[level-membership-for-anesthesiology-category]
Factors affecting cerebral blood flow
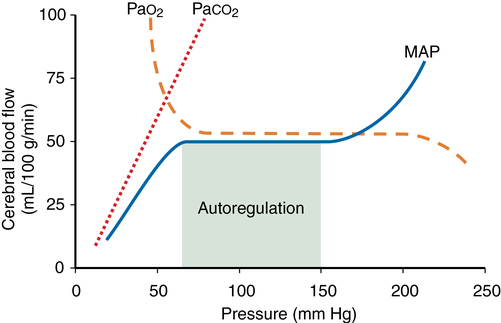
Cerebral metabolic rate (CMR), autoregulation, CO2 reactivity, and O2 reactivity are the main factors affecting cerebral blood flow (CBF). The relationships among the latter three are depicted in Figure 42-1. Temperature and anesthetic medications also each influence CBF.
Autoregulation
Autoregulation is defined as the maintenance of CBF over a range of mean arterial pressure (MAP) (see Figure 42-1). Cerebral vascular resistance (CVR) is adjusted to maintain constant CBF. Cerebral perfusion pressure equals MAP minus intracranial pressure (ICP). Because ICP (and therefore cerebral perfusion pressure) is not commonly available, MAP is used as a surrogate of cerebral perfusion pressure.
Autoregulation occurs when MAP is between 70 and 150 mm Hg in the normal brain (see Figure 42-1). This is a conservative estimate, given that considerable interindividual variation occurs. The lower limit of autoregulation (LLA) is the point at which the autoregulation curve deflects downward and CBF begins to decrease in proportion to MAP.
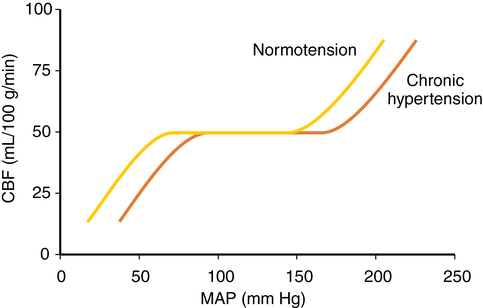
CVR varies directly with blood pressure to maintain flow, taking 1 to 2 min for flow to adjust after an abrupt change in blood pressure. In hypertensive patients, the autoregulatory curve is shifted to the right (Figure 42-2). A hypertensive patient may be at risk for developing brain ischemia at a MAP of 70 mm Hg, for example, because the LLA will be higher than in a nonhypertensive patient. Several weeks of blood pressure control may return the curve to normal. Following significant hypotension (lower than the LLA), autoregulation is impaired, and hyperemia may occur when MAP returns to the normal range. CO2 reactivity remains intact, and inducing hypocapnia may attenuate hyperemia.
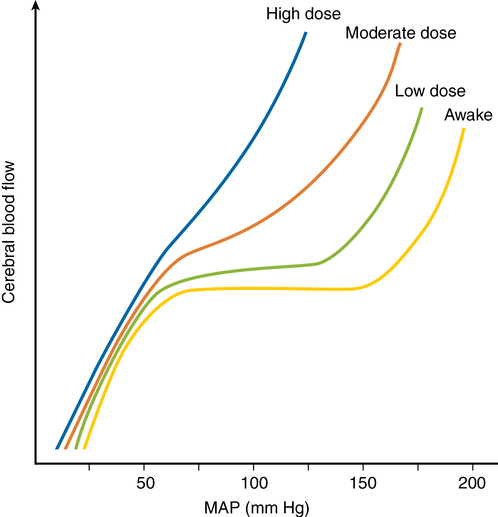
Autoregulation is impaired in areas of relative ischemia, surrounding mass lesions, after grand mal seizures, after head injury, and during episodes of hypercarbia or hypoxemia. Figure 42-3 shows how lost autoregulation may lead to dangerously low CBF. Regional or global ischemia may ensue.
CO2 reactivity
PaCO2 levels profoundly affect CBF by changing the H+ concentration in the extracellular fluid surrounding smooth muscle in arteriolar cell walls. CBF varies directly with PaCO2 (see Figure 42-1). The effect is greatest in the normal physiologic range of PaCO2. CBF changes 1 to 2 mL·100 g−1·min−1 for each 1-mm Hg change in PaCO2. As PaCO2 increases from 30 to 60 mm Hg, for example, CBF doubles. Below a PaCO2 level of 25, the response is attenuated.
O2 reactivity
PaO2 has little direct effect on CBF at values between 60 mm Hg and more than 300 mm Hg. A PaO2 level below 60 mm Hg markedly increases CBF if blood pressure is maintained (see Figure 42-1). This effect is not well understood. A variety of chemoreceptors and local humeral effects may be involved. At PaO2 levels above normal, up to 1 atm (760 mm Hg), only a very slight decrease in CBF has been measured.
Effects of anesthetic drugs
In general, anesthetic agents, except for ketamine and N2O, depress CMR.
Inhaled anesthetic agents
Inhaled anesthetic agents reduce CMRO2 (see Chapter 67, Central Nervous System Effects of the Inhalation Agents). Decreases in CMRO2 are dose dependent and nonlinear below 1 minimum alveolar concentration (MAC) of the agent; a precipitous drop is followed by a more gradual, linear decline as MAC is increased. Maximal reduction occurs with electroencephalographic suppression. Differences among the CMRO2 effects of isoflurane, desflurane, and sevoflurane are minor.
Autoregulation is impaired by volatile anesthetic agents in a dose-dependent manner (see Figure 42-3). Sevoflurane impairs autoregulation less than does isoflurane or desflurane, which do so less than halothane. (This pattern follows the vasodilatory potency of each gas.)
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Factors affecting cerebral blood flow
Cerebral metabolic rate (CMR), autoregulation, CO2 reactivity, and O2 reactivity are the main factors affecting cerebral blood flow (CBF). The relationships among the latter three are depicted in Figure 42-1. Temperature and anesthetic medications also each influence CBF.
Autoregulation
Autoregulation is defined as the maintenance of CBF over a range of mean arterial pressure (MAP) (see Figure 42-1). Cerebral vascular resistance (CVR) is adjusted to maintain constant CBF. Cerebral perfusion pressure equals MAP minus intracranial pressure (ICP). Because ICP (and therefore cerebral perfusion pressure) is not commonly available, MAP is used as a surrogate of cerebral perfusion pressure.
Autoregulation occurs when MAP is between 70 and 150 mm Hg in the normal brain (see Figure 42-1). This is a conservative estimate, given that considerable interindividual variation occurs. The lower limit of autoregulation (LLA) is the point at which the autoregulation curve deflects downward and CBF begins to decrease in proportion to MAP.
CVR varies directly with blood pressure to maintain flow, taking 1 to 2 min for flow to adjust after an abrupt change in blood pressure. In hypertensive patients, the autoregulatory curve is shifted to the right (Figure 42-2). A hypertensive patient may be at risk for developing brain ischemia at a MAP of 70 mm Hg, for example, because the LLA will be higher than in a nonhypertensive patient. Several weeks of blood pressure control may return the curve to normal. Following significant hypotension (lower than the LLA), autoregulation is impaired, and hyperemia may occur when MAP returns to the normal range. CO2 reactivity remains intact, and inducing hypocapnia may attenuate hyperemia.
Autoregulation is impaired in areas of relative ischemia, surrounding mass lesions, after grand mal seizures, after head injury, and during episodes of hypercarbia or hypoxemia. Figure 42-3 shows how lost autoregulation may lead to dangerously low CBF. Regional or global ischemia may ensue.
[/not-level-membership-for-anesthesiology-category]
