In addition to these highly penetrant hereditary syndromes, genome-wide association studies are identifying new loci that confer increased risk for either BCC or SCC, although the mechanistic details remain. Single-nucleotide polymorphisms (SNPs) within several well-characterized pigmentation loci—including
MC1R (melanocortin 1 receptor) and
OCA2 (oculocutaneous albinism II)—and a potential pigmentation locus
IRF4 (interferon regulatory factor 4) show linkage to increased risk for both BCC and SCC NMSC.
4–6 Other SNPs on 6p25 and 13q32 near the
EXOC2 and
UBAC2 genes also show linkage with increased risk of SCC,
6 whereas two functional SNPs in the nucleotide excision repair gene
ERCC6 are significantly associated with increased risk of BCC. A history of NMSC is also associated with increased risk for subsequent noncutaneous malignancies, and a previous SCC is associated with poor prognosis for cancers of the lung, colon, rectum, and breast and for non-Hodgkin lymphoma. Although the genetic basis of this is currently unknown, GWAS studies of patients
with NMSC and a noncutaneous malignancy will likely reveal additional loci conferring risk for cancer for which the appearance of NMSC may be a signal.
Basal Cell Carcinoma
BCC is a very common, slow-growing, locally invasive tumor that typically presents as a pink or pearly papule with superficial telangiectasia and occasional ulceration. BCC precursor lesions have not been identified, there is no evidence of neoplastic progression, phenotypic diversity even within the same tumor is common, and metastases are exceedingly rare. Chromosomal losses involving 9q mark both sporadic and inherited BCCs, leading to the discovery of mutations in
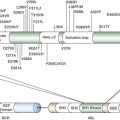
PTCH1,
3 a homologue of the
Drosophila ptc gene involved in embryonic development.
Ptch1 is a 12-pass transmembrane receptor for Sonic hedgehog (Shh), a secreted ligand involved in the proliferation and patterning of multiple tissues and organs during embryogenesis. Shh binds to Ptch and alleviates Ptch-mediated repression of Smo on responding cells (reviewed in Reference
1). Active Smo enters the primary cilium, where it functions to modify the expression and structure of a family of Gli transcription factors to alter the expression of Shh target genes. The complexity of the pathway is enhanced by the presence of additional vertebrate homologues of
Drosophila proteins, fused (a serine-threonine kinase), suppressor of fused (SuFu) that binds to Gli proteins, Cos 2 (Kif7), and Iguana (Dzip1), with SuFu, Cos2, and Iguana serving as inhibitors of Gli signaling.
1 Activation of the Shh pathway upregulates
Gli1 and
Ptch1, and these serve as markers for physiologic and pathologic Shh signaling. In human BCCs, inactivating mutation or deletion of PTCH1 results in constitutive signaling independent of SHH. In addition to loss-of-function
PTCH1 mutations, gain-of-function (oncogenic)
SMO mutations have been found in some BCCs where PTCH1 appears to be normal.
PTCH1 or
SMO mutations are implicated in about 80% of BCCs, whereas hedgehog signaling is active in all BCCs. Thus, additional mechanisms must exist for uncontrolled activation of this pathway. Rare mutations in SuFu have been reported in BCCs, and other syndromes are known to predispose to BCC, but the genetic bases for these have not been delineated. Although mutations in
p53 have been found in up to 50% of BCCs , most lesions fail to exhibit the genomic instability associated with other cancers where p53 function is compromised,
7 and the presence or absence of p53 mutations does not alter the histological phenotype, although loss of p53 function facilitates the eruption of BCC in animal models.
Gli activation is the driving force for tumor development in the setting of constitutive Hedgehog signaling. The three mammalian Gli proteins are zinc finger proteins that bind to a 9bp canonical binding site in the promoters of target genes following activation of hedgehog signaling (reviewed in Reference
1). The regulation of Gli activity by hedgehog signaling is complex; Gli2 and Gli3 proteins have both transcriptional repressor and activating domains, whereas Gli1 is strictly a transcriptional activator. Much of the regulation of Gli proteins is post-translational. Gli3 processing primarily generates a transcriptional repressor, whereas processed (phosphorylated) Gli2 is ubiquitinated and degraded. Shh signaling suppresses processing and degradation of Gli2 and stabilizes its transcriptional activation function. Genetic ablation studies in mice have displayed the consequences of Gli activity in vivo. Gli1-null mice are without a phenotype, whereas the developmental defects and impaired hair follicle growth caused by ablation of Gli2 indicate that Gli2 is the downstream effector of the Hh pathway.
8 Disruption of Gli3 produces a phenotype consistent with hedgehog activation, validating its action as a repressor of the pathway.
Genetically altered mouse models provide experimental evidence linking the hedgehog pathway to human BCC.
9 Targeting SHH or an activated SMO mutant to the epidermis and hair follicles upregulates Shh target genes and produces basal cell–like proliferations in newborn mouse skin. Overexpression of SHH in human keratinocytes followed by grafting onto severe combined immunodeficiency (SCID) mice produced BCC-like changes as well. Mouse models in which
Ptch gene function has been disrupted develop microscopic hair follicle–derived proliferations, with the appearance of a variety of macroscopic skin tumors, including BCCs, following exposure to ionizing or UV radiation.
10 Mice with skin targeted overexpression of human GLI1 or mouse Gli2 develop multiple BCCs or other tumors arising from hair follicles. The Hh pathway is regulated through primary cilia, present on most mammalian cells, which acts as an organizing structure for signaling and is essential for proteolytic processing of Gli proteins to activator and repressor forms. Inactivation of Kif3a, an essential component of the anterograde intraflagellar transport motor, blocks formation of BCC in mice expressing the activated form of SMO, but accelerates tumor formation driven by activated Gli2. Thus cilia play a dual role in both activating and inhibiting Shh signaling and tumorigenesis.
Taken together, these findings strongly support the concept that deregulated Shh signaling is central for BCC development. When Gli2 is targeted to mouse epidermis and hair follicles conditionally, BCCs develop from overexpression and regress when expression is discontinued. On reexpression of Gli2 in this model, tumors reemerge from a small residual population of precursor cells. What remains unclear is the identification of the hedgehog target genes
downstream from Gli that are essential for BCC formation. A number of candidates have been reported, including several cyclins, E2f1, N-Myc, Pdgfr, Bcl2, BEG4, FOXM1, and FOXE1, Mim, Hhip, Snail, Ptch1, and Gli1 (
Figure 41-1 ), but definitive studies on these genes are lacking. In both human and mouse BCC the Wnt pathway appears dysregulated due to elevated expression of β-catenin, and blockade of Wnt signaling in mice blocks hedgehog-driven tumor growth. Crosstalk between hedgehog and EGFR signaling is synergistic for BCC induction in animal models, suggesting that EGFR inhibitors could have a therapeutic role in the clinic. Small-molecule inhibitors of SMO have been developed based on the natural product cyclopamine, which causes BCC regression in animal models. In recent Phase III clinical trials, the SMO inhibitor vismodegib has caused significant tumor regression in patients with advanced sporadic BCC
11 and regression of preexisting lesions and prevention of new lesions in Gorlin syndrome patients.
12 Vismodegib was approved by the U.S. Food and Drug Administration (FDA) in January 2012 for adult patients with recurrent locally advanced or metastatic BCCs or who are not candidates for surgery or radiation. Other inhibitors are likely to follow, although target-based side effects may be an inherent limitation for long-term treatment. Because the Hedgehog pathway is aberrantly activated in malignancies arising in several internal organs,
1 these inhibitors are likely to have a significant impact on the treatment of a broad range of human neoplasms.
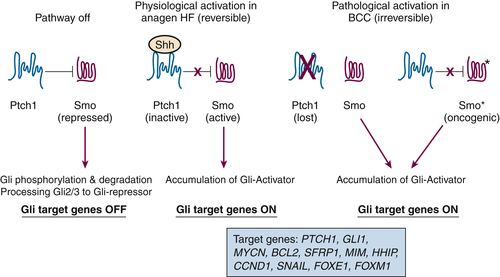
Figure 41-1 Simplified model depicting hedgehog signaling in skin physiology and BCC pathogenesis The major site of action for sonic hedgehog (SHH) is the hair follicle (HF). In resting hair follicles, SHH is not expressed, and PTCH1 dampens SMO activity. Under these conditions, Gli transcription factors are unstable and degraded. In addition, posttranslational processing of GLI2 and GLI3 processing yields a transcriptional repressor that suppresses SHH target gene expression. When SHH expression increases during HF growth (anagen), PTCH1 is inactivated, SMO is phosphorylated by casein kinase 1 and protein kinase A, and active SMO upregulates Gli proteins by posttranslational mechanisms, leading to accumulation of Gli transcriptional activators. One target of Gli transcription is PTCH1, which serves as a negative feedback on this reversible pathway. In BCC initiation, PTCH1 is genetically deleted (or SMO is mutationally activated to SMO∗), and the pathway is irreversibly activated. Gli2 appears to be the major driving factor to upregulate a number of effectors responsible for the neoplastic phenotype, including GLI1, MYCN, BCL2, SFRP1, MIM, HHIP, CCND1, SNAIL, FOXE1, and FOXM1.
Cutaneous Squamous Cell Carcinoma
Cutaneous SCC frequently presents as a firm, pink papule or nodule, with a conspicuous hyperkeratotic surface. Although SCCs represent only about 20% of nonmelanoma skin cancers, they are invasive and occasionally metastasize (1% to 2%). SCC is more frequent with higher cumulative sunlight exposure and as cancers associated with specific occupational exposures (coke oven and petroleum oil workers). There is roughly a 25- to 200-fold increase in SCC incidence in immunosuppressed organ transplant recipients, with a reversal of the BCC-to-SCC ratio; these tumors are more aggressive, occur in multiple locations within one patient, and are associated with increased morbidity and mortality. Conversely, a drug that activates local innate and adaptive immune responses through TLR-7 (imiquimod (1-(2-methylpropyl)-1
H-imidazo[4,5-
C]quinolin-4-amine) is highly effective in treating BCC, SCC, and its precursor lesion, actinic keratosis (AK).
13 Cutaneous SCC is usually preceded by a benign hyperproliferative-hyperkeratotic AK (
Figure 41-2 ,
A). These are sunlight-induced clonal lesions that frequently harbor
p53 mutations, particularly at codon 278 or other codons of the DNA binding domain of p53 that contain dipyrimidine sites. AKs often exhibit chromosomal changes, particularly loss of heterozygosity (LOH) at 3p, 13q, 17p, 17q, 9p, and 9q. Such changes are less frequent in SCC, clouding the direct relation of AK to SCC.
However, the frequency of evolution of AK to SCC is very low (0.1% to 10%), suggesting there may be a high-risk AK group with relevant genetic changes not yet documented. Activating mutations in the K-
RAS or Ha-
RAS gene are detected in approximately 10% of AK and SCC, and the RAS pathway is activated by non-mutational mechanisms in a much larger fraction of SCCs. Inactivating mutations or epigenetic silencing of p16
(INK4a) and activation of telomerase are other pathways associated with SCC development. Constitutive activation of the EGF receptor (EGFR) by amplification or increased expression of ligands with the formation of an autocrine loop is a frequent finding in SCC. Gene expression arrays have revealed several other genes whose expression is characteristic of SCC, but experimental validation of a causal relation remains to be determined.
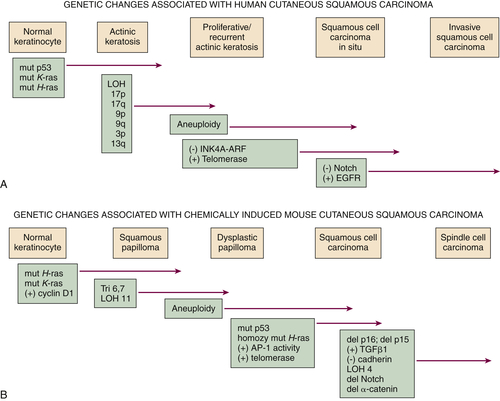
Figure 41-2 Genetic changes associated with (A) human cutaneous squamous carcinoma and (B) experimental mouse skin squamous carcinoma The multistage evolution of invasive squamous cell cancer is depicted schematically with frequently associated genetic changes detailed here. Although many common pathways exist in the two species, current understanding places p53 mutations early in human SCC development and ultraviolet light–induced mouse skin SCC (not shown), but p53 mutations occur late in chemically induced mouse skin SCC associated with malignant conversion. Increased activity of telomerase (deletion of inhibitor) or EGFR tyrosine kinase (gene amplification) and decreased signaling in the Notch pathway (mutation) may also result from epigenetic changes, as in the case of upregulation of EGFR ligand expression in mouse SCC development. This figure is modified from Dlugosz A, Merlino G, Yuspa SH. Progress in cutaneous cancer research. J Invest Dermatol Symp Proc 7:17-26, 2002, with permission of the Journal of Investigative Dermatology.
Constitutive activation of NFκB signaling is common in SCC,
14,15 and this is associated with upregulation of specific NFκB target genes associated with altered proliferation, invasion, angiogenesis, and inflammation. NFκB hyperactivation is a known inducer of cylindromatous skin tumors, because homozygous deletions of the CYLD gene, encoding a deubiquitinase that negatively regulates the NFκB pathway through targets such as TRAF2 and BCL-3, has been identified as the basis for familial cylindromatosis (see
Table 41-1). Homozygous deletion of CYLD in mice increases susceptibility to squamous skin tumors. However, studies with genetically modified human keratinocytes grafted to nude mice suggest that inhibition of NFκB together with activation of the
RAS oncogene is sufficient to convert normal keratinocytes into SCC, and a similar result has been obtained in the skin of transgenic mice. This controversy is unsettled at this time.
To prove causal relations among the various associations made by studying human SCC, model systems using
human and mouse keratinocytes in culture and animal models in vivo have been developed. The classical carcinogenesis model to induce SCC on mouse skin involves a limited application of a mutagenic agent such as a chemical carcinogen or UV light that “initiates” the cancer process in a subset of cells, followed by repeated applications of a nonmutagenic agent such as a phorbol ester that provides a microenvironment favorable for the clonal outgrowth of the initiated population. Tumor development proceeds through predictable stages, with the early emergence of benign squamous papillomas, the murine equivalent of AK, some of which progress to invasive SCC, mimicking human SCC with a low metastatic rate (see
Figure 41-2,
B). In some cases SCC will evolve into a more aggressive spindle cell cancer. This model has illuminated the genetic and biochemical changes that are permissive for SCC development under controlled experimental conditions. Heterozygous activating
Ras gene mutations are sufficient to “initiate” the target cells and produce squamous papillomas, and this is coupled to constitutive activation of the EGFR through overexpression of EGFR ligands (see
Figure 41-2,
B). Papillomas form spontaneously in transgenic mice overexpressing ErbB2 and TGFα in epidermis, and papilloma formation is reduced in mice where EGFR or SOS is ablated. Furthermore, tumor formation is completely inhibited in mice lacking the
Stat3 gene, a downstream target of the EGFR. The clinical relevance of this pathway is highlighted by the effectiveness of anti-EGFR drugs in head and neck SCC patients. The relevance of the RAS pathway in human skin cancer is emphasized by the development of multiple cutaneous SCC and keratoacanthoma expressing
Ha-RAS mutations in melanoma patients treated with the BRAF V600E inhibitor vemurafenib.
16 This drug also enhanced chemical carcinogenesis on mouse skin through stimulation of MAPK activity. Inactivation of PKCδ is also essential for papilloma development in mouse skin. PKCδ is inactivated by c-Src–mediated tyrosine phosphorylation in initiated mouse keratinocytes, whereas in human skin tumors the expression of PKCδ transcripts is greatly reduced. Transgenic mice overexpressing PKCδ in the skin do not form skin tumors. TGFβ plays a dual role in experimental SCC development, suppressing premalignant progression to SCC while enhancing phenotypic progression from SCC to a spindle cell phenotype.
17 However, topical inhibitors of the TGFβ type I receptor suppress papilloma formation, suggesting that TGFβ signaling is important in tumor outgrowth. Inactivation of p53 enhances malignant conversion of papillomas to SCC in chemical carcinogenesis but is an earlier event in UV light–induced skin carcinogenesis in both mice and humans. Members of the AP-1 transcription factor family also play a dual role in experimental skin tumor development, where c-Jun is essential for papilloma and c-Fos is essential for SCC development. Inhibition of AP-1 activity after the formation of benign papillomas prevents malignant conversion.
In contrast to murine models for BCC, the development of techniques to genetically alter mice greatly expanded the array of genes and pathways that influence SCC development on the skin (
Table 41-2 ), indicating a much more complex pathogenesis. Pathways and genes that can influence SCC development and progression to spindle cell tumors in mice include cyclin D1, ornithine decarboxylase, p16(ink4A), p15(ink4A), p63, E-cadherin, and TGFβ1, and these are frequently altered in human SCC. In several model systems, genetic alterations have resulted in the spontaneous development of SCC in the absence of a precursor lesion. For example, mice ablated for
Notch or β-
Catenin rapidly form SCCs even in the absence of carcinogenic exposure, and ablation of
Notch in epidermis promotes tumor formation by creating a barrier defect and promoting inflammation.
18 In fact, loss of function
Notch mutations is frequently found in cutaneous SCC.
19 Furthermore, clinical trials of γ-secretase inhibitors for the treatment of Alzheimer’s disease have been discontinued because of emergence of NMSC.
20 γ-Secretase is required for the activation of intracellular Notch action.
Defining the Cell of Origin for Cutaneous Cancers
Genetic profiling of both SCC and BCC indicate a monoclonal origin. However, the multiplicity of epithelial tumor phenotypes in skin (see
Table 41-1) and the divergent phenotypes within each suggest that either multiple target cells exist within the complex compartmentalization of the integument or that specific genetic lesions or tumor microenvironments acting on a multipotential target cell determine tumor phenotype and malignant potential.
21,22 Both human and murine skin contains several types of stem cells within different locations in the epidermis and hair follicle that maintain discrete epidermal compartments, with stem cells within the bulge region of the hair follicle the best characterized.
22 Gene expression analysis of isolated murine hair follicle stem cells and analysis of genetically altered mice has revealed specific gene expression patterns and signaling pathways that define stemness, and Lgr6+ cells residing above the follicle bulge may mark stem cells capable of generating all three self-renewing compartments of the epidermis.
23 Lineage tracing studies in mice and targeted activation of Hh signaling in different cells of the hair follicle have revealed that only cells originating in the interfollicular epidermis and upper infundibulum are capable of generating BCC in the absence of other factors, such as wounding hair growth cycle, irradiation, or supraphysiological expression of
pathway components. Targeting high levels of oncogenic Ras to basal and stem cell keratinocytes gives rise to squamous tumors with a high risk for malignant conversion, whereas targeting suprabasal keratinocytes produces only benign tumors. Lineage tracing with genetically altered mice shows that keratin 15 expressing cells of the hair follicle bulge region contribute to chemically induced squamous papillomas.
24 However, expression of physiological levels of oncogenic Ras in hair follicle bulge stem cells, hair germ, and outer root sheath or interfollicular keratinocytes only gives rise to benign papilloma, but SCC can develop in all cells when combined with p53 deletion. More committed hair matrix keratinocytes were unable to form tumors with Ras or Ras + p53 loss.
25,26 Although the cell of origin has not been identified for human cutaneous SCC, tumor-initiating or cancer stem cells (CSCs) have been isolated from cutaneous SCC based on expression of CD133 (prominin), which can regenerate tumors with similar histology and grade as the original in serial xenotransplants on immunocompromised mice.
27 In murine cutaneous SCC, 2 CSC populations have been identified differing in the expression of the hair follicle bulge marker CD34, and responsiveness to TGFβ1 signaling and FAK-mediated integrin signaling. Other studies have demonstrated dependence of CSC on β-catenin signaling and autocrine VEGF responses requiring the neuropilin receptor. Together these mouse models show that SCC can arise from multiple cells within the hair follicle and interfollicular epidermis, that distinct CSC populations exist within the same tumor, and that they have gene expression patterns distinct from normal hair follicle stem cells and require specific
microenvironmental stimuli and gene expression pathways to maintain the CSC phenotype.
Table 41-2
Genetically Modified Mouse Models for Cutaneous Squamous Cell Cancer
| Modification |
Enhancers |
Comments |
| Tg.AC (ζ globin-v-rasHa) |
Promoters, drugs |
Enhancers upregulate transgene |
| Tg.AC |
UVB |
p53 mutations are absent |
| K1-ras, K10-ras |
Promoters |
Predominantly papillomas |
| ΔK5-ras |
None |
Papillomas, KA, SCC |
| K6-ras |
Promoters |
SCC |
| K1-TGFα, K14-TGFα, MT-TGFα |
Promoters |
Predominantly papillomas that regress |
| Inv-c-MycER |
None |
Papilloma |
| K1-v-fos |
Promoters |
Papilloma |
| K5-E2F1 |
p53 deficiency |
Papilloma, SCC, BCC |
| K5-Igf1 |
Promoters |
Papilloma, SCC |
| K5-ErbB2 |
Promoters |
Papilloma, SCC |
| K5-SOS-F |
None |
Tumors inhibited by Egfr deficiency |
| K14-HPV16 |
FVB/N mouse strain |
Tumors inhibited by difluoromethylornithine |
| K6-ODC |
DMBA |
SCC, K-ras mutations |
| XP mutant models (A, C, D) |
Initiation/promotion/UVR |
Enhanced sensitivity |
| Egfr null mutant |
v-ras Ha |
Reduced tumor size |
| p53 null mutant |
DMBA/TPA |
Enhanced malignant conversion |
| p21waf1 null mutant |
DMBA/TPA |
Enhanced papilloma formation |
| c-fos null mutant |
Cross with Tg.AC |
Papilloma but no SCC |
| K14-PKCε |
DMBA/TPA |
Enhanced SCC, metastases |
| K14-PKCδ |
DMBA/TPA |
Reduced papilloma development |
| K5-src |
None or DMBA/TPA |
Enhanced spontaneous or induced SCC |
| K5-IκB mutant |
None |
Spontaneous SCC/NFκB inhibition |
| Notch null |
None |
Spontaneous SCC/nuclear β-catenin |
| α-catenin null |
None |
Spontaneous SCC/NFκB activation |
| Cyld null |
None or DMBA/TPA |
Enhanced papilloma/NFκB activation |
Importance of the Microenvironment in Cutaneous Cancer
Although much cutaneous cancer research has focused on cell-autonomous alterations in keratinocytes that contribute to cancer development, it is now clear that alterations in the cutaneous tissue environment are also critical. Changes in integrin distribution and expression occur during progression of human and chemically induced mouse SCC, and targeted changes in specific integrin complex expression can enhance or suppress malignant conversion. Similarly, mutations in the anchoring molecule collagen VII that block the interaction of collagen VII with laminin 5 predispose dystrophic epidermolysis bullosa to SCC.
Changes in the cutaneous immune microenvironment are also critical for tumor development. Inflammation and immunosuppression caused by UV irradiation are intimately linked to UV-induced skin cancer. Similarly, inflammation within the tumor microenvironment is associated with the progression of AK to SCC. Genetically altered mice lacking skin-resident γδ T cells have increased frequency of chemically induced papillomas and malignant conversion, suggesting that this resident T cell population is important in antitumor immunosurveillance. Surprisingly, Langerhans cells, the other epidermal resident immune cell, mediate chemical carcinogenesis through effects on carcinogen metabolism allowing DNA damage and mutation in keratinocytes. In contrast, mice with a TCRβ deletion (lacking all αβ T cells) have a significantly reduced carcinoma yield, and this may be due to a subset of tumor-promoting CD8
+ T cells.
28,29 Humoral immunity and B cells also play a tumor-promoting role in skin carcinogenesis through cutaneous deposition of IgG that enhances recruitment of proinflammatory myeloid cells. Factors that mediate inflammation such as prostaglandins, TNFα, and IFNγ enhance experimental cutaneous carcinogenesis. Likewise, chronic inflammatory skin conditions such as discoid lupus erythematosus, dystrophic epidermolysis bullosa, and chronic wounds are associated with increased susceptibility to human skin cancer.
Perspective
Advances in understanding the molecular basis of cutaneous cancer have reinforced the paradigm that particular genetic and epigenetic changes and the pathways they regulate contribute to skin cancer formation in a stage-specific manner. What benefit may come from these current insights? This is most clear in the case of BCC, where the molecular mechanism of pathogenesis is so precisely defined (perhaps better than in any other human cancer) that curative therapeutic targets are identified, and precise animal models have been developed for testing new therapeutics. Currently cyclopamine and derivatives with better therapeutic index are in clinical trials to block SMO, with remarkable results in advanced patients,
11 and tazarotene, a retinoid used successfully to treat BCC lesions in mouse models, is an inhibitor of GLI function.
30 Although BCC is generally not life threatening, the high frequency of these lesions on exposed skin favors a medical rather than the traditional surgical approach, an advance that is being achieved by translation of basic research. Recent advances in tools for large-scale expression and genomic analysis have been applied to SCC lesions and their precursor AK. Animal models and human tissue analyses have suggested that premalignant precursor lesions vary in risk for progression, and it is anticipated that molecular profiling will reveal markers to identify high-risk lesions for closer clinical scrutiny. Similarly, profiling of SCC will undoubtedly reveal signature markers associated with lesions at risk for metastatic spread. Currently two molecular therapeutic targets derived from basic research on SCC pathogenesis show promise for medical therapy of SCC. Inhibitors of the EGFR, in clinical use for several internal malignancies, show promise in animal models for the prevention of UV-induced mouse skin SCC. Ingenol-3-angelate (Picato), recently approved for treatment of AK, BCC, and SCC in situ, targets protein kinase C to induce an innate immune response and damage tumor vasculature.
31 Stimulation of innate immunity to destroy skin tumors is another paradigm for cancer therapy, first introduced into the clinic with the drug imiquimod (Aldara) that targets Toll-like receptor-7. Other targets identified from experimental studies that offer therapeutic potential are telomerase, TGFβ, Notch, and p53, because drugs targeting these molecules are in clinical trials for treating a number of epithelial cancers.
A developing concept anticipated from molecular analyses is that common gene or protein expression profiles would reveal similar pathogenic mechanisms for skin SCC and squamous tumors of the lung, head, and neck and other sites that pose a threat to life. Similarly, data already exist indicating that the hedgehog pathway is involved in internal malignancies such as pancreatic cancer. If such mechanisms are shared, then new drugs could be tested on skin tumors for therapeutic efficacy. The high frequency of skin cancers, their superficial location, and the capacity for topical testing suggest that the skin provides an excellent surrogate site for evaluating drug development for a variety of internal tumor sites. The skin is also a site that often predicts the presence of
internal tumors with such lesions as acanthosis nigricans, dermatomyositis, paraneoplastic pemphigus, and other dermatoses. Little is known of the pathogenesis of these premonitory lesions, but undoubtedly such knowledge would reveal important aspects of the host response to cancer. Thus, progress in skin cancer research will continue to provide important translational opportunities not just for these very prevalent lesions, but for the advancement of cancer treatment in general.
Acknowledgments
Because of the breadth of this review, we apologize for the unavoidable exclusion of references to work done by many outstanding investigators working in these areas. We acknowledge Professor Andrzej Dlugosz for advice on BCC pathogenesis.
References
1. Teglund S. , Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma . Biochim Biophys Acta . 1805 ; 181 : 2010 .
2. DiGiovanna J.J. , Kraemer K.H. Shining a light on xeroderma pigmentosum . J Invest Dermatol . 2012 ; 132 : 785 .
3. Johnson R.L. , Rothman A.L. , Xie J. et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome . Science . 1996 ; 272 : 1668 .
4. Han J. , Qureshi A.A. , Nan H. et al. A germline variant in the interferon regulatory factor 4 gene as a novel skin cancer risk locus . Cancer Res . 2011 ; 71 : 1533 .
5. Kosiniak-Kamysz A. , Pospiech E. , Wojas-Pelc A. et al. Potential association of single nucleotide polymorphisms in pigmentation genes with the development of basal cell carcinoma . J Dermatol . 2012 .
6. Nan H. , Xu M. , Kraft P. et al. Genome-wide association study identifies novel alleles associated with risk of cutaneous basal cell carcinoma and squamous cell carcinoma . Hum Mol Genet . 2011 ; 20 : 3718 .
7. Teh M.T. , Blaydon D. , Chaplin T. et al. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event . Cancer Res . 2005 ; 65 : 8597 .
8. Mill P. , Mo R. , Fu H. et al. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development . Genes Dev . 2003 ; 17 : 282 .
9. Kasper M. , Jaks V. , Are A. et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes . Proc Natl Acad Sci U S A . 2011 ; 108 : 4099 .
10. So P.L. , Lee K. , Hebert J. et al. Topical tazarotene chemoprevention reduces Basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation . Cancer Res . 2004 ; 64 : 4385 .
11. Sekulic A. , Migden M.R. , Oro A.E. et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma . N Engl J Med . 2012 ; 366 : 2171 .
12. Tang J.Y. , kay-Wiggan J.M. , Aszterbaum M. et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome . N Engl J Med . 2012 ; 366 : 2180 .
13. Eklind J. , Tartler U. , Maschke J. et al. Imiquimod to treat different cancers of the epidermis . Dermatol Surg . 2003 ; 29 : 890 .
14. Kobielak A. , Fuchs E. Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin . Proc Natl Acad Sci U S A . 2006 ; 103 : 2322 .
15. Loercher A. , Lee T.L. , Ricker J.L. et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma . Cancer Res . 2004 ; 64 : 6511 .
16. Su F. , Viros A. , Milagre C. et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors . N Engl J Med . 2012 ; 366 : 207 .
17. Glick A.B. TGFbeta1, Back to the Future: Revisiting its Role as a Transforming Growth Factor . Cancer Biol Ther . 2004 ; 3 : 276 .
18. Demehri S. , Turkoz A. , Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment . Cancer Cell . 2009 ; 16 : 55 .
19. Wang N.J. , Sanborn Z. , Arnett K.L. et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma . Proc Natl Acad Sci U S A . 2011 ; 108 : 17761 .
20. Extance A. Alzheimer’s failure raises questions about disease-modifying strategies . Nat Rev Drug Discov . 2010 ; 9 : 749 .
21. Singh A. , Park H. , Kangsamaksin T. et al. Keratinocyte Stem Cells and the Targets for Nonmelanoma Skin Cancer(dagger) . Photochem Photobiol . 2012 .
22. Beck B. , Blanpain C. Mechanisms regulating epidermal stem cells . EMBO J . 2012 ; 31 : 2067 .
23. Snippert H.J. , Haegebarth A. , Kasper M. et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin . Science . 2010 ; 327 : 1385 .
24. Li S,Park H,Trempus CS et al: A keratin 15 containing stem cell population from the hair follicle contributes to squamous papilloma development in the mouse. 2013;10:751-759.
25. Lapouge G. , Youssef K.K. , Vokaer B. et al. Identifying the cellular origin of squamous skin tumors . Proc Natl Acad Sci U S A . 2011 ; 108 : 7431 .
26. White A.C. , Tran K. , Khuu J. et al. Defining the origins of Ras/p53-mediated squamous cell carcinoma . Proc Natl Acad Sci U S A . 2011 ; 108 : 7425 .
27. Patel G.K. , Yee C.L. , Terunuma A. et al. Identification and characterization of tumor-initiating cells in human primary cutaneous squamous cell carcinoma . J Invest Dermatol . 2012 ; 132 : 401 .
28. Kwong B.Y. , Roberts S.J. , Silberzahn T. et al. Molecular Analysis of Tumor-Promoting CD8(+) T Cells in Two-Stage Cutaneous Chemical Carcinogenesis . J Invest Dermatol . 2009 ; 130 : 1726 .
29. Roberts S.J. , Ng B.Y. , Filler R.B. et al. Characterizing tumor-promoting T cells in chemically induced cutaneous carcinogenesis . Proc Natl Acad Sci U S A . 2007 ; 104 : 6770 .
30. So P.L. , Fujimoto M.A. , Epstein Jr. E.H. Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis . Mol Cancer Ther . 2008 ; 7 : 1275 .
31. Li L. , Shukla S. , Lee A. et al. The skin cancer chemotherapeutic agent ingenol-3-angelate (PEP005) is a substrate for the epidermal multidrug transporter (ABCB1) and targets tumor vasculature . Cancer Res . 2010 ; 70 : 4509 .