the total body magnesium content is 1000 mmol, and the plasma concentrations range from 0.70 to 0.95 mmol/l. The daily oral intake is 8–20 mmol (40% of which is absorbed) and the urinary loss, which is the major source of excretion of magnesium, varies from 2.5 to 8 mmol/day.40
HYPOMAGNESAEMIA
Hypomagnesaemia is caused by decreased intake or increased loss (Table 85.14). Clinical features include neurological signs of confusion, irritability, delirium tremors, convulsions and tachyarrhythmias. Hypomagnesaemia is often associated with resistant hypokalaemia and hypocalcaemia. Treatment consists of i.v. magnesium sulphate as a bolus of 10 mmol, administered over 5 minutes, followed by 20–60 mmol/day.
HYPERMAGNESAEMIA
Hypermagnesaemia is often caused by excessive administration of magnesium salts or conventional doses of magnesium in the presence of renal failure. Clinical features include drowsiness, hyporeflexia and coma, vasodilatation and hypotension, and conduction defects of sinoatrial and atrioventricular nodal block and asystole may occur. Treatment is directed towards increasing excretion of the ion, which may require dialysis. Intravenous calcium chloride may be used for rapidly treating the cardiac conduction defects.40
MAGNESIUM THERAPY
There are increasing reports of the use of magnesium as a therapy for a variety of conditions. A randomised control trial of over 10 000 women with pre-eclampsia demonstrated the efficacy of magnesium in the prevention of eclampsia,41 and it is also a recommended treatment for established eclampsia. It has been used to treat atrial fibrillation, to achieve both rate control and reversion to sinus rhythm in a number of settings, including post cardiac surgery, and in the emergency department.42,43 Magnesium, given either intravenously or nebulised, may be beneficial for patients with acute severe asthma.44,45 There are also preliminary trials to suggest that magnesium may prevent delayed cerebral ischaemia due to vasospasm in patients with subarachnoid haemorrhage.46
PHOSPHATE
While most of the body phosphate exists in bone, 15% is found in the soft tissues as ATP, red blood cell 2,3-DPG, and other cellular structural proteins, including phospholipids, nucleic acids and phosphoproteins. Phosphate also acts as a cellular and urinary buffer.40
HYPOPHOSPHATAEMIA
Hypophosphataemia may be caused by a decreased intake, increased excretion or intracellular redistribution (Table 85.15). While hypophosphataemia may besymptom-free, clinical features have been described which include paraesthesia, muscle weakness, seizures, coma, rhabdomyolysis and cardiac failure. Hypophosphataemia may be a prominent feature of the refeeding syndrome when it may be accompanied by other electrolyte disturbances such as hypokalaemia and hypomagnesaemia. Treatment consists of close monitoring and replacement as oral or i.v. sodium or potassium phosphate, 50–100 mmol/24 hours.
HYPERPHOSPHATAEMIA
Hyperphosphataemia is usually caused by an increased intake or decreased excretion (Table 85.16). Clinical features include ectopic calcification of nephrocalcinosis, nephrolithiasis and band keratopathy. Treatment may require haemodialysis; otherwise oral aluminium hydroxide and even hypertonic glucose solutions to shift ECF phosphate into the ICF can be used.
FLUID AND ELECTROLYTE REPLACEMENT THERAPY
GENERAL PRINCIPLES
In critical illness many of the body’s normal homeostatic mechanisms are deranged and basic life-preserving senses such as hunger and thirst may be abolished by disease processes or by treatments such as the use of sedation. As a result, the survival of critically ill patients depends on the administration of appropriate volumes of fluids, and appropriate quantities of electrolytes and nutrition by their medical and nursing attendants. Basal requirements for water, electrolytes and nutrients are discussed in Chapter 87. In addition to basal requirements, many critically ill patients have abnormal fluid and electrolyte losses that must be replaced; examples are discussed below.
GASTROINTESTINAL LOSSES
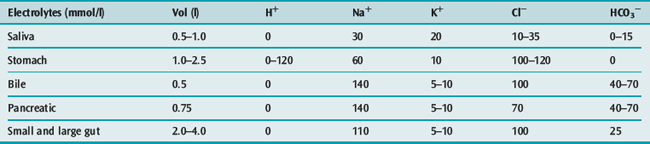
The daily volumes and composition of gastrointestinal tract (GIT) secretions in mmol/l are shown in Table 85.17. Clinical effects of fluid loss from the GIT are largely determined by the volume and composition of the fluid, and therapy is usually directed at replacing the losses. Gastric fluid loss (e.g. from vomiting and nasogastric suction) results in water, sodium, hydrogen ion, potassium and chloride depletion. Hence, metabolic alkalosis, hypokalaemia, hypotension and dehydration develop if the saline and potassium chloride losses are not correctly replaced.
RESUSCITATION FLUIDS
Systemic hypotension is a common feature of acute critical illness and first-line treatment is usually the administration of intravenous resuscitation fluid. The fluids available to clinicians to maintain or expand intravascular volume are crystalloids, colloids and blood products; the properties of colloid solutions and blood products are discussed in Chapter 89.
Whether the choice of resuscitation fluid influences patients’ outcomes has been the subject of long-running debate. The debate has been fuelled by the conflictingand inconclusive results of a number of meta-analyses.47–50 At present, the published data do not provide unequivocal support for either side of the debate. The only adequately powered trial to date, the Saline versus Albumin Fluid Evaluation Study, found that saline and albumin produced comparable outcomes in a heterogeneous population of adult patients.51 Whether specific fluids are beneficial or harmful in more highly selected subpopulations of critically ill patients is not yet clear. A number of investigator-initiated trials are currently underway and are expected to report their results in the next few years. These trials may provide clinicians with the data needed to make rational fluid choices.
1 Edelman IS, Leibman J. Anatomy of body water and electrolytes. Am J Med. 1959;27:25-77.
2 Gamble J. Chemical Anatomy, Physiology and Pathology of Extracellular Fluid. Cambridge, MA: Harvard University Press, 1954.
3 Moore FD, Olesen KH, McMurray JD. Body Composition in Health and Disease. Philadelphia: WB Saunders, 1963.
4 Bie P. Osmoreceptors, vasopressin, and control of renal water excretion. Physiol Rev. 1980;60:961-1048.
5 Humes HD. Disorders of water metabolism. In: Kokko JP, Tannen RL, editors. Fluid and Electrolytes. Philadelphia: WB Saunders; 1986:118-149.
6 Phillips PJ. Water metabolism. Anaesth Intensive Care. 1977;5:295-304.
7 Intersalt Cooperative Research Group. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ. 1988;297:319-328.
8 Weisberg LS. Pseudohyponatremia: a reappraisal. Am J Med. 1989;86:315-318.
9 Katz MA. Hyperglycemia-induced hyponatremia – calculation of expected serum sodium depression. N Engl J Med. 1973;289:843-844.
10 Arieff AI, Ayus JC. Endometrial ablation complicated by fatal hyponatremic encephalopathy. JAMA. 1993;270:1230-1232.
11 Gravenstein D. Transurethral resection of the prostate (TURP) syndrome: a review of the pathophysiology and management. Anesth Analg. 1997;84:438-446.
12 Hahn RG. Fluid and electrolyte dynamics during development of the TURP syndrome. Br J Urol. 1990;66:79-84.
13 Ghanem AN, Ward JP. Osmotic and metabolic sequelae of volumetric overload in relation to the TURP syndrome. Br J Urol. 1990;66:71-78.
14 Jensen V. The TURP syndrome. Can J Anaesth. 1991;38:90-96.
15 Agarwal R, Emmett M. The post-transurethral resection of prostate syndrome: therapeutic proposals. Am J Kidney Dis. 1994;24:108-111.
16 Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42:790-806.
17 Reynolds RM, Padfield PL, Seckl JR. Disorders of sodium balance. BMJ. 2006;332:702-705.
18 Cort JH. Cerebral salt wasting. Lancet. 1954;266:752-754.
19 Singh S, Bohn D, Carlotti AP, et al. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med. 2002;30:2575-2579.
20 Rabinstein AA, Wijdicks EF. Hyponatremia in critically ill neurological patients. Neurologist. 2003;9:290-300.
21 Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke. 1989;20:1156-1161.
22 Sterns RH, Riggs JE, Schochet SSJr. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med. 1986;314:1535-1542.
23 Martin RJ. Central pontine and extrapontine myelinolysis: the osmotic demyelination syndromes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 3):iii. 22–8
24 Bhardwaj A, Ulatowski JA. Hypertonic saline solutions in brain injury. Curr Opin Crit Care. 2004;10:126-131.
25 White H, Cook D, Venkatesh B. The use of hypertonic saline for treating intracranial hypertension after traumatic brain injury. Anesth Analg. 2006;102:1836-1846.
26 Cooper DJ, Myles PS, McDermott FT, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA. 2004;291:1350-1357.
27 Yeates KE, Morton AR. Vasopressin antagonists: role in the management of hyponatremia. Am J Nephrol. 2006;26:348-355.
28 Palm C, Pistrosch F, Herbrig K, et al. Vasopressin antagonists as aquaretic agents for the treatment of hyponatremia. Am J Med. 2006;119(7 Suppl 1):S87-92.
29 Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342:1493-1499.
30 Worthley LI. Hyperosmolar coma treated with intravenous sterile water. A study of three cases. Arch Intern Med. 1986;146:945-947.
31 Sterns RH, Cox M, Feig PU, et al. Internal potassium balance and the control of the plasma potassium concentration. Medicine (Baltimore). 1981;60:339-354.
32 Phelan DM, Worthley LI. Hypokalaemic coma. Intensive Care Med. 1985;11:257-258.
33 Stockigt JR. Potassium metabolism. Anaesth Intensive Care. 1977;5:317-325.
34 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 8: Stabilization of the patient with acute coronary syndromes. Circulation. 2005;112(24 Suppl):IV-89-110.
35 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 10.1: Life-threatening electrolyte abnormalities. Circulation. 2005;112(24 Suppl):IV-121-125.
36 Thomas DW. Calcium, phosphorus and magnesium turnover. Anaesth Intensive Care. 1977;5:361-371.
37 Slomp J, van der Voort PH, Gerritsen RT, et al. Albumin-adjusted calcium is not suitable for diagnosis ofhyper- and hypocalcemia in the critically ill. Crit Care Med. 2003;31:1389-1393.
38 Bushinsky DA, Monk RD. Electrolyte quintet: calcium. Lancet. 1998;352:306-311.
39 Ariyan CE, Sosa JA. Assessment and management of patients with abnormal calcium. Crit Care Med. 2004;32(4 Suppl):S146-154.
40 Weisinger JR, Bellorin-Font E. Magnesium and phosphorus. Lancet. 1998;352:391-396.
41 Altman D, Carroli G, Duley L, et al. Do women with pre-eclampsia, and their babies, benefit from magnesium sulphate? The Magpie Trial: a randomised placebo-controlled trial. Lancet. 2002;359:1877-1890.
42 Davey MJ, Teubner D. A randomized controlled trial of magnesium sulfate, in addition to usual care, for rate control in atrial fibrillation. Ann Emerg Med. 2005;45:347-353.
43 Henyan NN, Gillespie EL, White CM, et al. Impact of intravenous magnesium on post-cardiothoracic surgery atrial fibrillation and length of hospital stay: a meta-analysis. Ann Thorac Surg. 2005;80:2402-2406.
44 Rowe BH, Bretzlaff JA, Bourdon C, et al. Intravenous magnesium sulfate treatment for acute asthma in the emergency department: a systematic review of the literature. Ann Emerg Med. 2000;36:181-190.
45 Hughes R, Goldkorn A, Masoli M, et al. Use of isotonic nebulised magnesium sulphate as an adjuvant to salbutamol in treatment of severe asthma in adults: randomised placebo-controlled trial. Lancet. 2003;361:2114-2117.
46 van den Bergh WM, Algra A, van Kooten F, et al. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: a randomized controlled trial. Stroke. 2005;36:1011-1015.
47 Cochrane Injuries Group Albumin Reviewers. Human albumin administration in critically ill patients: systematic review of randomised controlled trials. BMJ. 1998;317:235-240.
48 Vincent JL, Navickis RJ, Wilkes MM. Morbidity in hospitalized patients receiving human albumin: a meta-analysis of randomized, controlled trials. Crit Care Med. 2004;32:2029-2038.
49 The Albumin Reviewers. Human albumin solution for resuscitation and volume expansion in critically ill patients. Cochrane Database Syst Rev. 2004;4:CD001208.. pub2
50 Roberts I, Alderson P, Bunn F, et al. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev. 2004:CD000567.
51 The SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med. 2004;350:2247-2256.