[level-membership-for-pathology-category]
1 EPITHELIUM
General classification of epithelia
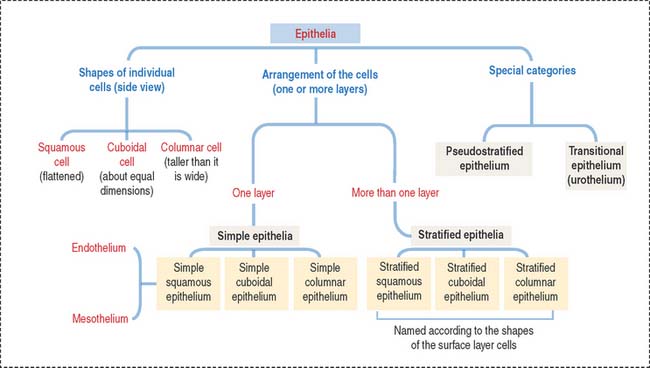
The epithelium is a tightly cohesive sheet of cells that covers or lines body surfaces (for example, skin, intestine, secretory ducts) and forms the functional units of secretory glands (for example, salivary glands, liver). The traditional classification and nomenclature of different types of epithelia are based on the shapes of individual cells and arrangement of the cells in one or more layers (Figure 1-1). The main characteristics of epithelia are listed in Box 1-A.
Box 1-A Main characteristics of epithelia
Epithelia are classified into:
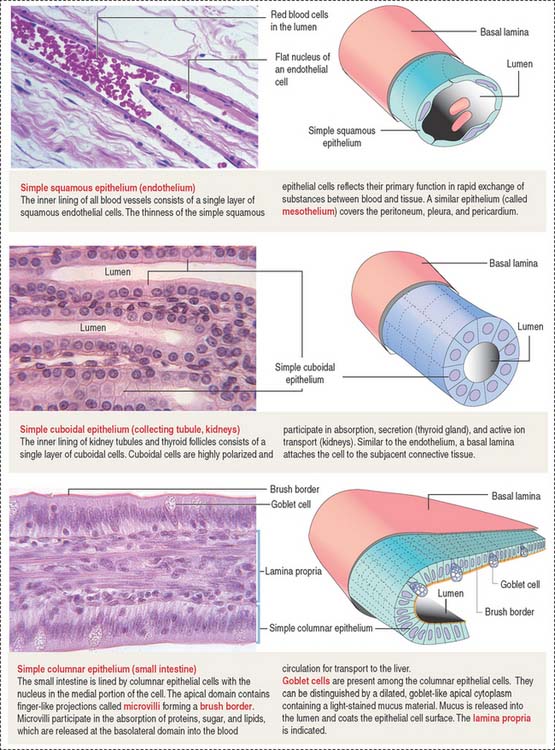
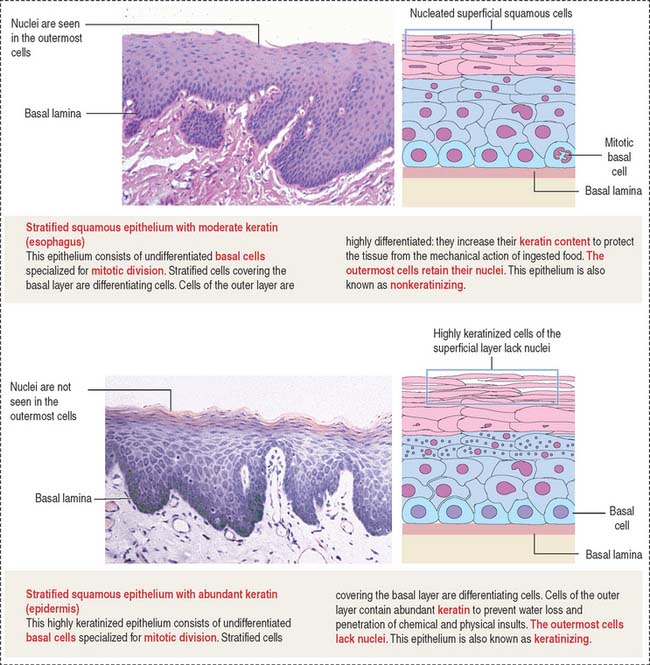
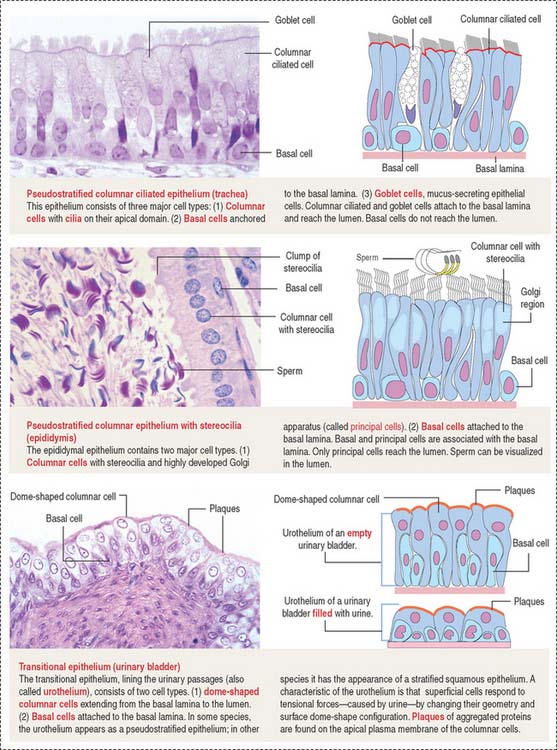
An important aspect of epithelia is its polarity. Most epithelial cells line surfaces and cavities and have three domains (Figure 1-5):
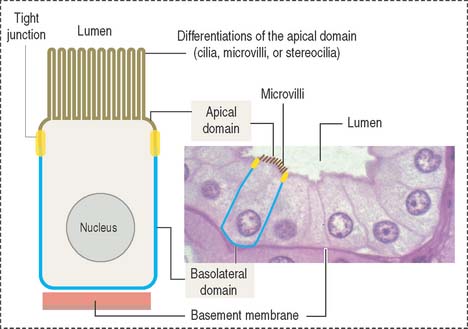
EPITHELIAL CELL POLARITY
Epithelial cells have two major domains (Figure 1-5):
Apical differentiations
The apical domain of some epithelial cells can display three types of differentiation:
Cilia (singular, cilium; Figure 1-6) are motile cell projections originating from basal bodies anchored by rootlets to the apical portion of the cytoplasm. A basal body contains nine triplet microtubules in a helicoid array without a central microtubular component. By contrast, a cilium consists of an assembly called an axoneme, formed by a central pair of microtubules surrounded by nine concentrically arranged microtubular pairs. This assembly is known as the 9 + 2 microtubular doublet arrangement. The axoneme is also a component of the sperm tail, or flagellum.
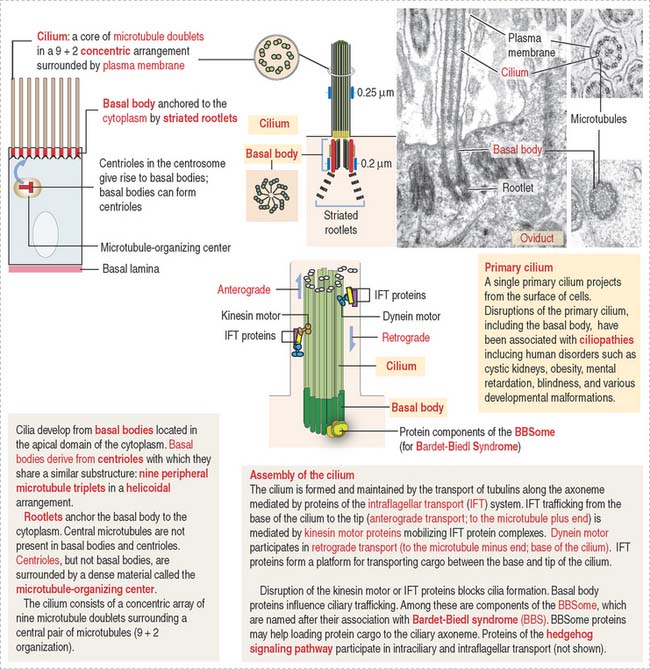
The trachea and the oviduct are lined by ciliated epithelial cells. In these epithelia, ciliary activity is important for the local defense of the respiratory system and for the transport of the fertilized egg to the uterine cavity.
Some cells have a primary cilium. The importance of primary cilia emerges from rare recessive human disorders known as ciliopathies caused by structural or functional abnormalities of cilia. The structure and mechanism of assembly of primary cilia are shown in Figure 1-6. The significant aspects of the primary cilium are: (1) they are non-motile; (2) they participate in the early stages of embryonic patterning leading to organogenesis; (3) many components of the hedgehog signaling pathway, essential at least in early development, are present in primary cilia; and (4) the position of the primary cilium, called kinocilium, of the hair cell of the organ of Corti in the inner ear determines the correct polarity of the actin-containing stereocilia (see Chapter 9, Sensory Organs: Vision and Hearing).
Microvilli (singular, microvillus; Figure 1-7) are finger-like cell projections of the apical epithelial cell surface containing a core of cross-linked microfilaments (a polymer of G-actin monomers). At the cytoplasmic end of the microvillus, bundles of actin and other proteins extend into the terminal web, a filamentous network of cytoskeletal proteins running parallel to the apical domain of the epithelial cell.
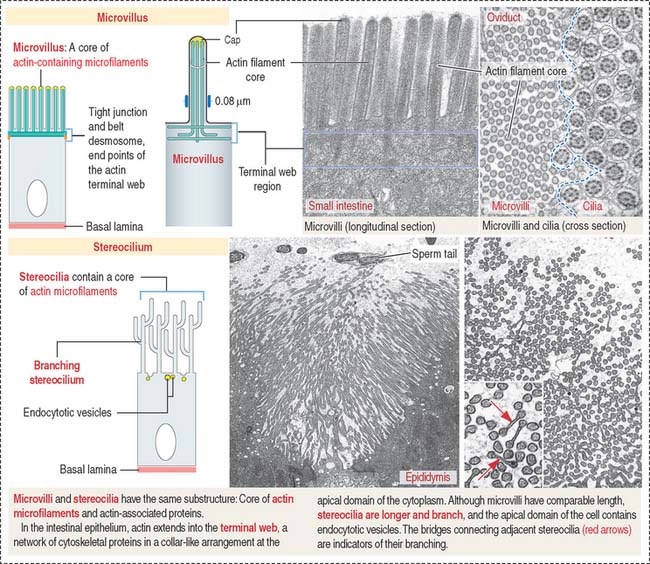
Stereocilia (singular, stereocilium; see Figure 1-7) are long and branching finger-like projections of the apical epithelial cell surface. Similar to microvilli, stereocilia contain a core of cross-linked actin with other proteins. Stereocilia do not have axonemes. Stereocilia are typical of the epithelial lining of the epididymis and contribute to the process of sperm maturation occurring in this organ.
CELL ADHESION MOLECULES
A sheet of epithelial cells results from the tight attachment of similar cells to each other and to the basal lamina, a component of the extracellular matrix. Cell adhesion molecules enable interepithelial cell contact, and this contact is stabilized by specialized cell junctions. A consequence of this arrangement is the apical and basolateral domain polarity of an epithelial sheet.
Although cell adhesion molecules and cell junctions are considered here within the framework of epithelia, nonepithelial cells also can use cell adhesion molecules and junctions to establish contact with each other, enabling cell-cell communication. A typical example of nonepithelial cells connected by specialized junctions is the cardiac muscle (see Chapter 7, Muscle Tissue).
There are two major classes of cell adhesion molecules (see Box 1-B):
Box 1-B Cell adhesion molecules
Cadherins (Figure 1-8) are a family of Ca2+-dependent molecules with a major role in cell adhesion and morphogenesis. A loss of cadherins is associated with the acquisition of invasive behavior by tumor cells (metastasis) (see Chapter 4, Connective Tissue).
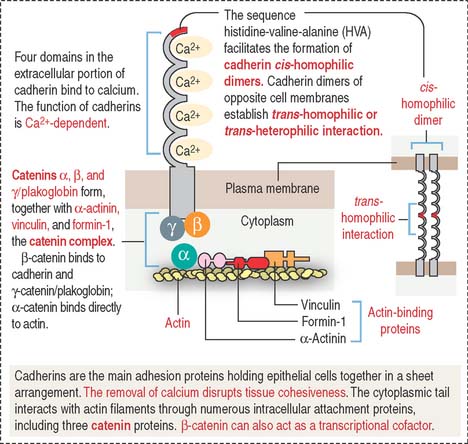
There are more than 40 different cadherins. E-cadherin is an epithelial cadherin found along the lateral cell surfaces and is responsible for the maintenance of most epithelial layers. The removal of calcium or the use of a blocking antibody to E-cadherin in epithelial cell cultures breaks down cell-cell attachment, and the formation of stabilizing junctions is disrupted. E-cadherin molecules form cis-homophilic dimers (“like-to-like”), which bind to dimers of the same or different class of cadherins in the opposite cell membrane (trans-homophilic or heterophilic [“like-to-unlike”] interaction). These forms of binding require the presence of calcium and result in a specialized zipper-like cell-cell adhesion pattern.
Members of the cadherin family also are present between cytoplasmic plaques of the zonula and the macula adherens. β-catenin plays a significant role in colorectal carcinogenesis (see Chapter 16, Lower Digestive Segment).
Selectins (Figure 1-9), similar to cadherins, are Ca2+-dependent cell adhesion molecules. In contrast to cadherins, selectins bind to carbohydrates and belong to the group of lectins (Latin lectum, to select). Each selectin has a carbohydrate-recognition domain (CRD) with binding affinity to a specific oligosaccharide attached to a protein (glycoprotein) or a lipid (glycolipid). The molecular configuration of the CRD is controlled by calcium.
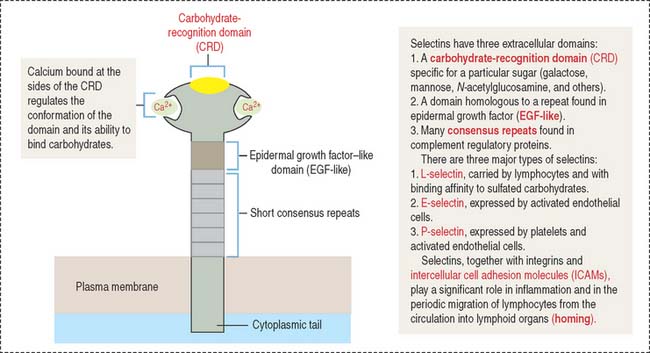
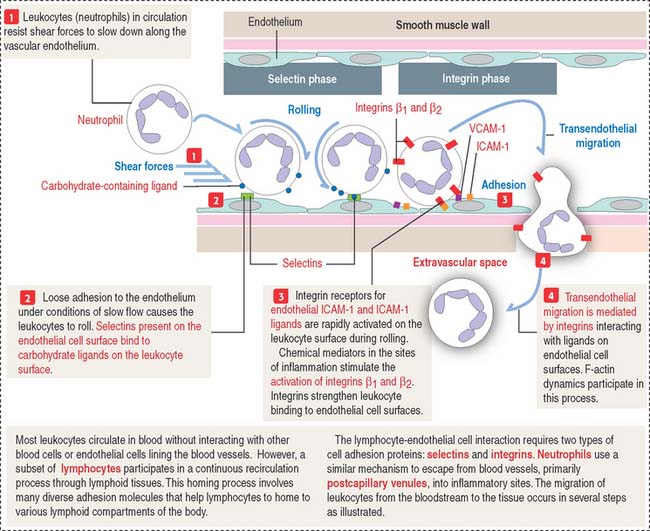
Selectins participate in the movement of leukocytes (Greek leukos, white, kytos, cell) circulating in blood (neutrophils, monocytes, B and T cells) toward tissues by extravasation. Extravasation is the essence of homing, a mechanism that enables leukocytes to escape from blood circulation and reach the sites of inflammation (see Figure 1-12). Homing also permits thymus-derived T cells to home in on peripheral lymph nodes (see Chapter 10, Immune-Lymphatic System).
The three major classes of cell surface selectins are as follows:
P-selectin is stored in cytoplasmic vesicles in endothelial cells. When endothelial cells are activated by inflammatory signaling, P-selectin appears on the cell surface. On their surface, leukocytes contain sialyl Lewis-x antigen, a specific oligosaccharide ligand for P-selectin. P-selectin binding to the antigen slows down streaming leukocytes in blood, and they begin to roll along the endothelial cell surfaces. P-selectins get additional help from members of the immunoglobulin (Ig) superfamily and integrins to stabilize leukocyte attachment, leading to extravasation (see Figure 1-12).
A conserved feature shared by all members of the Ig superfamily is an extracellular segment with one or more folded domains characteristic of immunoglobulins (Figure 1-10). Of particular interest is CD4, a member of the Ig superfamily and the receptor for the human immunodeficiency virus type 1 (HIV-1) in a subclass of lymphocytes known as T cells or helper cells. We discuss the significance of several members of the Ig superfamily in Chapter 10, Immune-Lymphatic System.
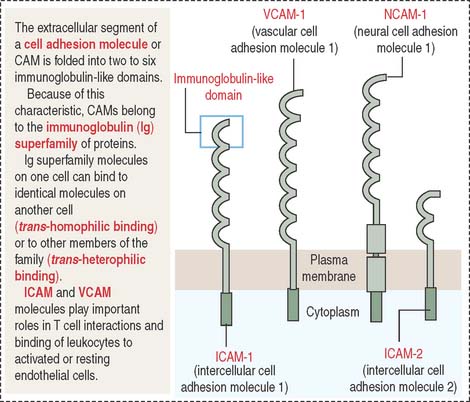
Other members of the Ig superfamily play important roles in the homing process during inflammation. Examples include intercellular adhesion molecules 1 and 2 (ICAM-1 and ICAM-2) on endothelial cell surfaces. ICAM-1 is expressed when an inflammation is in progress to facilitate the transendothelial migration of leukocytes (see Chapter 6, Blood and Hematopoiesis).
Integrins (Figure 1-11) differ from cadherins, selectins, and members of the Ig superfamily in that integrins are heterodimers formed by two associated α and β subunits encoded by different genes. There are about 22 integrin heterodimers consisting of 17 forms of α subunits and 8 forms of β subunits.
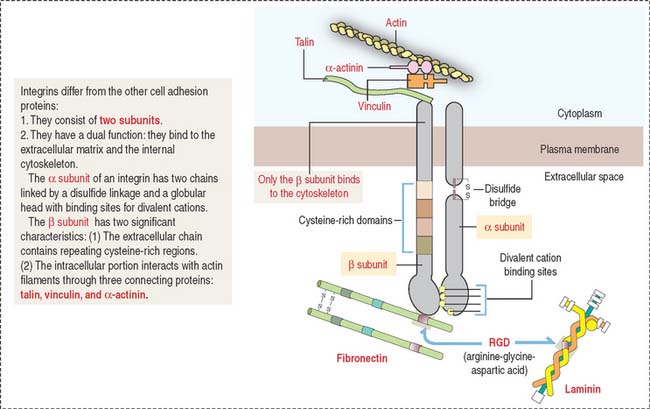
Almost every cell expresses one or several integrins. Similar to cadherins, the cytoplasmic domain of β integrins is linked to actin filaments through connecting proteins (talin, vinculin, and α-actinin).
The integrin–extracellular matrix relationship is critical for cell migration to precise sites during embryogenesis and can be regulated when cell motility is required. In addition to their role in cell-matrix interactions, integrins also mediate cell-cell interaction. Integrins containing β2 subunits are expressed on the surface of leukocytes and mediate cell-cell binding. An example is the α1β2 integrin heterodimer that binds to ligands on endothelial cell surfaces during the integrin phase (extravasation) of homing (Figure 1-12).
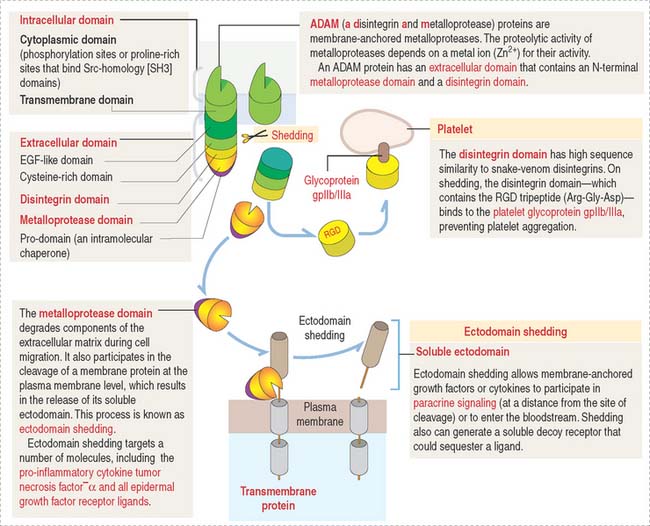
ADAM proteins
The reversal of integrin-mediated cell binding to the extracellular matrix can be disrupted by proteins called ADAM (for a disintegrin and metalloprotease). ADAMs have pivotal roles in fertilization, angiogenesis, neurogenesis, heart development, cancer and Alzheimer’s disease (see Chapter 8, Nervous Tissue).
A typical ADAM protein (Figure 1-13) contains an extracellular domain and an intracellular domain. The extracellular domain consists of several portions including a disintegrin domain and a metalloprotease domain.
CELL JUNCTIONS
Although cell adhesion molecules are responsible for cell-cell adhesion, cell junctions are necessary for providing stronger stability. In addition, the movement of solutes, ions, and water through an epithelial layer occurs across and between individual cell components. The transcellular pathway is controlled by numerous channels and transporters. The paracellular pathway is regulated by a continuous intercellular contact or cell junctions. A deficiency in the cell junctions accounts for acquired and inherited diseases caused by inefficient epithelial barriers.
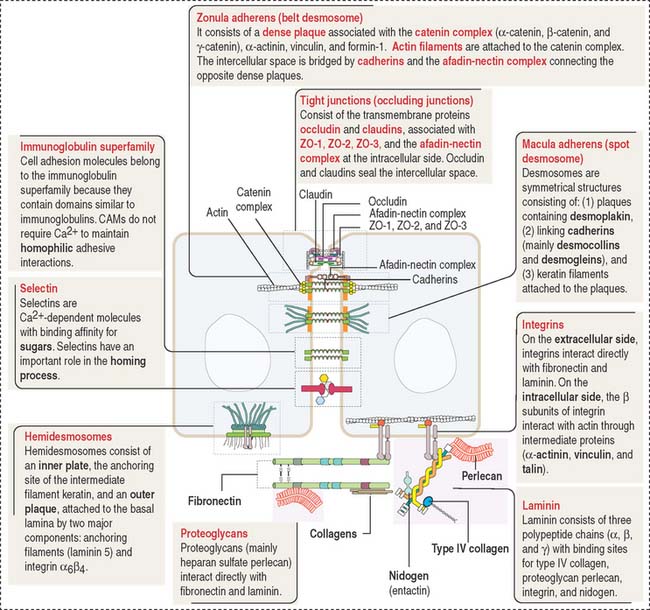
Cell junctions are symmetrical structures formed between two adjacent cells. There are three major classes of symmetrical cell junctions (Figure 1-14; see Box 1-C):
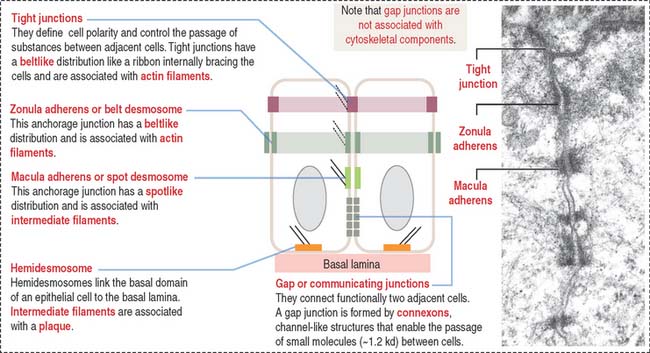
Box 1-C Cell junctions
Tight junctions (also called occluding junctions) (Figure 1-15) have two major functions:
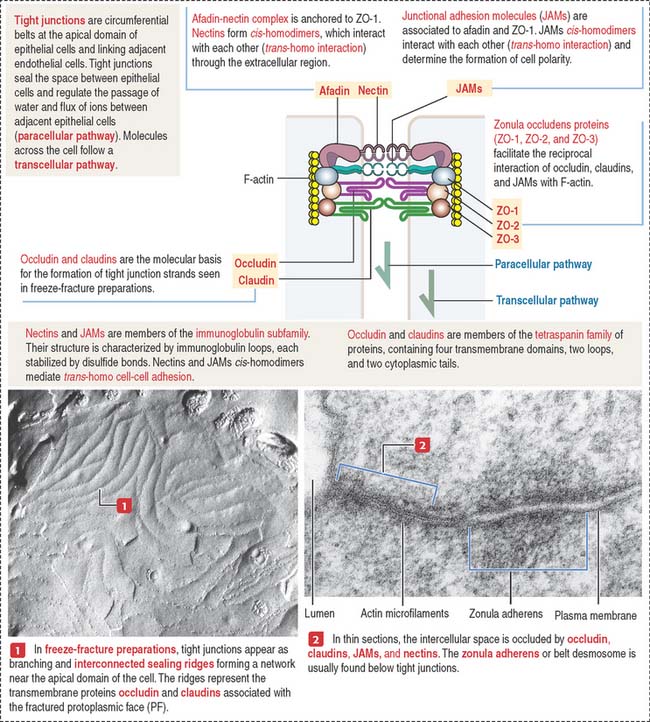
Occludin interacts with four major zonula occludin (ZO) proteins: ZO-1, ZO-2, ZO-3, and afadin. Claudin (Latin claudere, to close), a family of 16 proteins forming linear fibrils in the tight junctions, confers barrier properties on the paracellular pathway. A mutation in the gene encoding claudin 16 is the cause of a rare human renal magnesium wasting syndrome characterized by hypomagnesemia and seizures.
Tight junctions can be visualized by freeze-fracturing a network of branching and anastomosing sealing strands. We discuss in Chapter 2, Epithelial Glands, the procedure of freeze-fracturing for the study of cell membranes.
Anchoring junctions are found below the tight junctions, usually near the apical surface of an epithelium. There are three classes of anchoring junctions (see Figures 1-14, 1-16, 1-18, and 1-19):
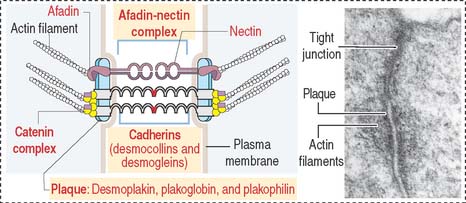
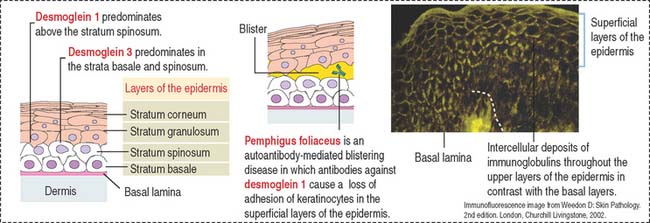
Similar to the tight junctions, the zonula adherens is a beltlike junction. The zonula adherens (Figure 1-16) is associated with actin microfilaments. This association is mediated by the interaction of cadherins (desmocollins and desmogleins) with catenins (α, β, and γ). The main desmogleins expressed in the epidermis of the skin are desmoglein 1 and desmoglein 3 (Figure 1-17).
The macula adherens (also called desmosome) is a spotlike junction associated with keratin intermediate filaments (also known as tonofilaments) extending from one spot to another on the lateral and basal cell surfaces of epithelial cells (Figure 1-18). Spot desmosomes provide strength and rigidity to an epithelial cell layer. Spot desmosomes are also present in the intercalated disks linking adjacent cardiocytes in heart (see Chapter 7, Muscle Tissue) and in the meninges lining the outer surfaces of the brain and spinal cord.
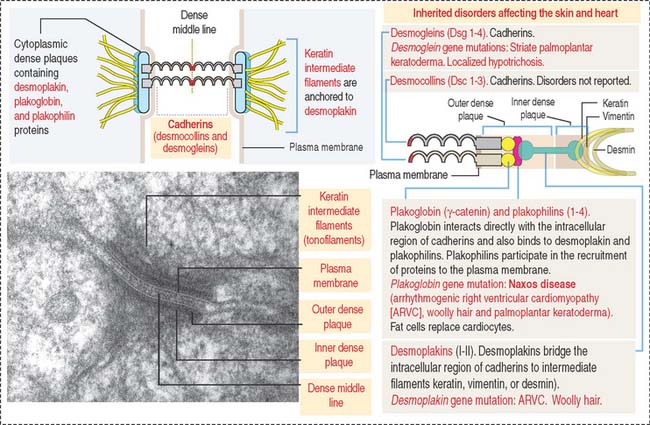
In contrast to occluding junctions, adjacent cell membranes linked by zonula and macula adherens are separated by a relatively wide intercellular space. This space is occupied by the glycosylated portion of proteins of the cadherin family, desmogleins and desmocollins, anchored to cytoplasmic plaques containing desmoplakin, plakoglobin (γ-catenin), and plakophilin. The cytoplasmic plaques are attached to the cytosolic face of the plasma membrane. The interlocking of similar cadherins binds two cells together by Ca2+-dependent homophilic or heterophilic interaction, as we have already seen. Inherited disorders of some of the desmosomal components are indicated in Figure 1-18.
The human desmosomal cadherins genes include four desmogleins and three desmocollins. Their cytoplasmic regions interact with plakoglobin and plakophilin. Desmoplakin interacts with the intermediate filaments keratin in epidermis, desmin in the intercalated disks, and vimentin in the meninges. Desmoglein 1 and desmoglein 3 maintain the cohesiveness of the epidermis, a stratified squamous epithelium. Autoantibodies to desmoglein 1 cause a blistering disease (disruption of cell adhesion) of the skin called pemphigus foliaceus (see Figure 1-17).
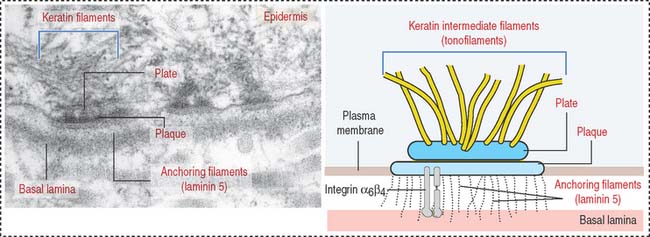
Hemidesmosomes are asymmetrical structures anchoring the basal domain of an epithelial cell to the underlying basal lamina (Figure 1-19).
Gap junctions are symmetrical communicating junctions formed by integral membrane proteins called connexins. Six connexin monomers associate to form a connexon, a hollow cylindrical structure that spans the plasma membrane. The end-to-end alignment of connexons in adjacent cells provides a direct channel of communication (1.5 to 2 nm in diameter) between the cytoplasm of two adjacent cells (Figure 1-20). Connexons have a clustering tendency and can form patches about 0.3 mm in diameter.
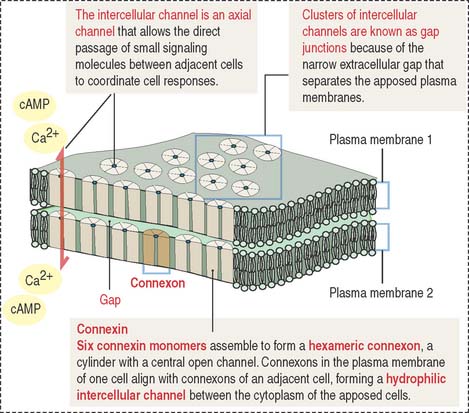
These junctions facilitate the movement of molecules 1.2 nm in diameter (for example, Ca2+ and cyclic adenosine monophosphate [cAMP]) between cells. The connexon axial channels close when the concentration of Ca2+ is high. This junction is responsible for the chemical and electrical “coupling” between adjacent cells. A typical example is cardiac muscle cells connected by gap junctions to enable the transmission of electrical signals.
Clinical significance: Connexin mutations in human disease
Mutations in the connexin 32 (Cx32) gene are found in X-linked Charcot-Marie-Tooth demyelinating neuropathy resulting in progressive degeneration of peripheral nerves, characterized by distal muscle weakness and atrophy and impairment of deep tendon reflexes. Connexin 32 protein is expressed in Schwann cells, which are involved in the production of rolled myelin tubes around the axons in the peripheral nervous system (see Chapter 8, Nervous Tissue). Gap junctions couple different parts of the rolled myelin tubes of the same Schwann cell, rather than different cells. A loss of the functional axial channels in myelin leads to the demyelinating disorder. Mutations in the connexin 50 (Cx50) gene are associated with congenital cataracts, leading to blindness.
BASEMENT MEMBRANE
Integrins mediate cell-matrix interactions by their binding affinity to the RGD domain in laminin and fibronectin (see Figure 1-11). Laminin and fibronectin are distinct proteins of the extracellular matrix and are associated with collagens, proteoglycans, and other proteins to organize a basement membrane, the supporting sheet of most epithelia.
The basement membrane consists of two components (Figure 1-21):
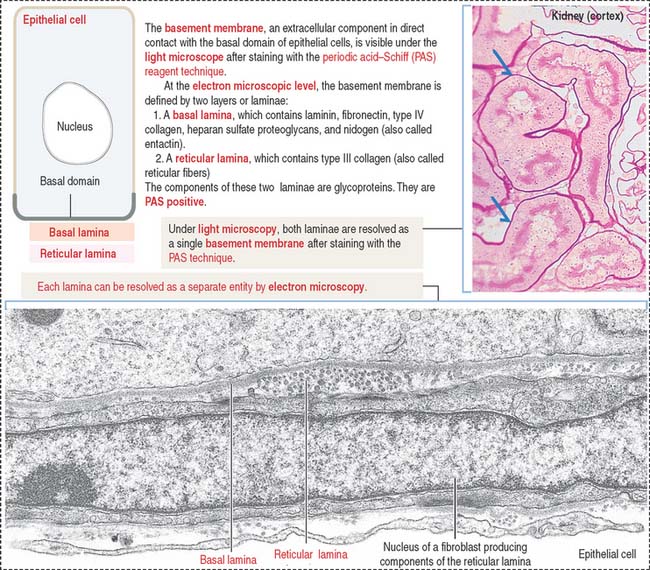
The basal and reticular laminae can be distinguished by electron microscopy. Under the light microscope, the combined basal and reticular laminae receive the name of basement membrane, which can be recognized by the periodic acid–Schiff (PAS) stain (see Figure 1-21; see Box 1-D).
Box 1-D Periodic acid–Schiff (PAS) reaction
The basal lamina has specific functions in different tissues. The double basal lamina of the renal corpuscle constitutes the most important element of the glomerular filtration barrier during the initial step in the formation of urine (see Chapter 14, Urinary System).
In skeletal muscle, the basal lamina maintains the integrity of the tissue, and its disruption gives rise to muscular dystrophies (see Chapter 7, Muscle Tissue).
Laminin (Figure 1-22) is a cross-shaped protein consisting of three chains: the α chain, the β chain, and the γ chain. Laminin molecules can associate with each other to form a meshlike polymer. Laminin and type IV collagen are the major components of the basal lamina, and both are synthesized by epithelial cells resting on the lamina.
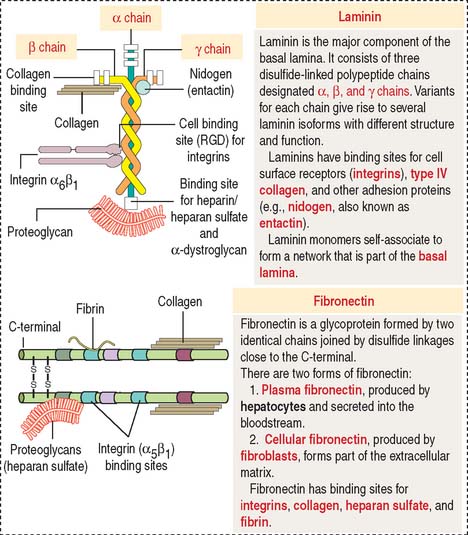
Laminin has binding sites for nidogen (also called entactin), proteoglycans (in particular, heparan sulfate perlecan), α-dystroglycan (see Chapter 7, Muscle Tissue), and integrins.
Fibronectin (see Figure 1-22) consists of two protein chains cross-linked by disulfide bonds. Fibronectin is the main adhesion molecule of the extracellular matrix of the connective tissue and is produced by fibroblasts. Fibronectin has binding sites for heparin present in proteoglycans, several types of collagens (types I, II, III, and V), and fibrin (derived from fibrinogen during blood coagulation).
Fibronectin circulating in blood is synthesized in the liver by hepatocytes. It differs from fibronectin produced by fibroblasts in that it lacks one or two repeats (designated EDA and EDB for extra domain A and extra domain B) as a result of alternative mRNA splicing. Circulating fibronectin binds to fibrin, a component of the blood clot formed at the site of blood vessel damage. The RGD domain of immobilized fibronectin binds to integrin expressed on the surface of activated platelets, and the blood clot enlarges. We return to the topic of blood coagulation or hemostasis in Chapter 6, Blood and Hematopoiesis.
How cells interact with one another and with the basal lamina
Figure 1-23 summarizes the highlights of cell adhesion molecules and cell junctions. An epithelium is a continuous sheet of polarized cells supported by a basement membrane. The polarized nature of an epithelium depends on the tight junctions that separate the polarized cells into apical and basolateral regions. Tight junctions control the paracellular pathway of solutes, ions, and water. Tight junctions form a belt around the circumference of each cell.
Endothelial cells, the constituents of a simple squamous epithelium, are linked by tight and spot desmosomes tightly regulated to maintain the integrity of the endothelium and protect the vessels from unregulated permeability, inflammation, and reactions leading to blood coagulation in the lumen (see Chapter 12, Cardiovascular System). Leukocytes reach the site of infection by attaching to endothelial cell surfaces and migrate across the endothelium into the underlying tissues by a mechanism called diapedesis. Leukocytes find their way through endothelial cell-cell junctions after docking to activated or resting endothelial cells by the endothelial cell adhesion molecules ICAM-1 and VCAM-1 (see Figure 1-10). ICAM-1 and VCAM-1 bind to β2 and β1 integrin subunits in leukocytes (see Figure 1-12).
Note in Figure 1-23 that:
CYTOSKELETON
The cytoskeleton has roles in:
Biochemical studies, involving the extraction of cytoskeletal proteins from cells with detergents and salts and in vitro translation of specific mRNA, showed that each class of filaments has a unique protein organization. When cytoskeletal proteins were purified, they were used as antigens for the production of antibodies. Antibodies are used as tools for the localization of the various cytoskeletal proteins in the cell. The immunocytochemical localization of cytoskeletal proteins (Figure 1-24) and cell treatment with various chemical agents disrupting the normal organization of the cytoskeleton have been instrumental in understanding the organization and function of the cytoskeleton.

Microfilaments
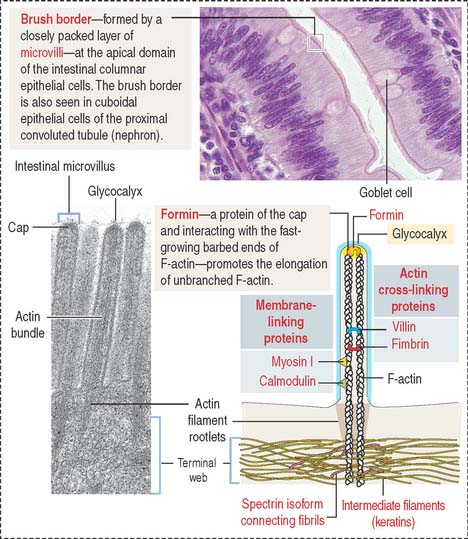
Actin is a versatile and abundant cytoskeletal component forming static and contractile bundles and filamentous networks specified by actin-binding proteins and their distinctive location and function in a cell. F-actin bundles are present in the microvilli of the intestinal (Figure 1-25) and renal epithelial cells (brush border) and the stereocilia from the hair cells of the inner ear.
We have already seen that the intracellular portion of the cell adhesion molecules cadherins and integrin β1 interacts with F-actin through linker proteins (see Figures 1-8 and 1-11). As discussed in Chapter 6, Blood and Hematopoiesis, actin—together with spectrin—forms a filamentous network on the inner face of the red blood cell membrane that is crucial for maintaining the shape and integrity of red blood cells. Spectrin is a tetramer consisting of two distinct polypeptide chains (α and β).
Growth of actin filaments may occur at both ends; however, one end (the “barbed end” or plus end) grows faster than the other end (the “pointed end” or minus end). The names correspond to the arrowhead appearance of myosin head bound at an angle to actin. Actin filaments can branch in the leading edge (lamellipodia) of cells involved in either motility or interaction with other cell types. F-actin branching is initiated from the side of a preexisting actin filament by Arp2/3 (for actin-related protein), an actin nucleating complex of seven proteins (Figure 1-26). Formin regulates the assembly of unbranched actin in cell protrusions such as the intestinal microvilli (see Figure 1-25).
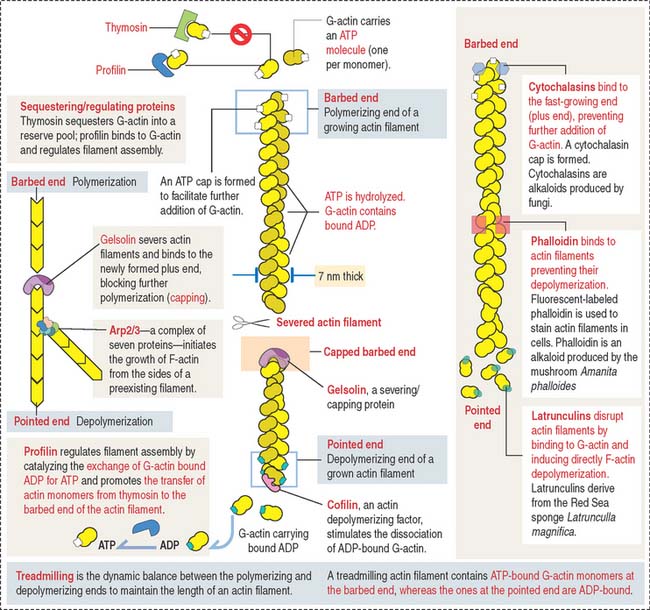
Actin monomers have a binding site for adenosine triphosphate (ATP), which is hydrolyzed to adenosine diphosphate (ADP) as polymerization proceeds. Actin polymerization is ATP-dependent (see Box 1-E).
Box 1-E Microfilaments
The kinetics of actin polymerization involves a mechanism known as treadmilling: G-actin monomers assembled at one end of the filament concurrently disassemble at the other end (see Figure 1-26). Four types of proteins control treadmilling (see Figure 1-26), as follows:
The assembly of G-actin monomers into filaments and the organization of these filaments into thick bundles are controlled by various types of actin-binding or actin-related proteins. A bundle of parallel nonbranching actin filaments, forming the core of the microvillus, is held together by actin-linking proteins, villin and fimbrin. Side arms of myosin-I and the Ca2+-binding protein calmodulin anchor the bundle to the plasma membrane (see Figure 1-25).
Arp2/3 and additional regulatory proteins form a nucleation complex for the assembly of branching actin filaments. Branching actin filaments assemble at the leading edge of a cell during cell motility. In the microvillus, formins (proteins with highly conserved formin-homology domains, FH1 and FH2), instead of the Arp2/3 complex, seem to regulate the elongation of nonbranching actin filaments, while remaining attached to the barbed end (see Box 1-E). Formins are located at the tip of the microvillus, the cap region (see Figure 1-25).
Male patients with defects in proteins that activate the Arp2/3 complex—in particular a protein of the Wiskott-Aldrich syndrome protein (WASP) family—display recurrent respiratory infections because of hereditary immunodeficiency, thrombocytopenia (low platelet count) present from birth on and eczema of the skin after the first month of life (see Box 1-F). The mutation is inherited from the mother, a healthy carrier of the defective gene.
Box 1-F Wiskott-Aldrich syndrome
Microvilli and stereocilia are comparable structures, although they differ in length and the number of actin filaments: intestinal microvilli are 1 to 2 μm long, 0.1 μm wide, and consist of 20 to 30 bundled actin filaments; stereocilia in hair cells of the inner ear have a tapered shape at their base, the length range is 1.5 to 5.5 μm, and each actin bundle contains up to 900 actin filaments. Hair cells are extremely sensitive to mechanical displacement, and a slight movement of the stereocilium is amplified into changes in electric potential transmitted to the brain. We study hair cells of the inner ear in Chapter 9, Sensory Organs: Vision and Hearing.
Microtubules
Microtubules are composed of tubulin dimers (Figure 1-27; see Box 1-G). Each tubulin dimer consists of two tightly bound tubulin molecules: α-tubulin and β-tubulin. Tubulin subunits are arranged in longitudinal rows called protofilaments. Thirteen protofilaments associate side by side with each other to form a cylinder of microtubules with a hollow core. The diameter of a microtubule is 25 nm.
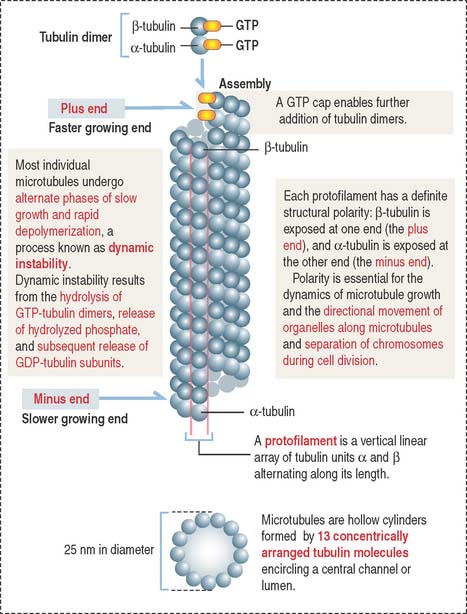
Box 1-G Microtubules
Similar to actin filaments, microtubules are structurally polarized. Microtubules have a plus end, which grows more rapidly than the minus end (see Figure 1-27).
The stability of microtubules can be modified by microtubule-associated proteins (MAPs). MAPs are classified into two groups: (1) classical MAPs, such as MAP1A, MAP1B, MAP2, and tau, and (2) nonclassical MAPs, including Lis1 and DCX family members. MAPs stabilize microtubules by phosphorylation/dephosphorylation. In Chapter 7, Nervous Tissue, we discuss the significance of tau phosphorylation and dephosphorylation in Alzheimer’s disease. A lack of expression of Lis1 causes a sever brain developmental disorder called lissencephaly.
Centrosome: A microtubule-organizing center
The centrosome has three major functions: (1) it nucleates the polymerization of tubulin subunits into microtubules, (2) it organizes microtubules into functional units, and (3) it duplicates once every cell cycle.
Centrosomes are part of the mitotic center, which, together with the mitotic spindle, constitutes the mitotic (or meiotic) apparatus (Figure 1-28). A centriole is a small cylinder (0.2 μm wide and 0.4 μm long) composed of nine microtubule triplets in a helicoid array. In contrast to most cytoplasmic microtubules, which display dynamic instability, the centriolar microtubules are very stable.
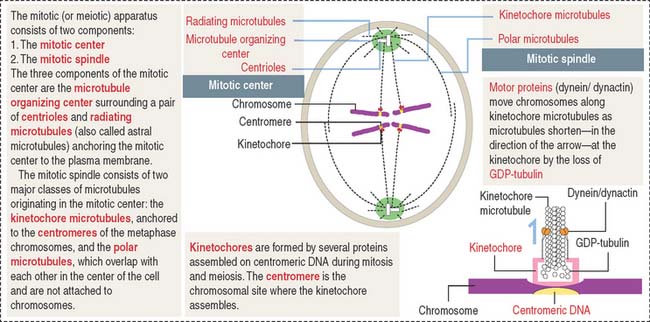
There are three types of microtubules extending from the centrosomes: radiating or astral microtubules, anchoring each centrosome to the plasma membrane; kinetochore microtubules, attaching the chromosome-associated kinetochore to the centrosomes; and polar microtubules, extending from the two poles of the spindle where opposite centrosomes are located (Figure 1-28). If kinetochores fail to assemble, chromosomes cannot segregate properly (see Box 1-H).
The pericentriolar material contains the γ-tubulin ring complex and numerous proteins, including pericentrin. Each γ-tubulin ring complex is the nucleation site or template for the assembly and growth of one microtubule. The centrioles do not have a direct role in the nucleation of microtubules in the centrosome. Tubulin dimers associate to the γ-tubulin ring by the α-tubulin subunit. Consequently, the minus end of each microtubule points to the centrosome; the plus end, the growing end, is oriented outward, free in the cytoplasm.
Microtubules in cilia and flagella
Centrioles give rise to structurally similar basal bodies, which are the outgrowth origin of cilia (see Figure 1-5) and flagella. A defect in the assembly of the basal body and cilia, caused by abnormal transport of ciliary proteins, results in the Bardet-Biedl syndrome (see Box 1-I). Cilia and flagella are motile cytoplasmic extensions containing a core of microtubules called the axoneme (Figure 1-29). The axoneme consists of nine peripheral microtubule doublets surrounding a central pair of microtubules. This arrangement is known as the 9 + 2 configuration (see Box 1-J).
Box 1-I Bardet-Biedl syndrome
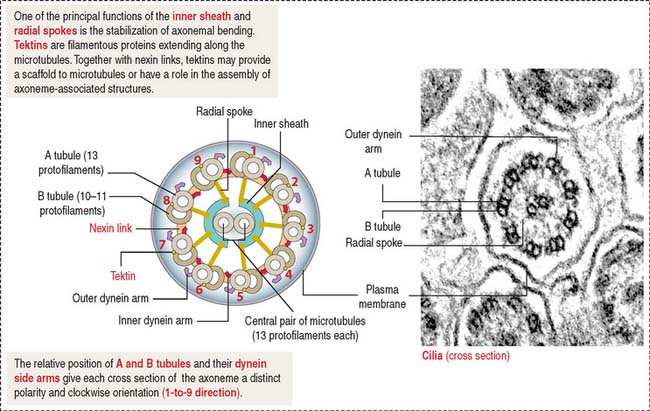
Box 1-J Major components of the ciliary and flagellar axonemes
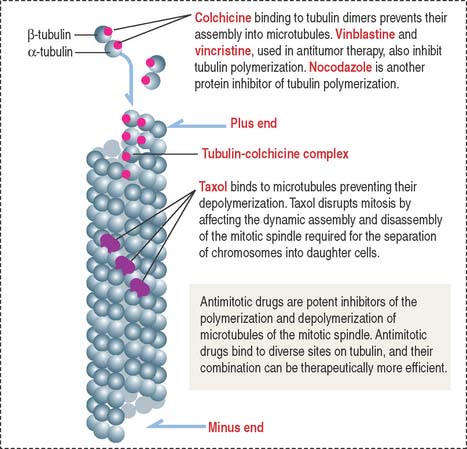
Clinical significance: Microtubule-targeted drugs and sterility
The first group includes colchicine, colcemid, vincristine, and vinblastine, which bind to tubulin and inhibit microtubule polymerization, blocking mitosis. Colchicine is used clinically in the treatment of gout. Vincristine and vinblastine, from Vinca alkaloids isolated from the leaves of the periwinkle plant, have been successfully used in childhood hematologic malignancies (leukemias). Neurotoxicity—resulting from the disruption of the microtubule-dependent axonal flow (loss of microtubules and binding of motor proteins to microtubules)–and myelosuppression are two side effects of microtubule-targeted drugs.
The second group includes taxol (isolated from the bark of the yew tree) with an opposite effect: It stabilizes microtubules instead of inhibiting their assembly (Figure 1-30). Paclitaxel (taxol) has been used widely to treat breast and ovarian cancers. Similar to Vinca alkaloids, its main side effects are neurotoxicity and suppression of hematopoiesis.
Microtubules–cytoskeletal tracks for cargo transport powered by motor proteins
The transport of vesicles and nonvesicle cargos occurs along microtubules and F-actin. Specific molecular motors associate to microtubules and F-actin to mobilize cargos to specific intracellular sites. Microtubule-based molecular motors include kinesin and cytoplasmic dynein for the long-range transport of cargos. F-actin–based molecular motors include unconventional myosin Va and VIla for the short-range transport of cargos. We discuss additional aspects of the F-actin–based cargo transport mechanism during the transport of melanosomes in Chapter 11, Integumentary System.
Three examples of microtubule-based cargo transport in mammalian systems are as follows (see Box 1-K):
Box 1-K Microtubule-based cargo transport by molecular motors
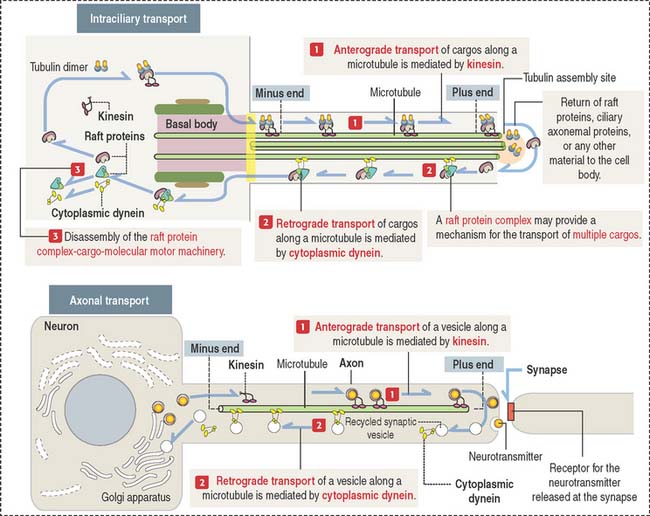
Axonal transport
Bundles of microtubules form tracks within the axon to carry these vesicles. Vesicles are transported by two motor proteins (see Figure 1-31):
Kinesins and cytoplasmic dyneins participate in two types of intracellular transport movements:
Kinesins and cytoplasmic dyneins have two ATP-binding heads and a tail. Energy derives from continuous ATP hydrolysis by ATPases present in the heads. The head domains interact with microtubules, and the tail binds to specific receptor binding sites on the surface of vesicles and organelles.
Myosin family associates with F-actin to form contractile structures
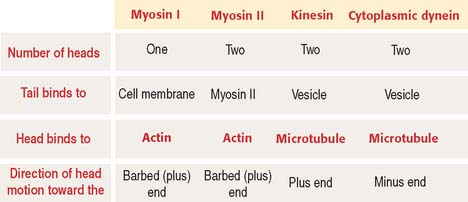
Members of the myosin family of proteins bind and hydrolyze ATP to provide energy for their movement along actin filaments from the pointed (minus) end to the barbed (plus) end. Myosin I and myosin II are the predominant members of the myosin family (Figure 1-32; see Box 1-L).
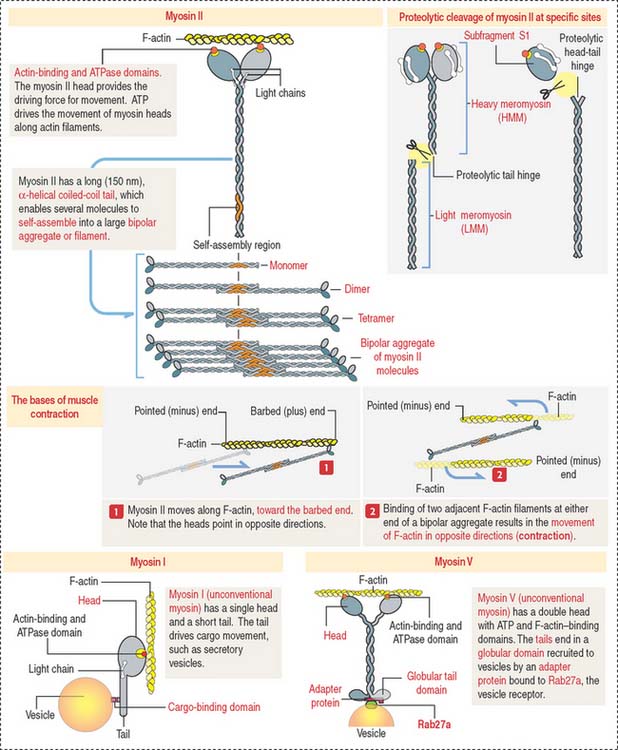
Box 1-L Types of myosins
Myosin II, a conventional myosin, is present in muscle and nonmuscle cells. Myosin II consists of a pair of identical molecules. Each molecule consists of an ATPase-containing head domain and a long rodlike tail. The tails of the dimer link to each other along their entire length to form a two-stranded coiled rod. The tail of myosin II self-assembles into dimers, tetramers, and a bipolar filament with the heads pointing away from the midline.
The two heads—linked together but pointing in opposite directions—bind to adjacent actin filaments of opposite polarity. Each myosin head bound to F-actin moves toward the barbed (positive) end. Consequently, the two actin filaments are moved against each other, and contraction occurs (see Figure 1-32).
Figure 1-33 summarizes the structural and functional characteristics of motor proteins.
Light-chain phosphorylation by myosin light-chain kinase
The self-assembly of myosin II and interaction with actin filaments in nonmuscle cells takes place in certain sites according to functional needs. These events are controlled by the enzyme myosin light-chain kinase (MLCK), which phosphorylates one of the myosin light chains (called the regulatory light chain) present on the myosin head. The activity of MLCK is regulated by the Ca2+-binding protein calmodulin (Figure 1-34).
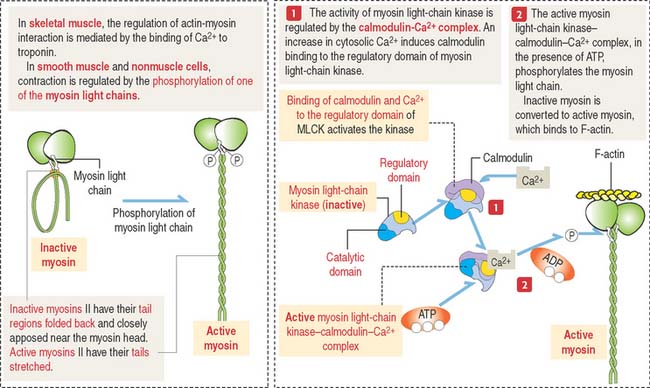
Phosphorylation of one of the myosin light chains results in two effects:
In smooth muscle cells, a phosphatase removes the phosphate group from myosin light chains. Skeletal muscle contraction does not require phosphorylation of the myosin light chains. We discuss additional details of muscle contraction when we study the muscle tissue (see Chapter 7, Muscle Tissue).
Intermediate filaments
Intermediate filaments (Figure 1-35) represent a heterogeneous group of structures so named because their diameter (10 nm) is intermediate between those of micro tubules (25 nm) and microfilaments (7 nm). Intermediate filaments are the most stable cytoskeletal structures.
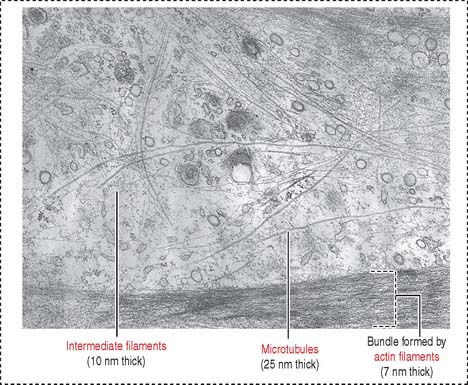
Detergent and salt treatments extract microfilament and microtubule components and leave intermediate filaments insoluble. All intermediate filaments have a common monomer consisting of a central α-helical rod flanked by head and tail domains (Figure 1-36).
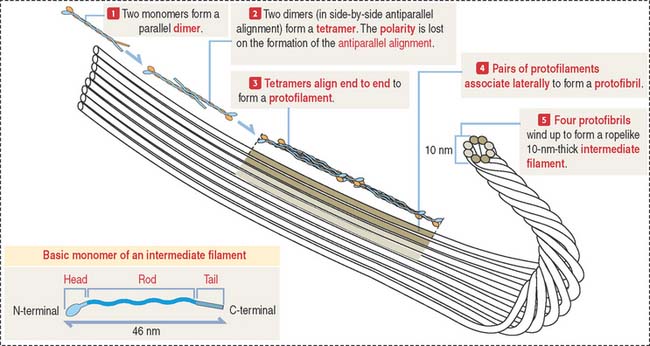
Intermediate filament protein monomers consist of three domains (see Figure 1-36): A central α-helical rod domain is flanked by a nonhelical N-terminal head domain and a C-terminal tail domain. During assembly, pairs of dimers—formed by the parallel alignment of monomers—associate into tetramers in a side-by-side but antiparallel orientation. About eight tetramers align end-to-end to form a protofilament. Pairs of protofilaments associate laterally to form a protofibril, and four protofibrils—a total of eight protofilaments—wind up to form a ropelike intermediate filament (see Figure 1-36). Intermediate filaments do not have the structural polarity seen in F-actin and microtubules. One end of an intermediate filament cannot be distinguished from another. Molecular motors associated to an intermediate filament would find it difficult to identify one direction from another.
The major function of intermediate filaments is to provide mechanical support for the cell. Five major types of intermediate filament proteins have been identified on the basis of sequence similarities in the rod domain. They are referred to as types I through V (see Box 1-M). About 50 intermediate filament proteins have been reported so far.
Box 1-M Types of intermediate filament proteins
Desmin (53 kd): A component of Z disks of striated muscle and smooth muscle cells.
Glial fibrillary acidic protein (GFAP 51 kd): Present in astrocytes.
Peripherin (57 kd): A component of axons in the peripheral nervous system.
Type I (acidic keratins) and type II (neutral to basic keratins). This class of proteins forms the intermediate filament cytoskeleton of epithelial cells (called cytokeratins to distinguish them from the keratins of hair and nails). Equal amounts of acidic (40 to 60 kd) and neutral-basic (50 to 70 kd) cytokeratins combine to form this type of intermediate filament protein. Type I and type II intermediate filament keratins form tonofilaments associated with molecules present in the cytoplasmic plaques of desmosomes and hemidesmosomes (see Figures 1-18 and 1-19). We come back to intermediate filament–binding proteins, such as filaggrins, when we discuss the differentiation of keratinocytes in the epidermis of the skin (Chapter 11, Integumentary System), and plectin, when we analyze the cytoskeletal protective network of skeletal muscle cells (Chapter 7, Muscle Tissue).
Type III. This group includes the following intermediate filament proteins:
Desmin (53 kd) is a component of skeletal muscle cells and is localized to the Z disk of the sarcomere (see Chapter 7, Muscle Tissue). This intermediate filament protein keeps individual contractile elements of the sarcomeres attached to the Z disk and plays a role in coordinating muscle cell contraction. Desmin is also found in smooth muscle cells.
Glial fibrillary acidic protein (GFAP) (51 kd) is observed in astrocytes and some Schwann cells (see Chapter 8, Nervous Tissue).
Peripherin (57 kd) is a component of neurons of the peripheral nervous system and is coexpressed with neurofilament proteins (see Chapter 8, Nervous Tissue).
Type IV. Neurofilaments are the main components.
A group of human diseases, known as laminopathies, are linked to defects in proteins of the nuclear envelope, including lamins (see Box 1-N). Numerous laminopathies affect cardiac and skeletal muscle, adipose tissue (lipodystrophies), and motor and sensory peripheral nerves.
Box 1-N Clinical features of laminopathies
Two hypotheses concerning the pathogenic mechanism of laminopathies have been considered:
During mitosis, the phosphorylation of lamin serine residues causes a transient disassembly of the meshwork, followed by a breakdown of the nuclear envelope into small fragments. At the end of mitosis, lamins are dephosphorylated, and the lamin meshwork and the nuclear envelope reorganize. See the cell nucleus section concerning the mechanism of phosphorylation and dephosphorylation of lamins during the cell cycle.
Hemidesmosomes and intermediate filaments
Hemidesmosomes are specialized junctions observed in basal cells of the stratified squamous epithelium attaching to the basement membrane (Figure 1-37). Inside the cell, the proteins BPAG1 (for bullous pemphigoid antigen 1) and plectin (members of the plakin family of cross-linker proteins) are associated to intermediate filaments (also called tonofilaments). Plectin connects intermediate filaments to the integrin subunit β4.
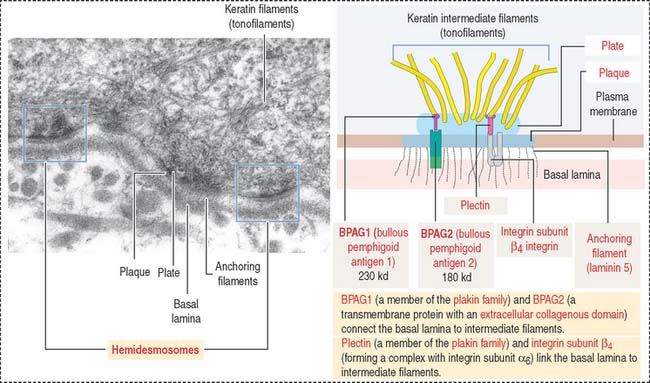
Clinical significance: Intermediate filaments and blistering diseases
Bullous pemphigoid is an autoimmune blistering disease similar to pemphigus vulgaris (called “pemphigoid”). Blisters or bullae develop at the epidermis-dermis junction when circulating immunoglobulin G (IgG) cross-reacts with bullous pemphigoid antigen 1 or 2. IgG-antigen complexes lead to the formation of complement complexes (C3, C5b, and C9), which damage the attachment of hemidesmosomes and perturb the synthesis of anchoring proteins by basal cells (Figure 1-38).
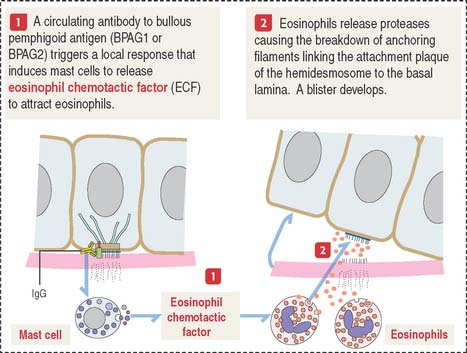
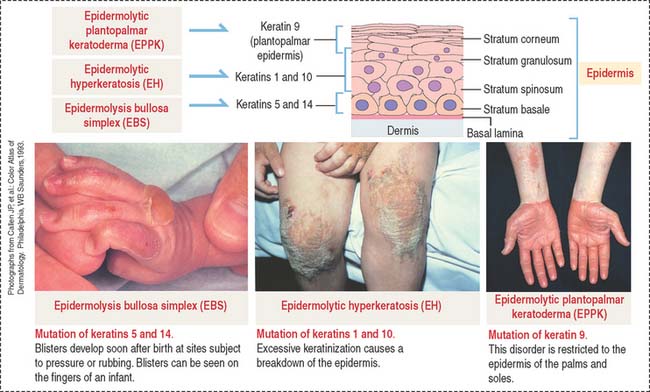
Intermediate filaments strengthen the cellular cytoskeleton. The expression of mutant keratin genes results in the abnormal assembly of keratin filaments, which weakens the mechanical strength of cells and causes inherited skin diseases, as shown in Figure 1-39:
CELL NUCLEUS
Nuclear envelope and nuclear pore complex
The mammalian cell nucleus consists of three major components: (1) the nuclear envelope, (2) chromatin, and (3) the nucleolus. The nuclear envelope consists of two concentric membranes separated by a perinuclear space. The inner nuclear membrane is associated with the nuclear lamina (see Box 1-O), chromatin, and ribonucleoproteins. The outer nuclear membrane is continuous with the membranes of the endoplasmic reticulum and can be associated with ribosomes.
Box 1-O Nuclear lamina
The nuclear pore complex has a tripartite structure, composed of a central cylindrical body placed between inner and outer octagonal rings, each consisting of eight protein particles. The central cylinder consists of a central plug and eight radiating spokes (Figure 1-40). The exact role of individual nuclear pore complex proteins in nucleocytoplasmic trafficking is unclear.
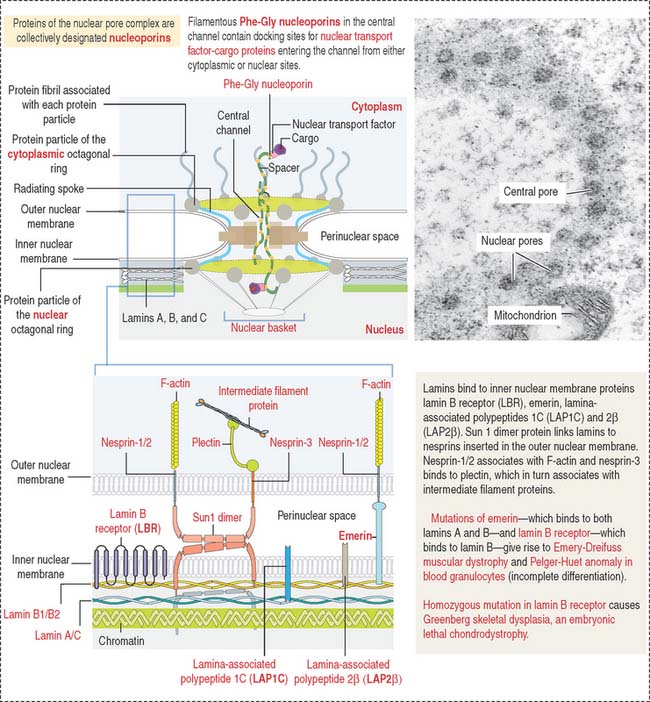
Nuclear pore complexes embedded in the nuclear envelope establish bidirectional communication gates for the trafficking of macromolecules between the cytoplasm and the nucleus. Small molecules (less than 40 to 60 kd) can diffuse through the nuclear pore complex. Proteins of any size, containing a nuclear localization amino acid sequence (NLS, Pro-Lys-Lys-Lys-Arg-Lys-Val), can be imported into the nucleus, however, by an energy-dependent mechanism (requiring ATP and GTP).
Nucleocytoplasmic transport: Ran-GTPase
Ran shuttles across the nuclear pores and accumulates inside the nucleus by an active transport mechanism (Figure 1-41).
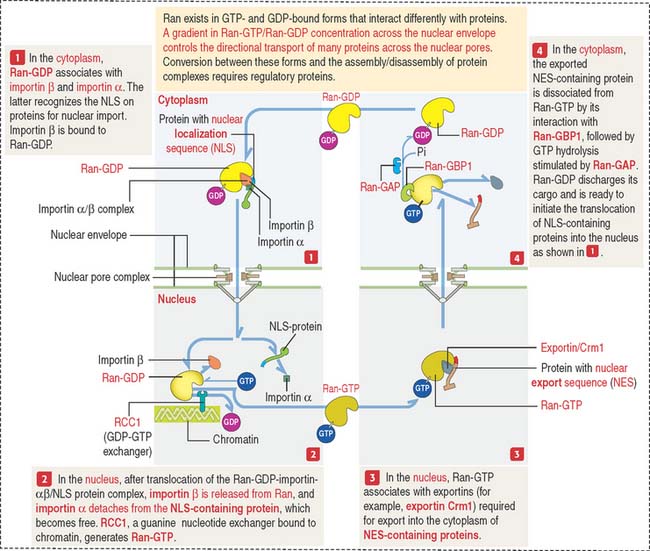
Ran-GTPase also has a role in the assembly of the mitotic spindle.
Chromatin
Chromatin is defined as particles or “beads” (called nucleosomes) on a double-stranded DNA string (Figure 1-42). Each nucleosome consists of a histone octamer core and about two turns of DNA wound around the histone core. The histone octamer contains two molecules each of H2A, H2B, H3, and H4 histones. H1 histone cross-links the DNA molecule wrapped around the octamer.
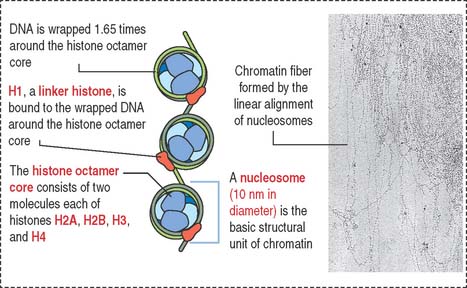
Chromatin is packed in separate chromosomes that can be visualized during mitosis (or meiosis). During interphase (phases G1, S, and G2 of the cell cycle), individual chromosomes cannot be identified as such, but are present in a diffuse or noncondensed state.
Diffuse chromatin, called euchromatin (“good chromatin”), is transcriptionally (RNA synthesis) active and represents about 10% of total chromatin. Euchromatin is the site of synthesis on nonribosomal RNAs, including mRNA and transfer RNA (tRNA) precursors. Condensed chromatin, called heterochromatin (“different chromatin”), is transcriptionally inactive and represents about 90% of total chromatin (Figure 1-43).
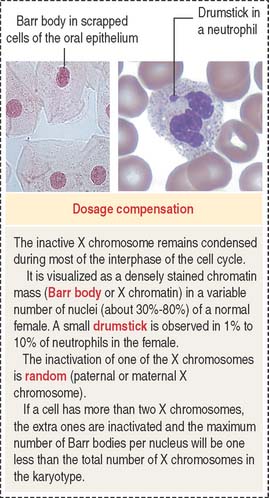
Dosage compensation: Inactivation of one of the X chromosomes
In humans, the inactivated X chromosome is recognized by the presence of the Barr body, a heterochromatin mass observed adjacent to the nuclear envelope or in the form of a drumstick in polymorphonuclear leukocytes (see Figure 1-43). If a cell has more than two X chromosomes, the extra X chromosomes are inactivated, and more than one Barr body is visualized.
Nucleolus
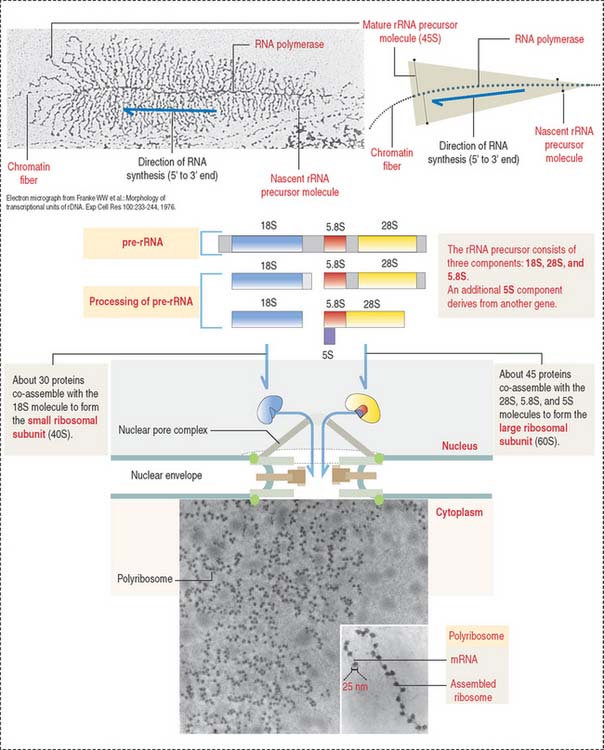
The nucleolus is the site of synthesis of ribosomal RNA (rRNA) and assembly or ribosomal subunits. The nucleolus houses several proteins, including fibrillarin and nucleolin, required for pre-rRNA processing. In addition, the nucleolus contains nucleostemin, a protein unrelated to ribosomal biogenesis. Nucleolin and nucleostemin are shuttling proteins; they relocalize from the nucleolus to the nucleoplasm where they interact with protein p53, a protector of DNA damage by preventing DNA replication in response to genomic stress. We come back to p53 later (see Figure 1-54).
Structurally, the nucleolus consists of three major components (Figure 1-44; see Box 1-P):
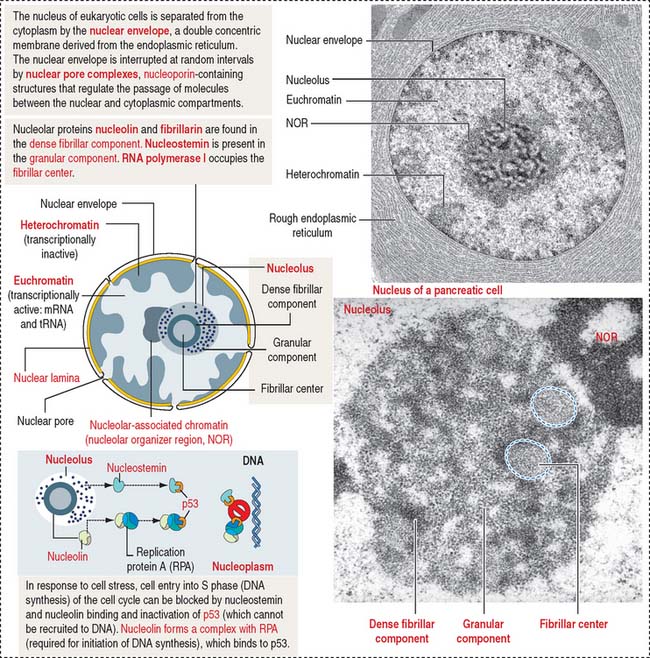
Box 1-P Nucleolus
The active process of rRNA synthesis can be visualized at the electron microscopic level (Figure 1-45) by spreading the contents of nuclei of cells with hundreds of nucleoli (e.g., amphibian oocytes). rRNA genes can be seen as repeating gene units along the chromatin axis, like “Christmas trees,” pointing in the same direction and separated by nontranscribed spacers. The entire rRNA gene region is covered by more than 100 RNA polymerase I molecules synthesizing an equivalent number of fibrils, each with a terminal granule.
Localization of nucleic acids
Cytochemistry and autoradiography (Figure 1-46) provide information about the cellular distribution and synthesis of nucleic acids. The Feulgen reaction is specific for the localization of DNA (see Box 1-Q). Basic dyes, such as toluidine blue, stain DNA and RNA (see Box 1-R). Pretreatment with deoxyribonuclease (DNAse) and ribonuclease (RNAse) defines the distribution sites of DNA and RNA by selective removal of one of the nucleic acids.
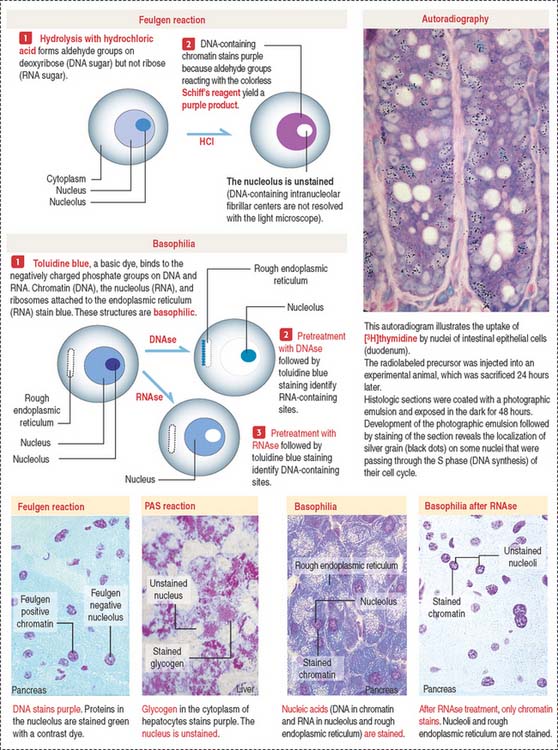
Box 1-Q PAS and Feulgen reactions
Box 1-R Basophilia and acidophilia
Many cytologie stains use acidic and basic dyes.
Autoradiography and radiolabeled precursors for one of the nucleic acids can determine the timing of their synthesis. In this technique, a radioactive precursor of DNA ([3H]thymidine) or RNA ([3H]uridine) is exposed to living cells. As a result of exposure to the radiolabel, any synthesized DNA or RNA contains the precursor. The radioactivity is detected by coating the cells with a thin layer of a photographic emulsion. Silver-containing crystals of the emulsion are exposed to structures of the cell containing radioactive DNA or RNA. After development of the emulsion, silver grains indicate the location of the labeled structures. This approach has been used extensively for determining the duration of several phases of the cell cycle.
CELL CYCLE
The cell cycle is defined as the interval between two successive mitotic divisions resulting in the production of two daughter cells (Figure 1-47). The cell cycle is traditionally divided into two major phases: (1) interphase and (2) mitosis (also known as the M phase).
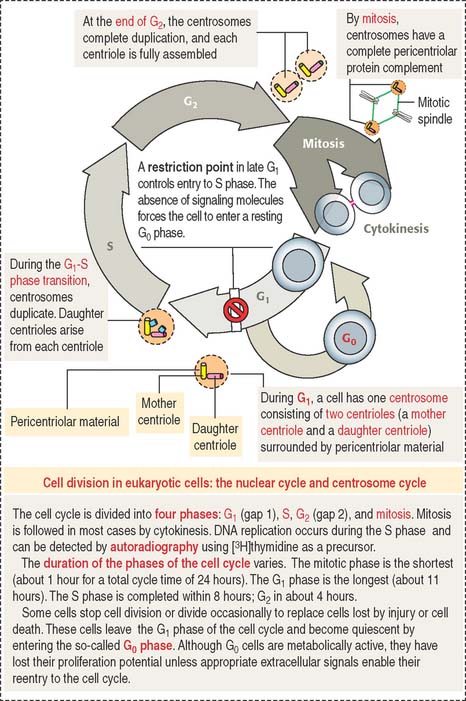
The activities of cyclin-dependent protein kinases–cyclin complexes coordinate the timed progression of the nuclear and centrosome cycles (see Box 1-S). Figure 1-48 provides additional details.
Box 1-S Cell cycle
Breakdown and reassembly of the nuclear envelope
The disassembly of the nuclear envelope occurs at the end of the mitotic and meiotic prophase. It involves the fragmentation of the nuclear envelope, the dissociation of the nuclear pore complexes, and the depolymerization of the nuclear lamina (Figure 1-49).
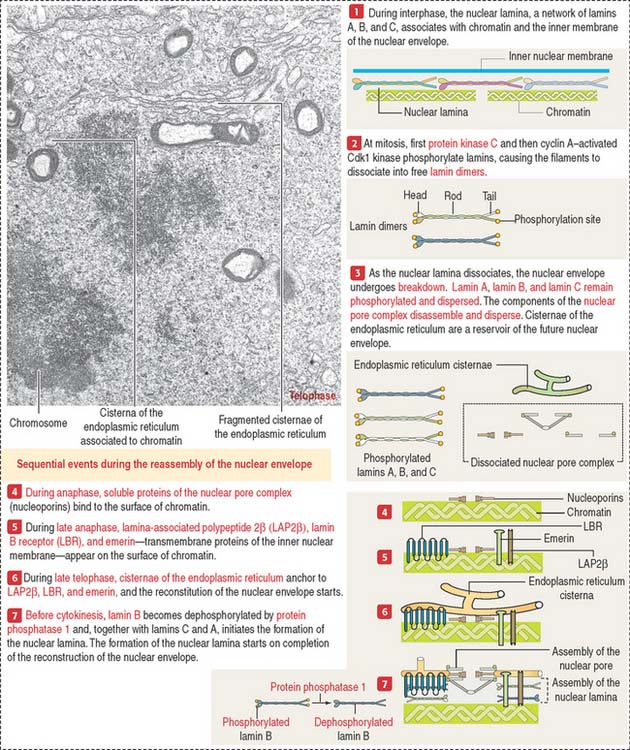
The nuclear lamina is composed of type V intermediate filament proteins, lamins A, B, and C, which associate with each other to form the nuclear lamina. Phosphorylation of lamins—catalyzed first by protein kinase C and later by cyclin A–activated Cdk1 kinase—results in the disassembly of the nuclear lamina. In addition, the components of the nuclear pore complex, the nucleoporins, and the membranous cisternae of the endoplasmic reticulum also disperse. The endoplasmic reticulum is the nuclear membrane reservoir for nuclear envelope reassembly.
A final step in the reconstruction of the nuclear envelope is the dephosphorylation of lamin B by protein phosphatase 1. Dephosphorylated lamin B associates with lamins A and C to form the nuclear lamina before cytokinesis. This sequence of events stresses the impact of gene mutations affecting the expression of lamin A or lamin-binding proteins (see Box 1-M) as causes of laminopathies.
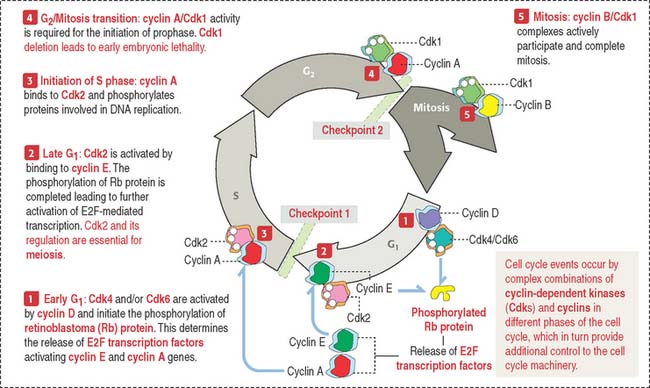
Tumor-suppressor genes
The retinoblastoma model provides important clues on how suppressor genes work (Figure 1-50). Each cell has duplicate copies of the retinoblastoma (Rb) gene as a safety backup. When the two copies of the Rb gene are mutated, an abnormal Rb protein induces cancerous growth of retinal cells.
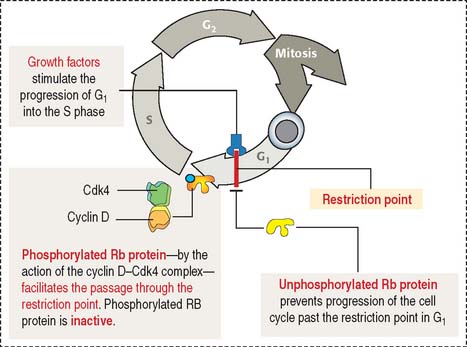
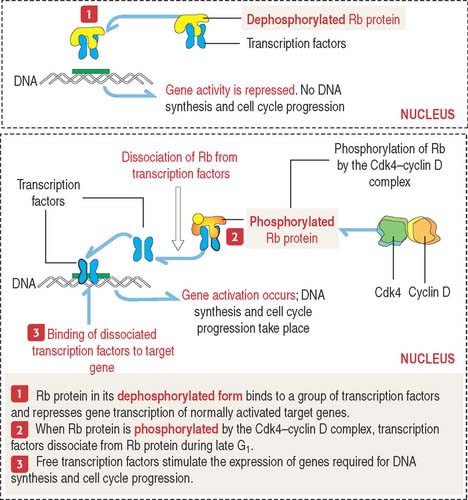
When Rb protein is phosphorylated by the Cdk4–cyclin D complex, it dissociates from the transcription factor complex, which activates specific gene expression (Figure 1-51). Phosphorylated Rb protein switches transcription factors from suppression to activation required for DNA synthesis and progression of the cell cycle.
Clinical significance: Retinoblastoma gene and other suppressor genes
A second type of retinoblastoma, the sporadic form, is seen in children whose parents have no history of the disease. Once cured, these patients, as adults, do not transmit the disease to the next generation. Children with the sporadic retinoblastoma are genetically normal at fertilization, but during embryonic development two somatic mutations occur in a cell lineage, giving rise to the photoreceptors of the retina: the rods and cones. The resulting double-mutated Rb genes induce cells to proliferate into a retinoblastoma.
In Chapter 16, Lower Digestive Segment, we study the tumor-suppressor adenomatous polyposis coli (APC) gene responsible for a hereditary form of colon cancer (familial adenomatous polyposis) derived from the malignant transformation of some of the many polyps (benign tumors) observed in individuals affected by this condition.
MITOSIS
Mitosis is preceded by the duplication of a pair of centrioles, each of which moves toward opposite sites of the nucleus to organize a centrosome. The primary function of the centrosome is the formation and maintenance of the mitotic spindle consisting of microtubules. Because of this function, the centrosome is also called the microtubule-organizing center (MOC). About 1000 new microtubules can be generated per minute on each centrosome using a pool of tubulin dimers derived from disassembled cytoplasmic microtubules.
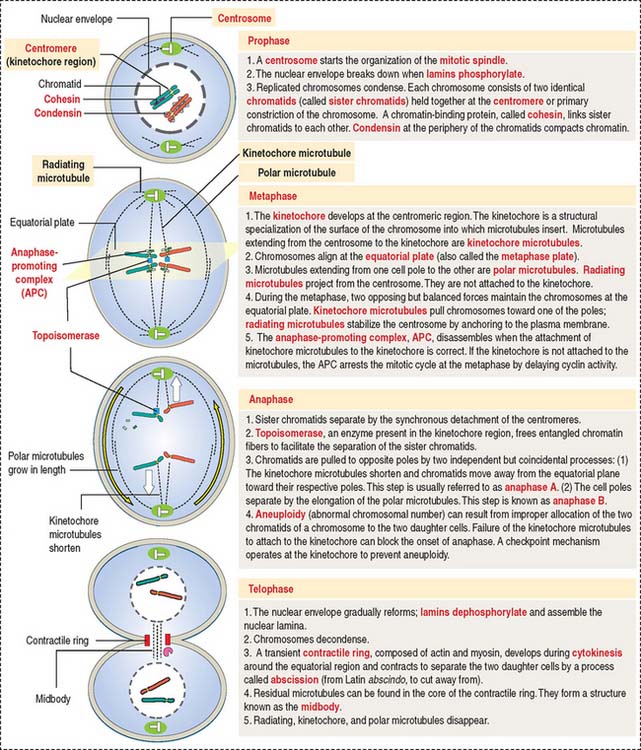
Mitosis is divided into four substages: prophase, metaphase, anaphase, and telophase. The highlights of mitosis are summarized in Figure 1-53.
Telomerase, senescence, and cancer
The telomeres are the ends of chromosomes formed by a stretch of repeated nucleotide sequences (see Figure 1-52). Telomeres are responsible for maintaining chromosomal integrity and represent the cellular biological clock. When DNA polymerases fail to copy the chromosomal ends, telomeres decrease in size with every cell division. Cellular senescence occurs when the telomeres shorten to a point at which the integrity of a chromosome cannot be maintained.
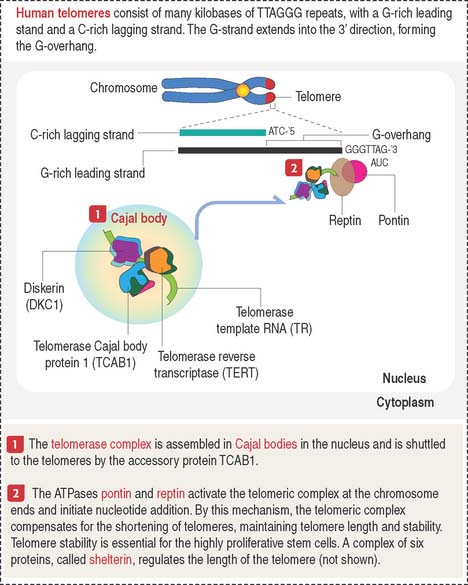
Most tumor cells express high levels of telomerase. The telomerase complex (see Figure 1-52) consists of the catalytic telomerase reverse transcriptase (TERT), the RNA sub unit telomerase template RNA (TR), which provides the template for repeat synthesis of chromosome ends, and dyskerin (DKC1), an auxiliary protein. This complex is assembled in Cajal bodies in the nucleus and is transported to the telomeres by an accessory protein, telomerase Cajal protein 1 (TCAB1). Two ATPases, pontin and reptin, activate the telomerase complex at the chromosomal end and initiate nucleotide addition.
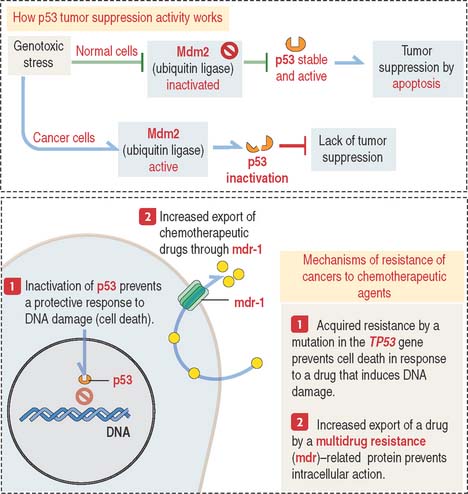
Clinical significance: Role of protein p53 in chemotherapy
There are two kinds of resistance of tumors to chemotherapeutic agents:
One form of acquired resistance is caused by genes of the multidrug-resistance (mdr) gene family (Figure 1-54). These genes encode ATP-dependent pumps involved in the transport of large organic compounds. We see the mdr gene family of proteins again in Chapter 17, Digestive Glands, when we discuss the mechanism of bile secretion by hepatocytes.
DNA damage induced by chemotherapy and radiotherapy—genotoxic stress—triggers the activation of p53, a transcription factor tetramer that destroys terminally damaged cells through the activation of a cell death program, or apoptosis (see Box 1-T). In normal cells, genotoxic stress leads to the inhibition of Mdm2 (for mouse double minute 2) allowing activation of p53 and continuation of normal growth and development (see Figure 1-54).
Box 1-T p53, a tumor-suppressor protein
Mdm2 is a ubiquitin ligase that binds to p53 and facilitates its ubiquitin-dependent degradation in the cytoplasm by the 26S proteasome (see Figure 3-14 in Chapter 3, Cell Signaling). Mdm2 inhibition (for example, by ARF—for alternate reading frame—a 14-kd protein) allows p53 to activate its tumor-suppressor functions. Mdm2 exerts a similar inhibitory effect on retinoblastoma tumor-suppressor protein (Rb protein). The protein levels of ARF, Mdm2, and p53 are not abundant in genotoxic stress-free cells. The half-life of p53 is only 10 to 15 minutes.
Mutations of the TP53 gene, which encodes the p53 protein, are observed in 50% of human cancers. The loss of TP53 gene expression by an autosomal dominant mutation is responsible for a multicancer phenotype known as Li-Fraumeni syndrome (see Box 1-U). p53 is a tumor-suppressor gene. The inactivation of p53 activity is disrupted in drug-resistant cancer cells (see Figure 1-54). Loss of p53 expression is observed in human cancer cells, and clinical studies suggest that inactivation of p53 expression correlates with resistance to chemotherapeutic agents.
Box 1-U Li-Fraumeni syndrome
Pharmacologic agents binding to Mdm2 could stabilize and increase the levels of p53 in cancer cells to exert a tumor-suppressor activity through its death-inducing functions. We discuss in detail the mechanism of programmed cell death or apoptosis in Chapter 3, Cell Signaling.
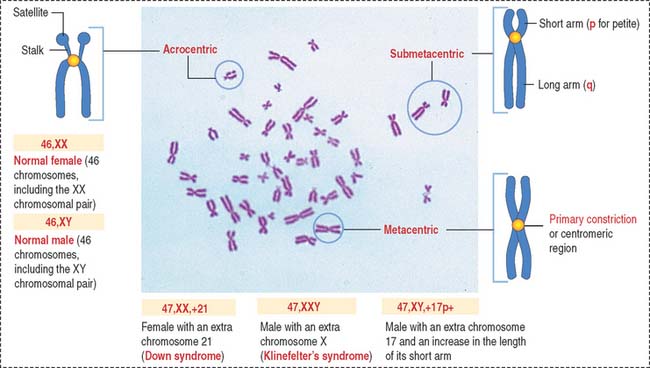
Karyotyping
In the notation of human cytogenetics, the total number of chromosomes (46) is followed by the total number of sex chromosomes (Figure 1-55). A normal male is identified as 46,XY (46 chromosomes, including the XY chromosomal pair) and a female as 46,XX (46 chromosomes, including the XX chromosomal pair).
Microtubules provide tracks for motor protein transporting vesicle and nonvesicle cargos within the cell. Molecular motors, such as kinesin and cytoplasmic dynein, mediate the transport of cargos. There are three major microtubule-based transport systems: axonemal transport, which includes intraciliary and intraflagellar transport; axonal transport; and intramanchette transport. Manchette is a transient structure involved in sperm development.
Breakdown of the nuclear envelope occurs at the end of prophase. It involves the fragmentation of the nuclear envelope, dissociation of nuclear pore complexes, and phosphorylation of lamins (depolymerization). Reassembly of the nuclear envelope involves the dephosphorylation of lamins by a protein phosphatase.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
1 EPITHELIUM
General classification of epithelia
The epithelium is a tightly cohesive sheet of cells that covers or lines body surfaces (for example, skin, intestine, secretory ducts) and forms the functional units of secretory glands (for example, salivary glands, liver). The traditional classification and nomenclature of different types of epithelia are based on the shapes of individual cells and arrangement of the cells in one or more layers (Figure 1-1). The main characteristics of epithelia are listed in Box 1-A.
Box 1-A Main characteristics of epithelia
Epithelia are classified into:
An important aspect of epithelia is its polarity. Most epithelial cells line surfaces and cavities and have three domains (Figure 1-5):
EPITHELIAL CELL POLARITY
Epithelial cells have two major domains (Figure 1-5):
Apical differentiations
The apical domain of some epithelial cells can display three types of differentiation:
Cilia (singular, cilium; Figure 1-6) are motile cell projections originating from basal bodies anchored by rootlets to the apical portion of the cytoplasm. A basal body contains nine triplet microtubules in a helicoid array without a central microtubular component. By contrast, a cilium consists of an assembly called an axoneme, formed by a central pair of microtubules surrounded by nine concentrically arranged microtubular pairs. This assembly is known as the 9 + 2 microtubular doublet arrangement. The axoneme is also a component of the sperm tail, or flagellum.
The trachea and the oviduct are lined by ciliated epithelial cells. In these epithelia, ciliary activity is important for the local defense of the respiratory system and for the transport of the fertilized egg to the uterine cavity.
Some cells have a primary cilium. The importance of primary cilia emerges from rare recessive human disorders known as ciliopathies caused by structural or functional abnormalities of cilia. The structure and mechanism of assembly of primary cilia are shown in Figure 1-6. The significant aspects of the primary cilium are: (1) they are non-motile; (2) they participate in the early stages of embryonic patterning leading to organogenesis; (3) many components of the hedgehog signaling pathway, essential at least in early development, are present in primary cilia; and (4) the position of the primary cilium, called kinocilium, of the hair cell of the organ of Corti in the inner ear determines the correct polarity of the actin-containing stereocilia (see Chapter 9, Sensory Organs: Vision and Hearing).
Microvilli (singular, microvillus; Figure 1-7) are finger-like cell projections of the apical epithelial cell surface containing a core of cross-linked microfilaments (a polymer of G-actin monomers). At the cytoplasmic end of the microvillus, bundles of actin and other proteins extend into the terminal web, a filamentous network of cytoskeletal proteins running parallel to the apical domain of the epithelial cell.
Stereocilia (singular, stereocilium; see Figure 1-7) are long and branching finger-like projections of the apical epithelial cell surface. Similar to microvilli, stereocilia contain a core of cross-linked actin with other proteins. Stereocilia do not have axonemes. Stereocilia are typical of the epithelial lining of the epididymis and contribute to the process of sperm maturation occurring in this organ.
CELL ADHESION MOLECULES
A sheet of epithelial cells results from the tight attachment of similar cells to each other and to the basal lamina, a component of the extracellular matrix. Cell adhesion molecules enable interepithelial cell contact, and this contact is stabilized by specialized cell junctions. A consequence of this arrangement is the apical and basolateral domain polarity of an epithelial sheet.
Although cell adhesion molecules and cell junctions are considered here within the framework of epithelia, nonepithelial cells also can use cell adhesion molecules and junctions to establish contact with each other, enabling cell-cell communication. A typical example of nonepithelial cells connected by specialized junctions is the cardiac muscle (see Chapter 7, Muscle Tissue).
There are two major classes of cell adhesion molecules (see Box 1-B):
Box 1-B Cell adhesion molecules
Cadherins (Figure 1-8) are a family of Ca2+-dependent molecules with a major role in cell adhesion and morphogenesis. A loss of cadherins is associated with the acquisition of invasive behavior by tumor cells (metastasis) (see Chapter 4, Connective Tissue).
There are more than 40 different cadherins. E-cadherin is an epithelial cadherin found along the lateral cell surfaces and is responsible for the maintenance of most epithelial layers. The removal of calcium or the use of a blocking antibody to E-cadherin in epithelial cell cultures breaks down cell-cell attachment, and the formation of stabilizing junctions is disrupted. E-cadherin molecules form cis-homophilic dimers (“like-to-like”), which bind to dimers of the same or different class of cadherins in the opposite cell membrane (trans-homophilic or heterophilic [“like-to-unlike”] interaction). These forms of binding require the presence of calcium and result in a specialized zipper-like cell-cell adhesion pattern.
Members of the cadherin family also are present between cytoplasmic plaques of the zonula and the macula adherens. β-catenin plays a significant role in colorectal carcinogenesis (see Chapter 16, Lower Digestive Segment).
Selectins (Figure 1-9), similar to cadherins, are Ca2+-dependent cell adhesion molecules. In contrast to cadherins, selectins bind to carbohydrates and belong to the group of lectins (Latin lectum, to select). Each selectin has a carbohydrate-recognition domain (CRD) with binding affinity to a specific oligosaccharide attached to a protein (glycoprotein) or a lipid (glycolipid). The molecular configuration of the CRD is controlled by calcium.
Selectins participate in the movement of leukocytes (Greek leukos, white, kytos, cell) circulating in blood (neutrophils, monocytes, B and T cells) toward tissues by extravasation. Extravasation is the essence of homing, a mechanism that enables leukocytes to escape from blood circulation and reach the sites of inflammation (see Figure 1-12). Homing also permits thymus-derived T cells to home in on peripheral lymph nodes (see Chapter 10, Immune-Lymphatic System).
The three major classes of cell surface selectins are as follows:
P-selectin is stored in cytoplasmic vesicles in endothelial cells. When endothelial cells are activated by inflammatory signaling, P-selectin appears on the cell surface. On their surface, leukocytes contain sialyl Lewis-x antigen, a specific oligosaccharide ligand for P-selectin. P-selectin binding to the antigen slows down streaming leukocytes in blood, and they begin to roll along the endothelial cell surfaces. P-selectins get additional help from members of the immunoglobulin (Ig) superfamily and integrins to stabilize leukocyte attachment, leading to extravasation (see Figure 1-12).
A conserved feature shared by all members of the Ig superfamily is an extracellular segment with one or more folded domains characteristic of immunoglobulins (Figure 1-10). Of particular interest is CD4, a member of the Ig superfamily and the receptor for the human immunodeficiency virus type 1 (HIV-1) in a subclass of lymphocytes known as T cells or helper cells. We discuss the significance of several members of the Ig superfamily in Chapter 10, Immune-Lymphatic System.
Other members of the Ig superfamily play important roles in the homing process during inflammation. Examples include intercellular adhesion molecules 1 and 2 (ICAM-1 and ICAM-2) on endothelial cell surfaces. ICAM-1 is expressed when an inflammation is in progress to facilitate the transendothelial migration of leukocytes (see Chapter 6, Blood and Hematopoiesis).
Integrins (Figure 1-11) differ from cadherins, selectins, and members of the Ig superfamily in that integrins are heterodimers formed by two associated α and β subunits encoded by different genes. There are about 22 integrin heterodimers consisting of 17 forms of α subunits and 8 forms of β subunits.
Almost every cell expresses one or several integrins. Similar to cadherins, the cytoplasmic domain of β integrins is linked to actin filaments through connecting proteins (talin, vinculin, and α-actinin).
The integrin–extracellular matrix relationship is critical for cell migration to precise sites during embryogenesis and can be regulated when cell motility is required. In addition to their role in cell-matrix interactions, integrins also mediate cell-cell interaction. Integrins containing β2 subunits are expressed on the surface of leukocytes and mediate cell-cell binding. An example is the α1β2 integrin heterodimer that binds to ligands on endothelial cell surfaces during the integrin phase (extravasation) of homing (Figure 1-12).
ADAM proteins
The reversal of integrin-mediated cell binding to the extracellular matrix can be disrupted by proteins called ADAM (for a disintegrin and metalloprotease). ADAMs have pivotal roles in fertilization, angiogenesis, neurogenesis, heart development, cancer and Alzheimer’s disease (see Chapter 8, Nervous Tissue).
A typical ADAM protein (Figure 1-13) contains an extracellular domain and an intracellular domain. The extracellular domain consists of several portions including a disintegrin domain and a metalloprotease domain.
CELL JUNCTIONS
Although cell adhesion molecules are responsible for cell-cell adhesion, cell junctions are necessary for providing stronger stability. In addition, the movement of solutes, ions, and water through an epithelial layer occurs across and between individual cell components. The transcellular pathway is controlled by numerous channels and transporters. The paracellular pathway is regulated by a continuous intercellular contact or cell junctions. A deficiency in the cell junctions accounts for acquired and inherited diseases caused by inefficient epithelial barriers.
Cell junctions are symmetrical structures formed between two adjacent cells. There are three major classes of symmetrical cell junctions (Figure 1-14; see Box 1-C):
Box 1-C Cell junctions
Tight junctions (also called occluding junctions) (Figure 1-15) have two major functions:
Occludin interacts with four major zonula occludin (ZO) proteins: ZO-1, ZO-2, ZO-3, and afadin. Claudin (Latin claudere, to close), a family of 16 proteins forming linear fibrils in the tight junctions, confers barrier properties on the paracellular pathway. A mutation in the gene encoding claudin 16 is the cause of a rare human renal magnesium wasting syndrome characterized by hypomagnesemia and seizures.
Tight junctions can be visualized by freeze-fracturing a network of branching and anastomosing sealing strands. We discuss in Chapter 2, Epithelial Glands, the procedure of freeze-fracturing for the study of cell membranes.
Anchoring junctions are found below the tight junctions, usually near the apical surface of an epithelium. There are three classes of anchoring junctions (see Figures 1-14, 1-16, 1-18, and 1-19):
Similar to the tight junctions, the zonula adherens is a beltlike junction. The zonula adherens (Figure 1-16) is associated with actin microfilaments. This association is mediated by the interaction of cadherins (desmocollins and desmogleins) with catenins (α, β, and γ). The main desmogleins expressed in the epidermis of the skin are desmoglein 1 and desmoglein 3 (Figure 1-17).
The macula adherens (also called desmosome) is a spotlike junction associated with keratin intermediate filaments (also known as tonofilaments) extending from one spot to another on the lateral and basal cell surfaces of epithelial cells (Figure 1-18). Spot desmosomes provide strength and rigidity to an epithelial cell layer. Spot desmosomes are also present in the intercalated disks linking adjacent cardiocytes in heart (see Chapter 7, Muscle Tissue) and in the meninges lining the outer surfaces of the brain and spinal cord.
In contrast to occluding junctions, adjacent cell membranes linked by zonula and macula adherens are separated by a relatively wide intercellular space. This space is occupied by the glycosylated portion of proteins of the cadherin family, desmogleins and desmocollins, anchored to cytoplasmic plaques containing desmoplakin, plakoglobin (γ-catenin), and plakophilin. The cytoplasmic plaques are attached to the cytosolic face of the plasma membrane. The interlocking of similar cadherins binds two cells together by Ca2+-dependent homophilic or heterophilic interaction, as we have already seen. Inherited disorders of some of the desmosomal components are indicated in Figure 1-18.
The human desmosomal cadherins genes include four desmogleins and three desmocollins. Their cytoplasmic regions interact with plakoglobin and plakophilin. Desmoplakin interacts with the intermediate filaments keratin in epidermis, desmin in the intercalated disks, and vimentin in the meninges. Desmoglein 1 and desmoglein 3 maintain the cohesiveness of the epidermis, a stratified squamous epithelium. Autoantibodies to desmoglein 1 cause a blistering disease (disruption of cell adhesion) of the skin called pemphigus foliaceus (see Figure 1-17).
Hemidesmosomes are asymmetrical structures anchoring the basal domain of an epithelial cell to the underlying basal lamina (Figure 1-19).
Gap junctions are symmetrical communicating junctions formed by integral membrane proteins called connexins. Six connexin monomers associate to form a connexon, a hollow cylindrical structure that spans the plasma membrane. The end-to-end alignment of connexons in adjacent cells provides a direct channel of communication (1.5 to 2 nm in diameter) between the cytoplasm of two adjacent cells (Figure 1-20). Connexons have a clustering tendency and can form patches about 0.3 mm in diameter.
These junctions facilitate the movement of molecules 1.2 nm in diameter (for example, Ca2+ and cyclic adenosine monophosphate [cAMP]) between cells. The connexon axial channels close when the concentration of Ca2+ is high. This junction is responsible for the chemical and electrical “coupling” between adjacent cells. A typical example is cardiac muscle cells connected by gap junctions to enable the transmission of electrical signals.
Clinical significance: Connexin mutations in human disease
Mutations in the connexin 32 (Cx32) gene are found in X-linked Charcot-Marie-Tooth demyelinating neuropathy resulting in progressive degeneration of peripheral nerves, characterized by distal muscle weakness and atrophy and impairment of deep tendon reflexes. Connexin 32 protein is expressed in Schwann cells, which are involved in the production of rolled myelin tubes around the axons in the peripheral nervous system (see Chapter 8, Nervous Tissue). Gap junctions couple different parts of the rolled myelin tubes of the same Schwann cell, rather than different cells. A loss of the functional axial channels in myelin leads to the demyelinating disorder. Mutations in the connexin 50 (Cx50) gene are associated with congenital cataracts, leading to blindness.
BASEMENT MEMBRANE
Integrins mediate cell-matrix interactions by their binding affinity to the RGD domain in laminin and fibronectin (see Figure 1-11). Laminin and fibronectin are distinct proteins of the extracellular matrix and are associated with collagens, proteoglycans, and other proteins to organize a basement membrane, the supporting sheet of most epithelia.
The basement membrane consists of two components (Figure 1-21):
The basal and reticular laminae can be distinguished by electron microscopy. Under the light microscope, the combined basal and reticular laminae receive the name of basement membrane, which can be recognized by the periodic acid–Schiff (PAS) stain (see Figure 1-21; see Box 1-D).
Box 1-D Periodic acid–Schiff (PAS) reaction
The basal lamina has specific functions in different tissues. The double basal lamina of the renal corpuscle constitutes the most important element of the glomerular filtration barrier during the initial step in the formation of urine (see Chapter 14, Urinary System).
In skeletal muscle, the basal lamina maintains the integrity of the tissue, and its disruption gives rise to muscular dystrophies (see Chapter 7, Muscle Tissue).
Laminin (Figure 1-22) is a cross-shaped protein consisting of three chains: the α chain, the β chain, and the γ chain. Laminin molecules can associate with each other to form a meshlike polymer. Laminin and type IV collagen are the major components of the basal lamina, and both are synthesized by epithelial cells resting on the lamina.
Laminin has binding sites for nidogen (also called entactin), proteoglycans (in particular, heparan sulfate perlecan), α-dystroglycan (see Chapter 7, Muscle Tissue), and integrins.
Fibronectin (see Figure 1-22) consists of two protein chains cross-linked by disulfide bonds. Fibronectin is the main adhesion molecule of the extracellular matrix of the connective tissue and is produced by fibroblasts. Fibronectin has binding sites for heparin present in proteoglycans, several types of collagens (types I, II, III, and V), and fibrin (derived from fibrinogen during blood coagulation).
Fibronectin circulating in blood is synthesized in the liver by hepatocytes. It differs from fibronectin produced by fibroblasts in that it lacks one or two repeats (designated EDA and EDB for extra domain A and extra domain B) as a result of alternative mRNA splicing. Circulating fibronectin binds to fibrin, a component of the blood clot formed at the site of blood vessel damage. The RGD domain of immobilized fibronectin binds to integrin expressed on the surface of activated platelets, and the blood clot enlarges. We return to the topic of blood coagulation or hemostasis in Chapter 6, Blood and Hematopoiesis.
How cells interact with one another and with the basal lamina
Figure 1-23 summarizes the highlights of cell adhesion molecules and cell junctions. An epithelium is a continuous sheet of polarized cells supported by a basement membrane. The polarized nature of an epithelium depends on the tight junctions that separate the polarized cells into apical and basolateral regions. Tight junctions control the paracellular pathway of solutes, ions, and water. Tight junctions form a belt around the circumference of each cell.
Endothelial cells, the constituents of a simple squamous epithelium, are linked by tight and spot desmosomes tightly regulated to maintain the integrity of the endothelium and protect the vessels from unregulated permeability, inflammation, and reactions leading to blood coagulation in the lumen (see Chapter 12, Cardiovascular System). Leukocytes reach the site of infection by attaching to endothelial cell surfaces and migrate across the endothelium into the underlying tissues by a mechanism called diapedesis. Leukocytes find their way through endothelial cell-cell junctions after docking to activated or resting endothelial cells by the endothelial cell adhesion molecules ICAM-1 and VCAM-1 (see Figure 1-10). ICAM-1 and VCAM-1 bind to β2 and β1 integrin subunits in leukocytes (see Figure 1-12).
Note in Figure 1-23 that:
CYTOSKELETON
The cytoskeleton has roles in:
Biochemical studies, involving the extraction of cytoskeletal proteins from cells with detergents and salts and in vitro translation of specific mRNA, showed that each class of filaments has a unique protein organization. When cytoskeletal proteins were purified, they were used as antigens for the production of antibodies. Antibodies are used as tools for the localization of the various cytoskeletal proteins in the cell. The immunocytochemical localization of cytoskeletal proteins (Figure 1-24) and cell treatment with various chemical agents disrupting the normal organization of the cytoskeleton have been instrumental in understanding the organization and function of the cytoskeleton.
Microfilaments
Actin is a versatile and abundant cytoskeletal component forming static and contractile bundles and filamentous networks specified by actin-binding proteins and their distinctive location and function in a cell. F-actin bundles are present in the microvilli of the intestinal (Figure 1-25) and renal epithelial cells (brush border) and the stereocilia from the hair cells of the inner ear.
We have already seen that the intracellular portion of the cell adhesion molecules cadherins and integrin β1 interacts with F-actin through linker proteins (see Figures 1-8 and 1-11). As discussed in Chapter 6, Blood and Hematopoiesis, actin—together with spectrin—forms a filamentous network on the inner face of the red blood cell membrane that is crucial for maintaining the shape and integrity of red blood cells. Spectrin is a tetramer consisting of two distinct polypeptide chains (α and β).
Growth of actin filaments may occur at both ends; however, one end (the “barbed end” or plus end) grows faster than the other end (the “pointed end” or minus end). The names correspond to the arrowhead appearance of myosin head bound at an angle to actin. Actin filaments can branch in the leading edge (lamellipodia) of cells involved in either motility or interaction with other cell types. F-actin branching is initiated from the side of a preexisting actin filament by Arp2/3 (for actin-related protein), an actin nucleating complex of seven proteins (Figure 1-26). Formin regulates the assembly of unbranched actin in cell protrusions such as the intestinal microvilli (see Figure 1-25).
Actin monomers have a binding site for adenosine triphosphate (ATP), which is hydrolyzed to adenosine diphosphate (ADP) as polymerization proceeds. Actin polymerization is ATP-dependent (see Box 1-E).
Box 1-E Microfilaments
The kinetics of actin polymerization involves a mechanism known as treadmilling: G-actin monomers assembled at one end of the filament concurrently disassemble at the other end (see Figure 1-26). Four types of proteins control treadmilling (see Figure 1-26), as follows:
The assembly of G-actin monomers into filaments and the organization of these filaments into thick bundles are controlled by various types of actin-binding or actin-related proteins. A bundle of parallel nonbranching actin filaments, forming the core of the microvillus, is held together by actin-linking proteins, villin and fimbrin. Side arms of myosin-I and the Ca2+
[/not-level-membership-for-pathology-category]
