CHAPTER 29 Extracellular Matrix Molecules
Although the extracellular matrix is composed of only five classes of macromolecules—collagens, elastin, proteoglycans, hyaluronan, and adhesive glycoproteins—it can take on a rich variety of different forms with vastly different mechanical properties. This is possible for two reasons. First, each of these classes of macromolecule comes in a number of variants (encoded by different genes or produced by alternative splicing), each with distinctive properties. Second, the cells that constitute the extracellular matrix are versatile with respect to secreting different proportions of these isoforms in different geometrical arrangements. As a result, the extracellular matrix in different tissues is adapted to particular functional requirements, which vary as widely as tendons, blood vessel walls, cartilage, bone, the vitreous body of the eye, and subcutaneous fat. Beyond providing mechanical support, the extracellular matrix also strongly influences embryonic development, provides pathways for cellular migration, provides essential survival signals, and sequesters important growth factors. This chapter introduces the macromolecules of the extracellular matrix.
Collagen
The defining feature of collagens is a rod-shaped domain composed of a triple helix of polypeptides (Fig. 29-1). Each polypeptide folds into a left-handed helix that repeats every third residue with the side chains on the outside. Three of these helices associate to form a triple helix that may be up to 420 nm long. The triple helical domains have a repeating amino acid sequence: glycine-X-Y, where X is most often proline and Y is most often hydroxyproline. The small glycine residues allow tight contact between the polypeptides in the core of the triple helix. Larger residues, even alanine, interfere with packing. Poly-l-proline has a strong tendency to form a left-handed helix like individual collagen chains but does not form a triple helix, owing to steric interference. The triple helix is most stable if all X residues are proline and all Y residues are hydroxyproline, but other residues at some of these positions are essential for collagen to assemble higher-order structures. (Despite their name, α-chains, the collagen polypeptides do not form α-helices.)
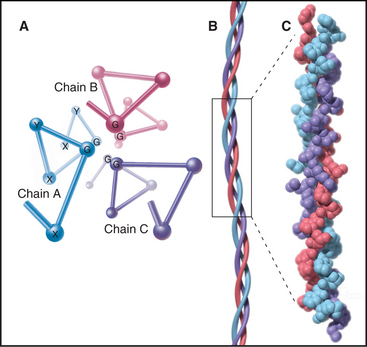
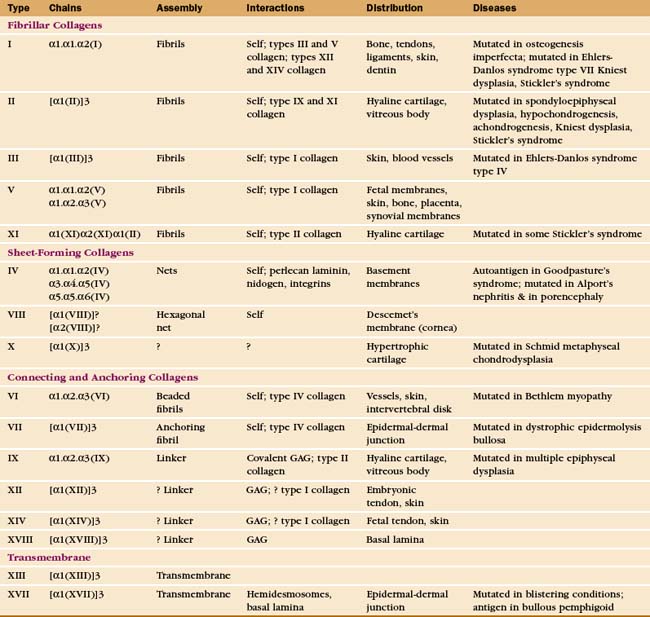
The collagen family is remarkably diverse. Humans have about 100 genes with collagen triple repeats, and more than 20 specialized collagen proteins have been characterized (Fig. 29-2 and Appendix 29-1).
Other proteins, including the extracellular enzyme acetylcholine esterase (see Fig. 11-8) and some cell surface receptors, have similar triple helical domains but are not classified as collagens. To be a collagen, a protein must also form fibrils or other assemblies in the extracellular matrix. Nematodes, which lack connective tissue, seem to have lost the genes for fibrillar collagens but have elaborated a family of 160 genes for collagens that form their cuticle.
The size and shape of collagens vary according to function. Collagens are named numerically (type I, type II, etc.) in the order of their discovery, a nomenclature that bears no relationship to their function. Appendix 29-1 groups collagens according to function. Polypeptides are called α-chains, and Roman numerals in their names correspond to their type number. Some collagens are homotrimers of three identical α-chains. Others are heterotrimers of two or three different α-chains. Some chains (e.g., [a1(II)]) are used in more than one type of collagen.
Fibrillar Collagens
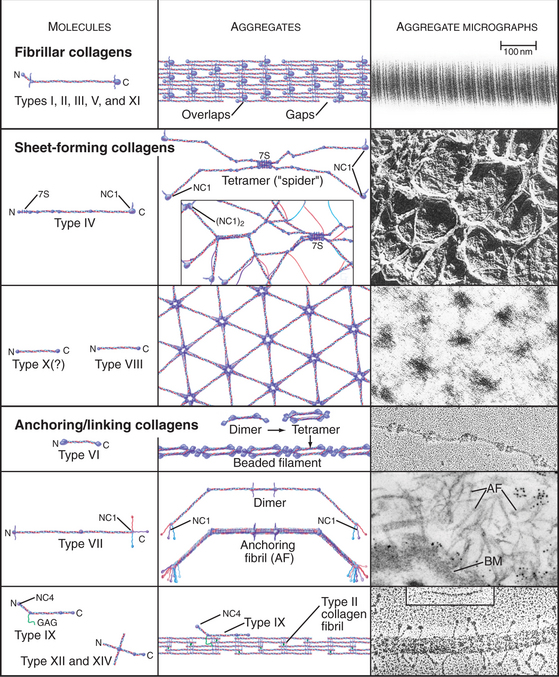
Triple helical rod-shaped collagen molecules about 300 nm long self-associate to form banded fibrils (Fig. 29-2). Collagen fibrils provide tensile strength to tendons, ligaments, bones, and dense connective tissue, thus reinforcing most organs. They also form the scaffolding for cartilage and the vitreous body in the eye. Fibrillar collagens are widespread in nature and have been highly conserved during evolution, so the homologs from sponges to vertebrates are similar. Each fibrillar collagen can form homopolymers in vitro; but in vivo, most form heteropolymers with at least one other type of fibrillar collagen (Appendix 29-1). This mix of the fibrillar collagen subunits is one factor that regulates the size of collagen fibers. Proteoglycans also participate (Appendix 29-2).
The biosynthesis and assembly of fibrillar collagens involve a remarkable number of posttranslational modifications, including several rounds of precise proteolytic cleavage, glycosylation, catalyzed folding, and chemical cross-linking (Fig. 29-4). The final product is a smooth fibril with staggered molecules that are cross-linked to their neighbors. These strong but flexible collagen fibrils reinforce all the tissues of the body, where they form a variety of higher-order structures. Loose connective tissue (see Fig. 32-1A) has an open network of individual fibrils or small bundles of fibrils that support the cells. In many tissues, the fibrils of type I and associated collagens aggregate to form the so-called collagen fibers that are visible by light microscopy (Fig. 29-3A). In extreme cases, such as in tendons, the extracellular matrix consists almost exclusively of tightly packed, parallel bundles of collagen fibers (see Fig. 32-1B). Layers of orthogonal collagen fibers make the transparent cornea through which one sees (Fig. 29-3C). In bone, type I collagen fibrils form regular layers reinforced by calcium phosphate crystals (see Fig. 32-5). In cartilage and the vitreous body of the eye, type II collagen fibrils trap glycosaminoglycans and proteoglycans, which retain enough water for the matrix to resist compression (see Fig. 32-3) and, in the case of the eye, to provide an optically clear path for light.

Biosynthesis and Assembly of Fibrillar Collagens
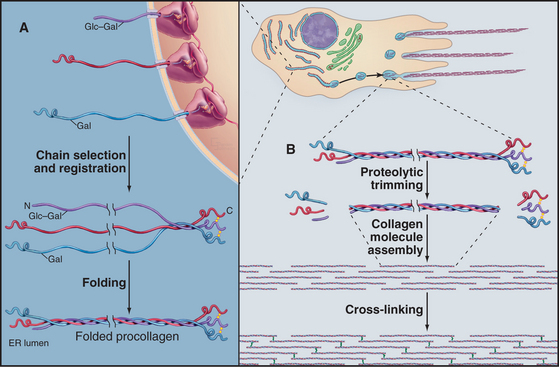
All fibrillar collagens are most likely to be produced by similar mechanisms, but type I collagen has been studied the most extensively. Type I collagen is synthesized and secreted by fibroblasts, using the exocytic pathway that is employed for other secretory proteins (see Chapter 21), but the biosynthesis of collagen is noteworthy for the extensive number of processing steps required to prepare the protein for assembly in the extracellular matrix.
The initial transcript, referred to as preprocollagen, translocates into the lumen of the rough endoplasmic reticulum, where intracellular processing begins (Fig. 29-4). First, removal of the N-terminal signal sequence yields procollagen with unfolded α-chains with N- and C-terminal nonhelical propeptides. Second, enzymes hydroxylate some prolines and lysines. Third, enzymes add sugars (gal-glu or gal) to the delta-carbon of some lysines, by a mechanism distinct from the typical glycosylation of asparagine or serine.
Procollagen passes through the Golgi apparatus and moves in vesicles to the cell surface, where it is secreted. Some cells have specialized collagen assembly sites (Fig. 29-4). Like ships laying down communication cables on the ocean floor, fibroblasts help to determine the arrangement of collagen fibrils as they move through tissues (Fig. 29-3C).
Outside the cell, proteolytic enzymes—procollagen proteases—cleave the propeptides from the triple helical domain, forming the mature collagen molecule (formerly called tropocollagen). Relieved of its inhibitory propeptides, collagen self-assembles into fibrils by a classical entropy-driven process (Fig. 29-5). Adjacent collagen molecules are staggered by 67 nm, so a 35-nm gap is required between the ends of the collagen molecules (five staggers at 67 nm = 335 nm = one molecular length of 300 nm + a 35-nm gap).
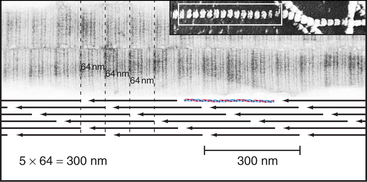
Figure 29-5 structure of collagen fibrils. Electron micrographs and drawing of molecular packing.
(Micrographs courtesy of Alan Hodges, Marine Biological Laboratory, Woods Hole, Massa-chusetts.)
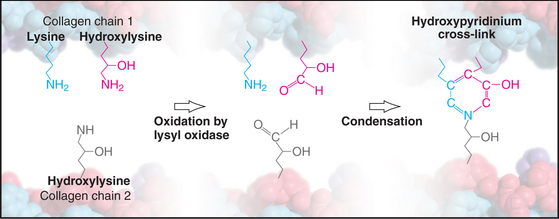
Weak, noncovalent bonds between collagen molecules specify the self-assembly of fibrils but provide little tensile strength, so covalent cross-linking is required for reinforcement. For most fibrillar collagens, the enzyme lysyl oxidase catalyzes the formation of covalent bonds between the ends of collagen molecules (Figs. 29-4 and 29-6). The enzyme oxidizes the e amino groups of selected lysines and hydroxylysines to aldehydes. These aldehydes react spontaneously with nearby lysine and hydroxylysine side chains to form a variety of covalent cross-links between two or three polypeptides. Disulfide bonds, rather than modified lysine side chains, cross-link type III collagen fibrils. Covalent bonds between the inextensible triple helices give mature collagen fibrils their great tensile strength.
Point mutations or deletions in collagen genes or lack of function of one of the enzymes that processes collagen (lysyl hydroxylase, lysyl oxidase, or procollagen proteases) can each cause defective collagen fibrils (Appendix 29-1). These defects cause a remarkable variety of deforming and even lethal human diseases: brittle bones (osteogenesis imperfecta), fragile cartilage (several forms of dwarfism), and weak connective tissue (Ehlers-Danlos syndrome). Chapter 34 covers these diseases in more detail.
Sheet-Forming Collagens
A second group of collagens polymerizes into sheets rather than fibrils (Fig. 29-2). These sheets surround organs, epithelia, or even whole animals. Six different human genes for type IV collagen encode proteins that form net-like polymers that assemble into the basal lamina beneath epithelia (Fig. 29-7) and around muscle and nerve cells. The concluding section of this chapter provides details about basal lamina structure, function, and diseases. Hexagonal nets of type VIII collagen form a special basement membrane (Descemet’s membrane) under the endothelium of the cornea. Related collagens form the cuticle of earthworms and the organic skeleton of sponges.
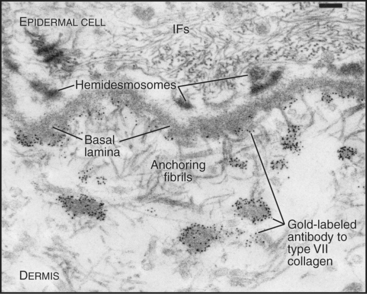
Figure 29-7 anchoring fibrils of type vii collagen. Electron micrograph of a thin section of human skin reacted with a gold-labeled antibody to the C-terminal domain of type VII collagen. Top to bottom, Basal epithelial cell with keratin intermediate filaments (IFs) attached to hemidesmosomes, which link to the basal lamina. Short fibrils of type VII collagen link the basal lamina to plaques in the dermis. Both ends of these bipolar fibrils (Fig. 29-2) are labeled with gold. Bar is 0.1 mm.
(Courtesy of D. R. Keene, Portland Shriners Hospital, Oregon.)
Linking Collagens
Connecting and anchoring collagens link fibrillar and sheet-forming collagens to other structures (Fig. 29-2). The type VII collagen homotrimer has an exceptionally long triple-helix domain with nonhelical domains at the N-terminus of each chain. Type VII molecules self-associate tail to tail to form antiparallel dimers. In the process, proteases remove the C-terminal globular domain. Several dimers associate laterally to form so-called anchoring fibrils that link type IV collagen of the basal lamina of stratified epithelia to plaques in the underlying connective tissue (Fig. 29-7). Mutations in type VII collagen cause both the dominant and recessive forms of a severe blistering disease, dystrophic epidermolysis bullosa. In heterozygotes, mutated chains interfere with the assembly of anchoring fibrils by normal type VII collagen chains. Without anchoring fibrils, the basal lamina adheres weakly to the connective tissue matrix. Even mild physical trauma to the skin causes the epithelium to pull away from the connective tissue, forming a blister. Related diseases are caused by mutations in intermediate filaments (see Fig. 35-6).
Type IX collagen links glycosaminoglycans to type II collagen fibrils (Fig. 29-2). This collagen heterotrimer has a serine modified with a glycosaminoglycan chain of variable length. Type IX collagens do not polymerize, but they associate laterally with type II collagen fibrils. The N-terminal helical segment and associated glycosaminoglycan project from the surface of the type II collagen fibril. In the vitreous body of the eye, these polysaccharides fill most of the extracellular space.
Elastic Fibers
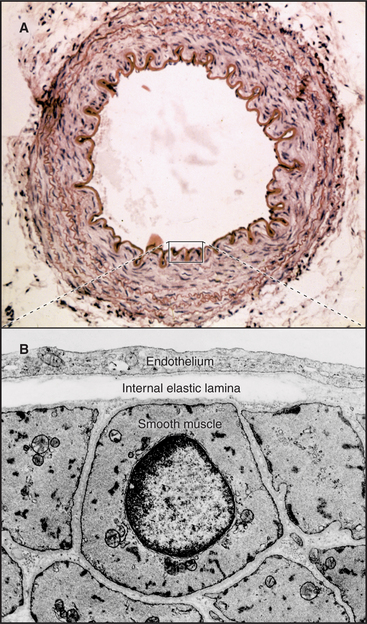
In contrast to inextensible collagen fibrils, elastic fibers are similar to rubber. They are found throughout the body but are prominent in the connective tissue of skin, the walls of arteries (Fig. 29-8), and the lung. They recoil passively after tissues are stretched. Every time the heart beats, pressurized blood flows into and stretches the large arteries. Energy stored in elastic fibers pushes blood through the circulation between heartbeats.
Elastic fibers are a composite material: A network of fibrillin microfibrils is embedded in an amorphous core of cross-linked elastin, which makes up 90% of the organic mass (Fig. 29-9). Fibroblasts produce both components. Loose bundles of microfibrils initiate assembly. A third protein, called fibulin, is required for elastin subunits to assemble between the micro-fibrils.
Fibrillin is the primordial component of elastic fibers, having arisen in Cnidarians (see Fig. 2-9). It is a long, floppy protein consisting of a tandem array of domains (Fig. 29-10). Humans have two fibrillin genes, and both fibrillin-1 and fibrillin-2 are components of 10-nm microfibrils, along with several glycoproteins. In microfibrils, fibrillin molecules interact head to tail with a reinforcing disulfide bond, but their arrangement is still being investigated. Microfibrils are about 100 times stiffer than elastin, and they stretch by rearrangement of molecules and domains rather than unfolding.
Elastin subunits are a family of closely related 60-kD proteins called tropoelastins, the products of alternative splicing from a single elastin gene. They have long sequences that are rich in hydrophobic residues interrupted by short sequences with pairs of lysines separated by two or three small amino acids (Fig. 29-11). Lysine-rich sequences are thought to form α-helices with pairs of lysines adjacent on the surface.
As tropoelastin assembles on the surface of elastic fibers, lysyl oxidase oxidizes paired lysines of tropoelastin to aldehydes. Oxidized lysines condense into a desmosine ring that covalently cross-links tropoelastin molecules to each other (Fig. 29-11). The four-way cross-links, involving pairs of lysines from two tropoelastin molecules, are unique to elastin. The same enzyme catalyzes the cross-linking of collagen, but it forms only two- and three-way cross-links.
Elastic fibers are similar to rubber except that elastic fibers require water as a lubricant. Hydrophobic segments between the cross-links are thought to form extensible random coils that account for the elastic properties of the fibril (Fig. 29-12). A difference in entropy of the polypeptide in the contracted and stretched states is thought to be the physical basis for the elasticity. The birefringence of elastic fibers increases when they are stretched, presumably as a result of alignment of polypeptide chains. Stretched fibers store energy, owing to ordering (low entropy) of the polypeptide chains. Fibers shorten when the resistance is reduced, because the polypeptide chains return to their disordered, lower-energy, higher-entropy state. Unfolding of fibrillin domains may contribute to the elasticity similar to titin in muscle cells (see Fig. 39-7), but this has not been studied.
Collagens are found across the phylogenetic tree, but only vertebrates are known to produce elastin. Invertebrates evolved two completely different elastic proteins. Mollusks have elastic fibers composed of the protein abductin. Insects use another protein, called resilin, to make elastic fibers.
Marfan syndrome, the disease caused by dominant mutations in the fibrillin-1 gene, illustrates the physiological functions of elastic fibers. Elastic fibers of patients with Marfan syndrome are poorly formed, accounting for most of the pathological changes that are observed. Most dangerously, weakness of elastic fibers in the aorta leads to an enlargement of the vessel, called an aneurysm, which is prone to rupture, with fatal consequences. Prophylactic replacement of the aorta with a synthetic graft and medical treatment with drugs that block β-adrenergic receptors (see Fig. 27-3) allow patients a nearly normal life span. In some patients, a floppy mitral valve in the heart causes regurgitation of blood from the left ventricle back into the left atrium. Weak elastic fibers that suspend the lens of the eye result in dislocation of the lens and impaired vision. Weak elastic fibers result in lax joints and curvature of the spine. Most affected patients are tall, with long limbs and fingers, but the connection of these features to fibrillin is not known. The manifestations of the disease are quite variable, even within one family, for reasons that are not understood. Mutations in fibrillin-2 cause congenital contractural arachnodactyly, a disease characterized by joint stiffness. New fibrillin mutations arise spontaneously, and most families that are tested have different mutations, including both point mutations and deletions. All patients are heterozygotes. Most of the known fibrillin-1 mutations make the protein unstable and susceptible to proteolysis. Other point mutations interfere with folding.
Glycosaminoglycans and Proteoglycans
Glycosaminoglycans (GAGs, formerly called mucopolysaccharides) are long polysaccharides made up of repeating disaccharide units, usually a hexuronic acid and a hexosamine (Fig. 29-13). With one important exception—hyaluronan—GAGs are synthesized as covalent, posttranslational modifications of a large family of proteins called proteoglycans. These proteins vary in structure and function, but their associated GAGs confer some common features.
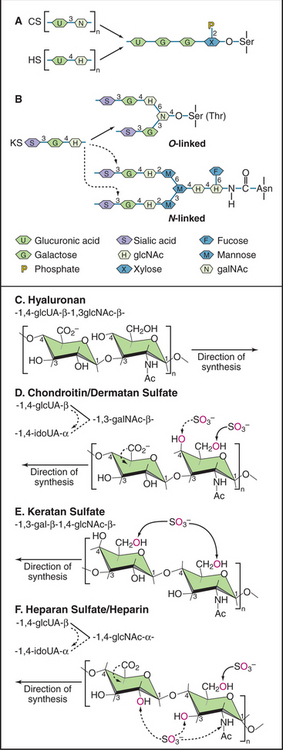
Of the known GAGs, hyaluronan (formerly called hyaluronic acid) is exceptional in two regards. First, enzymes on the cell surface synthesize the alternating polymer of [d-glucuronic acid β (1 → 3) D-N-acetyl glucosamine β (1 → 4)]n (Fig. 29-13). Other GAGs are synthesized as posttranslational modifications of a core protein. Second, hyaluronan is not modified postsynthetically, as are all other GAGs. The linear polymer, often exceeding 20,000 disaccharide repeats (a length >20 mm) is released into the extracellular space.
In contrast to proteins, nucleic acids, and even N-linked oligosaccharides, which are precisely determined macromolecular structures, the GAG chains of proteoglycans appear to vary both in length and the sequence of the sugar groups. The four-step synthesis of GAGs (Fig. 29-13) explains this variability:
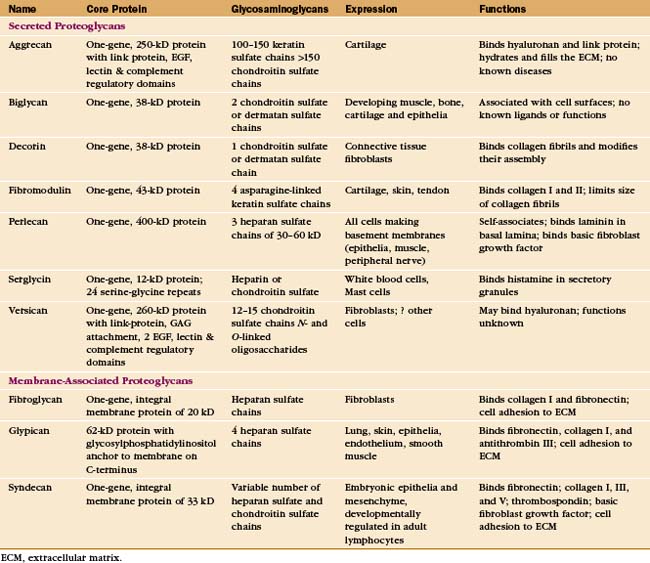
The nomenclature for proteoglycans is in flux, so specific proteoglycans may have multiple names. The old nomenclature was based on the identity of the GAGs that are bound to the protein. For example, the major proteoglycan of basement membranes was called heparan sulfate proteoglycan. This nomenclature is imprecise, as more than one type of proteoglycan carries heparan sulfate. Once the core proteins were characterized, it was reasonable to develop a nomenclature based on these core proteins. Consequently, the basement membrane proteoglycan is now known as perlecan, the name of its core protein. The weakness of this system is that the protein name reveals nothing about the associated GAGs. This information is important because various cells add different GAGs to the same core protein or can modify the same GAG in different ways.
Cells secrete many proteoglycans into the extracellular matrix, but they retain some types on the plasma membrane through transmembrane polypeptides or a glycosylphosphatidylinositol anchor (Appendix 29-2 and Fig. 29-14). The core proteins vary in size from 100 to 4000 amino acids. Many are modular, consisting of familiar structural domains: EGF, complement regulatory protein, leucine-rich repeats, or lectin. Three collagens carry GAG side chains: Types IX and XII have chondroitin sulfate chains, and type XVII has heparin sulfate chains.
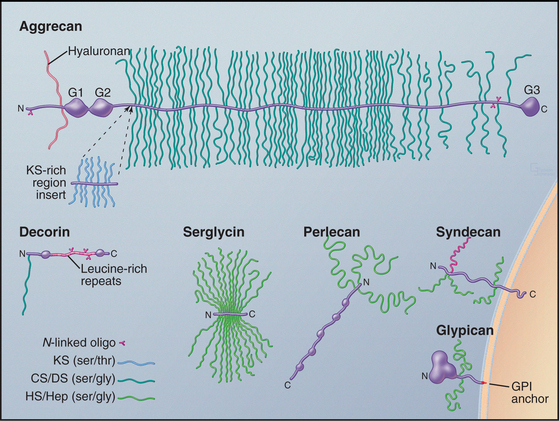
The number of GAGs attached to the core protein varies from one (decorin) to more than 200 (aggrecan) (Fig. 29-14). A particular core protein can have identical (fibroglycan, glypican, versican) or different (aggrecan, serglycin, syndecan) types of GAGs. Some cell types can add different GAGs to the same core protein or secrete a core protein without GAGs.
Given their physical properties and distribution among the fibrous elements of the extracellular matrix, proteoglycans and hyaluronan are thought to be elastic space-fillers. Each hydrophilic disaccharide unit bears a carboxyl or sulfate group or both, so GAGs are highly charged polyanions that extend themselves by electrostatic repulsion in solution and attract up to 50 g of water per gram of proteoglycan. Hyaluronan, the largest GAG, occupies a vast volume. A single hydrated molecule of 25,000 kD occupies a volume similar to that of a small organelle with a diameter of 200 nm. Retention of water by hyaluronan and aggrecan-keratan sulfate/chondroitin sulfate proteoglycan is essential in cartilage (see Fig. 32-3). In the extracellular matrix of other tissues, networks of densely charged hyaluronan restrict water flow, limit diffusion of solutes (especially macromolecules), and impede the passage of microorganisms. Hyaluronan and proteoglycans also act as lubricants in joint cavities and as an optically transparent, space-filling medium in the vitreous body of the eye.
Adhesive Glycoproteins
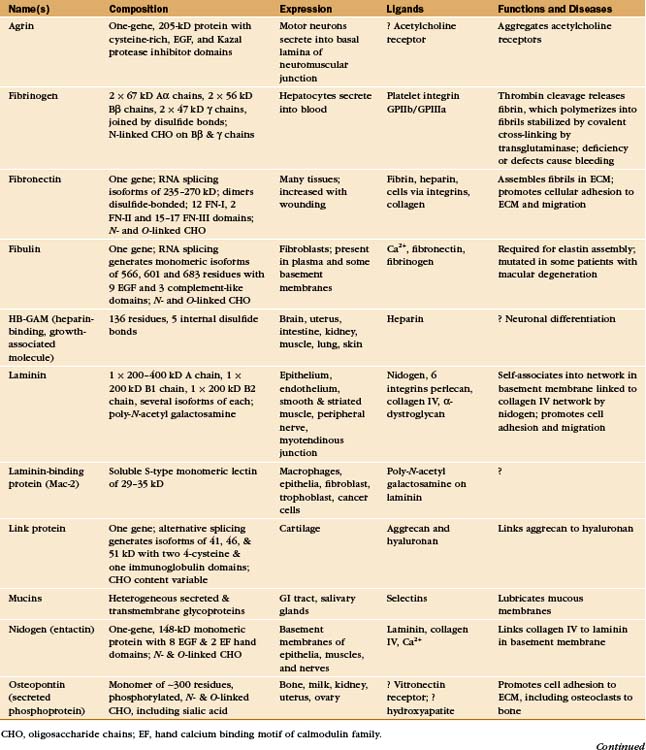
In principle, the macromolecules of the extracellular matrix and the constituent cells might interact relatively nonspecifically, but the evidence suggests that specific molecular interactions mediate virtually all of the interactions that organize the matrix and the associated cells. Most interactions are between proteins. Some are between proteins and sugars. Although some of these interactions are direct (with some cell surface receptors binding collagen directly), adapters called adhesive glycoproteins mediate many of the interactions (Appendix 29-3).
Adhesive glycoproteins were discovered by using biochemical assays for factors that favor particular interactions, such as adherence of cells to a matrix component. Further work revealed that adhesive glycoproteins are more than molecular glue; they also provide cells with signals required for the development and repair of tissues. Cells receive these signals when they bind to the matrix components. Chapter 30 focuses on their receptors.
Adhesive glycoproteins provide specific molecular interactions in the matrix by binding to cells, matrix macromolecules, or both. Adhesive proteins with multiple binding sites for cell surface receptors link cells together. For example, fibrinogen aggregates platelets during blood clotting (see Fig. 30-14). Other adhesive proteins link cells to the extracellular matrix. For instance, fibronectin mediates the attachment of cells to fibrin and collagen (Fig. 29-15). A third group of adhesive proteins link matrix macromolecules together. For example, nidogen attaches laminin to collagen and link protein attaches aggrecan-proteoglycan to hyaluronan.
The variety of tasks requires numerous adhesive proteins. In fact, the diversity exists beyond the named proteins (Appendix 29-3), as multiple genes or, more commonly, alternative splicing of the product of a single gene (see Fig. 16-6), generate multiple, functionally distinct isoforms of most of the named proteins. Particular isoforms are often expressed in specific tissues at predictable times during development.
Most adhesive glycoproteins are constructed of a series of compact modules (see Fig. 3-13 and Appendix 29-3). During evolution, duplication and recombination of the coding sequences for the domains produced the genes for these large proteins. In addition to the domains, each of these proteins also contains a significant fraction of unique sequences.
Most adhesive glycoproteins that interact with cells bind to heterodimeric transmembrane receptors called integrins (see Fig. 30-9). Remarkably, the integrin-binding sites of many adhesive proteins include the simple tripeptide arginine-glycine-aspartic acid (RGD [Fig. 29-15]).
Fibronectin
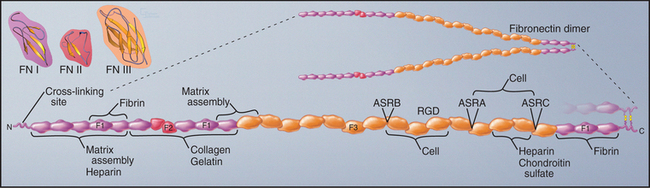
Fibronectins are large proteins that consist of two polypeptides of about 235 kD linked by disulfide bonds near their C-termini (Fig. 29-15). In electron micrographs, fibronectin appears as a V-shaped pair of long, flexible rods connected at one end. In solution, the molecule is probably more compact. Each polypeptide is a linear array of three types of domains called FN-I, FN-II, and FN-III. All three types of fibronectin domains consist of antiparallel b strands with conserved residues in their hydrophobic cores. Two disulfide bonds stabilize FN-I and FN-II domains, whereas FN-III domains have no disulfide bonds. FN-I and FN-II domains consist of about 45 residues; FN-III domains are twice as large. FN-I and FN-II domains are present in a few other proteins, whereas the human genome contains about 170 genes with FN-III domains, including proteins in the extracellular matrix (Appendix 29-3), on the cell surface (human growth hormone receptor; see Fig. 24-6), and inside cells (titin; see Fig. 39-7).
Fibronectin binds a variety of ligands, including cell surface receptors, collagen, proteoglycans, and fibrin (another adhesive protein). Thus, it contributes to adhesion of cells to the extracellular matrix and may also cross-link matrix molecules. Ligand-binding sites in the various domains were identified by isolating proteolytic fragments and by expressing fibronectin fragments (Fig. 29-15). Some of these binding sites are cryptic and are exposed only when the protein is stretched. The RGD sequence that contributes to the integrin-binding site of fibronectin is located on an exposed loop of FN-III domain 10. The variably spliced V domain included in plasma fibronectin has a second integrin-binding site. Chapter 30 provides additional details on integrins.
Two pools of fibronectin have different distributions and solubility properties. Tissue fibronectin forms insoluble fibrils in connective tissues throughout the body, especially in embryos and healing wounds. Fibroblasts use an integrin-dependent process to assemble fibronectin dimers into fibrillar aggregates large enough to visualize by light microscopy (Fig. 29-16). The structure of these microscopic fibrils is not known. Denaturing agents and disulfide reduction are required to solubilize these fibrils. Disulfide bonds between the two subunits of fibronectin are also essential to form this continuous protein network, so fibronectin with deletions from the C-terminus cannot assemble into fibrils. Although difficult to study because of their large size and insolubility, fibronectin fibrils seem to bind cells more efficiently than soluble fibronectin, and may have additional activities important for biological functions.
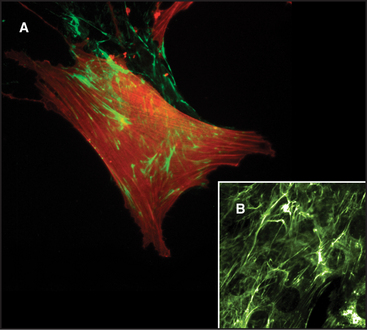
Soluble plasma fibronectin dimers circulate in the body fluids. The protein differs from tissue fibronectin as a result of alternate splicing of the mRNA. In blood clots, the enzyme transglutaminase covalently couples plasma fibronectin to fibrin, forming a provisional matrix for wound repair (see Fig. 32-11).
Thus, it was predicted that deletion of the single fibronectin gene in mice would be lethal, owing to devastating effects very early in embryogenesis. It is true that homozygous null mutant mice die during embryogenesis as a result of failure to form mesodermal structures, including the notochord, muscles, heart, and blood vessels. However, the surprise was how far the embryos developed without fibronectin. In fact, up to about day 8 (when the basic body plan is already determined), the embryos appeared to be almost normal. (Mice with null mutations in the main fibronectin receptor, integrin a5, have similar but slightly milder defects.) One interpretation is that fibronectin and its receptor are less important for early development than was anticipated. Changes in the expression of other adhesive glycoproteins or receptors may compensate for the fibronectin defects during the first few days of devel-opment.
Tenascin
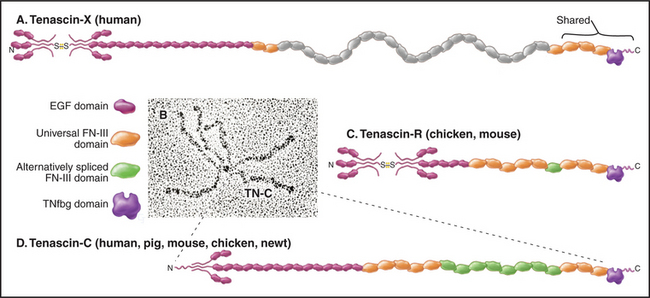
Tenascins are a family of giant proteins with six arms (Fig. 29-17), found in the extracellular matrix of many embryonic tissues, wounds, and tumors. The N-terminal ends of three subunits self-associate through a triple helical coiled-coil. Disulfides covalently link two of these three-chain units to make the hexameric molecule. The arms of the four isoforms consist of different numbers of EGF and FN-III domains, terminated by three similar FN-III domains and the fibrinogen-like domains.
The Basal Lamina
The basal lamina, a thin, planar assembly of extracellular matrix proteins, supports all epithelia, muscle cells, and nerve cells outside the central nervous system (Fig. 29-18). This two-dimensional network of protein polymers forms a continuous rug under epithelia and a sleeve around muscle and nerve cells. In addition, basal laminae are semipermeable filters for macromolecules, a particularly important role that they play in the conversion of blood plasma into urine in the kidney. The genes for basal lamina components are very ancient, having arisen in early metazoans.
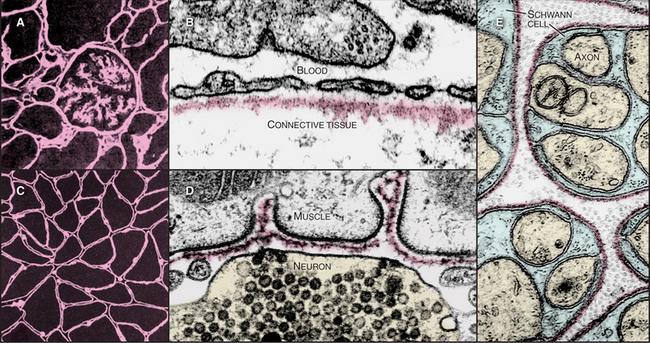
In electron micrographs of thin sections of tissues prepared by chemical fixation, the basal lamina is a homogenous, finely fibrillar material that is separated from the adjacent cell by a clear gap (Fig. 29-18D). This gap is not present when the tissue is prepared by rapid freezing, so it might be an artifact. This would reconcile biochemical evidence that plasma membrane proteins connect cells directly to the basal lamina. In some tissues, fine type VII collagen fibrils connect the lamina to underlying connective tissue. The basal lamina and associated collagen fibrils form the “basement membrane” that is observed in histologic preparations of epithelia. A basal lamina alone cannot be seen by light microscopy without special labels, such as those used in Figure 29-18A and C.
Although many proteins contribute to the stability of the basal lamina (Fig. 29-19), only the adhesive glycoprotein laminin is essential for the initial assembly of basal laminae during embryogenesis. The C-terminal end of the cross-shaped laminin molecule binds to cell surface receptors (integrins, dystroglycan; see Fig. 39-7). Laminins self-assemble into continuous, two-dimensional networks through noncovalent interactions of their short arms. Mouse embryos that lack dystroglycan or laminin die early in development, owing to failure to make basal laminae.
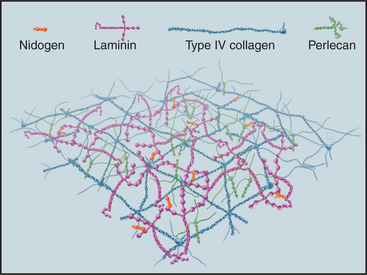
The subsequent addition of other proteins reinforces the laminin network. A two-dimensional network of collagen IV self-assembles through head-to-head interactions of the N-termini of four molecules and tail-to-tail interactions of the C-terminal NC1 domains of two molecules (Fig. 29-2). Mouse embryos that lack collagen IV make nascent basal laminae composed of laminin but eventually die from defects in basal lamina.
Other proteins reinforce the collagen IV and laminin in basal laminae. The rod-shaped protein nidogen cross-links laminin to type IV collagen. Perlecan, a heparan sulfate proteoglycan, provides additional cross-links, as it binds to itself in addition to laminin, nidogen, and collagen IV. These cross-links help to determine the porosity of basal lamina and thus the size of molecules that can filter through it. Fibrillin and an associated protein, fibulin, are also present.
Restricted human tissues express four additional type IV collagens; remarkably, each has been implicated in human disease (Table 29-1). Patients with Alport’s X-linked familial nephritis all have mutations in the a5(IV) collagen gene. More than 200 different point mutations and deletions are known. Other patients with autosomally inherited Alport’s syndrome have mutations in their a3(IV) or a4(IV) collagen genes. These mutations generally interfere with folding of the collagen molecule and disrupt the basement membranes that form the blood filtration barrier in the glomerulus of the kidney, causing progressive kidney failure that is usually fatal in males. These mutations also cause defects in the eye and ear, other places where the a5(IV) collagen gene is expressed. In Goodpasture’s syndrome, the immune system produces autoantibodies to the C-terminal NC1 domain of a3(IV) collagen. The protein sequences that elicit autoantibody production are buried in the NC1 domain, so they may be exposed by bacterial infections or organic solvents, predisposing events in the syndrome. Antibodies bound to basement membranes in the kidney and lung cause inflammation that leads to kidney failure and bleeding in the lungs.
Table 29-1 INHERITED DISEASES OR MUTANT PHENOTYPES OF BASAL LAMINA COMPONENTS
| Protein Subunit | Distribution | Disease or Mutant Phenotype |
|---|---|---|
| Collagen α3IV | Many tissues | Human autoantibodies cause Goodpasture’s syndrome of renal failure. |
| Collagen α5IV | Kidney, muscle | Human mutation causes Alport’s syndrome of renal failure. |
| Laminin α1 | Many tissues | Fly null mutation is lethal during embryogenesis. |
| Laminin α2 | Muscle, heart | Mouse dy mutation causes muscular dystrophy. |
| Laminin γ2 | Epidermis | Human mutation causes Herlitz’s junctional epidermolysis bullosa. |
| Perlecan | Many tissues | Worm unc-52 mutation disrupts myofilament attachment to membrane. |
Matrix Metalloproteinases
Many physiological processes depend on the controlled degradation of the extracellular matrix. Examples include tissue remodeling during embryogenesis (e.g., resorption of a tadpole tail), wound healing, involution (massive shrinkage secondary to loss of cells and extracellular matrix) of the uterus after childbirth, shedding of the uterine endometrium during menstruation, and invasion of the uterine wall by the embryonic trophoblast during implantation. Conversely, uncontrolled destruction of extracellular matrix contributes to degenerative diseases, such as emphysema and arthritis. In addition to their roles in remodeling, many of these enzymes cleave and release biologically active fragments from matrix or membrane proteins. Three classes of Zn-depended proteases account for both the physiological and pathological degradation of diverse extracellular matrix and cell surface proteins.
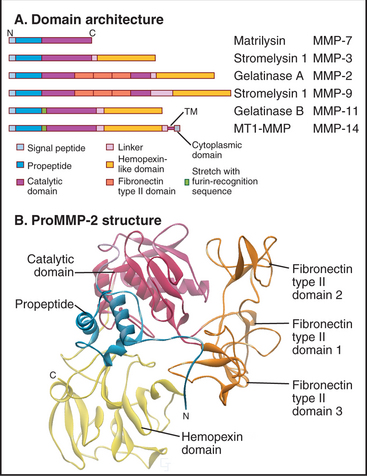
The first class is called matrix metalloproteinases (MMPs). These 24 homologous enzymes share a zinc-protease domain (Fig. 29-20) similar to bacterial thermolysin. Gelatinases have three FN-II domains inserted into the sequence of the catalytic domain. All have an N-terminal signal sequence and are processed through the secretory pathway. Between the signal sequence and catalytic domain, all MMPs have an autoinhibitory propeptide, including a conserved cysteine that binds to the zinc ion in the catalytic site. A C-terminal transmembrane domain anchors several MMPs to the plasma membrane. All other MMPs are secreted. Most MMPs have a C-terminal regulatory domain that influences the substrate specificity of the catalytic domain. After secretion, inactive pro-MMPs bind directly or indirectly to cell surface receptors.
The second class of Zn-dependent proteases consists of about 35 proteases called ADAMs (a disintegrin and metalloproteinase). These enzymes are anchored to the plasma membrane by a single transmembrane sequence. Like other metalloproteinases, they are inhibited by TIMPs. ADAMs cleave and release extracellular domains of cell surface proteins, some of which are important informational molecules (e.g., tumor necrosis factor [TNF]-a; transforming growth factor [TGF]-a). ADAM-17 null mutations are lethal during embryogenesis, owing to a lack of TGF-a or other ligands for EGF receptors. A polymorphism in the ADAM-33 gene is strongly associated with human asthma, although the mechanism is not yet understood.
Baum J, Brodsky B. Folding of peptide models of collagen and misfolding in disease. Curr Opin Struct Biol. 1999;9:122-128.
Burgeson RE, Christiano AM. The dermal-epidermal junction. Curr Opin Cell Biol. 1997;9:651-658.
Capila I, Linhardt RJ. Heparin-protein interactions. Angewandte Chemie Int Ed. 2002;41:390-412.
Hacker U, Nybakken K, Perrimon N. Heparan sulphate proteoglycans: The sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530-541.
Hudson BG, Tryggvason K, Sundraamoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome and Type IV collagen. New Engl J Med. 2003;348:2543-2556.
Hutter H, Vogel BE, Plenefisch JD, et al. Conservation and novelty in the evolution of cell adhesion and extracellular matrix genes. Science. 2000;287:989-994.
Iozzo RV. Basement membrane proteoglycans: From cellar to ceiling. Nat Rev Mol Cell Biol. 2005;6:646-656.
Kielty CM, Sherrat MJ, Marson A, Baldock C. Fibrillin microfibrils. Adv Protein Chem. 2005;70:405-436.
Kreis T, Vale R, editors. Guidebook to the Extracellular Matrix and Adhesion Proteins, 2nd ed., Oxford, UK: Oxford University Press, 1999.
Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis: A cell-mediated matrix assembly process. Matrix Biol. 2005;24:389-399.
Mithieux SM, Weiss AS. Elastin. Adv Prot Chem. 2005;70:437-461.
Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16:558-564.
Ricard-Blum S, Dublet B, van der Rest M. Unconventional Collagens Types VI, VII, VIII, IX, XIV, XVI and XIX. Oxford, UK: Oxford University Press, 2000.
Rosenbloom J, Abrams WR, Mecham R. Extracellular matrix 4: The elastic fiber. FASEB J. 1993;7:1208-1218.
Schenk S, Quaranta V. Tales from the crypt[ic] sites of the extracellular matrix. Trends Cell Biol. 2003;13:366-375.
Sugahara K, Mikami T, Uyama T, et al. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612-620.
Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: Host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2005;20:9-22.
Timpl R, Sasaki T, Kostka G, Chu ML. Fibulins: A versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol. 2003;4:479-489.
White JM. ADAMs: Modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. 2003;15:598-606.
Yurchenco PD, Wadsworth WG. Assembly and tissue functions of early embryonic laminins and netrins. Curr Opin Cell Biol. 2004;16:572-579.