Chapter 75 Evaluation and Management of Arrhythmias in the Pediatric Population
Supraventricular Tachycardia
General Presentation and Evaluation
Supraventricular tachycardias (SVTs), which are arrhythmias that originate above the ventricles and involve the atria, the atrioventricular (AV) node, or the accessory bypass tracts, represent the most common tachycardias seen in infants and children, with an incidence of 1 in 250 to 1000 in the United States.1 SVTs are classified by identifying those components of the heart that are involved in maintaining the tachycardia. SVTs include primary atrial tachycardias (those in which the AV node and the ventricles are not involved), AV reciprocating tachycardias (those that involve both the atrium and the ventricle), and AV nodal re-entrant tachycardias (those that do not involve the atria as part of the circuit). The presentation of the pediatric patient with SVT varies from palpitations to signs of marked cardiac failure. This variability is related to the patient’s age at presentation and his or her ability to communicate symptoms, the duration and rate of the tachycardia, as well as the mechanism of SVT. Parents of infants with SVT occasionally note changes in feeding habits and irritability as symptoms. Symptoms in those old enough to voice complaints include palpitations (noted as skipped or rapid heart beats, or as “heart beeping”), chest pain, shortness of breath, fatigue, dizziness, and syncope.
The evaluation of pediatric patients includes a 12- or 15-lead electrocardiogram (ECG), which may show Wolff-Parkinson-White (WPW) syndrome or other abnormalities such as an ectopic atrial rhythm. While the ECG is often normal when SVT is not present, an ECG during SVT can be very helpful in distinguishing the mechanism of the tachycardia. A recent study suggested that leads V1 and III were the most helpful in this diagnosis.2 Twenty-four-hour ambulatory (Holter) monitoring is useful to screen for occult arrhythmias or to assess heart rate ranges and variability if the patient is having frequent symptoms (i.e., on a daily basis). Transtelephonic event monitors are often helpful in those with fleeting symptoms, allowing the patient or parent to record symptoms as they occur with simultaneous ECG recordings. Exercise tolerance testing is useful in those with palpitations or symptoms associated with activities. More invasive testing such as a transesophageal or intracardiac electrophysiological study (EPS) is used to induce arrhythmia and determine the particular mechanism and often as a prelude to catheter ablation.
Mechanisms
Three primary mechanisms are involved in SVT: (1) enhanced automaticity, (2) triggered automaticity, and (3) re-entry. Automaticity is the ability of cardiac myocytes to spontaneously depolarize, which leads to myocardial contractions. Sinus and AV nodes are the primary sites of automaticity in the heart. Enhanced automaticity occurs when myocytes outside the sinus or the AV node depolarize spontaneously, which leads to atrial or ventricular ectopic beats or SVT and to ventricular tachycardias (VTs) when repetitive ventricular ectopic beats occur.3
Triggered automaticity results from spontaneous myocardial contractions that occur secondary to oscillations during repolarization reaching threshold and leading to a depolarization.4,5 These oscillations are referred to as after-depolarizations. Arrhythmias arising from triggered automaticity have characteristics shared by both enhanced automaticity and re-entry. They are highly catecholamine sensitive and have warm-up and cool-down phases with a wide variation in heart rate, similar to other forms of automaticity. They can be induced and terminated with pacing maneuvers and respond to direct-current cardioversion, as do re-entrant arrhythmias. Triggered automaticity is thought to play a role in the arrhythmias seen in digoxin toxicity.6
Re-entry occurs when a wavefront of electrical activation travels through tissue for a distance and then re-enters the original tissue and propagates through the circuit again. Re-entrant SVT is the most common type of tachycardia seen in pediatrics. To have a re-entrant circuit, at least two pathways with distinct conduction properties and refractory periods must be present. The sequence of events in a re-entrant circuit is as follows. A stimulus encounters two distinct pathways, with one pathway refractory to conduction (the pathway with the longer refractory period) and one ready for conduction (the pathway with the shorter refractory period). The impulse is conducted along the pathway, with the shorter refractory period having enough conduction delay to allow the first pathway to recover and to conduct the impulse back to the original site of entry into the circuit. The impulse then re-enters the original pathway and transverses the circuit again. Re-entry can occur in the sinus node, the atrial tissue, the AV node, and the ventricular tissue or between the atrium and the ventricle. Those re-entry circuits between the atrium and the ventricle involve specialized conduction tissue referred to as accessory pathways. In AV nodal re-entrant tachycardia (AVNRT), the accessory pathway is in the AV node or in the AV nodal region. Moe and colleagues initially described the evidence of dual AV node physiology in 1956.7 Re-entry tachycardias have characteristics that are distinct from those seen in automatic tachycardias. They often have sudden onset and termination, with patients frequently feeling that they have been “switched” on and off. The tachycardia is regular, with little variation in rate. Re-entry tachycardias often respond to vagal maneuvers by slowing or terminating the tachycardia. These tachycardias are easily induced and terminated with pacing protocols ranging from single premature stimuli to burst pacing or by direct-current cardioversion.
Specific Mechanisms in Children
The most common specific mechanism of SVT in children, representing 75% to 90% of arrhythmias in children without other cardiac conditions, is re-entry through an accessory pathway between the atrium and the ventricle or within the AV node.8,9 Over two thirds of these pathways are concealed, and one third are manifested as WPW or other forms of pre-excitation. AV nodal re-entry represents around 15% of pediatric SVTs, most commonly seen in adolescents, with up to one third of SVTs presenting in adolescence, the cause being AVNRT.9,10 Automatic atrial ectopic tachycardia represents 10% to 18% of SVTs in children. Atrial flutter and atrial fibrillation (AF) are responsible for 10% and 40% in non-postoperative and postoperative children, respectively, and junctional ectopic tachycardia responsible for less than 1% in non-postoperative patients.
Atrioventricular Reciprocating Tachycardias
Presentation and Treatment
Tachycardia may be seen as early as during fetal development. The diagnosis of fetal SVT may be made by auscultation of the baby’s heart rate during maternal examination and is confirmed by fetal ultrasonography or fetal magnetocardiography.11 See Chapter 76 for detailed information on these arrhythmias.
It has been reported that 50% to 60% of SVTs in children present in the first year of life, with accessory pathway–mediated re-entrant tachycardia being the most common mechanism.12,13 The heart rate in infants with SVT is usually 220 to 320 beats/min. Neonates and infants with tachycardia are often very ill, presenting with congestive heart failure, which occurs if the tachycardia has been present for more than 48 to 72 hours before the patient receives medical care. This occurs because of the infant’s inability to communicate and the family’s lack of awareness that the child could have a significant medical condition. Parents may note that the infant is acting somewhat different from normal, is more irritable, or is not eating well. This is often interpreted as colic or some other “normal” childhood problem. At presentation, these babies often are acidotic from decreased cardiac output and may need aggressive resuscitation, including artificial ventilation and rapid termination of the tachycardia. Intravenous (IV) adenosine is effective in the acute termination of SVT in this population, but IV access is often difficult in a 3- to 4-kg baby with congestive heart failure. It has been suggested that in the treatment of infants, the starting doses of adenosine should be higher than those generally recommended as the average effective dose in infants, which is around 200 µg/kg.14 If IV access is not possible, transesophageal overdrive pacing has proven to be effective. Once the rhythm is restored to normal and the cardiac function has begun to recover, IV access becomes easier, and IV medications can be administered. If the infant is hemodynamically compromised and the rhythm cannot be medically converted, electrical synchronized cardioversion is indicated.
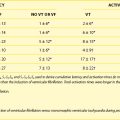
Medications for the acute treatment of SVT are shown in Table 75-1. Digoxin is a first-line medication used in the treatment of SVT in infants with decreased myocardial performance. It is contraindicated as a chronic treatment in patients with WPW syndrome but can be used acutely in those with decreased ventricular function with careful monitoring. The authors of this chapters do not discharge patients with WPW syndrome on digoxin because of its potential effect to enhance conduction of atrial impulses to the ventricle but switch them to another medication once the cardiac function has normalized. Other IV medications used acutely to treat SVT include β-blockers such as esmolol, amiodarone, and procainamide. These medications must be used with caution because of their negative inotropic effects, which can lead to worsening of cardiac function. The use of IV calcium channel blockers is contraindicated in infants and children younger than 1 year because as sudden death has been reported in some cases.15,16 In a long-term study from Sweden, the SVT was managed with a single drug in 62% of patients.17
Table 75-1 Pharmacologic Agents for Acute Treatment of Supraventricular Tachycardia
| AGENT | INITIAL TREATMENT (IV) |
|---|---|
| Adenosine | 50–100 µg/kg, increase by 50-µg/kg increments |
| Every 2 min to 400 µg/kg or 12 mg maximal dose | |
| Amiodarone | IV: 5 mg/kg over 1 hour, followed by IV bolus of 2.5 mg/kg q4–6h |
| Digoxin | Dose is age dependent |
Give in 3 doses (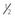 TDD, TDD, 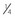 TDD, TDD, 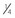 TDD) TDD) |
|
| Preterm infant: 10–20 µg/kg TDD | |
| Term newborn to adolescent: 30–40 µg/kg TDD oral to maximal TDD of 1–1.5 mg (IV 3/4 PO) | |
| Oral maintenance: 10 µg/kg/day q12h | |
| Esmolol | IV Load: 200–500 µg/kg/min over 2–4 min. May increase in 50–100 µg/kg/min increments (maximum dose = 1000 µg/kg/min) |
| Maintenance infusion: 50–200 µg/kg/min | |
| Phenylephrine | 100 µg/kg bolus |
| 10 µg/kg/min infusion | |
| Procainamide | 5 mg/g over 5–10 min or 10–15 mg/kg over 30–45 min |
| 20–100 µg/kg/min infusion | |
| Propranolol | 0.05–0.1 mg/kg over 5 min q6h |
| Verapamil | 0.05–0.30 mg/kg over 3–5 min |
| Maximal dose: 10 mg |
IV, Intravenous; TDD, total digitalizing dose.
Prognosis
While the majority of SVTs will resolve around 1 to 2 years of age, one third will recur by adolescence. Recurrence is only 10% for AVRT secondary to concealed accessory pathway but more than 31% if a WPW pattern persists.17
Presentation in Older Children
After the peak of presentation in the first year of life, the other two common ages of presentation are early childhood (6 to 8 years of age) and adolescence.12,18 SVT in children is predominantly caused by concealed or manifested accessory pathways (WPW) throughout early childhood, with the proportion of patients with AVNRT increasing with age.8 The heart rate in older children is generally in the range of 160 to 280 beats/min. Older children often present with palpitations or dizziness. It is not uncommon to hear a 3- or 4-year-old complain that the heart is “beeping” too fast. These patients are generally not in SVT long enough to develop congestive heart failure, as seen in infants, because they can communicate to their caregivers that they are experiencing something abnormal or unusual. With the exception of those with WPW syndrome with rapid conduction down their accessory pathway during an atrial tachycardia, patients rarely present with syncope during SVT.
Treatment
The long-term treatment of infants with AV reciprocating tachycardia includes the use of oral preparations of the medications listed in Table 75-2, with digoxin or propranolol/β-blocker medications being the most commonly used agents. During initiation of therapy with oral antiarrhythmics in infants, the authors of this chapter monitor the patients in the hospital for at least five half-lives of the medication, allowing a steady state to be reached, watching for side effects, and educating the family about administering the medication and recognizing the tachycardia. The family can also be taught about simple vagal mechanisms to interrupt the tachycardia. When starting therapy with oral β-blockers in infants and young children, the patient must be monitored for hypoglycemia and the caregivers must know how to recognize the symptoms of hypoglycemia. The authors of this chapter ask the family to notify medical personnel if their child is not tolerating the medication or if side effects occur. It is not the authors’ practice to discharge patients with heart rate monitors. Families can assess their children for recurrent SVT without resorting to continuous monitoring. Most infants are treated with oral medications for 10 to 12 months, with the dosage adjusted on the basis of weight gain. At 10 months to 1 year of age, if no recurrences of the SVT have occurred, the patient will be weaned from the medication unless the WPW pattern is still present on the ECG. Approximately one third of all patients who develop SVT in the first 3 months of life will outgrow it by 1 year of age.12,18,19,20
The short- and long-term medical management of children and adolescents is similar to that of infants. The exception is that catheter ablation becomes more of a therapeutic option around 4 to 5 years of age and is more commonly used after 8 years of age. Ablation can and has been performed in younger children, but the overall consensus is that it should only be used for patients 4 years of age or younger who have medically refractory arrhythmias, as the risks of the procedure are higher under 4 years of age.21,22 See Chapter 78 for detailed information on catheter ablation in children.
Wolff-Parkinson-White Syndrome
Presentation
WPW syndrome in children is very similar to that in adults. This syndrome involves an accessory pathway between the atrium and the ventricle that usually has bi-directional conduction properties. The reported incidence of WPW syndrome ranges from 1 to 4 per 1000 live births.23 Of all patients who present with WPW syndrome in childhood, one fifth to one third will have associated cardiac abnormalities. The most common congenital lesions include Ebstein’s anomaly of the tricuspid valve and L-transposition of the great arteries.18 Patients with WPW syndrome present any time from fetal life through adolescence. The patients who present during fetal life and early infancy present with re-entrant SVT. Children and adolescents also present with re-entrant SVT but can occasionally present with atrial fibrillation (AF) with rapid conduction down the accessory pathway. Approximately 10% of children will present with an antidromic tachycardia that uses the accessory pathway as the antegrade limb, with approximately half of these patients having multiple accessory pathways.24 A number of asymptomatic patients are noted to have the pattern of WPW syndrome on ECGs obtained for other reasons. Sudden death can be the first manifestation of WPW, leading some authors to suggest that risk stratification, including an EPS, should be done for all patients with WPW beyond infancy and early childhood.25 The presence of a short refractory period or multiple pathways increases the risk of a subsequent life-threatening event, and catheter ablation is advised.26 Occasionally, an infant presents with a narrow-complex tachycardia with no evidence of a WPW pattern on a baseline ECG, but pre-excitation becomes obvious when the patient is treated with a medication (e.g., digoxin) that slows AV nodal conduction.
Treatment
A special consideration in the pediatric population is the patient who presents with asymptomatic WPW syndrome following an ECG obtained for some other indication such as chest pain or for a school physical examination. The authors recommend that patients older than 6 to 8 years of age have the evaluation mentioned above to assess whether they have rapid conduction down their accessory pathways. Generally, these patients are taken to the electrophysiology laboratory to measure their minimum cycle length of pre-excitation both with atrial pacing and during AF. A minimum cycle length of pre-excitation of less than 220 ms is used during AF as a marker that the patient may have a significant risk of sudden death. This is based on studies performed by Bromberg et al and Paul et al, which demonstrated that these values are helpful, though not fully predictive of cardiac arrest and syncope.27,28 Additional studies by Pappone and Santinelli show an increased risk for sudden death with WPW with short accessory pathway effective refractory periods and with multiple pathways and recommend that catheter ablation be performed.26,29,30
Activity Recommendations
Activity restriction is generally not required in individuals with controlled SVT. If WPW is present, risk stratification with EPS and possible catheter ablation are recommended before participation in any activity, according to the Bethesda Guidelines.31
Primary Atrial Tachycardia
Ectopic Atrial Tachycardia
Ectopic atrial tachycardia (EAT), or automatic atrial tachycardia, is an arrhythmia arising from both atria with inappropriately fast atrial rates. This tachycardia represents approximately 10% of the SVT seen in the overall population.32 The heart rates seen in EAT vary on the basis of the patient’s age and catecholamine state during the tachycardia. The heart rates in automatic atrial tachycardias will be inappropriately fast for the patient’s level of activity and are generally in the 130 to 250 beats/min range. In general, the rates are not as fast as those seen in re-entrant tachycardias. The pulse rate may not be reflective of the atrial rate because of variable atrial conduction through the AV node.
Tachycardias arising from foci of increased automaticity can be found in all areas of the atria. The automatic foci are more likely to be located in certain areas than in others. In the right atrium, the foci are frequently found in the right atrial appendage, and in the left atrium, they are often mapped to the orifices of the pulmonary veins.33–35
Presentation
EAT is not seen frequently in young infants, but the overall incidence of automatic atrial tachycardia reported by Gillette was 18% of all SVT seen in children.36 EATs usually have a “warm-up” phase in which the heart rate will gradually increase to its maximum rate. During termination, it will “cool down” with a gradual slowing of the rate and become indistinguishable from that of sinus rhythm. This gradual increase in heart rate and termination, which is markedly different from the sudden onset and termination seen in re-entrant SVT, makes it difficult for the patient to recognize the tachycardia. Failure to perceive the arrhythmia and its incessant nature can lead to significant depression of myocardial function, which results in a tachycardia-mediated cardiomyopathy. Many patients with EAT present with signs of congestive heart failure with decreased left ventricular (LV) contractility, AV valve regurgitation, and atrial dilation.37 If the tachycardia is not treated aggressively, the myocardial function can continue to decline, resulting in an irreversible cardiomyopathy. Resolution of the cardiomyopathy can take 6 to 12 months after the tachycardia has been terminated.
Evaluation
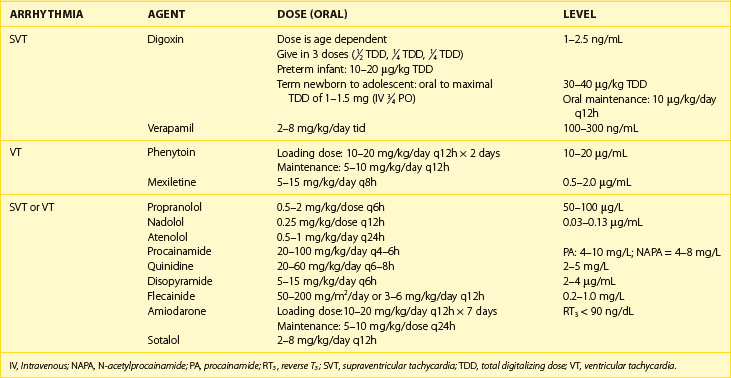
ECGs in patients with an EAT generally show a P-wave axis distinct from sinus rhythm. When the focus arises from the left atrium, the P wave is negative in lead I; those with the focus in the low right atrium show a negative P-wave axis in lead aVF and a positive P wave in lead I. Occasionally, the focus is in an area close to the sinus node or in the high right atrium with the P-wave axis similar to sinus tachycardia (0 to 90 degrees in the frontal plane). This can lead to a delay in diagnosis and institution of therapy, when the rhythm is thought to be sinus tachycardia. EAT, as well as other primary atrial tachycardias, can show variable degrees of AV conduction on ECG and Holter monitoring, which is often helpful in establishing the diagnosis. Figure 75-1 represents an ECG of a patient with EAT.
Treatment
The medical management of EAT may be problematic, as complete control of the tachycardia is difficult to attain. Two strategies are used in the treatment of EAT. One strategy is an attempt to slow the ventricular rate by slowing conduction through the AV node. The primary medication used in this effort is digoxin, which slows the conduction through the AV node by enhancement of vagal activity. Digoxin is a positive inotropic agent and helps improve ventricular performance. Calcium channel blockers can increase AV block but are negative inotropes and should be used with caution. Medications that have the potential to slow the tachycardia include β-blockers and class I and class III antiarrhythmics. β-Blockers have the potential to slow the tachycardia by blocking the effect of catecholamines on the ectopic focus. Class IC medications such as flecainide and propafenone have shown some success in the management of EAT.38,39 Amiodarone and sotalol (class III) have been used with modest success by slowing conduction throughout the myocardium as well as by slowing AV conduction.40–42 Class IA antiarrhythmic medications such as procainamide decrease automaticity, prolong refractoriness, and slow conduction velocity.43,44 All of these medications have the potential to decrease myocardial performance and must be used with caution in patients with decreased LV function. Similarly, the side effects must be carefully monitored. The second strategy, nonpharmacologic methods of treatment, is gaining increasing interest because of the difficulty in the medical management of EAT and the sequelae of uncontrolled tachycardia. These procedures include surgical ablation procedures, which have reasonable success rates but require an open chest procedure, frequently including cardiac bypass.45,46 Catheter ablation, using radiofrequency or cryothermal energy, has become the treatment of choice for patients with poorly controlled EAT.35,34,47 Certain technical aspects may make catheter ablation of EAT challenging. It is difficult to induce EAT. If the arrhythmia does not occur spontaneously, it cannot be mapped and ablated in the electrophysiology laboratory. EAT is considerably catecholamine sensitive. It is not uncommon for the tachycardia to “go to sleep” when the patient does, making sedation an issue in the laboratory. See Chapter 78 for information on ablation.
Multifocal Atrial Tachycardia or Chaotic Atrial Tachycardia
Multifocal atrial tachycardia (MAT), or chaotic atrial tachycardia (CAT), is a primary atrial tachycardia arising from multiple areas of enhanced automaticity in the atria. By definition, the tachycardia must have at least three distinct P-wave morphologies to be considered a MAT. These tachycardias are similar to other primary atrial tachycardias in that there may be variable conduction from the atrium to the ventricle. MAT is occasionally confused with AF in that multiple P-wave morphologies and variable P-R and R-R intervals are present. The most common presentation occurs in the newborn, with a large percentage resolving spontaneously over time.48
Presentation and Management
Yeager et al reported a 17% incidence of sudden death in patients with MAT.49 Two of these deaths were thought to be bradycardia mediated. The bradycardia that is encountered may be related to the aggressive medical therapy required to control the rapid heart rates.
Atrial Flutter
Neonates present with atrial flutter in utero, at the time of birth, or shortly thereafter. In most cases, the patient has a structurally normal heart, although congenital heart disease may be present. Atrial flutter represents 15% to 50% of all fetal SVTs, often resulting in fetal hydrops (a form of congestive heart failure).50–52 Treatment with sotalol has been effective in 80% of fetuses with atrial flutter.53 See Chapter 76 for additional information on fetal atrial arrhythmias.
Atrial flutter seen in the newborn presents most commonly in the first month of life, often within 2 days of birth. A report of 50 cases indicated that heart failure was present in 20% of these infants. The atrial rate range was 340 to 580 beats/min.54 Twenty-two percent developed additional atrial or supraventricular arrhythmias during the follow-up. AF is generally converted to sinus rhythm with medication, transesophageal pacing, or direct current cardioversion. In the remainder of these patients, recurrence is rare.
Patients with congenital heart disease may develop atrial flutter before or following surgical interventions. This is most commonly seen in those lesions with extensive atrial surgery or prior postoperative atrial dilation. The multiple atrial flutter circuits seen after surgery for congenital heart defects results in slower rates and unusual flutter wave axis and morphology. These different “flutter” characteristics have led many experts to refer to this as intra-atrial re-entry tachycardia rather than as atrial flutter. Refer to Chapter 80 for additional information on atrial flutter in association with congenital heart disease.
Atrial Fibrillation
AF has been studied in individuals with WPW syndrome. Inducible AF has been associated with spontaneous clinical AF in 73% and subsequent ventricular fibrillation in 27%.29 Additional studies of subjects with WPW syndrome reported that AVRT degenerated to AF in 51% and to AF with rapid ventricular response in 34%.56
Junctional Ectopic Tachycardia
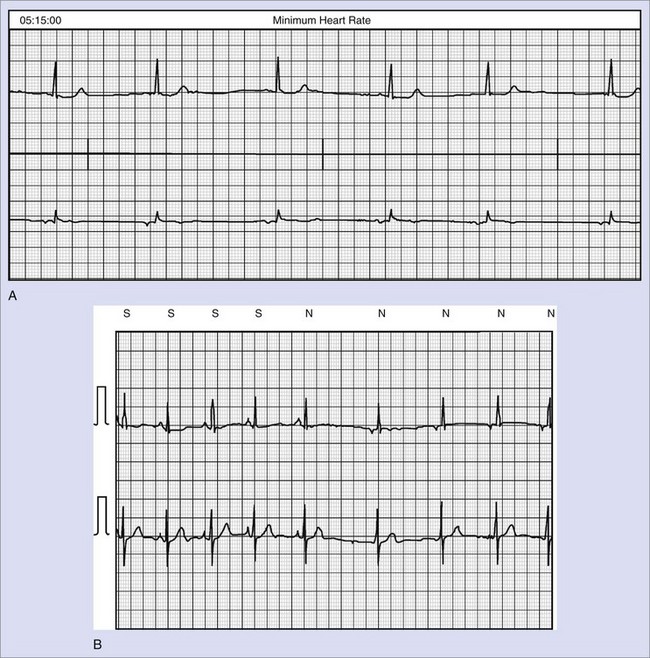
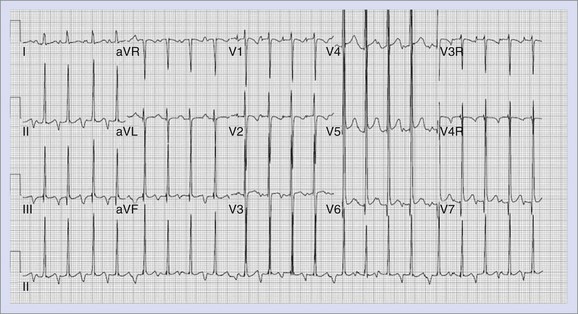
Junctional ectopic tachycardia (JET) is an automatic tachycardia that arises from the AV junction. Two distinct types of JET are seen in childhood. The first is a familial form that occurs in early infancy and may be associated with congenital heart defects in up to 50% of patients.57 The second type is seen in the early postoperative period following repair of congenital heart disease (see Chapter 80).58 In both forms, the tachycardia appears to be secondary to enhanced automaticity. In those patients who present with the familial type of JET, the heart rates will range from 180 to 240 beats/min. The ECG findings in JET show a tachycardia with a ventricular rate that is faster than the atrial rate, with a narrow QRS complex similar to that seen in the patient’s normal sinus rhythm. The ECG findings of JET are shown in Figure 75-2. Rarely, patients with JET develop rate-related aberrancy, and some postoperative patients have a pre-existing bundle branch block that will lead to a wide-complex tachycardia. If the QRS is wide, the diagnosis of VT must be considered and ruled out by comparison with the QRS in normal sinus rhythm or by pacing the atrium faster than the ventricular rate to demonstrate conduction through the His-Purkinje system with persistent wide-complex QRS morphology.
JET tends to be faster and more incessant when it manifests before 6 months of age.59 Patients with JET can present with signs and symptoms of congestive heart failure secondary to the persistently elevated heart rate. Sudden death has been reported in this patient population.59,60
Treatment
Treatment strategies for the familial form of JET include digoxin to slow the rhythm and provide inotropic support. Digoxin alone may not be sufficient to manage this arrhythmia, and the addition of a class IA, class IC, class II, or class III antiarrhythmic agent may be required. It has been the chapter authors’ experience that a combination of these medications is required to control the tachycardia. Class III agents such as IV amiodarone are quickly added to treat individuals who have rapid rates and poor ventricular function, while carefully monitoring blood pressure and cardiac output. The amount of antiarrhythmic medication needed to control the rate to prevent decompensation of cardiac function may suppress the sinus node considerably, necessitating a pacemaker. Amiodarone appears to be the most effective agent in the largest group reported with a 60% success rate. Patients with JET have been treated with catheter ablation with a 80% to 85% success rate but with a risk of AV block, which has led to the more recent recommendations of the use of cryothermal energy (see Chapter 78).61,62 Because of the proximity of the AV node and His bundle to the area of enhanced automaticity that is responsible for JET, catheter ablation in this setting carries a relatively high risk of causing complete heart block. It is possible that, over time, the junctional rate will slow to a point that the patient could be weaned from the chronic medications. Refer to Chapter 80 for additional information on postoperative JET.
Ventricular Arrhythmias
Ventricular arrhythmias include premature ventricular contractions (PVCs), couplets, nonsustained ventricular tachycardia (NS-VT), sustained VT, and ventricular fibrillation (VF). PVCs may be seen in 15% of normal newborns, one third of normal adolescents, and two thirds of adolescents and adults with repaired congenital heart disease.63 PVCs may occur without identifiable cause in children and are often benign. PVCs may be associated with acute or more chronic conditions. A marked difference in prognosis for PVCs is seen between children with normal hearts and those with abnormal hearts, so investigation of children with PVCs for associated conditions should be undertaken.
Prognosis
Long-term follow-up suggests that PVCs and VT disappear over time in 37% to 65% of patients with normal hearts.64 Sudden death is rare in children with PVCs with otherwise normal health, but a few cases have been reported in the literature. In children with abnormal hearts, PVCs may be precursors of more serious arrhythmias, especially if they are complex-multiform, coupled, or associated with VT.
Ventricular Tachycardia
As with PVCs, VT occurs in both acute and chronic situations. Ventricular arrhythmias are less common than supraventricular arrhythmias in children but appear to be occurring more frequently in recent years or are being recognized to a greater extent. An increase in VT in patients after congenital heart surgery is seen, as survival after complex surgery has increased (see Chapter 80).65 Improved methods of surveillance and diagnosis of arrhythmias have made it possible to recognize various etiologies of VT in children, with the most common being congenital LQTS, cathecholaminergic polymorphic ventricular tachycardia (CPVT), hypertrophic and dilated cardiomyopathies, ARVC, myocarditis, abnormal foci or circuits in structurally normal hearts, and idiopathic etiologies.
Electrocardiographic Manifestations
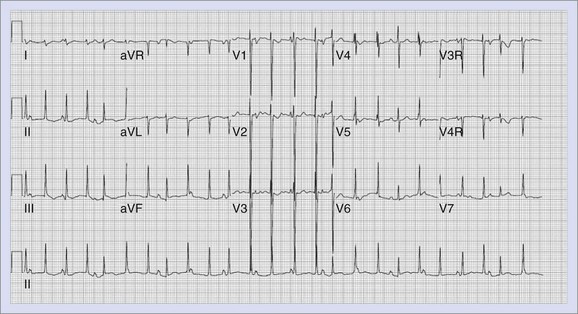
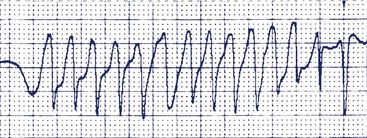
Further differentiation is made according to the morphology, with VT being described as monomorphic or polymorphic. Two types of polymorphic VT have been described, torsades de pointes and bi-directional VT (Figure 75-3). Torsades de pointes is associated with LQTS and so named because of its twisting, undulating nature. Bi-directional VT, with beat-to-beat variation in the QRS axis on ECG, has been associated with digoxin toxicity, familial hyperkalemic paralysis, or CPVT.66
Etiology of Ventricular Tachycardia
Causes of acute VT not associated with congenital heart defects are shown in Box 75-1. These most common causes include metabolic and electrolyte abnormalities; infectious processes such as myocarditis, which may cause LV microaneurysms67; human immunodeficiency virus (HIV) infections68; blunt cardiac trauma, including commotio cordis69–71; coronary ischemia, especially in association with Kawasaki disease; and drugs such as caffeine, inhalation anesthetics, and recreational drugs, including amphetamines and cocaine.
The causes of chronic or recurrent VT include congenital heart disease, both preoperatively and postoperatively; acquired heart diseases; metabolic disorders, including disorders of fatty acid metabolism72; neuromuscular disorders such as Duchenne’s muscular dystrophy; cardiomyopathies, including ARVC; hypertrophic cardiomyopathy (HCM); tumors and infiltrates; left ventricular noncompaction (LVNC), VT originating in both the right ventricle and the left ventricle, associated with structurally normal hearts; VT associated with LQTS; and other primary electrophysiological abnormalities such as Brugada syndrome and CPVT. A detailed list is provided in Table 75-3. While failure to identify a specific cause for VT in children is not unusual, persistence in evaluations often identifies the pathology in patients initially thought to have an unidentifiable etiology.73–77
| Congenital Heart Disease | Ebstein anomaly |
| Tetralogy of Fallot, absent PV leaflets | |
| Aortic valve disease, AI/AS | |
| Mitral valve prolapse | |
| Hypertrophic cardiomyopathy/IHSS | |
| Coronary artery anomalies | |
| Eisenmenger syndrome, pulmonary hypertension | |
| Postoperative CHD | Tetralogy of Fallot, DORV |
| Ventricular septal defects | |
| AV canal defects | |
| Aortic valve disease, stenosis, and insufficiency | |
| Single ventricle complexes status post–Fontan repair D-TGA status post–intra-atrial repair | |
| Acquired heart disease | Rheumatic heart disease |
| Lyme disease | |
| Myocarditis | |
| Kawasaki disease | |
| Cardiomyopathies | Hypertrophic |
| RV cardiomyopathy/dysplasia | |
| Dilated cardiomyopathy | |
| Postviral | |
| Connective tissue disease: SLE | |
| Marfan syndrome | |
| Muscular dystrophy, Friedrich ataxia | |
| Tumors and infiltrates | Rhabdomyoma |
| Hemosiderosis: Thalassemia, sickle cell disease | |
| Oncocytic cardiomyopathy | |
| Leukemia | |
| Idiopathic/structurally normal heart | RV outflow tract VT |
| LV septal VT/fascicular tachycardia | |
| Primary arrhythmias | LQTS |
| Congenital complete heart block | |
| Familial VT | |
| Other | Myocardial ischemia/infarction |
AI, Aortic insufficiency; AS, aortic stenosis; AV, atrioventricular; CHD, congenital heart disease; DORV, double outlet left ventricle; D-TGA, D-transposition of the great arteries; IHSS, idiopathic hypertrophic subaortic stenosis; LQTS, long QT syndrome; LV, left ventricular; PV, pulmonary valve; RV, right ventricular; SLE, systemic lupus erythematosus; VT, ventricular tachycardia.
Mechanism of Ventricular Tachycardia
VT has been reported to result from re-entry, triggered automaticity, and abnormal automaticity. An EPS is helpful in differentiating these mechanisms. The mechanisms of VT in children include re-entry in 60% and abnormal automaticity in 40%.78 Re-entry is most often the mechanism in patients with congenital heart disease after surgical repairs, related to re-entry circuits that develop around suture lines and ventriculotomy scars. Triggered automaticity is thought to be the mechanism in CPVT.
Clinical Correlations
Presentation of patients with VT varies and depends, to a large extent, on the underlying etiology and clinical status with regard to myocardial function and structure. In one study of patients with VT with structurally normal hearts, presentation was most common in infancy (48%), with 58% being younger than 6 months.73 Associated findings were heart failure in 30%, hemodynamic compromise or collapse in 23%, and in utero diagnosis in 18%. Diagnosis was incidental in 30%. No specific etiology was found in 50%, with cardiomyopathy or myocarditis (20%) being the most common etiology identified.
Clinical Signs and Symptoms
The type and degree of symptoms appear to be rate related, with symptoms being most common in patients with rates greater than 150 beats/min. Except for those patients with underlying cardiac disease or ventricular dysfunction, patients with VT have symptoms similar to those of SVT, with the severity of symptoms relating more to the rate than to the mechanism of tachycardia. Symptoms include dyspnea, shortness of breath, chest or abdominal pain, palpitations, dizziness, syncope, and cardiac arrest or sudden death. Older children may exhibit exercise intolerance or easy fatigability. Infants may feed poorly and be irritable or lethargic. Patients with VT and heart disease usually have symptoms, whereas only one third with normal hearts and VT have symptoms. The type of symptom relates to both the tachycardia rate and the underlying state of the myocardium. Sudden death occurs most commonly in the presence of an abnormal heart but has also been reported in patients with normal hearts.74,79,80 Children younger than 5 years or those in incessant tachycardia may not have a perception of a fast heart rate or be able to accurately express what they are feeling. Signs include palpitations, sensation of a rapid heart rate, tachypnea, or hypotension with accompanying pallor and diaphoresis as well as signs of congestive heart failure. Although VT usually has a sudden onset, it may occur during exercise and be difficult to perceive. It may gradually “warm up” or increase in rate.
Specific Associated Conditions
Accelerated Ventricular Rhythm
An accelerated ventricular rhythm is a rhythm originating from the ventricle with all the characteristics of VT but with a rate that is only slightly more rapid than the underlying sinus rhythm, usually less than 120 beats/min. It is often seen in children with normal hearts. This arrhythmia is not uncommon in neonates and has been reported in two patients with fetal tachycardia. It is self-limited, resolving in 2 weeks to 3 months after birth.81 These early ventricular arrhythmias are probably related to developmental factors associated with the autonomic nervous system. In older children, these arrhythmias may be related to unidentified viral infections with myocarditis that affects only the conduction system. This arrhythmia is seen around puberty and probably relates to autonomic and hormonally mediated factors. In addition, accelerated ventricular rhythms have been reported in association with metabolic abnormalities, medication, ARVC, and myocardial infarction.82 In pediatric patients, this arrhythmia is generally thought to be benign, even in the occasional patient who has congenital heart disease.82,83 It has been suggested that those rhythm disturbances arising from the right ventricular outflow tract (RVOT) may be a marker for future development of ARVC in some patients.82–84
Idiopathic Ventricular Tachycardia
VT in the absence of underlying heart or known genetic disease is unusual in childhood. Of that population, 27% present in infancy, with the mean age of presentation being 5 years.85 Severe heart failure was uncommon but did occur in 12%, with 36% having some evidence of LV dysfunction. Resolution can be expected in two thirds of patients over time. The most favorable prognosis is in patients with right ventricular tachycardia and in infants.
Arrhythmogenic Right Ventricular Cardiomyopathy
An unusual cause of VT known as arrhythmogenic right ventricular cardiomyopathy or arrhythmogenic right ventricular dysplasia (ARVD) was first described in 1978.86 The VT has a LBBB pattern in most instances. One pediatric series reported ARVC in 30% of its patients with VT and an apparently normal heart, although it is much less common in most other pediatric series. ARVC is a familial form of right ventricular (RV) cardiomyopathy associated with sudden death.87 It has an autosomal dominant genetic pattern with variable penetrance and variable expression. The pathologic lesion involves massive replacement of the RV wall by fibrous tissue, fatty tissue, or both. Focal myocardial changes may be present and include necrosis, degeneration, or hypertrophy and chronic inflammatory infiltrates. This is a progressive process that starts with the epicardium or midmyocardium and extends to become transmural. Although many cases are familial, sporadic cases have been reported as well. ARVC has not been commonly reported in young patients, as it usually presents in the second to fourth decade, with a male predominance, but should be considered in previously healthy children or adolescents who present with VT.
Evaluation
Patients who are suspected of having this condition should have an ECG, echocardiogram, and MRI. Because of the localized nature of this condition, echocardiography may not be diagnostic. MRI may be more helpful by demonstrating thinning of the RV myocardium replaced by fatty tissue or showing localized areas of hypokinesis in the infundibulum, free wall, or RV apex, accompanied by RV dilation and decreased contractility. LV free wall and septal involvement in this process has been noted.88 Although the above findings have been helpful in making the diagnosis in adults, standard MRI criteria applied to 81 pediatric patients suspected of having ARVD provided a low diagnostic yield.89 Other potential diagnostic modalities include exercise testing, contrast ventriculography, signal-averaged ECG, and single photon emission computed tomography (SPECT) analysis.90–96 Research on genetic identification is still under way. Immunohistochemical analysis of myocardial biopsies has demonstrated reduced levels of plakoglobin at intercalated discs in patients with ARVC and not in other forms of heart-muscle disease.97 This may prove helpful in establishing a diagnosis of ARVC. Currently, no single gold standard exists, and the best strategy consists of combining information from several diagnostic tests.89 Children in affected families should be evaluated by using ECGs, 24-hour Holter monitors, echocardiograms, and MRIs.77,98
Treatment and Follow-Up
Variable medical therapies, including β-blockers and sotalol, have been suggested for patients with frequent, symptomatic, or potentially life-threatening ventricular arrhythmias. Automatic implantable cardioverter-defibrillators (ICDs) have been used and can be life saving in these patients.99,100 Extensive surgical procedures, including a complete electrical disconnection of the RV free wall, have been reported.101 The prognosis and clinical course reported in these patients have been quite variable. Continued surveillance with periodic Holter monitoring and exercise stress testing are important in the follow-up of this patient group, as the incidence of serious arrhythmias increases with age.
Long QT Syndrome
Congenital LQTS is an inherited condition characterized by syncope, seizures, and sudden death, associated in most individuals with a prolongation of the Q-T interval on the ECG.102 An example of the ECG in LQTS is shown in Figure 75-4. In addition to the prolongation of the QTc, these patients often have bizarre or notched T-wave morphology with prominent U waves or T-wave alternans. They develop life-threatening VT, known as torsades de pointes, or VF. This syndrome includes Jervell and Lange-Nielson syndrome (JLNS), described in 1957, associated with congenital deafness caused by an autosomal recessive inheritance and Romano-Ward syndrome, described in 1963 and 1964, demonstrating autosomal dominant inheritance, without hearing deficit.103–105
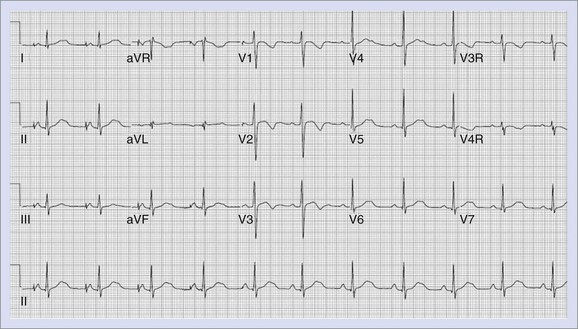
FIGURE 75-4 Electrocardiogram of patient with long QT syndrome. The QTc measures 490 ms. Note the long, notched T waves.
The prevalence of the disorder is estimated to be 1 : 2000 to 1 : 10,000.106–108 This condition has variable expression and penetrance within families, leading to a spectrum of severely affected members with repeated cardiac events and arrest to those with the identical mutation but who are totally asymptomatic.109
In 1993, statistics from a group of 287 children were compiled from a number of medical centers.110 The initial presentation was cardiac arrest (9%), syncope (26%), seizures (10%), presyncope, or palpitations (6%). Sixty-seven percent with symptoms had exercise-related symptoms. Thirty-nine percent were identified because of family history or the identification of other family members with the syndrome. Thirty-nine percent of the patients were asymptomatic at presentation; of these, 4% experienced sudden death compared with 8% overall. The strongest predictors of sudden death were QTc longer than 0.60 seconds and noncompliance with taking recommended medication.
Bradycardia is commonly seen in these patients, and some may develop or present with second-degree AV block. This is more common in neonates who may have second-degree or third-degree AV block, but it may be seen in older children, especially during exercise.111
One series reported sudden death occurring in 73% without treatment, and others have reported sudden death in 21% of symptomatic patients in the first year after presenting with syncope.112,113
Diagnosis and Evaluation
The diagnosis of this syndrome is made from a variety of criteria. Schwartz et al provided some criteria and suggested a scale for identifying these patients by categorizing them into high-risk, intermediate-risk, or low-risk groups.114 All criteria involve measurement of the Q-T interval and taking a careful history of syncope, seizures, and arrhythmias in the patients and their families. Commonly, a complete history may reveal syncope or seizures associated with exercise or emotional stress or a family history of sudden death in young relatives, including unexplained automobile accidents or drowning.
Additional studies such as 24-hour monitoring and exercise stress testing may provide helpful information in the form of significantly prolonged Q-T intervals, especially during recovery from exercise, or the occurrence of polymorphic ventricular arrhythmias during or after exercise. The use of provocative tests such as isoproterenol or epinephrine infusions can identify some subclinical cases, especially LQT1 with a prolongation or paradoxical response in these individuals.115,116
A high level of suspicion is needed to diagnose LQTS. Any patient who presents with syncope during or immediately after exercise or with VT, especially of the polymorphic or torsades de pointes type, or in association with physical or emotional stress should have an ECG, with determination of QTc intervals. Evaluation of a resting ECG may not be sufficient as more than 10% to 15% of gene carriers may have normal ECGs.117 A more worrisome study by Priori showed an even lower penetrance of the gene in Italian families, with probands initially thought to have sporadic occurrences of LQTS. Genetic studies revealed multiple family members who were genetic carriers but with normal ECGs.118 ECGs should be obtained in all family members of identified individuals with LQTS. When genetic mutations are identified, this can be used to screen family members more specifically.
Clinical Implications of Molecular Genetics
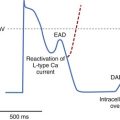
The molecular genetic understanding of LQTS began in 1991.119 Many other genetic discoveries have resulted in the current and rapidly expanding knowledge of the etiology of LQTS.120–125 Genetic studies have identified repeated mutations in at least 12 genes that encode for proteins that modulate the ion channels, primarily potassium or sodium channels, causing LQTS by altering cardiac repolarization and increasing the risk for ventricular arrhythmias. Mutations in a few other genes have been found in only isolated individuals. It is estimated that 25% of the gene mutations have not yet been identified. The mutations are primarily in the coding regions of the genes. These mutations result in a decrease in repolarizing potassium currents prolonging repolarization or in late entry of sodium or calcium into the cardiac cell prolonging depolarization or repolarization. Both of these mechanisms prolong the Q-T interval. These genes that involve sodium and potassium currents include KCNQ1(IKs), KCNH2(IKr), minK, and MiRP1, and SCN5A and represent LOT1, 2, 5, 6, and 3, respectively.122–127 Syndromic LQT genetic disorders include LQT7, otherwise known as Andersen-Tawil syndrome, associated with skeletal malformations and periodic paralysis, caused by mutations in the KCNJ2 gene with reduced Kir2.1 currents and LQT8, or Timothy syndrome, associated with syndactyly, and mutations in the CACNA1C gene with increase in the Cav1.2 current. LQT9 is associated with mutations in the cavelolin-3 gene, and LQT10 is caused by a mutation in the SCN4B gene, both with late increases in sodium currents. Other mutations have been found in the ankyrin-B gene causing malfunction in a cytoskeletal membrane adapter (LQT4). LQT11 is associated with mutations in AKAP9 and LQT12 with mutations in SNTA1. In addition to these genes that affect ionic channels altering the repolarization phase of the cardiac action potential and resulting in the development of ventricular arrhythmias, an imbalance or oversensitivity of the myocardium to sympathetic stimulation appears to play a role in the development of ventricular arrhythmias. The trigger for arrhythmia in the LQTS is believed to be spontaneous secondary depolarizations that arise during or just following the prolonged plateau phase of action potentials, early after-depolarizations (EADs). Increased sympathetic tone may increase EADs, with these spontaneous repolarizations triggering a sustained arrhythmia. The implications of the location of the mutations, coding type, and the resulting biophysical malfunction has been well described for LQT1 mutations.128 Mutations located in the transmembrane portion of the ion channel protein and the degree of channel malfunction independently affect the clinical course of the individual.
Specific Genetic Defects
KvLQT1 (LQT1) and MinK (LQT5)
The KCNQ1 gene encodes the voltage-gated potassium channel α-subunits.126,129 MinK (KCNE1) encodes a much smaller potassium channel β-subunit. MinK subunits assemble with KCNQ1 subunits to form cardiac IKs channels.124,130,131 Abnormalities of either or both of these genes inhibit channel function and prolong repolarization by affecting IKs by a greater than 50% reduction in channel function, which is a dominant-negative effect. Other mutations may reduce repolarizing potassium currents by causing trafficking defects and interfering with the transport of the subunits to the cell membrane, resulting in an up to 50% reduction in channel function (haplo-insufficiency). Current evidence suggests that more than 40% of affected LQTS families have KCNQ1 mutation.132 Approximately 5% of mutations identified to date have involved minK.132,133 It has been reported that a homozygous mutation of KCNQ1 or minK causes JLNS.134,135 Both KCNQ1 and minK are expressed in the inner ear. Homozygous mutations of KCNQ1 or minK have no functional IKs channels. This leads to inadequate endolymph production and deterioration of the organ of Corti with neural deafness.136
KCNH2 (LQT2) and MiRP1 (LQT6)
In 1994, Jiang et al identified the human ether-a-go-go-related (HERG) gene, now referred to as KCNH2.127 These mutations represent 45% of the total number of LQTS mutations found to date. This gene encodes for the α-subunits that form the cardiac potassium channel delayed rectifier IKr channel, the second of two channels responsible for the termination of the plateau phase of the action potential.137,138 These mutations result in decreased outward potassium current, preventing termination of the plateau phase of the action potential. KCNH2 subunits assemble with the MiRP1 (minK-related protein1), also known as KCNE2, to form cardiac IKr channels.125 MiRP1 mutations represent 2% of the identified LQTS mutations. Multiple drugs are known to prolong the Q-T interval and potentially induce arrhythmias. The structure of KCNH2 channels appears to be predisposed to blockage by multiple drugs, making this the channel most commonly blocked by drugs.139
SCN5A (LQT3)
Jiang et al reported an additional group of LQTS families with mutations in the human cardiac sodium channel gene that encodes the subunit of sodium channel responsible for initiating cardiac action potentials.122,140,141 Gain of function mutations, especially in the inactivation gate between domains III and IV of the sodium channel, have been reported with abnormal gain of function, resulting in continued inward sodium current prolonging the action potential and predisposing to ventricular arrhythmias.133,142
Special Considerations in Long QT Syndrome
Vincent’s study in 1992 indicated that some patients may be genetic carriers for this syndrome without significant prolongation of the Q-T interval.117 Some noncarriers (15%) have abnormal prolongation of the Q-T interval above 0.44 seconds. A QTc of more than 0.47 seconds had a 100% positive predictive value in gene carriers. Above 0.47 seconds, no false-positives were seen in this study. Only 6% of gene carriers had a QTc of less than 0.44 seconds.117 These genetic tests may make it possible to identify more specifically many patients with LQTS. Priori identified families with only a 25% penetrance, and 33% of family members considered to be unaffected on clinical grounds were found to be gene carriers.118
Another interesting association that has come from the International Registry relates to the association of asthma and LQTS. The occurrence of asthma in LQTS patients increases with QTc duration. Asthma comorbidity in LQTS patients is associated with an increased risk of cardiac events. This risk is diminished after initiation of β-blocker therapy.144
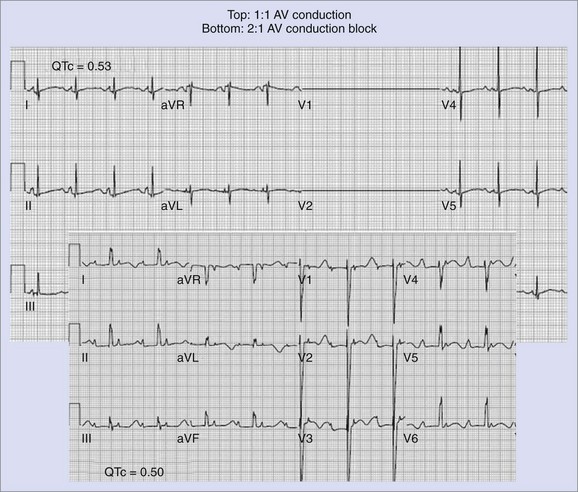
A special group of LQTS patients are newborns. Studies have suggested that sinus bradycardia is more likely associated with LQT1 and 2 : 1 AV block with LQT2 or LQT3.145,146 2 : 1 AV block has been associated with a high mortality rate,145,147 but more recent studies show this can be moderated.147a An example of 2 : 1 AV block in association with LQTS is shown in Figure 75-5.
Jervell Lange-Nielsen Syndrome
JLNS is the general descriptor applied to LQTS associated with a hearing deficit. It is caused by homozygous or compound heterozygous mutations in the KCNQ1 or KCNE1 genes, resulting in a reduced IK current and associated sensorineural deafness.134,148 Most (85% to 93%) of these patients experience cardiac events, and 50% are symptomatic by age 3 years.149 Events are generally triggered by emotions or exercise. Many have events despite β-blocker therapy, and early consideration of defibrillators has been advised.
Gene-Specific Clinical Correlations
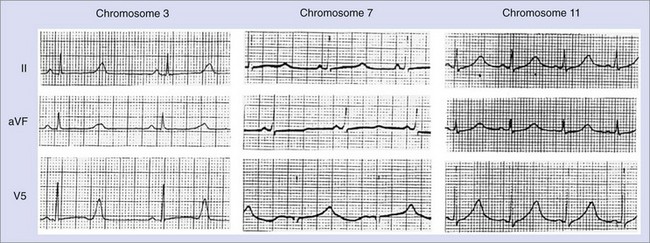
ST-T wave changes have been associated with specific genetic mutations, as shown in Figure 75-6. Because of an overlap, the specificity of this finding is limited.150,151 The influence of genotype on clinical course is being elucidated.150 The frequency of cardiac events is higher among subjects with LQT1 (63%) or LQT2 (46%) than among patients with LQT3 (18%). The likelihood of dying during a cardiac event is higher among patients with LQT3 (20%) than among patients with LQT1 or LQT2 (4%). Cardiac events in patients with LQT1 occur frequently during exercise (62%), especially swimming.152–154 Only 3% occurred during sleep. Among patients with LQT1, around 53% experience the first event by age 15 years, with 86% becoming symptomatic by 20 years of age. The probability of a cardiac event increases during adolescence in all three of the major genotypes (LQT1, LQT2, and LQT3), with the risk being higher in males before puberty and higher in females after puberty. In those with LQT2, symptoms are most commonly triggered by emotions (43%) or auditory stimuli (26%). While some events do occur with exercise (37%), more occur during rest (49%) or sleep (29%). The average age of first manifestation of symptoms is 16 years. Pregnancy increases the risk, particularly in women with LQT2, for up to 9 months after delivery. In those with LQT3, symptoms are less commonly triggered by exercise (13%) and occur primarily at rest (39%). The median age of presentation is 16 years. The percentage of patients who are free of recurrence with β-blocker therapy is higher, and the death rate is lower among patients with LQT1 (81% and 4%, respectively) than among those with LQT2 (59% and 4%) and LQT3 (50% and 17%).152 Patients with LQT3 have more cardiac events at rest or during sleep, whereas patients with LQT2 experience more events during exercise or stress. LQT2 events are more likely to be stimulated by loud noises.155
Risk Factors for Long QT Syndrome
High-risk factors for patients with LQTS include a QTc greater than 0.50 seconds to greater than 0.53 seconds, aborted cardiac arrest, torsades de pointes or complex ventricular arrhythmia, recent syncope (>2 times in past 2 years), male gender and ages 10 to 12 years, and noncompliance with prescribed LQTS medications. While younger males with LQT1 have the highest risk, no gender difference exists between young individuals with LQT2 and those with LQT3. In adulthood, a higher risk of cardiac events is present in females than in males with LQT1 and LQT2. The risks of lethal events are highest in males with LQT3 (19% risk) and females with LQT3 (18% risk), higher in males with LQT1 and LQT2 (5% and 6% risk, respectively) than in females with LQT1 and LQT2 (2% risk for either). Additionally, risks are higher in individuals with a QTc greater than 0.53 seconds, in individuals with recent syncope (within 2 years), and in individuals with frequent syncope (>2 times).152,156 The location of the mutation in LQT2 imparts higher risk to those with pore region mutations, whereas the site of the mutation appears to have less impact in LQT1.157 A study evaluating the risks of aborted cardiac arrest and sudden cardiac death in children found that a QTc greater than 0.50 seconds and syncope in males and syncope in females conferred the greatest risks.157
The probability of a first cardiac event before the age of 40 years and before therapy can be classified as high-risk, intermediate-risk, and low-risk by the characteristics illustrated in Figure 75-7.158 In LQT1 and LQT2, the risk of dying during a cardiac event is 4%, with the risk of death being 4% overall. In LQT3, the risk of dying is 17%, with the risk of dying during an event being 20%.
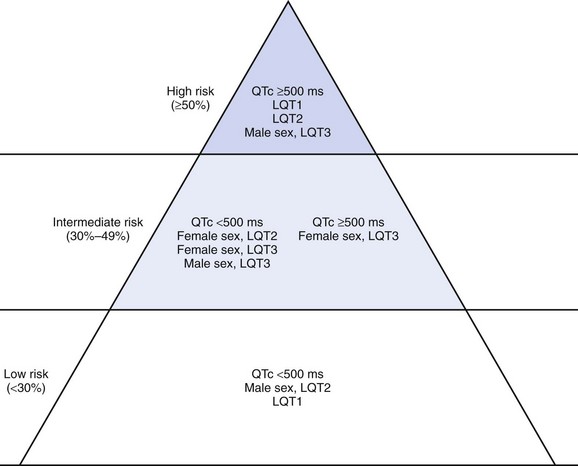
FIGURE 75-7 Risk stratification by gender, genotype and QTc.
(From Prior SG, Schwartz PJ, Napolitano C, et al: Risk stratification in the long-QT syndrome, N Engl J Med 348:1866–1873, 2003.)
Sudden Infant Death Syndrome
Schwartz reported on ECGs on 34,442 Italian babies on days 3 or 4 of life; 24 subsequently died because of sudden infant death syndrome (SIDS) and 12 had prolongation of the Q-T interval greater than 0.44 seconds.159 Additional reports have confirmed SCNA5 and HERG mutations in a few babies who died from SIDS.133–135 Postmortem molecular studies in infants and children with autopsy-negative sudden unexplained death found that 10% to 30% had genetic mutations for one of the electrical conditions associated with SCA.160
Evaluation and Diagnosis of LQTS
Careful evaluation of the ECG and 24-hour Holter monitoring is essential. The QTc is measured manually by using Bazett’s formula.161 The longest Q-T interval in any lead is divided by the square root of the preceding R-R interval. The measurement should be made manually and calculated, as computerized values are frequently incorrect. The authors consider a value greater than 0.46 seconds in any lead in children as abnormal on the resting or exercise ECG. Although a number of methods have been proposed for calculating the QTc in the presence of sinus arrhythmia, the authors make every attempt to record an ECG not in sinus arrhythmia.162 In addition to the Q-T interval, each lead of a standard 12- or 15-lead ECG should be evaluated for abnormal T-wave morphology and ST morphology. As different filters are used on the Holter monitor, the authors consider a value greater than 0.50 seconds as abnormal. In addition to Q-T interval prolongation, the Holter monitor may be helpful in illustrating T-wave abnormalities, R-on-T phenomenon, short runs of nonsustained VT, sustained VT, or torsades de pointes.
Provocative testing should include exercise stress testing in children who are able to exercise. Exercise will generally obliterate sinus arrhythmia; a strip can be obtained and a reasonable QTc calculation made. The recovery period with heart rates around 120 to 130 beats/min seems to demonstrate the greatest degree of QTc prolongation in many patients. Exercise may uncover abnormal T waves, polymorphic PVCs, or VT. If suspicion for LQTS is high and other testing has not been definitive, isoproterenol or epinephrine infusion may help identify LQTS. During these provocative tests, T-wave abnormalities may occur in addition to QTc prolongation or the development of ventricular arrhythmias.157
Efforts to identify patients at high risk for syncope and sudden death continue. High-risk ECG markers have included a QTc greater than 0.53 seconds, T-wave alternans, and QTc dispersion.163,164 Dispersion of the Q-T interval has been correlated with high risk in patients with LQTS.163,165,166 QT dispersion, which indicates heterogeneity of repolarization, could predispose to the development of torsades de pointes. In Priori’s study, patients not responding to β-blockers had a significantly higher dispersion of repolarization than did responders.163 In Shah’s study, patients with LQTS at high risk for developing critical ventricular arrhythmias had a QT or JT dispersion more than 55 ms.166 Little information is available on microvolt T-wave alternans in patients with LQTS, although visible T-wave alternans is known to be a high-risk factor.
Treatment
Emergent treatment of these patients includes lidocaine and cardioversion. Magnesium may be used to treat torsades de pointes.167,168 Intravenous propranolol and phenytoin have also been successfully used in these patients.169 Class I agents, which are known to prolong the Q-T interval in normal patients, should be avoided in patients with LQTS. This is felt to be related to QTc prolongation with associated bradycardia, ventricular arrhythmias, or both. Temporary pacing and removal of the offending agent are effective measures in this situation. Sudden death is secondary to the ventricular arrhythmias (torsades de pointes) of the type shown in Figure 75-8, which frequently degenerate to VF. The standard long-term treatment in this condition is the use of β-blockers. Those most commonly used are propranolol and nadolol. Some authors have suggested long-acting propranolol or atenolol. A concern about once-daily dosing relates to the lowest levels of the medication being present in the early morning hours, a particularly high-risk time for some patients. Therefore, we suggest twice-daily dosing at least in this group of patients. One study did suggest a less-than-favorable result with atenolol.170 In patients with compliance issues, once-daily β-blocker would be preferable to no medication. The dose of β-blocker required is variable and is usually greater per kilogram in younger patients. Most teenagers only require 10 to 20 mg of nadolol twice daily. The authors titrate the appropriate dose on the basis of the heart rate response to maximal exercise testing, aiming for a blunted maximal heart rate response of 150 to 160 beats/min on therapy. The prevalence of sudden death decreases from more than 50% to 1% to 2% with β-blockers.171,172 Other series indicate that β-blockers are not protective in every segment of this population and are most effective in patients with LOT1.173 While β-blockers are less effective in LQT2 and even less in LQT3, as no harm has been proven with this regimen, the authors continue to use them in all patients, with ICD backup when indicated clinically. The authors have added mexiletine to the therapeutic regimen of many of their patients with LQT3. Their patients are followed up yearly or twice yearly with exercise stress tests and Holter monitoring to establish the adequacy of treatment, the development of significant ventricular arrhythmias, or both. Patients who do not respond to β-blockers may be treated with mexiletine, phenytoin, or pacing. Rarely, other antiarrhythmics may be used, but those known to prolong the Q-T interval should be avoided. Potassium therapy may be helpful, especially in patients with LQT2, in which case potassium should be maintained above 4 mmol/L.
Left cervico-thoracic sympathetic denervation (LCSD) is a controversial treatment with variable success but may be useful in individuals whose condition is refractory to drug or ICD therapy.174–177 Permanent pacing, often in association with an ICD, has been shown to be an effective adjunctive treatment in these patients, especially those with severe bradycardia either from the syndrome itself or from the β-blocker therapy.178,179 The rate of the pacing should be at least 10% to 20% higher than the sinus rate and in severe cases should control the rhythm as much of the time as possible. Pauses should be avoided. Episodes of torsades de pointes may be reduced or eliminated with this treatment. In patients known to have had a cardiac arrest or frequent or significant syncope associated with ventricular arrhythmias, the chapter authors recommend the implantation of an automatic ICD. These devices can recognize VT or VF according to programmed criteria and provide a series of shocks to convert the patient’s rhythm to normal (sinus rhythm). Some devices can provide backup pacing, but at present, these are not appropriate for continuous higher rate pacing, as the battery is depleted prematurely. The sizes of the devices led to limited use in smaller children, but improved technology now allows even small children to benefit from this technology. This is not a therapy to be undertaken lightly at this time, as the need for constant follow-up and inappropriate discharges from the device can significantly affect a child’s life and lifestyle.180 Groh reported on 35 patients with LQTS who had ICDs and were followed up for a mean of 31 months.94 The major indication was aborting of sudden death. Sixty percent of patients had at least one appropriate discharge in the follow-up period. Two patients had multiple discharges and required additional therapies. None of the patients died. These results were similar to those reported earlier by Silka.181
A greater understanding of the molecular mechanisms of LQTS has prompted a number of studies to identify more specific or gene-directed therapies.182 The sodium channel blocker mexiletine has been used in some patients.183 The Q-T interval was noted to decrease with mexiletine treatment in patients with LQT3. Sato et al had reported using a potassium channel opener, nicorandil, in a patient with LQTS.184 Trials are under way with potassium supplementation and spironolactone. An LCSD has been recommended in patients with recurrent cardiac events and repeated appropriate ICD discharges. The benefit of this is still being debated.
In the past, it was considered that adults were at extremely low risk and did not need medication, but this has been shown to be incorrect. Therapy is now recommended in most adults, especially in those who have had symptoms throughout their lives.185,186 It is recommended that patients with LQTS avoid caffeine, adrenergic stimulants such as epinephrine, and over-the-counter stimulants such as decongestants. Medications that prolong the Q-T interval should be avoided. A list of these medications can be found at the website www.qtdrugs.org.
Short QT Syndrome
In 2000, Gussak reported a familial short Q-T interval associated with AF. Subsequent reports associated a short Q-T interval with sudden death. In 2004, Brugada found mutations in KCNH2.187 Short QT syndrome (SQTS) is diagnosed by a short Q-T interval of less than 0.32 seconds or QTc less than 0.34 seconds associated with AF, syncope, and sudden death. Additionally, tall symmetric T waves are present, as illustrated in Figure 75-9. Five forms of SQTS have been found with mutations in KCNH2, KCNQ1, and KCNJ2 predominantly resulting in gain of function and shortened repolarization. Additional mutations in the CACNB2B and CACN1C genes encoding the subunits of cardiac L-type calcium channels have been reported. ICD implantation has been recommended as the treatment.187–189 Quinidine has been suggested as an effective therapy, particularly in the young.190
Brugada Syndrome
The association of an ECG pattern of RBBB and ST segment elevation in ECG leads V1 to V3 with sudden death, which was reported in 1992, has been labeled Brugada syndrome.191 Patients die during sleep, which is presumed to occur secondary to VF. The average age of diagnosis is 44 years, although this condition has been reported in a 2-day-old and linked to SIDS.192,193 The syndrome is both sporadic and inherited, with an autosomal dominant mode. A mutation of SCNA5 causes loss of function with slowing of conduction velocity.194 Mutations in the SCN5A gene leading to loss of function account for approximately 18% to 30% of Brugada syndrome cases.192,195 A newly identified gene called glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L), which results in a partial reduction of INa, has been associated with the syndrome.196 The syndrome occurs most commonly in males (3 : 1) and Asians. Presentation in childhood is uncommon, with a series of 30 children finding symptoms in only one third, most identified during a family evaluation.197,198 Fever was the most common precipitating factor. Patients with unexplained syncope or aborted sudden death or a family history of these occurrences should be evaluated for this condition. More recent reports indicate that approximately 20% of patients with Brugada syndrome develop supraventricular arrhythmias.198a A meta-analysis by Gehi suggested that a prior history of syncope or aborted sudden cardiac death, male gender, and a spontaneous typical type I Brugada ECG (coved ST-segment elevation >2 mm) are predictors of high risk.198b
Evaluation and Treatment
Patients with this condition have normal Q-T intervals, ST-segment elevation with a “coved” appearance, RV conduction delay, and a propensity for sudden death. Sodium channel blocking agents such as procainamide or flecainide have been used to unmask this condition.197,199 Placement of the precordial leads several intercostal spaces higher than usual may unmask the condition as well. In children, the condition also may be unmasked by fever; vagotonic agents or events; or medications, including tricyclics or β-blockers; and recreational drugs such as cocaine.192,198 Antiarrhythmics have not decreased the incidence of sudden death in these patients, and implanted defibrillators are often recommended, although this remains controversial, as does the use of inducible VT in the electrophysiology laboratory.192,195,200,201 Some evidence exists that quinidine may be effective in children too young for ICD implantation who present with potentially threatening symptoms.202
Cathecholaminergic Polymorphic Ventricular Tachycardia
Cathecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic disorder associated with stress-induced, bi-directional VT that may degenerate into VF and result in sudden death. This condition usually occurs in childhood, adolescence, or young adulthood, with an estimated prevalence of 1 : 10,000.203 Reports of mutation in the cardiac ryanodine receptor gene RyR2 with autosomal dominant inheritance have been found in 70% of families with catecholamine-induced VT.204 Abnormalities of this gene would result in an abnormality of intracellular calcium handling, which leads to calcium overload and tachyarrhythmias. An autosomal recessive form of CPVT has been reported to be associated with mutations in the CASQ2 gene, which encodes calsequestrin, a protein that is the major calcium (Ca++) reservoir within the sarcoplasmic reticulum.205 Mutations can be identified in 50% to 70% of cases.206 The proposed mechanism is triggered automaticity. The initial symptom is usually syncope triggered by emotional or physical stress. The mean age of onset is between 7 and 9 years.206 Approximately 30% have a positive family history of syncope or sudden death. Over 70% also have supraventricular arrhythmias.203 Patients with CPVT frequently have associated sinus node dysfunction and inducible atrial tachyarrhythmias, which indicates that the pathogenesis of CPVT is associated with not only the ventricular myocardium but also with broad regions of the heart, including the sinus node and atrial muscle.207
Evaluation and Treatment
Treatment includes β-blockers and ICD implantation in patients who have had significant symptomatic episodes or have threatening arrhythmias seen on Holter monitoring or exercise stress tests.203 Verapamil has been proposed as an additional therapeutic agent.208 Left cardiac sympathetic denervation (LCSD) has been suggested for refractory cases.209
Myocarditis
Patients with myocarditis present another special problem. Of patients with ventricular ectopy, 14% to 50% have been shown to have histologic evidence of myocarditis.210,211 The most common causes of viral myocarditis include coxsackie A and B viruses and adenovirus. A large number of these patients present with ventricular arrhythmias, usually single PVCs or nonsustained VT and only mild or no impairment of ventricular function.
Evaluation and Treatment
Steroid therapy has been beneficial in some patients.212 The use of IV immunoglobulin may help in the recovery of LV function and improve survival in the first year after presentation in these patients.213 In instances of simple ventricular arrhythmias, no treatment may be needed. Patients with more complex or frequent arrhythmias may need treatment. The chapter authors have used β-blockers or mexiletine successfully in these patients. Some patients have slow rates from sinus node dysfunction or AV block and develop rapid ventricular arrhythmias as the heart rate slows. Temporary pacing may be necessary in these patients and may result in the control of the ventricular arrhythmia without the use of pharmacologic agents. When ventricular arrhythmias in these patients are potentially life threatening or impair ventricular function, immediate treatment may be needed. Often, these patients with diminished myocardial function require inotropic support to maintain cardiac output. Although each patient has unique sensitivities, a supportive agent with the least arrhythmogenic characteristics should be chosen, if possible. For example, dobutamine is usually less arrhythmogenic than dopamine, which, in turn, is less arrhythmogenic than epinephrine or isoproterenol. Ventricular arrhythmias may occur in patients with myocarditis and associated complete heart block and slow escape rhythms. In these instances, an increase in the heart rate with a temporary transvenous pacemaker may be all that is needed to control the ventricular arrhythmia. In general, pressor agents should not be used just to increase the heart rate because of their arrhythmogenic potential in this subset of patients.
Postoperative Ventricular Tachycardia
With few exceptions, patients who undergo intracardiac surgery risk the development of postoperative arrhythmias and conduction defects.214 A history of previous surgery on the ventricle or previous elevation of LV pressure before surgery is seen in most patients with VT after surgery for congenital heart disease. Most of the instances of sudden death in these postoperative patients are seen after repair of aortic stenosis, coarctation, transposition of the great arteries, or tetralogy of Fallot.215
Evaluation
All postoperative patients, especially those noted earlier to be at the highest risk, should have periodic follow-up (usually yearly) with standard ECGs. Holter monitoring should be performed every 2 to 3 years in those without known arrhythmias and every year in those with identified arrhythmias. Those being treated for arrhythmias may need more frequent monitoring. Patients with arrhythmias on Holter monitoring who are old enough to exercise should perform an exercise stress test. Those with complex arrhythmias (nonsustained VT, polymorphic PVCs, or polymorphic VT) or monomorphic VT should undergo further testing. This would include an EPS and possible catheter ablation as described in Chapter 76. The EPS has been used to evaluate the propensity of these patients to develop VT, evaluate the efficacy of specific pharmacologic therapies, and locate the site of origin of the arrhythmia in patients who are candidates for ablative therapy.216–218
Management and Treatment of Patients with Postoperative Ventricular Tachycardia
Because of their high incidence in postoperative patients, the precise role of ventricular arrhythmias in the occurrence of sudden death is unclear. It is known that after repair of tetralogy of Fallot, exercise stress testing or Holter monitoring will uncover a 25% to 70% incidence of ventricular arrhythmias. In 1985, Garson reported that treatment of more than 10 PVCs per hour on Holter monitoring decreased the incidence of sudden death in their study population.218a In contrast, Sullivan and others have reported no increase in the incidence of sudden death by not treating similar patients.219 A definitive conclusion is still pending as no large controlled study of these patients has been performed yet. The presence of frequent or complex ventricular ectopy probably identifies a high-risk group, but at present, the ability to further identify patients at highest risk is limited. It appears that patients with QRS duration above 180 ms and severe pulmonary regurgitation represent the high-risk group. The best time for valve replacement in children has not yet been determined.
Ventricular Tachycardia and Tumors
An association is known to exist between VT and cardiac rhabdomyomas. An incessant form of VT has been reported in infants and young children secondary to myocardial hamartomas. These patients can be treated successfully with surgical ablation.220 The chapter authors have found that aggressive medical management, including combination drug therapy, may be used to control this type of VT. These tumors and rhabdomyomas may regress over time. A more diffuse infiltrative type of disease known as histiocytoid or oncocytic cardiomyopathy of infancy is known to manifest as incessant VT in infancy and is usually fatal.221 Any patient with a diagnosis of tuberous sclerosis, which is known to be associated with cardiac rhabdomyoma, should have an ECG and echocardiogram done. If tumors are noted, 24-hour Holter monitoring is necessary.
Ventricular Tachycardia and Mitral Valve Prolapse
In adults, the incidence of sudden death associated with mitral valve prolapse (MVP) is 1.4%.222 Sudden death has been reported in children and adolescents, especially in athletes with MVP.223 The sudden death is proposed to be secondary to ventricular arrhythmias.224 One study found MVP in 25% of patients with idiopathic VT. On follow-up, sudden death did not occur in any patient.224a Twenty-four-hour monitoring has revealed PVCs or more complex ventricular arrhythmias in as many as 46% of children with MVP.225 Twenty-three percent of these arrhythmias were considered life threatening. Exercise stress testing revealed serious ventricular arrhythmias in 20% of patients. β-Blockers have been the drugs of choice for the control of these arrhythmias. The incidence of these events was 0.32% per year. Predictors of adverse outcome included an enlarged left ventricle or left atrium, MVP with thickened mitral leaflets, or cardiovascular symptoms.225
Although no large or long-term follow-up studies have been performed in pediatric patients with MVP, studies in relatively young adults have been performed and have shown a small but significant risk. Four hundred four U.S. Air Force aviators with a diagnosis of MVP were followed up for a mean of 8.6 years and evaluated for “suddenly incapacitating events” described as sudden cardiac death, syncope, pre-syncope, and cerebral ischemic episodes.225a
Left Ventricular Aneurysms
Aneurysms or diverticula of the left ventricle have been reported in children, especially newborns, and may be associated with ventricular arrhythmias. Treatment is determined by the clinical condition of the patient. Many of these diverticula will regress over time.226
Ventricular Tachycardia and Cardiomyopathies
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy is the most common inherited cardiovascular disorder affecting 1 in 500 individuals in the general population.227,228 A high incidence of sudden death is seen in children with hypertrophic cardiomyopathy (HCM), also known as idiopathic hypertrophic subaortic stenosis (IHSS), which is presumed secondary to ventricular arrhythmias.229–232 HCM is the most common cause of sudden death in young competitive athletes.229,233 The clinical manifestations are a result of cellular disarray, fibrosis, and hypertrophy of the myocyte leading to diastolic dysfunction, myocardial ischemia, and arrhythmias.234,235
Clinical Correlations
A family history of ventricular arrhythmias leading to sudden death, including nonsustained VT on 24-hour Holter monitors, previous syncope, extensive generalized hypertrophy, or ventricular systolic or diastolic dysfunction, may identify a high-risk population.232 Asymptomatic VT on Holter monitoring may or may not be predictive.236 Abnormal blood pressure response to exercise may predict sudden death. Exercise stress testing may uncover ST-segment depression or an abnormal systolic blood pressure response, including a small difference between peak and resting systolic blood pressure. Predictors of sudden cardiac death include higher left ventricular outflow tract (LVOT) pressure gradient at rest and failure of systolic blood pressure to increase during exercise testing.237 Two events that predict subsequent sudden death include a combination of inducible sustained VT at EPS and a history of cardiac arrest or syncope.235 A multi-center study evaluating 1511 consecutive patients with HCM demonstrated a relative risk of sudden death of 1.78 in patients with unexplained syncope and 0.91 with neurally mediated syncope compared with patients without syncope. A fivefold increased risk was seen in patients with unexplained syncope within 6 months of their initial evaluation. Of the 1511 subjects, 147 were younger than 18 years of age. During 6.5 years of follow-up, 15 sudden deaths occurred, that is, an incidence of 15.7 per 1000 person-years. Seven of the 147 patients had experienced unexplained syncope before their initial evaluation, with 3 dying suddenly. In those with syncope, the mortality rate was 120 per 1000 person-years compared with 13 per 1000 person-years in those without unexplained syncope.236 More than one third of patients who experience syncope with VT/VF or resuscitated VF will die within 7 years from sudden cardiac death or progressive heart failure.237 Unfortunately, many high-risk patients will not have inducible VT at EPS but may still be at high risk. It has been suggested that fractionation of electrograms with programmed stimulation may be a marker for high-risk patients.238 Recent studies suggest that myocardial bridging of the left anterior descending coronary arteries may cause myocardial ischemia in children with HCM.239 The primary problem with regard to treatment is the divergence of opinion regarding the bridging or compression of branches from LV hypertrophy.240
HCM is a genetic disease caused by mutations in contractile sarcomeric proteins. The penetrance is often incomplete and 20% to 30% of adults with these gene mutations do not express the phenotype.241 It is hoped that the recent genetic identification of HCM mutations will increase understanding and help identify high-risk patients.240 The first gene identified for HCM is the β-myosin heavy chain MHC located on the long arm of chromosome 14.242 The MHC gene (MyHC) is responsible for 35% of familial HCM.243 Multiple other genes that code the proteins of the sarcomere have been found to be responsible for HCM. These include the gene of cardiac T-troponin (cTnT),241 the gene of α-tropomyosin, and the gene of binding protein C (MyBP-C).244 Specific mutations have been reported to be associated with a higher incidence of sudden death, but this is not fully agreed upon.242
Evaluation
All family members and identified patients should be monitored with periodic ECGs and echocardiograms. ECG findings have been identified as major criteria in the diagnosis of hypertrophic cardiomyopathy, including left ventricular overload, deep Q waves more than 40 ms in the LV inferior lateral wall, and T-wave inversion 3 mm or more in V3 to V6, L1, and aVL and 5 mm or more in L2, L3, and aVF (Figure 75-10).244 In identified patients, yearly Holter monitoring should be performed. Exercise stress testing should be performed in those who are of an appropriate age. EPS should be performed in those who have had syncope, documented complex arrhythmias, or symptoms suggesting ventricular arrhythmias. It has been noted that atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are elevated up to 5 and 85 times the normal levels, respectively, in patients with HCM.245 These values may be useful in the initial evaluation as well as follow-up in those with a diagnosis of HCM.
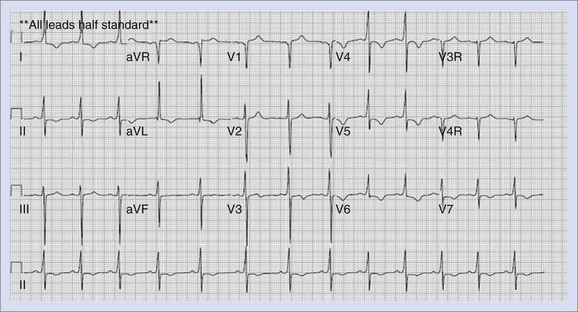
Treatment
Treatment includes antiarrhythmic therapy, “pacemakers,” myotomy or myectomy, and ICDs.170,246–251 Maron reported a retrospective multi-center study of efficacy of ICDs in preventing sudden death in 128 patients with HCM who were judged to be at higher risk for sudden death and had ICDs implanted. Twenty-three percent had appropriate shocks or anti-tachycardia pacing, and 7% had inappropriate shocks per year. In those in whom secondary prevention was provided after cardiac arrest or VT, 11% had appropriate activation. The interval between implantation and the first appropriate discharge was substantially prolonged by 4 to 9 years in six patients. The defibrillators were highly effective in terminating life-threatening ventricular arrhythmias.251a The recommendations of the 36th Bethesda Conference are that athletes with a diagnosis of HCM or probable HCM abstain from competitive sports and vigorous training, with the exception of low-intensity activities.252
Dilated Cardiomyopathy
The most common etiology of dilated cardiomyopathy (DCM) in children is idiopathic,253 but other causes include antecedent myocarditis, familial or genetic associations, and immune regulatory abnormalities.253–256 The prognosis for children with DCM is poor, and the reported mortality has been estimated at 24% to 50%. In a study of 62 children and adolescents (followed up for 3.9 to 4.5 years), 50% died, 16% recovered, 27% had residual decreased LV function, and 7% underwent orthotopic heart transplantation.257 In another study of 41 children followed up for a median of 2.5 years, 30% died, 32% recovered, and 38%survived with decreased LV function.258 Mortality rates at follow-up were 24% at 1 year and 29% at 5 years. Poorer prognosis was associated with clinical severity at presentation, lower mean shortening fraction, and severe arrhythmias.258 Little information is available on risk stratification in children with dilated cardiomyopathy.
Clinical Correlations
In adults, a variety of high-risk factors have been suggested, including the presence of ventricular arrhythmia, LV end-diastolic diameter of more than 70 mm, and nonsustained VT on Holter monitoring. Low ejection fraction of less than 30% and nonsustained VT are high-risk factors as well.253
Treatment
Treatment includes antiarrhythmic therapy, biventricular pacing, and implantable ICDs. Ten percent of patients will require heart transplantation. In the past decade, a number of pediatric studies have looked at the effect of β-blocker therapy in the treatment of pediatric patients with dilated cardiomyopathy.260–262 These studies have primarily evaluated the safety and effectiveness of carvedilol in this population. Each of these has demonstrated a significant improvement in systemic ventricular function as assessed by shortening, ejection fraction, or both. Carvedilol was added to other standard therapeutic modalities in each of these studies. The use of carvedilol, other β-blockers, or both in patients who develop significant arrhythmias may result in an interaction with other antiarrhythmics such as amiodarone.
Biventricular pacing has been reported recently as a successful treatment modality in adults with symptomatic drug-refractory heart failure secondary to dilated cardiomyopathy and associated interventricular conduction delay.263,264 Several electrophysiologists are recommending a combination of biventricular pacing, implantable ICDs, or both for adults.251,265 The usefulness of biventricular pacing in pediatric patients with a dilated cardiomyopathy, interventricular conduction delays, ventricular asynchrony, and congestive heart failure has not been studied in large clinical trials but may be a new treatment option for these patients. Several large multi-center trials of biventricular pacing are ongoing, including trials to evaluate the effects of biventricular pacing on ventricular arrhythmias.263,264,266 Although early results in adults are encouraging, many questions still remain to be answered.
Ventricular Tachycardia and Structurally Normal Hearts
Two sites have been associated with VT and structurally normal hearts. The first is located in the right ventricular outflow tract (RVOT), usually in the anterior portion, most commonly anteroseptal, but also in the anterolateral and anterior regions.267 The morphology of VT on ECG is LBBB, most commonly with an inferior axis but also with a superior or rightward axis (Figure 75-11). An RBBB pattern with superior or rightward axis is also seen. Both sustained and nonsustained VT are seen clinically. It has been suggested that RVOT tachycardias may resolve over time. However, Drago reported biopsies positive for acute myocarditis, ARVD fatty infiltration, and other histologic abnormalities in 67% of patients without obvious heart disease and RVOT tachycardia.269 The tachycardia may be induced as nonsustained or sustained VT by programmed electrical stimulation and isoproterenol in 40% to 80% of selected patients. Pacemapping can help identify the specific site of VT in the RVOT.
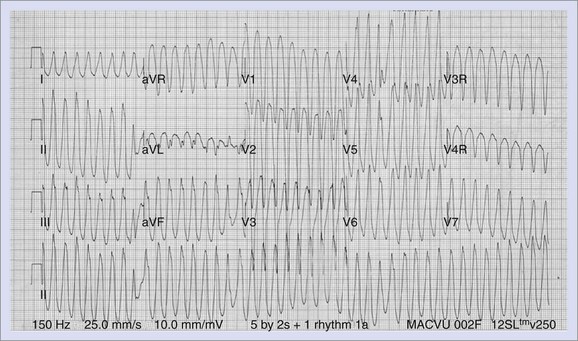
The second form of VT in patients without heart disease originates from the left ventricle, most commonly in the septum in the mid-to-inferior position. The morphology of the VT is generally RBBB with left axis deviation (Figure 75-12). It can be induced in a similar fashion to VT in the RVOT. These VTs are often sensitive to verapamil. VT from the basal aspect of the superior LV septum can appear as repetitive monomorphic VT, revealing a dominant R-wave pattern in V1, an inferior axis, and a precordial R-wave transition at or before lead V2.270 These VTs may be responsive to verapamil or adenosine, which suggests a triggered mechanism.271 Some LV VTs originate in the LVOT with an LBBB pattern.
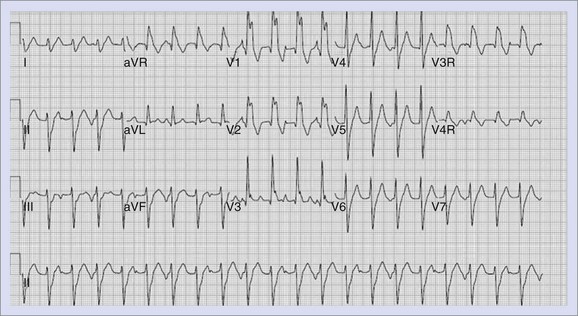
Treatment
Both forms of VT are amenable to treatment with catheter ablation.259–261270 Those with an LBBB pattern, inferior axis, and early precordial transition may be ablated from the left or noncoronary aortic sinus of Valsalva.262 For LV septal VTs, ablation at the pre-Purkinje potential recording site during VT seems to be most successful.272
Acute Treatment of Ventricular Tachycardia
Procainamide IV has been used to treat acute episodes of VT. Because of the associated negative inotropic effects, patients must be monitored very carefully during the infusion. More recently, amiodarone has been shown to be effective for VT when given intravenously. Other drugs used in acute therapy are listed in Table 75-4. Rapid ventricular pacing may be used to convert the rhythm to sinus if pharmacologic therapy fails or is contraindicated.
| INITIAL TREATMENT | DOSAGE | LEVEL |
|---|---|---|
| Lidocaine | 1-2 mg/kg IV bolus q5-15 min | 1.5-5.0 mg/L |
| IV infusion: 20-50 µg/kg/min | ||
| Cardioversion | 1-5 watt-sec/kg; double if ineffective | |
| SECONDARY TREATMENT | IV DOSAGE | LEVEL |
| Amiodarone | 5 mg/kg over 1 hour, followed by 2.5 mg/kg IV bolus q4-6 h | |
| Magnesium | 0.25 mEq/kg over 1 min, followed by 1 mEq/kg over 5 hours to achieve Mg++ level of 3-4 mg/dL | |
| Phenytoin | 3-5 mg/kg over 5 min, not to exceed 1 mg/kg/min | 10-20 µg/mL |
| Procainamide | 5 mg/kg over 5-10 min or 15 mg/kg over 30-45 min | NAPA: 4-10 mg/L |
| Infusion: 20-100 µg/kg/min | PA: 4-8 mg/L | |
| Propranolol | 0.05-0.1 mg/kg over 5 min q6h |
NAPA, N-acetylprocainamide; PA, procainamide.
Long-Term Treatment of Ventricular Tachycardia
Once the arrhythmia has been converted, choice of an appropriate long-term regimen is essential to maintaining the stability of the patient. The drugs most commonly used are listed in Table 75-1. If lidocaine has been successful, it may be continued until adequate levels of a chronic regimen have been reached or the acute causative agent is no longer present. When switching to mexiletine, the lidocaine should be gradually weaned as the mexiletine is initiated to prevent the combined toxicity of these two drugs, as their side effects are similar. Propranolol or other β-blockers are especially effective in patients whose arrhythmia is sensitive to adrenergic stimuli.274 Class I agents and amiodarone are effective in more refractory cases.274,275 A combination of amiodarone and propranolol has been effective in infants and children.276
As sudden death occurs in up to 30% of patients with VT and congenital heart defects, these patients should be placed on a long-term regimen. This type of arrhythmia is poorly tolerated by patients in the immediate postoperative period, but the arrhythmia generally responds to lidocaine or IV amiodarone and to correction of other underlying hemodynamic and metabolic abnormalities. Studies have shown that patients with early postoperative VT are likely to develop this arrhythmia in the late postoperative period, so long-term therapy is often recommended. The patient who presents months to years after surgery also requires long-term therapy. A thorough investigation is needed to rule out underlying hemodynamic abnormalities, as VT is tolerated less well by this group of patients. Residual or new hemodynamic lesions may be contributing to the arrhythmia. Mexiletine has been shown to be an effective long-term drug in patients after tetralogy of Fallot repair, as is the β-blocker class of drugs.261 Class I agents as well as amiodarone may be effective in refractory cases. The EPS may be a helpful guide to medical therapy.216,217 In cases refractory to drug therapy, the EPS can determine the site of origin of the tachycardia and direct treatment by surgical or catheter ablation. Patients with life-threatening episodes or those not responding to medical or ablative therapy may require ICDs. See Chapter 80 for more details.
Electrophysiological Study of Ventricular Tachycardia
Specific indications for the performance of EPS in children with VT are included in Box 75-2. Intracardiac recordings during VT show the absence of His-bundle deflections consistently preceding ventricular depolarizations. Atrial capture at rates more rapid than the tachycardia normalizes the QRS complex. One of the most significant differences between adults and children is the mechanism responsible for VT.78 In adults, more than 90% have inducible VT. In children, only 30% to 60% have inducible or re-entrant VT, and the rest have triggered or automatic VT. Inducible re-entrant VT in children is more commonly associated with structural heart disease.
Box 75-2 Indications for EPS in Ventricular Tachycardia
Indications for EPS
EPS, Electrophysiological study; VT, ventricular tachycardia; VF, ventricular fibrillation.
Catheter Ablation of Ventricular Tachycardia
In the pediatric population, the categories of VT that are amenable to catheter ablation include those in structurally normal hearts that originate primarily in the RVOT or in the left ventricle, especially in the mid-to-inferior septum. The other category of patients who may have VT that is amenable to catheter ablation are those who have postoperative VT, particularly in the RVOT, commonly those with tetralogy of Fallot.217 Scarring from right ventriculotomy or infundibular resection allows a zone of slow conduction producing the substrate for re-entry.
Prognosis of Ventricular Tachycardia
The prognosis of VT depends on the underlying condition. Reviews of VT in children have reported a high incidence of death: 10% to 47%.65 The higher incidence occurs in those patients with underlying structural or postoperative heart disease and poor hemodynamic results. Sudden death has been reported in patients with normal hearts at an incidence of 6% to 8%.277
In the absence of structural heart disease, especially in the neonate or young child, spontaneous resolution of VT may occur. The outlook for infants and children with VT is excellent if the VT can be controlled with treatment.73
Sinus Node Dysfunction
Atrioventricular Block
First-Degree and Second-Degree Atrioventricular Block
First-degree and second-degree heart block may be congenital or acquired. Acquired etiologies include traumatic damage associated with surgery for congenital heart defects and inflammatory processes such as rheumatic heart disease or Lyme carditis. The EPS in pediatric patients has generally localized the delay to the AV node.269 Delay in the His-Purkinje system with prolongation of the H-V interval may be more significant, indicating a predisposition to the development of complete heart block.279,280 Patients with first-degree and second-degree AV block are generally asymptomatic unless the ventricular rate is significantly decreased. It is not uncommon to see first-degree and Mobitz I AV block during 24-hour Holter monitoring in teenagers, especially during sleep. The low heart rates are especially significant in patients with compromised myocardial function, and thus the cardiac output may be insufficient to meet the patient’s needs.
Treatment
If a higher rate is needed, pharmacologic agents such as atropine may be helpful, especially if the block is in the AV node and partially mediated by vagal influences. Isoproterenol may increase the heart rate by increasing the rate of the escape pacemaker. As mentioned earlier, oral medications such as caffeine, theophylline, and antivagolytics are often not very useful in changing the heart rate or AV conduction. Acutely, temporary transcutaneous or transvenous pacing may be necessary. For persistent symptomatic or high-grade AV block, permanent pacing may be needed. In some patients with second-degree AV block, progression to complete AV block may occur.279,280
Complete Heart Block
Complete heart block is the most common cause of significant bradycardia in children. The ventricular rate is usually 40 to 80 beats/min, depending on the patient’s age. The QRS morphology and heart rate are related to the location of the escape pacemaker. The higher the origin of the pacemaker within the AV conduction system, the faster is the ventricular rate and the narrower the QRS complex (Figure 75-13). Wider QRS escape complexes usually originate from the His bundle or below. The usual rate in the neonate is 60 to 80 beats/min.
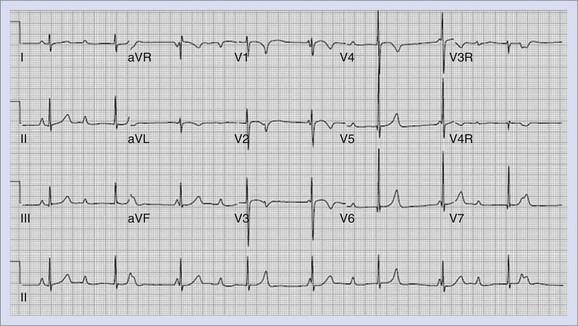
Complete heart block may be either congenital or acquired and occurs in 1 per 20,000 live births. The same causes of acquired heart block that cause second-degree AV block discussed earlier can cause complete heart block. In a series of 599 patients with congenital complete heart block, followed up for more than 10 years, 92% survival was seen in patients without associated structural heart disease.281 The greatest risk of mortality was during the first weeks of life, especially in those delivered prematurely secondary to fetal hydrops, with half of the deaths occurring during the first year. The highest risk was indicated by associated cardiac anomalies, cardiomegaly, ventricular bradycardia less than 55 beats/min, and atrial tachycardia greater than 140 beats/min in infants.
A strong association has been noted with connective tissue disease in the mother and congenital complete heart block. The prevalence has been reported to be as high as 85% in mothers of affected infants.282,283 Only one half of these affected mothers are symptomatic, whereas the other half, although asymptomatic, are serologically positive. A mother with systemic lupus erythematosus has a 1 : 20 risk of having a child with complete heart block if she is anti-Ro positive.284 Maternal immunoglobulin G antibodies to soluble tissue ribonucleoprotein antigens, found in the cytoplasm or nucleus of human cells (anti-Ro:SS-A and anti-La:SS-B), cross the placenta after 12 to 16 weeks of gestation. This results in an inflammatory response in the fetal heart, particularly in the conduction system, leading to the destruction of the AV node.285–287
Buyon demonstrated an increased risk when the maternal antibodies target specific portions of the ribonuclear complex, particularly the 48-kd La (SS-B), the 52-kd Ro (SS-A), and the 60-kd Ro (SS-A). The highest risk was reactivity to both the 48-kd La (SS-B) and the 52-kd Ro (SS-A) components.288 Because of this immunologic factor, dexamethasone and plasmapheresis have been recommended as effective treatments for the mother of an affected fetus.288,289 Although the heart block has not been shown to resolve with these treatments, the clinical course of the fetus has improved, with decreased pleuro-pericardial effusions and other signs of fetal hydrops.
Presentation
Structural heart disease is present in one third to one fourth of infants with congenital heart block. The associated congenital heart defects most commonly include those with L-transposition of the great arteries (L-TGA) (physiologically corrected transposition) or the heterotaxy syndromes. With associated heart disease, mortality in the first year has been reported to be 29% in one study.281 Complex postoperative congenital heart lesions or those with an unusual course of the AV conduction system may develop postoperative block.291 After ventricular septal defect repair, the presence of RBBB and left anterior hemiblock is 7% to 17%, with a 1% incidence of complete heart block.292 Complete heart block has been reported to occur as late as 14 years after surgical repair.293 Complete heart block occurs more commonly after repair of AV canal defects, probably because of the unusual course of the conduction system in these lesions, and may be seen in up to 7% of patients.294 Physiologically corrected L-TGA is associated with AV conduction disturbances ranging from first-degree to complete heart block in 30% to 60% of patients.
In a study of patients with congenital heart block who were followed up for more than 30 years, only 10% did not require a pacemaker, and a 20% incidence of sudden death was noted. Mitral regurgitation and prolonged QTc were poor prognostic findings. Pacemakers were recommended even in asymptomatic adults.295
Evaluation and Treatment
In emergencies, immediate transthoracic pacing can be accomplished. The transcutaneous pacemaker may be effective in short-term emergency situations but should be replaced with another pacing method as soon as possible. Placement of a temporary transvenous pacemaker, either through the umbilical vein or the femoral vein under direct fluoroscopic observation, is preferred. Although infants with rates less than 50 beats/min or slightly higher rates and associated congenital heart defects or cardiomyopathies may require pacing, this decision should not be made on the basis of heart rate alone. Older children with congenital heart block may develop evidence of exercise intolerance, easy fatigability, or syncope. Not all patients with congenital complete heart block need a pacemaker, and many do not require one until they reach an older age.295
Pacemakers may be placed because of associated ventricular arrhythmias, either during sleep or exercise, easy fatigability or exercise intolerance, syncope, or presyncope.296–298 Other indicators for pacing that have been associated with sudden death include severe bradycardia, ventricular ectopy especially with exercise, prolonged pauses, increased QRS width, prolongation of QTc, and a junctional recovery time of more than 3 seconds.299,300 Another important indicator for pacer placement is cardiac enlargement shown by chest x-ray or echocardiogram. With cardiac output being the product of heart rate and stroke volume, the LV will dilate, indicating that the heart rate is not adequate to meet the metabolic demand.
With postoperative heart block, temporary pacing is usually performed 7 to 10 days after surgery. Permanent pacing should be performed if sinus rhythm does not return because of the high incidence of sudden death in this group of postoperative patients if they are not paced.301 As the return of sinus rhythm does not ensure that heart block will not recur at a later time, close follow-up is indicated.
Abnormalities in intraventricular conduction may be congenital or acquired. Congenital RBBB may be inherited or found in association with Ebstein’s anomaly of the tricuspid valve.302,303 Children with Kearns-Sayre syndrome are likely to develop RBBB that progresses to complete heart block.304 Prophylactic placement of a pacemaker is recommended in this group of patients to prevent sudden death. Postoperative RBBB is common after repair of many cardiac anomalies and is either central or peripheral.305,306
Implantable Pacemakers
The use of permanent pacemakers in children was first reported in the early 1960s and predated many recent advances in lead and generator technology.307–310 These advances, including programmability and miniaturization, have significantly increased pacemaker use in the pediatric population.311–314 See Chapter 79 for more detailed information on the use of pacemakers in children.
Key References
Benson DWJr, Smith WM, Dunnigan A, et al. Mechanisms of regular, wide QRS tachycardia in infants and children. Am J Cardiol. 1982;49(7):1778-1788.
Collins KK, Van Hare GF, Kertesz NJ, et al. Pediatric nonpost-operative junctional ectopic tachycardia medical management and interventional therapies. J Am Coll Cardiol. 2009;53(8):690-697.
Daubert JP, Zareba W, Rosero SZ, et al. Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am Heart J. 2007;153(4 Suppl):53-58.
Davis AM, Gow RM, McCrindle BW, Hamilton RM. Clinical spectrum, therapeutic management, and follow-up of ventricular tachycardia in infants and young children. Am Heart J. 1996;131(1):186-191.
Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363(9424):1881-1891.
Garson AJr, Dick M, Fournier A, et al. The long QT syndrome in children. An international study of 287 patients. Circulation. 1993;87(6):1866-1872.
Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51(24):2291-2300.
Hanisch D. Pediatric arrhythmias. J Pediatr Nurs. 2001;16(5):351-362.
Ko JK, Deal BJ, Strasburger JF, Benson DWJr. Supraventricular tachycardia mechanisms and their age distribution in pediatric patients. Am J Cardiol. 1992;69(12):1028-1032.
Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45(8):1340-1345.
Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24(21):1965-1991.
Michaelsson M, Jonzon A, Riesenfeld T. Isolated congenital complete atrioventricular block in adult life. A prospective study. Circulation. 1995;92(3):442-449.
Pfammatter JP, Paul T. Idiopathic ventricular tachycardia in infancy and childhood: A multicenter study on clinical profile and outcome. Working Group on Dysrhythmias and Electrophysiology of the Association for European Pediatric Cardiology. J Am Coll Cardiol. 1999;33(7):2067-2072.
Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358(2):169-176.
Santinelli V, Radinovic A, Manguso F, et al. The natural history of asymptomatic ventricular pre-excitation: A long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53(3):275-280.
Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783-790.
Vetter VL. Ventricular arrhythmias in pediatric patients with and without congenital heart disease. In: Horowitz LN, editor. Current management of arrhythmias. Philadelphia: BC Decker, 1990.
Wilde AA, Bezzina CR. Genetics of cardiac arrhythmias. Heart. 2005;91(10):1352-1358.
Zareba W. Genotype-specific ECG patterns in long QT syndrome. J Electrocardiol. 2006;39(4 Suppl):S101-S106.
Zipes DP, Ackerman MJ, Estes NAIII, et al. Task Force 7: Arrhythmias. J Am Coll Cardiol. 2005;45(8):1354-1363.
1 Ludomirsky A, Garson AJ. Supraventricular tachycardia. In: Garson AJ, Gillette PC, editors. Pediatric arrhythmias: Electrophysiology and pacing. Philadelphia: Saunders, 1990.
2 Liberman L, Pass RH, Starc TJ. Optimal surface electrocardiogram lead for identification of the mechanism of supraventricular tachycardia in children. Pediatr Emerg Care. 2008;24(1):28-30.
3 Akhtar M, Tchou PJ, Jazayeri M. Mechanism of clinical tachycardias. Am J Cardiol. 1988;61:9A-19A.
4 Damiano BP, Rosen MR. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation. 1984;69(5):1013-1025.
5 Gilmour RFJr, Heger JJ, Prystowsky EN, Zipes DP. Cellular electrophysiologic abnormalities of diseased human ventricular myocardium. Am J Cardiol. 1983;51(1):137-144.
6 Ferrier GR. Digitalis arrhythmias: Role of oscillatory afterpotentials. Prog Cardiovasc Dis. 1977;19:459.
7 Moe GK, Preston JB, Burlington H. Physiologic evidence for a dual AV transmission system. Circulation. 1956;4:357.
8 Ko JK, Deal BJ, Strasburger JF, Benson DWJr. Supraventricular tachycardia mechanisms and their age distribution in pediatric patients. Am J Cardiol. 1992;69(12):1028-1032.
9 Hanisch D. Pediatric arrhythmias. J Pediatr Nurs. 2001;16(5):351-362.
10 Kugler JD, Danford DA. Management of infants, children, and adolescents with paroxysmal supraventricular tachycardia. J Pediatr. 1996;129(3):324-338.
11 Jaeggi ET, Nii M. Fetal brady- and tachyarrhythmias: New and accepted diagnostic and treatment methods. Semin Fetal Neonatal Med. 2005;10(6):504-514.
12 Perry JC, Garson AJr. Supraventricular tachycardia due to Wolff-Parkinson-White syndrome in children: Early disappearance and late recurrence. J Am Coll Cardiol. 1990;16(5):1215-1220.
13 Sanatani S, Hamilton RM, Gross GJ. Predictors of refractory tachycardia in infants with supraventricular tachycardia. Pediatr Cardiol. 2002;23(5):508-512.
14 Dixon J, Foster K, Wyllie J, Wren C. Guidelines and adenosine dosing in supraventricular tachycardia. Arch Dis Child. 2005;90(11):1190-1191.
15 Epstein ML, Kiel EA, Victorica BE. Cardiac decompensation following verapamil therapy in infants with supraventricular tachycardia. Pediatrics. 1985;75(4):737-740.
16 Abinader E, Borochowitz Z, Berger A. A hemodynamic complication of verapamil therapy in a neonate. Helv Paediatr Acta. 1981;36(5):451-455.
17 Gilljam T, Jaeggi E, Gow RM. Neonatal supraventricular tachycardia: Outcomes over a 27-year period at a single institution. Acta Paediatr. 2008;97(8):1035-1039.
18 Mantakas ME, McCue CM, Miller WW. Natural history of Wolff-Parkinson-White syndrome discovered in infancy. Am J Cardiol. 1978;41(6):1097-1103.
19 Wolff GS, Han J, Curran J. Wolff-Parkinson-White syndrome in the neonate. Am J Cardiol. 1978;41(3):559-563.
20 Nadas AS, Daeschner CW, Roth A, Blumenthal SL. Paroxysmal tachycardia in infants and children: Study of 41 cases. Pediatrics. 1952;9(2):167-181.
21 Van Hare GF, Witherell CL, Lesh MD. Follow-up of radiofrequency catheter ablation in children: Results in 100 consecutive patients. J Am Coll Cardiol. 1994;23(7):1651-1659.
22 Van Hare GF, Javitz H, Carmelli D, et al. Prospective assessment after pediatric cardiac ablation: Recurrence at 1 year after initially successful ablation of supraventricular tachycardia. Heart Rhythm. 2004;1(2):188-196.
23 Chung KY, Walsh TJ, Massie E. Wolff-Parkinson-White syndrome. Am Heart J. 1965;69:116-133.
24 Benson DWJr, Smith WM, Dunnigan A, et al. Mechanisms of regular, wide QRS tachycardia in infants and children. Am J Cardiol. 1982;49(7):1778-1788.
25 Pappone C, Manguso F, Santinelli R, et al. Radiofrequency ablation in children with asymptomatic Wolff-Parkinson-White syndrome. N Engl J Med. 2004;351(12):1197-1205.
26 Santinelli V, Radinovic A, Manguso F, et al. The natural history of asymptomatic ventricular pre-excitation: A long-term prospective follow-up study of 184 asymptomatic children. J Am Coll Cardiol. 2009;53(3):275-280.
27 Bromberg BI, Lindsay BD, Cain ME, Cox JL. Impact of clinical history and electrophysiologic characterization of accessory pathways on management strategies to reduce sudden death among children with Wolff-Parkinson-White syndrome. J Am Coll Cardiol. 1996;27(3):690-695.
28 Paul T, Guccione P, Garson AJr. Relation of syncope in young patients with Wolff-Parkinson-White syndrome to rapid ventricular response during atrial fibrillation. Am J Cardiol. 1990;65(5):318-321.
29 Pappone C, Santinelli V, Rosanio S, et al. Usefulness of invasive electrophysiologic testing to stratify the risk of arrhythmic events in asymptomatic patients with Wolff-Parkinson-White pattern: Results from a large prospective long-term follow-up study. J Am Coll Cardiol. 2003;41(2):239-244.
30 Pappone C, Radinovic A, Santinelli V. Sudden death and ventricular preexcitation: Is it necessary to treat the asymptomatic patients? Curr Pharm Des. 2008;14(8):762-765.
31 Zipes DP, Ackerman MJ, Estes NAIII, et al. Task Force 7: Arrhythmias. J Am Coll Cardiol. 2005;45(8):1354-1363.
32 Gillette PC. The mechanisms of supraventricular tachycardia in children. Circulation. 1976;54:133.
33 Mehta AV, Sanchez GR, Sacks EJ, et al. Ectopic automatic atrial tachycardia in children: Clinical characteristics, management and follow-up. J Am Coll Cardiol. 1988;11:379.
34 Walsh EP, Saul JP, Hulse JE, et al. Transcatheter ablation of ectopic atrial tachycardia in young patients using radiofrequency current. Circulation. 1992;86:1138.
35 Silka MJ, Gillette PC, Garson AJ, et al. Transvenous catheter ablation of a right atrial automatic ectopic tachycardia. J Am Coll Cardiol. 1997;5:999.
36 Gillette PC, Garson AJ. Electrophysiologic and pharmacologic characteristics of automatic ectopic atrial tachycardia. Circulation. 1977;56:571.
37 Gillette PC, Smith RT, Garson AJ, et al. Chronic supraventricular tachycardia: A curable cause of congestive cardiomyopathy. JAMA. 1985;253:391.
38 Perry JC, McQuinn R, Smith RT. Flecainide acetate for resistant arrhythmias in the young: Efficacy and pharmacokinetics. J Am Coll Cardiol. 1989;14:185.
39 Zeigler VL, Gillette PC, Ross B, et al. Flecainide for supraventricular and ventricular arrhythmias in children and young adults. Clin Prog Electrophysiol Pacing. 1986;4:328.
40 Coumel P, Fidelle J. Amiodarone in the treatment of cardiac arrhythmias in children: One hundred thirty-five cases. Am Heart J. 1992;100:1063.
41 Bernuth GV, Engelhardt W, Kramer HH, et al. Atrial automatic tachycardia in infancy and childhood. Eur Heart J. 1992;13:1410.
42 Colloridi V, Perri C, Ventriglia F, et al. Oral sotalol in pediatric atrial ectopic tachycardia. Am Heart J. 1992;123:254.
43 Düren DR, Liem DL, Lie KI. Effects of digitalis in patients with paroxysmal atrioventricular nodal tachycardia. Circulation. 1975;52:779.
44 Rosen MR, Merker C, Gelband H, et al. Effects of procainamide on the electrophysiologic properties of the canine ventricular conduction system. J Pharmacol Exp Ther. 1973;185:438.
45 Gillette PC, Garson AJ, Hesslein PS, et al. Successful surgical treatment of atrial, junctional and ventricular tachycardia unassociated with accessory connections in infants and children. Am Heart J. 1981;102:984.
46 Ott DA, Gillette PC, Garson AJ, et al. Surgical management of refractory supraventricular tachycardia in infants and children. J Am Coll Cardiol. 1985;5:124.
47 Margolis PO, Roman CA, Moulton KP, et al. Radiofrequency catheter ablation of left and right ectopic atrial tachycardia [abstract]. Circulation. 1991;82:718.
48 Liberthson RR, Colan SD. Multifocal or chaotic atrial rhythm. Pediatr Cardiol. 1982;2:179.
49 Yeager SB, Hougen TJ, Levy AM. Sudden death in infants with chaotic atrial rhythm. Am J Dis Child. 1984;138:689.
50 Dunnigan A, Benson WJr. Benditt DG: Atrial flutter in infancy: Diagnosis, clinical features, and treatment. Pediatrics. 1985;75(4):725-729.
51 Mendelsohn A, Dick M, Serwer GA. Natural history of isolated atrial flutter in infancy. J Pediatr. 1991;119(3):386-391.
52 Soyeur DJ. Atrial flutter in the human fetus: Diagnosis, hemodynamic consequences, and therapy. J Cardiovasc Electrophysiol. 1996;7(10):989-998.
53 Cuneo BF. Outcome of fetal cardiac defects. Curr Opin Pediatr. 2006;18(5):490-496.
54 Texter KM, Kertesz NJ, Friedman RA, Fenrich ALJr. Atrial flutter in infants. J Am Coll Cardiol. 2006;48(5):1040-1046.
55 Lee PC, Hwang B, Chen SA, et al. The results of radiofrequency catheter ablation of supraventricular tachycardia in children. Pacing Clin Electrophysiol. 2007;30(5):655-661.
56 Harahsheh A, Du W, Singh H, Karpawich PP. Risk factors for atrioventricular tachycardia degenerating to atrial flutter/fibrillation in the young with Wolff-Parkinson-White. Pacing Clin Electrophysiol. 2008;31(10):1307-1312.
57 Gillette PC, Garson AJ. Junctional ectopic tachycardia in children: Electrocardiography, electrophysiology and pharmacologic response. Am J Cardiol. 1979;44:298.
58 Case CL, Gillette PC. Automatic atrial and junctional tachycardias in the pediatric patient: Strategies for diagnosis and management. Pacing Clin Electrophysiol. 1993;16(6):1323-1335.
59 Collins KK, Van Hare GF, Kertesz NJ, et al. Pediatric nonpost-operative junctional ectopic tachycardia medical management and interventional therapies. J Am Coll Cardiol. 2009;53(8):690-697.
60 Villain E, Vetter VL, Garcia JM, et al. Evolving concepts in the management of congenital junctional ectopic tachycardia. A multicenter study. Circulation. 1990;81(5):1544-1549.
61 Van Hare GF, Velvis H, Langberg JJ. Successful transcatheter ablation of congenital junctional ectopic tachycardia in a ten-month-old infant using radiofrequency energy. Pacing Clin Electrophysiol. 1990;13(6):730-735.
62 Rychik J, Marchlinski FE, Swersky SH, et al. Transcatheter radiofrequency ablation of congenital junctional ectopic tachycardia in a neonate. Pediatr Cardiol. 1996;17:220-222.
63 Alexander ME, Berul CI. Ventricular arrhythmias: When to worry. Pediatr Cardiol. 2000;21(6):532-541.
64 Tsuji A, Nagashima M, Hasegawa S, et al. Long-term follow-up of idiopathic ventricular arrhythmias in otherwise normal children. Jpn Circ J. 1995;59(10):654-662.
65 Vetter VL. Ventricular arrhythmias in pediatric patients with and without congenital heart disease. In: Horowitz LN, editor. Current management of arrhythmias. Philadelphia: BC Decker, 1990.
66 Benson DWJr, Gallagher JJ, Sterba R, et al. Catecholamine induced double tachycardia: Case report in a child. Pacing Clin Electrophysiol. 1980;3(1):96-103.
67 Chimenti C, Calabrese F, Thiene G, et al. Inflammatory left ventricular microaneurysms as a cause of apparently idiopathic ventricular tachyarrhythmias. Circulation. 2001;104(2):168-173.
68 Saidi AS, Moodie DS, Garson AJ, et al. Electrocardiography and 24-hour electrocardiographic ambulatory recording (Holter monitor) studies in children infected with human immunodeficiency virus type 1. The Pediatric Pulmonary and Cardiac Complications of Vertically Transmitted HIV-1 Infection Study Group. Pediatr Cardiol. 2000;21(3):189-196.
69 Perron AD, Brady WJ, Erling BF. Commotio cordis: An underappreciated cause of sudden cardiac death in young patients. Assessment and management in the ED. Am J Emerg Med. 2001;19(5):406-409.
70 Kohl P, Nesbitt AD, Cooper PJ, Lei M. Sudden cardiac death by commotio cordis: Role of mechano-electric feedback. Cardiovasc Res. 2001;50(2):280-289.
71 Link MS, Wang PJ, Maron BJ, Estes NA. What is commotio cordis? Cardiol Rev. 1999;7(5):265-269.
72 Bonnet D, Martin D, Pascale De Lonlay, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248-2253.
73 Davis AM, Gow RM, McCrindle BW, Hamilton RM. Clinical spectrum, therapeutic management, and follow-up of ventricular tachycardia in infants and young children. Am Heart J. 1996;131(1):186-191.
74 Fulton DR, Chung KJ, Tabakin BS, Keane JF. Ventricular tachycardia in children without heart disease. Am J Cardiol. 1985;55(11):1328-1331.
75 Rocchini AP, Chun PO, Dick M. Ventricular tachycardia in children. Am J Cardiol. 1981;47(5):1091-1097.
76 Noh CI, Gillette PC, Case CL, Zeigler VL. Clinical and electrophysiological characteristics of ventricular tachycardia in children with normal hearts. Am Heart J. 1990;120(6 Pt 1):1326-1333.
77 Corrado D, Basso C, Thiene G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc Res. 2001;50(2):399-408.
78 Vetter VL, Josephson ME, Horowitz LN. Idiopathic recurrent sustained ventricular tachycardia in children and adolescents. Am J Cardiol. 1981;47(2):315-322.
79 Garson AJr, Smith RT, Moak JP, et al. Ventricular arrhythmias and sudden death in children. J Am Coll Cardiol. 1985;5(6 Suppl):130B-133B.
80 Deal BJ, Miller SM, Scagliotti D, et al. Ventricular tachycardia in a young population without overt heart disease. Circulation. 1986;73(6):1111-1118.
81 Freire G, Dubrow I. Accelerated idioventricular rhythm in newborns: A worrisome but benign entity with or without congenital heart disease. Pediatr Cardiol. 2008;29(2):457-462.
82 MacLellan-Tobert SG, Porter CJ. Accelerated idioventricular rhythm: A benign arrhythmia in childhood. Pediatrics. 1995;96(1 Pt 1):122-125.
83 Nakagawa M, Yoshihara T, Matsumura A, et al. Accelerated idioventricular rhythm in three newborn infants with congenital heart disease. Chest. 1993;104(1):322-323.
84 Martini B, Nava A, Thiene G, et al. Accelerated idioventricular rhythm of infundibular origin in patients with a concealed form of arrhythmogenic right ventricular dysplasia. Br Heart J. 1988;59(5):564-571.
85 Pfammatter JP, Paul T. Idiopathic ventricular tachycardia in infancy and childhood: A multicenter study on clinical profile and outcome. Working Group on Dysrhythmias and Electrophysiology of the Association for European Pediatric Cardiology. J Am Coll Cardiol. 1999;33(7):2067-2072.
86 Frank R, Fontaine G, Vedel J, et al. Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia [translation]. Arch Mal Coeur Vaiss. 1978;71(9):963-972.
87 Dungan WT, Garson AJr. Gillette PC: Arrhythmogenic right ventricular dysplasia: A cause of ventricular tachycardia in children with apparently normal hearts. Am Heart J. 1981;102(4):745-750.
88 Lobo FV, Silver MD, Butany J, Heggtveit HA. Left ventricular involvement in right ventricular dysplasia/cardiomyopathy. Can J Cardiol. 1999;15(11):1239-1247.
89 Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373(9671):1289-1300.
90 Casset-Senon D, Babuty D, Alison D, et al. Delayed contraction area responsible for sustained ventricular tachycardia in an arrhythmogenic right ventricular cardiomyopathy: Demonstration by Fourier analysis of SPECT equilibrium radionuclide angiography. J Nucl Cardiol. 2000;7(5):539-542.
91 Sekiguchi K, Miya Y, Kaneko Y, et al. Evaluation of signal-averaged electrocardiography for clinical diagnosis in arrhythmogenic right ventricular dysplasia. Jpn Heart J. 2001;42(3):287-294.
92 Turrini P, Angelini A, Thiene G, et al. Late potentials and ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 1999;83(8):1214-1219.
93 Toyofuku M, Takaki H, Sunagawa K, et al. Exercise-induced ST elevation in patients with arrhythmogenic right ventricular dysplasia. J Electrocardiol. 1999;32(1):1-5.
94 Groh WJ, Silka MJ, Oliver RP, et al. Use of implatable cardioverter-defibrillators in the congenital long QT syndrome. Am J Cardiol. 1996;78(6):703-706.
95 Fontaine G, Fontaliran F, Hebert JL, et al. Arrhythmogenic right ventricular dysplasia. Annu Rev Med. 1999;50:17-35.
96 Casset-Senon D, Philippe L, Babuty D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy by Fourier analysis of gated blood pool single-photon emission tomography. Am J Cardiol. 1998;82(11):1399-1404.
97 Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075-1084.
98 Li D, Ahmad F, Gardner MJ, et al. The locus of a novel gene responsible for arrhythmogenic right-ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet. 2000;66(1):148-156.
99 Link MS, Wang PJ, Haugh CJ, et al. Arrhythmogenic right ventricular dysplasia: Clinical results with implantable cardioverter defibrillators. J Interv Card Electrophysiol. 1997;1(1):41-48.
100 Tavernier R, Gevaert S, De Sutter J, et al. Long term results of cardioverter-defibrillator implantation in patients with right ventricular dysplasia and malignant ventricular tachyarrhythmias. Heart. 2001;85(1):53-56.
101 Guiraudon GM, Klein GJ, Gulamhusein SS, et al. Total disconnection of the right ventricular free wall: Surgical treatment of right ventricular tachycardia associated with right ventricular dysplasia. Circulation. 1983;67(2):463-470.
102 Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109(2):399-411.
103 Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54(1):59-68.
104 Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare delleceta pediatrica. Clin Pediatr. 1963;45:565.
105 Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103-106.
106 Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3:18.
107 Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51(24):2291-2300.
108 Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358(2):169-176.
109 Moss AJ. Long QT syndrome. JAMA. 2003;289(16):2041-2044.
110 Garson AJr, Dick M, Fournier A, et al. The long QT syndrome in children. An international study of 287 patients. Circulation. 1993;87(6):1866-1872.
111 Scott WA, Dick M. Two : one atrioventricular block in infants with congenital long QT syndrome. Am J Cardiol. 1987;60(16):1409-1410.
112 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: A prospective international study. Circulation. 1985;71(1):17-21.
113 Moss AJ, Robinson JL. Long QT syndrome. Heart Dis Stroke. 1992;1(5):309-314.
114 Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation. 1993;88(2):782-784.
115 Vetter VL, Berul CI, Sweeten TL. Response of corrected QT intervals to exercise, pacing and isoproterenol. Cardiol Young. 1993;3(Suppl 1):63.
116 Callans DJ, Schwartzman D, Marchlinski FE. Is an isoproterenol induced QT abnormality specific to patients with the long QT syndrome? [abstract]. Circulation. 1996;94(Suppl 1):1-432.
117 Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med. 1992;327(12):846-852.
118 Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: Clinical impact. Circulation. 1999;99(4):529-533.
119 Keating M, Dunn C, Atkinson D, et al. Consistent linkage of the long-QT syndrome to the Harvey ras-1 locus on chromosome 11. Am J Hum Genet. 1991;49(6):1335-1339.
120 Towbin JA, Li H, Taggart RT, et al. Evidence of genetic heterogeneity in Romano-Ward long QT syndrome. Analysis of 23 families. Circulation. 1994;90(6):2635-2644.
121 Vincent GM. Heterogeneity in the inherited long QT syndrome. J Cardiovasc Electrophysiol. 1995;6(2):137-146.
122 Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet. 1994;8(2):141-147.
123 Schott JJ, Charpentier F, Peltier S, et al. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am J Hum Genet. 1995;57(5):1114-1122.
124 Barhanin J, Lesage F, Guillemare E, et al. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78-80.
125 Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97(2):175-187.
126 Keating M, Atkinson D, Dunn C, et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252(5006):704-706.
127 Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80(5):795-803.
128 Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115(19):2481-2489.
129 Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17-23.
130 Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80-83.
131 Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96(1):195-215.
132 Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104(4):569-580.
133 Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102(10):1178-1185.
134 Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause Jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17(3):267-268.
135 Splawski I, Tristani-Firouzi M, Lehmann MH, et al. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17(3):338-340.
136 Vetter DE, Mann JR, Wangemann P, et al. Inner ear defects induced by null mutation of the isk gene. Neuron. 1996;17(6):1251-1264.
137 Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81(2):299-307.
138 Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269(5220):92-95.
139 Mitcheson JS, Chen J, Lin M, et al. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A. 2000;97(22):12329-12333.
140 Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80(5):805-811.
141 Gellens ME, George ALJr, Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89(2):554-558.
142 Bennett PB, Yazawa K, Makita N, George ALJr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376(6542):683-685.
143 Reference deleted in proofs.
144 Rosero SZ, Zareba W, Moss AJ, et al. Asthma and the risk of cardiac events in the long QT syndrome. Long QT Syndrome Investigative Group. Am J Cardiol. 1999;84(12):1406-1411.
145 Lupoglazoff JM, Denjoy I, Villain E, et al. Long QT syndrome in neonates: Conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol. 2004;43(5):826-830.
146 Lupoglazoff JM, Cheav T, Baroudi G, et al. Homozygous SCN5A mutation in long-QT syndrome with functional two-to-one atrioventricular block. Circ Res. 2001;89(2):E16-E21.
147 Trippel DL, Parsons MK, Gillette PC. Infants with long-QT syndrome and 2:1 atrioventricular block. Am Heart J. 1995;130(5):1130-1134.
147a Aziz PF, Tanel RE, Zelster IJ, et al. Congenital long QT syndrome and 2 : 1 atrioventricular block: An optimistic outcome in the current era. Heart Rhythm. 2010;78:1-5.
148 Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15(2):186-189.
149 Schwartz PJ, Spazzolini C, Crotti L, et al. The Jervell and Lange-Nielsen syndrome: Natural history, molecular basis, and clinical outcome. Circulation. 2006;113(6):783-790.
150 Zareba W. Genotype-specific ECG patterns in long QT syndrome. J Electrocardiol. 2006;39(4 Suppl):S101-S106.
151 Zhang L, Timothy KW, Vincent GM, et al. Spectrum of ST-T-wave patterns and repolarization parameters in congenital long-QT syndrome: ECG findings identify genotypes. Circulation. 2000;102(23):2849-2855.
152 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89-95.
153 Ackerman MJ, Tester DJ, Porter CJ. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin Proc. 1999;74(11):1088-1094.
154 Choi G, Kopplin LJ, Tester DJ, et al. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110(15):2119-2124.
155 Wilde AA, Jongbloed RJ, Doevendans PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQTS2) patients from KVLQT1-related patients (LQTS1). J Am Coll Cardiol. 1999;33(2):327-332.
156 Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296(10):1249-1254.
157 Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117(17):2184-2191.
158 Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348(19):1866-1874.
159 Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338(24):1709-1714.
160 Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007;49(2):240-246.
161 Bazett HC. An analysis of the time-relations of electrocardiograms. Heart. 1920;7:353-370.
162 Garson AJr. How to measure the QT interval—what is normal? Am J Cardiol. 1993;72(6):14B-16B.
163 Priori SG, Napolitano C, Diehl L, Schwartz PJ. Dispersion of the QT interval. A marker of therapeutic efficacy in the idiopathic long QT syndrome. Circulation. 1994;89(4):1681-1689.
164 Moss AJ. Measurement of the QT interval and the risk associated with QTc interval prolongation: A review. Am J Cardiol. 1993;72(6):23B-25B.
165 Malfatto G, Beria G, Sala S, et al. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23(2):296-301.
166 Shah MJ, Wieand TS, Rhodes LA, et al. QT and JT dispersion in children with long QT syndrome. J Cardiovasc Electrophysiol. 1997;8(6):642-648.
167 Banai S, Tzivoni D. Drug therapy for torsade de pointes. J Cardiovasc Electrophysiol. 1993;4(2):206-210.
168 Kothari SS, Krishnaswami S. Magnesium sulfate therapy in torsades de pointes. Indian Heart J. 1988;40(3):210-211.
169 Crawford MH, Karliner JS, O’Rourke RA, Friedman WF. Prolonged Q-T interval syndrome. Successful treatment with combined ventricular pacing and propanolol. Chest. 1975;68(3):369-371.
170 Trippel DL, Gillette PC. Atenolol in children with ventricular arrhythmias. Am Heart J. 1990;119(6):1312-1316.
171 Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: Clinical implications. J Intern Med. 2006;259(1):39-47.
172 Moss AJ, Robinson J. Clinical features of the idiopathic long QT syndrome. Circulation. 1992;85(1 Suppl):I140-I144.
173 Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101(6):616-623.
174 Locati EH, Schwartz PJ, Moss AJ, et al. Long-term survival after left cervico-thoracic sympathectomy in high risk long QT syndrome patients with refractory ventricular arrhythmias. J Am Coll Cardiol. 1986;7:235A.
175 Packer DL, Coltorti F, Smith MS, et al. Sudden death after left stellectomy in the long QT syndrome. Am J Cardiol. 1984;54(10):1365-1366.
176 Ouriel K, Moss AJ. Long QT syndrome: An indication for cervicothoracic sympathectomy. Cardiovasc Surg. 1995;3(5):475-478.
177 Schwartz PJ, Locati EH, Moss AJ, et al. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation. 1991;84(2):503-511.
178 Eldar M, Griffin JC, Abbott JA, et al. Permanent cardiac pacing in patients with the long QT syndrome. J Am Coll Cardiol. 1987;10(3):600-607.
179 Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84(4):1524-1529.
180 Daubert JP, Zareba W, Rosero SZ, et al. Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am Heart J. 2007;153(4 Suppl):53-58.
181 Silka MJ, Kron J, Dunnigan A, Dick M. Sudden cardiac death and the use of implantable cardioverter-defibrillators in pediatric patients. The Pediatric Electrophysiology Society. Circulation. 1993;87(3):800-807.
182 Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94(8):1996-2012.
183 Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92(12):3381-3386.
184 Sato T, Hata Y, Yamamoto M, et al. Early afterdepolarization abolished by potassium channel opener in a patient with idiopathic long QT syndrome. J Cardiovasc Electrophysiol. 1995;6(4):279-282.
185 Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49(3):329-337.
186 Sze E, Moss AJ, Goldenberg I, et al. Long QT syndrome in patients over 40 years of age: Increased risk for LQTS-related cardiac events in patients with coronary disease. Ann Noninvasive Electrocardiol. 2008;13(4):327-331.
187 Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109(1):30-35.
188 Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: A new clinical syndrome? Cardiology. 2000;94(2):99-102.
189 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: A familial cause of sudden death. Circulation. 2003;108(8):965-970.
190 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: Pharmacological treatment. J Am Coll Cardiol. 2004;43(8):1494-1499.
191 Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391-1396.
192 Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29(10):1130-1159.
193 Skinner JR, Chung SK, Montgomery D, et al. Near-miss SIDS due to Brugada syndrome. Arch Dis Child. 2005;90(5):528-529.
194 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293-296.
195 Brugada J, Brugada R, Brugada P. Channelopathies: A new category of diseases causing sudden death. Herz. 2007;32(3):185-191.
196 Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105(6):707-713.
197 Naccarelli GV, Antzelevitch C. The Brugada syndrome: Clinical, genetic, cellular, and molecular abnormalities. Am J Med. 2001;110(7):573-581.
198 Skinner JR, Chung SK, Nel CA, et al. Brugada syndrome masquerading as febrile seizures. Pediatrics. 2007;119(5):e1206-e1211.
198a Schimpf R, Giustetto C, Eckardt L, et al. Prevalence of supraventricular tachyarrhythmias in a cohort of 115 patients with Brugada syndrome. Ann Noninvasive Electrocardiol. 2008;13(3):266-269.
198b Gehi AK, Duong TD, Metz LD, et al. Risk stratification of individuals with the Brugada electrocardiogram: A meta-analysis. J Cardiovasc Electrophysiol. 2006;17(6):577-583.
199 Brugada R. Use of intravenous antiarrhythmics to identify concealed Brugada syndrome. Curr Control Trials Cardiovasc Med. 2000;1(1):45-47.
200 Priori SG, Napolitano C. Management of patients with Brugada syndrome should not be based on programmed electrical stimulation. Circulation. 2005;112:285-291.
201 Brugada P, Brugada R, Brugada J. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation. 2005;112(2):279-292.
202 Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11(11):1277-1280.
203 Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51(1):23-30.
204 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196-200.
205 Wilde AA, Bezzina CR. Genetics of cardiac arrhythmias. Heart. 2005;91(10):1352-1358.
206 Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106(1):69-74.
207 Sumitomo N, Sakurada H, Taniguchi K, et al. Association of atrial arrhythmia and sinus node dysfunction in patients with catecholaminergic polymorphic ventricular tachycardia. Circ J. 2007;71(10):1606-1609.
208 Celiker A, Erdogan I, Karagoz T, Ozer S. Clinical experiences of patients with catecholaminergic polymorphic ventricular tachycardia. Cardiol Young. 2009;19(1):45-52.
209 Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358(19):2024-2029.
210 Balaji S, Wiles HB, Sens MA, Gillette PC. Immunosuppressive treatment for myocarditis and borderline myocarditis in children with ventricular ectopic rhythm. Br Heart J. 1994;72(4):354-359.
211 Wiles HB, Gillette PC, Harley RA, Upshur JK. Cardiomyopathy and myocarditis in children with ventricular ectopic rhythm. J Am Coll Cardiol. 1992;20(2):359-362.
212 Ino T, Okubo M, Akimoto K, et al. Corticosteroid therapy for ventricular tachycardia in children with silent lymphocytic myocarditis. J Pediatr. 1995;126(2):304-308.
213 Drucker NA, Colan SD, Lewis AB, et al. Gamma-globulin treatment of acute myocarditis in the pediatric population. Circulation. 1994;89(1):252-257.
214 Vetter VL, Horowitz LN. Electrophysiologic residua and sequelae of surgery for congenital heart defects. Am J Cardiol. 1982;50(3):588-604.
215 Silka MJ, Hardy BG, Menashe VD, Morris CD. A population-based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol. 1998;32(1):245-251.
216 Garson AJr, Porter CB, Gillette PC, McNamara DG. Induction of ventricular tachycardia during electrophysiologic study after repair of tetralogy of Fallot. J Am Coll Cardiol. 1983;1(6):1493-1502.
217 Horowitz LN, Vetter VL, Harken AH, Josephson ME. Electrophysiologic characteristics of sustained ventricular tachycardia occurring after repair of tetralogy of Fallot. Am J Cardiol. 1980;46(3):446-452.
218 Deal BJ, Scagliotti D, Miller SM, et al. Electrophysiologic drug testing in symptomatic ventricular arrhythmias after repair of tetralogy of Fallot. Am J Cardiol. 1987;59(15):1380-1385.
218a Garson AJr, Randall DC, Gillette PC, et al. Prevention of sudden death after repair of tetralogy of Fallot: Treatment of ventricular arrhythmias. J Am Coll Cardiol. 1985;6(1):221-227.
219 Sullivan ID, Presbitero P, Gooch VM, et al. Is ventricular arrhythmia in repaired tetralogy of Fallot an effect of operation or a consequence of the course of the disease? A prospective study. Br Heart J. 1987;58(1):40-44.
220 Garson AJr, Gillette PC, Titus JL, et al. Surgical treatment of ventricular tachycardia in infants. N Engl J Med. 1984;310(22):1443-1445.
221 Malhotra V, Ferrans VJ, Virmani R. Infantile histiocytoid cardiomyopathy: Three cases and literature review. Am Heart J. 1994;128(5):1009-1021.
222 Jeresaty RM. Sudden death in the mitral valve prolapse-click syndrome. Am J Cardiol. 1976;37(2):317-318.
223 Maron BJ, Roberts WC, McAllister HA, et al. Sudden death in young athletes. Circulation. 1980;62(2):218-229.
224 Swartz MH, Teichholz LE, Donoso E. Mitral valve prolapse: A review of associated arrhythmias. Am J Med. 1977;62(3):377-389.
224a La Vecchia L, Ometto R, Centofante P, et al. Arrhythmic profile, ventricular function, and histomorphometric findings in patients with idiopathic ventricular tachycardia and mitral valve prolapse: Clinical and prognostic evaluation. Clin Cardiol. 1998;21:731-735.
225 Kavey RE, Sondheimer HM, Blackman MS. Detection of dysrhythmia in pediatric patients with mitral valve prolapse. Circulation. 1980;62(3):582-587.
225a Osswald SS, Gaffney FA, Kruyer WB, et al. Military aviators with mitral valve prolapse: Long-term follow-up and aeromedical endpoints. Aviat Space Environ Med. 2007;78(9):845-851.
226 Papagiannis J, Van PR, Schwint O, et al. Congenital left ventricular aneurysm: Clinical, imaging, pathologic, and surgical findings in seven new cases. Am Heart J. 2001;141(3):491-499.
227 Poliac LC, Barron ME, Maron BJ. Hypertrophic cardiomyopathy. Anesthesiology. 2006;104(1):183-192.
228 Franz WM, Muller OJ, Katus HA. Cardiomyopathies: From genetics to the prospect of treatment. Lancet. 2001;358(9293):1627-1637.
229 Maron BJ, Roberts WC, Epstein SE. Sudden death in hypertrophic cardiomyopathy: A profile of 78 patients. Circulation. 1982;65(7):1388-1394.
230 McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: Role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47(3):532-538.
231 Maron BJ, Savage DD, Wolfson JK, Epstein SE. Prognostic significance of 24 hour ambulatory electrocardiographic monitoring in patients with hypertrophic cardiomyopathy: A prospective study. Am J Cardiol. 1981;48(2):252-257.
232 McKenna WJ, England D, Doi YL, et al. Arrhythmia in hypertrophic cardiomyopathy. I: Influence on prognosis. Br Heart J. 1981;46(2):168-172.
233 Maron BJ, Fananapazir L. Sudden cardiac death in hypertrophic cardiomyopathy. Circulation. 1992;85(1 Suppl):I57-I63.
234 Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24(21):1965-1991.
235 Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363(9424):1881-1891.
236 McKenna WJ, Franklin RC, Nihoyannopoulos P, et al. Arrhythmia and prognosis in infants, children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1988;11(1):147-153.
237 Yetman AT, Hamilton RM, Benson LN, McCrindle BW. Long-term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1998;32(7):1943-1950.
238 Saumarez RC. Electrophysiological investigation of patients with hypertrophic cardiomyopathy. Evidence that slowed intraventricular conduction is associated with an increased risk of sudden death. Br Heart J. 1994;72(6 Suppl):S19-S23.
239 Mattos BP, Torres MA, Freitas VC. Diagnostic evaluation of hypertrophic cardiomyopathy in its clinical and preclinical phases. Arq Bras Cardiol. 2008;91(1):51-62.
240 Marian AJ. Sudden cardiac death in patients with hypertrophic cardiomyopathy: From bench to bedside with an emphasis on genetic markers. Clin Cardiol. 1995;18(4):189-198.
241 Thierfelder L, Watkins H, MacRae C, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell. 1994;77(5):701-712.
242 Watkins H, Rosenzweig A, Hwang DS, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326(17):1108-1114.
243 McKenna WJ, Spirito P, Desnos M, et al. Experience from clinical genetics in hypertrophic cardiomyopathy: Proposal for new diagnostic criteria in adult members of affected families. Heart. 1997;77(2):130-132.
244 Marian AJ, Roberts R. The molecular genetic basis for hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33(4):655-670.
245 Elliott PM, Reith S, McKenna WJ. Hypertrophic cardiomyopathy. In: Crawford MH, DiMarco JP, Paulus WJ, editors. Cardiology. London: Mosby, 2004.
246 McKenna WJ, Oakley CM, Krikler DM, Goodwin JF. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53(4):412-416.
247 Watkins H, Rosenzweig A, Hwang DS, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326(17):1108-1114.
248 Rosing DR, Condit JR, Maron BJ, et al. Verapamil therapy: A new approach to the pharmacologic treatment of hypertrophic cardiomyopathy: III. Effects of long-term administration. Am J Cardiol. 1981;48(3):545-553.
249 Jeanrenaud X, Goy JJ, Kappenberger L. Effects of dual-chamber pacing in hypertrophic obstructive cardiomyopathy. Lancet. 1992;339(8805):1318-1323.
250 Cohn LH, Trehan H, Collins JJJr. Long-term follow-up of patients undergoing myotomy/myectomy for obstructive hypertrophic cardiomyopathy. Am J Cardiol. 1992;70(6):657-660.
251 Gaita F, Bocchiardo M, Porciani MC, et al. Should stimulation therapy for congestive heart failure be combined with defibrillation backup? Am J Cardiol. 2000;86(9A):165K-168K.
251a Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342(6):365-373.
252 Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45(8):1340-1345.
253 Elliott P. Cardiomyopathy. Diagnosis and management of dilated cardiomyopathy. Heart. 2000;84(1):106-112.
254 Bione S, D’Adamo P, Maestrini E, et al. A novel X-linked gene, G4.5 is responsible for Barth syndrome. Nat Genet. 1996;12(4):385-389.
255 Keeling PJ, McKenna WJ. Clinical genetics of dilated cardiomyopathy. Herz. 1994;19(2):91-96.
256 Ortiz-Lopez R, Li H, Su J, et al. Evidence for a dystrophin missense mutation as a cause of X-linked dilated cardiomyopathy. Circulation. 1997;95(10):2434-2440.
257 Arola A, Jokinen E, Ruuskanen O, et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol. 1997;146(5):385-393.
258 Nogueira G, Pinto FF, Paixao A, Kaku S. Idiopathic dilated cardiomyopathy in children: Clinical profile and prognostic determinants. Rev Port Cardiol. 2000;19(2):191-200.
259 Reference deleted in proofs.
260 Blume ED, Canter CE, Spicer R, et al. Prospective single-arm protocol of carvedilol with ventricular dysfunction. Pediatr Cardiol. 2006;27:336-342.
261 Bajcetic M, Kokic AN, Djukic M, et al. Effects of carvedilol on left ventricular function and oxidative stress in infants and children with idiopathic dilated cardiomyopathy: A 12-month, two-center, open-label study. Clin Ther. 2008;30:702-714.
262 Bruns LA, Chrisant MK, Lamour JM, et al. Carvedilol as therapy in pediatric heart failure: An initial multicenter experience. J Pediatr. 2001;138:505-511.
263 Cazeau S, Ritter P, Lazarus A, et al. Multisite pacing for end-stage heart failure: Early experience. Pacing Clin Electrophysiol. 1996;19(11 Pt 2):1748-1757.
264 Blanc JJ, Etienne Y, Gilard M, et al. Evaluation of different ventricular pacing sites in patients with severe heart failure: Results of an acute hemodynamic study. Circulation. 1997;96(10):3273-3277.
265 Lozano I, Bocchiardo M, Achtelik M, et al. Impact of biventricular pacing on mortality in a randomized crossover study of patients with heart failure and ventricular arrhythmias. Pacing Clin Electrophysiol. 2000;23(11 Pt 2):1711-1712.
266 Gras D, Cazeau S, Mabo P, et al. Long-term benefit of cardiac resynchronization in heart failure patients: The 12 month results of the InSyncTM trial [abstract]. J Am Coll Cardiol. 2000;35(Suppl A):230.
267 Klein LS, Shih HT, Hackett FK, et al. Radiofrequency catheter ablation of ventricular tachycardia in patients without structural heart disease. Circulation. 1992;85(5):1666-1674.
268 Reference deleted in proofs.
269 Gillette PC, Reitman MJ, Gutgesell HP, et al. Intracardiac electrography in children and young adults. Am Heart J. 1975;89(1):36-44.
270 Callans DJ, Menz V, Schwartzman D, et al. Repetitive monomorphic tachycardia from the left ventricular outflow tract: Electrocardiographic patterns consistent with a left ventricular site of origin. J Am Coll Cardiol. 1997;29(5):1023-1027.
271 Lauer MR, Liem LB, Young C, et al. Cellular and clinical electrophysiology of verapamil-sensitive ventricular tachycardias. J Cardiovasc Electrophysiol. 1992;3:500-514.
272 Aiba T, Suyama K, Aihara N, et al. The role of Purkinje and pre-Purkinje potentials in the reentrant circuit of verapamil-sensitive idiopathic LV tachycardia. Pacing Clin Electrophysiol. 2001;24(3):333-344.
273 Kornbluth A, Frishman WH, Ackerman M. Beta-adrenergic blockade in children. Cardiol Clin. 1987;5(4):629-649.
274 Garson AJr, Gillette PC, McVey P, et al. Amiodarone treatment of critical arrhythmias in children and young adults. J Am Coll Cardiol. 1984;4(4):749-755.
275 Moak JP, Smith RT, Garson AJr. Newer antiarrhythmic drugs in children. Am Heart J. 1987;113(1):179-185.
276 Drago F, Mazza A, Guccione P, et al. Amiodarone used alone or in combination with propranolol: A very effective therapy for tachyarrhythmias in infants and children. Pediatr Cardiol. 1998;19(6):445-449.
277 Vetter VL. Ventricular arrhythmias in patients with congenital heart disease. In: Greenspon AJ, Waxman HL, editors. Contemporary management of ventricular arrhythmias. Philadelphia: FA Davis, 1991.
278 Walsh CA, McAlister HF, Andrews CA, et al. Pacemaker implantation in children: A 21-year experience. Pacing Clin Electrophysiol. 1988;11(11 Pt 2):1940-1944.
279 Kelly DT, Brodsky SJ, Krovetz LJ. Mobitz type II atrioventricular block in children. J Pediatr. 1971;79(6):972-976.
280 Young D, Eisenberg R, Fish B, Fisher JD. Wenckebach atrioventricular block (Mobitz type I) in children and adolescents. Am J Cardiol. 1977;40(3):393-399.
281 Michaelsson M, Engle MA. Congenital complete heart block: An international study of the natural history. Cardiovasc Clin. 1972;4(3):85-101.
282 Chameides L, Truex RC, Vetter V, et al. Association of maternal systemic lupus erythematosus with congenital complete heart block. N Engl J Med. 1977;297(22):1204-1207.
283 McCue CM, Mantakas ME, Tingelstad JB, Ruddy S. Congenital heart block in newborns of mothers with connective tissue disease. Circulation. 1977;56(1):82-90.
284 Ramsey-Goldman R, Hom D, Deng JS, et al. Anti-SS-A antibodies and fetal outcome in maternal systemic lupus erythematosus. Arthritis Rheum. 1986;29(10):1269-1273.
285 Scott JS, Maddison PJ, Taylor PV, et al. Connective-tissue disease, antibodies to ribonucleoprotein, and congenital heart block. N Engl J Med. 1983;309(4):209-212.
286 Taylor PV, Scott JS, Gerlis LM, Esscher E, Scott O. Maternal antibodies against fetal cardiac antigens in congenital complete heart block. N Engl J Med. 1986;315(11):667-672.
287 Litsey SE, Noonan JA, O’Connor WN, et al. Maternal connective tissue disease and congenital heart block. Demonstration of immunoglobulin in cardiac tissue. N Engl J Med. 1985;312(2):98-100.
288 Buyon JP, Winchester RJ, Slade SG, et al. Identification of mothers at risk for congenital heart block and other neonatal lupus syndromes in their children. Comparison of enzyme-linked immunosorbent assay and immunoblot for measurement of anti-SS-A/Ro and anti-SS-B/La antibodies. Arthritis Rheum. 1993;36(9):1263-1273.
289 Buyon JP, Winchester R. Congenital complete heart block. A human model of passively acquired autoimmune injury. Arthritis Rheum. 1990;33(5):609-614.
290 Reference deleted in proofs.
291 Fryda RJ, Kaplan S, Helmsworth JA. Postoperative complete heart block in children. Br Heart J. 1971;33(4):456-462.
292 Kulbertus HE, Coyne JJ, Hallidie-Smith KA. Conduction disturbances before and after surgical closure of ventricular septal defect. Am Heart J. 1969;77(1):123-131.
293 Moss AJ, Klyman G, Emmanouilides GC. Late onset complete heart block. Newly recognized sequela of cardiac surgery. Am J Cardiol. 1972;30(8):884-887.
294 Levy MJ, Cuello L, Tuna N, Lillehei CW. Atrioventricularis communis: Clinical aspects and surgical treatment. Am J Cardiol. 1964;14:587-598.
295 Michaelsson M, Jonzon A, Riesenfeld T. Isolated congenital complete atrioventricular block in adult life. A prospective study. Circulation. 1995;92(3):442-449.
296 Karpawich PP, Gillette PC, Garson AJr, et al. Congenital complete atrioventricular block: Clinical and electrophysiologic predictors of need for pacemaker insertion. Am J Cardiol. 1981;48(6):1098-1102.
297 Dewey RC, Capeless MA, Levy AM. Use of ambulatory electrocardiographic monitoring to identify high-risk patients with congenital complete heart block. N Engl J Med. 1987;316(14):835-839.
298 Levy AM, Camm AJ, Keane JF. Multiple arrhythmias detected during nocturnal monitoring in patients with congenital complete heart block. Circulation. 1977;55(2):247-253.
299 Sholler GF, Walsh EP. Congenital complete heart block in patients without anatomic cardiac defects. Am Heart J. 1989;118(6):1193-1198.
300 Benson DWJr, Spach MS, Edwards SB, et al. Heart block in children. Evaluation of subsidiary ventricular pacemaker recovery times and ECG tape recordings. Pediatr Cardiol. 1982;2(1):39-45.
301 Lillehei CW, Sellers RD, Bonnabeau RC, Eliot RS. Chronic postsurgical complete heart block. With particular reference to prognosis, management, and a new P-wave pacemaker. J Thorac Cardiovasc Surg. 1963;46:436-456.
302 Esscher E, Hardell LI, Michaelsson M. Familial, isolated, complete right bundle-branch block. Br Heart J. 1975;37(7):745-747.
303 Giuliani ER, Fuster V, Brandenburg RO, Mair DD. Ebstein’s anomaly: The clinical features and natural history of Ebstein’s anomaly of the tricuspid valve. Mayo Clin Proc. 1979;54(3):163-173.
304 Gallastegui J, Hariman RJ, Handler B, et al. Cardiac involvement in the Kearns-Sayre syndrome. Am J Cardiol. 1987;60(4):385-388.
305 Krongrad E. Prognosis for patients with congenital heart disease and postoperative intraventricular conduction defects. Circulation. 1978;57(5):867-870.
306 Horowitz LN, Alexander JA, Edmunds LHJr. Postoperative right bundle branch block: Identification of three levels of block. Circulation. 1980;62(2):319-328.
307 Kugler JD, Danford DA. Pacemakers in children: An update. Am Heart J. 1989;117(3):665-679.
308 Taber RE, Estoye LR, Green ER, Gahagan T. Treatment of congenital and acquired heart block with an implantable pacemaker. Circulation. 1964;29:182-185.
309 Kangos JJ, Griffiths SP, Blumenthal S. Congenital complete heart block. A classification and experience with 18 patients. Am J Cardiol. 1967;20(5):632-638.
310 Glenn WW, De LN, Van Heeckeren DW, et al. Heart block in children. Treatment with a radiofrequency pacemaker. J Thorac Cardiovasc Surg. 1969;58(3):361-373.
311 Furman S, Young D. Cardiac pacing in children and adolescents. Am J Cardiol. 1977;39(4):550-558.
312 Young D. Permanent pacemaker implantation in children: Current status and future considerations. Pacing Clin Electrophysiol. 1981;4(1):61-67.
313 Beder SD, Hanisch DG, Cohen MH, et al. Cardiac pacing in children: A 15-year experience. Am Heart J. 1985;109(1):152-156.
314 Goldman BS, Williams WG, Hill T, et al. Permanent cardiac pacing after open heart surgery: Congenital heart disease. Pacing Clin Electrophysiol. 1985;8(5):732-739.