Epithelial Neoplasms of the Large Intestine
Mark Redston
David K. Driman
Introduction
The most common neoplasms of the large intestine are adenomas (see Chapter 22). Adenomas are the precursor of most primary malignant epithelial neoplasms of the large intestine. Although abundant clinical, morphologic, and genetic evidence suggests that primary epithelial malignant neoplasms are a heterogeneous group of tumors, most clinicians consider these neoplasms together to be “colorectal carcinoma.”1,2 Much of the discussion in this chapter refers to colorectal carcinoma (CRC) as a generic, single disease, with the recognition that this is an oversimplification. In practice, approximately 85% of CRCs are typical adenocarcinomas; relatively distinct histologic subtypes form the remainder (Box 27.1). Recent advances in genetic testing for hereditary colorectal cancer syndromes and for sensitivity to targeted therapies also highlight the clinical relevance of emerging molecular classification systems.
Clinical Features
Early-stage CRC is typically diagnosed only at the time of screening or other, unrelated colonoscopy and does not usually manifest with symptoms, signs, or other laboratory findings. Advanced cancers are more likely to result in clinical symptoms, including a change in bowel habits, constipation, abdominal distention, hematochezia, or tenesmus (rectosigmoid lesions). It is important to appreciate that these “colorectal-type” symptoms and signs apply predominantly to left-sided cancers; right-sided cancers often manifest insidiously with nonspecific systemic symptoms and signs such as fatigue, weight loss, and anemia. Only approximately 40% of patients have localized disease at presentation. Approximately 40% have regional metastases, and approximately 20% have distant metastases.3 Endoscopy with biopsy is the standard diagnostic approach. Computed tomography and magnetic resonance imaging (MRI) are used to assess depth of invasion, regional spread, and distant metastases; rectal MRI is the gold standard to assess the extent of local spread in rectal cancers, with transrectal ultrasound an alternative for early lesions.
A variety of screening recommendations have been proposed and endorsed by the American Gastroenterological Association, American Medical Association, and American Cancer Society (Table 27.1). Standard guidelines are modified in individuals with a personal or family history of colorectal adenoma or carcinoma.4 Screening is clinically effective and cost-effective, yet its use remains relatively low.5 Screening colonoscopy provides the added benefit of polyp removal, and it is well established that polypectomy can prevent CRC.6 An estimated 90% of CRC deaths could potentially be prevented by implementing a combination of (1) strategies aimed at improving screening, polyp management, and early diagnosis of CRC; (2) lifestyle modifications, including dietary change and increased exercise; and (3) chemoprevention.3,7–9
Table 27.1
Guidelines for Early Detection of Adenomas and Colorectal Cancer in Average-Risk Individuals
| Method | Interval* |
| Tests That Detect Adenomatous Polyps and Cancer† | |
| Flexible sigmoidoscopy | Every 5 yr |
| Colonoscopy | Every 10 yr |
| Double-contrast barium enema | Every 5 yr |
| Computed tomography colonography | Every 5 yr |
| Tests That Primarily Detect Cancer | |
| Guaiac-based fecal occult blood test | Annual |
| Fecal immunochemical test | Annual |
| Stool DNA test | Interval uncertain |
* Beginning at age 50 yr.
† These tests should be encouraged if resources are available and the patient is willing to undergo an invasive procedure.
From Levin B, et al. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology. 2008;134:1570-1596.
Incidence
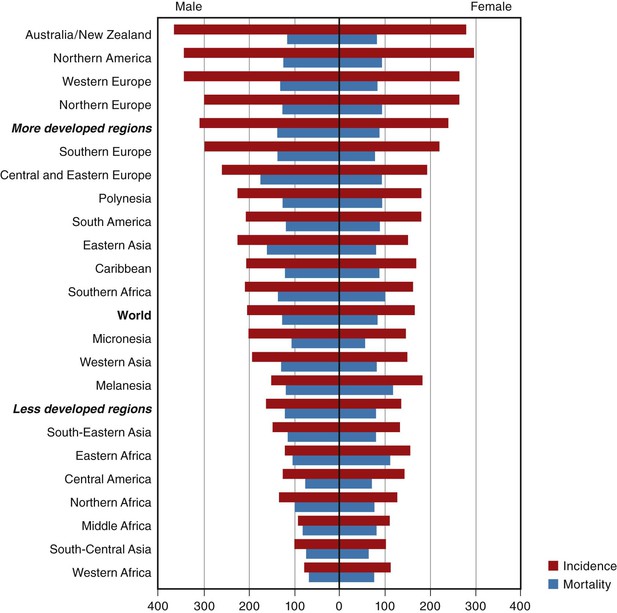
Malignant epithelial tumors of the colon and rectum are the fourth most common cancer in men worldwide (after lung, prostate, and stomach), and the third most common in women (after breast and uterine cervix), accounting for 9.4% of all cancers in 2008, with more than 1 million new cases diagnosed each year.3,10,11 There is marked variation in the age-standardized incidence, with a 25-fold difference between high-risk regions (affluent countries including Australia, New Zealand, Europe, the Americas, and Japan) and low-risk regions (developing countries including Africa, India, and other parts of southeast Asia) (Fig. 27.1).11 The likely role of environmental and lifestyle influences, particularly diet, alcohol intake, and physical activity, in the genesis of these differences is supported by abundant data. There are also significant global differences in the age at onset of CRC, with a mean age of only 50 years in developing countries.
In the United States, there were an estimated 103,170 new cases of colon cancer and 40,290 new cases of rectal cancer in 2012.10 CRC is the third most common cancer in both men and women and the fourth most common cancer overall, representing about 9% of all cancers.3 Overall, it is the second leading cause of cancer death, behind only lung cancer, and it is the leading cause of cancer death among nonsmokers.3 The lifetime risk for development of CRC is estimated at about 5%.3,12 U.S. Surveillance, Epidemiology, and End Results (SEER) Program statistics reveal an incidence of 30.45 per 100,000 for colon carcinoma and 12.16 per 100,000 for rectal carcinoma in 2009.4,13 CRC is significantly more common in men (combined age-adjusted incidence, 54.0 per 100,000, versus 40.2 per 100,000 in women); this difference is more striking for rectal cancer than for colonic cancer, 3,4 and the increased incidence in men is apparent only after the age of 50 years. Despite the higher incidence in men, women live longer, so there are a similar number of total cases and cancer deaths in men and women.3 Since 1987, the incidence of colon cancer in the United States has been slowly falling.14 The incidence increases with age, with approximately 9% of cases occurring before age 50 years and only approximately 1% before age 35 years.3,4
In addition to the global variation in CRC incidence, there are significant regional and ethnic differences in incidence within the United States. The incidence varies by approximately 1.5-fold between high-risk regions (predominantly the northeast Atlantic coast) and low-risk regions (predominantly the South and Midwest).3 Table 27.2 presents the differences related to racial and ethnic backgrounds. The incidence is highest among African Americans and lowest among individuals of Native American origin.3
Table 27.2
Incidence and Mortality Rates of Colorectal Cancer by Race and Ethnicity, United States, 2004-2008*
| Race/Ethnic Group | Incidence | Mortality | ||
| Male | Female | Male | Female | |
| White | 54.6 | 40.3 | 20.1 | 14.0 |
| African American | 66.9 | 49.7 | 30.5 | 20.4 |
| Asian American/Pacific Islander | 42.4 | 32.7 | 13.3 | 9.9 |
| American Indian/Alaska Native | 51.5 | 41.5 | 19.8 | 14.0 |
| Hispanic/Latino | 48.6 | 34.2 | 15.5 | 10.3 |

* Rates per 100,000, age-adjusted to the 2004 U.S. standard population.
From American Cancer Society. Cancer Facts and Figures 2012. Atlanta: American Cancer Society; 2012:44.
Epidemiology
The risk of CRC is influenced by both endogenous (constitutional) and exogenous (environmental) factors (Table 27.3).15 For the practicing surgical pathologist, genetic predisposition and long-standing inflammatory bowel disease (IBD) have the most direct clinical impact, and these topics are discussed later. Age, as discussed previously, is the most powerful risk factor. CRC is predominantly a disease of late middle-aged and elderly individuals.3 The increased risk in males is thought to be related to differences in hormonal milieu.16
Table 27.3
Risk Factors for Colorectal Cancer*
| Factor | Relative Risk |
| Family history (first-degree relative) | 1.8 |
| Physical inactivity (<3 hr/wk) | 1.7 |
| Inflammatory bowel disease (physician-diagnosed Crohn’s disease, ulcerative colitis, or pancolitis) | 1.5 |
| Obesity | 1.5 |
| Red meat | 1.5 |
| Smoking | 1.5 |
| Alcohol (>1 drink/day) | 1.4 |
| High vegetable consumption (≥5 servings/day) | 0.7 |
| Oral contraceptive use (≥5 yr) | 0.7 |
| Estrogen replacement (≥5 yr) | 0.8 |
| Multivitamins containing folic acid | 0.5 |
* Modifiable factors are in bold text.
From American Cancer Society. Cancer Facts and Figures 2002. Atlanta: American Cancer Society; 2002:20. [Data from Colditz et al., 2000.15]
Remaining risk factors are largely related to lifestyle and, importantly, are modifiable, suggesting the potential for interventions aimed at significantly reducing the incidence of CRC.17 The five most convincingly implicated lifestyle factors are obesity, physical activity, and ingestion of red meat, processed meat, and alcohol.18 Of these modifiable risk factors, diet has been the most extensively studied, and although there is little doubt that elevated risk is consistently associated with a “Western” type of diet, it has been difficult to determine which components are most important. Diets with a high calorie intake and those rich in meat, particularly animal fat, have been implicated in many studies.19–21 Possible mechanisms for this effect include the production of heterocyclic amines, stimulation of higher levels of fecal bile acids, production of reactive oxygen species, and elevated insulin levels.22,23 In addition to high-risk factors, there are inverse associations with vegetable and fiber consumption, although fiber falls out of some multivariate analyses when adjusted for other dietary risk factors.24–26 This effect could be related to anticarcinogens, antioxidants, folate, induction of detoxifying enzymes, binding of luminal carcinogens, fiber fermentation to produce volatile fatty acids, or reduced contact time with epithelium because of faster transit.24,25 Several studies, including a large pooled multivariate analysis, have found that high folate intake is associated with a decreased risk of CRC, providing some of the most direct evidence of dietary risk factor relationships.27,28 Finally, alcohol intake has been associated with an increased risk of CRC.29
There is an inverse association between use of nonsteroidal antiinflammatory drugs and CRC risk.7 Smoking exposure is associated with CRC, although the relative risk is less than for many other tobacco-related cancers,30 and some studies consider cigarette smoking only a suggestive risk factor.18 Sedentary lifestyle,31 long-standing IBD32 (see Colitis-Associated Neoplasia), pelvic irradiation,33 and ureterosigmoidostomy are also associated with an increased risk of CRC.
Finally, there is evidence that there are important differences in the epidemiologic risk factors associated with different subtypes of CRC. There has been a trend in recent years toward the development of more proximal cancers,13 which may relate to changes in epidemiologic risk factors. In fact, most of the lifestyle factors are associated specifically with increased risks of colon cancer, and only age and gender are predictive of an increased risk of rectal cancer.34 Furthermore, there are molecular biologic differences between right- and left-sided CRCs that would support different epidemiologic associations.35 More recently, research has focused on the complex interactions of hormones, energy balance, intestinal flora, and inflammation.21
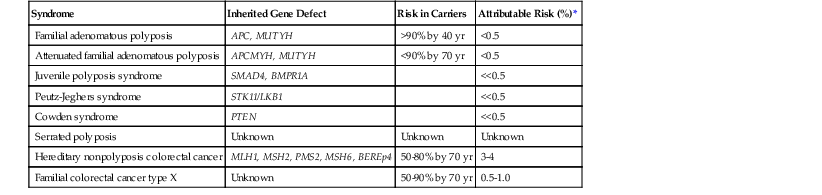
Genetic polyposis syndromes account for fewer than 0.5% of all incident CRCs (Table 27.4). Nonpolyposis forms of hereditary colorectal carcinoma have a much higher overall contribution to the causation of CRC and are discussed in detail later in this chapter.
Table 27.4
Classification of Genetic Syndromes That Predispose to Colorectal Cancer
| Syndrome | Inherited Gene Defect | Risk in Carriers | Attributable Risk (%)* |
| Familial adenomatous polyposis | APC, MUTYH | >90% by 40 yr | <0.5 |
| Attenuated familial adenomatous polyposis | APCMYH, MUTYH | <90% by 70 yr | <0.5 |
| Juvenile polyposis syndrome | SMAD4, BMPR1A | <<0.5 | |
| Peutz-Jeghers syndrome | STK11/LKB1 | <<0.5 | |
| Cowden syndrome | PTEN | <<0.5 | |
| Serrated polyposis | Unknown | Unknown | Unknown |
| Hereditary nonpolyposis colorectal cancer | MLH1, MSH2, PMS2, MSH6, BEREp4 | 50-80% by 70 yr | 3-4 |
| Familial colorectal cancer type X | Unknown | 50-90% by 70 yr | 0.5-1.0 |
* Proportion of colorectal cancer attributed to this syndrome.
Pathogenesis
Progression from Adenoma to Carcinoma
Most, if not all CRCs arise from adenomas, either conventional adenomas, sessile serrated adenomas/polyps (SSA/Ps), or traditional serrated adenomas (in decreasing order of frequency).36,37 Residual adenoma is identified in approximately 10% to 30% of CRCs; in the remainder, the adenomas are presumably overgrown by cancer.38 There are distinct associations between the histologic type of precursor lesion and the subsequent pathogenesis of the derived CRC. These indicate that there are two broad pathways involved in neoplastic progression in the colorectum: the conventional adenoma pathway and the serrated adenoma/polyp pathway.
The conventional adenoma pathway accounts for approximately 70% to 80% of all CRCs and is much more prevalent in the left colon and rectum than in the right colon. Conventional adenomas typically precede cancer by approximately 15 years.39 The prevalence of conventional adenomas in the U.S. population is approximately 25% by age 50 years, and 50% by age 70 years, and these adenomas have a high lifetime risk of progression if not removed.40 Exact risks of progression are not known; however, one study estimated a 10% to 15% chance of progression during 10 years for a 1-cm conventional adenoma.41 Endoscopic removal of conventional adenomas decreases the incidence of subsequent CRC in treated patients.6,42
The serrated pathway has been increasingly recognized in the past 10 years, and it is estimated to account for approximately 20% to 30% of all CRCs.43–47 Most CRCs arising in the serrated pathway develop from SSA/Ps, particularly those located in the right colon. CRC is typically preceded by the development of foci of true dysplasia within these polyps.45,48,49 Historically, conventional endoscopic screening programs have been less effective at reducing right-sided CRC, in large part because the risk of progression of serrated polyps, almost all of which were previously diagnosed as hyperplastic polyps, was not recognized.50 The exact risk of progression of these polyps is yet to be fully characterized, although the malignant potential appears sufficient to warrant complete endoscopic removal.37 Challenges remain for endoscopic surveillance programs, because serrated polyps are difficult to recognize and completely remove endoscopically,51 and because the underlying genetic instabilities that drive these lesions have the propensity to result in CRCs in relatively small polyps (see later discussion). Traditional serrated adenomas may also precede CRC; however, these lesions are much less common, and the risk of progression appears to be lower. Conventional hyperplastic polyps, particularly those of the left colon, rarely, if ever, progress to cancer.
Aberrant crypt foci represent the earliest stages of colorectal neoplasms and are present before the development of grossly apparent adenomatous polyps.39 Aberrant crypt foci are microscopic lesions most readily identified by examination of methylene blue–stained, stripped mucosal sheets under a dissecting microscope. They are characterized by a localized collection of crypts that show an increase in crypt diameter and an increased number of lining epithelial cells, which imparts a serrated or slitlike appearance. Histologic sections of aberrant crypt foci reveal a range of findings, such as normal or only mildly hyperplastic epithelium, features more typical of serrated polyps, or, rarely, true dysplasia (the latter being similar to microscopic adenomas incidentally identified in patients with familial adenomatous polyposis).52 Aberrant crypt foci may also be visualized endoscopically, although the technical challenges of these methods have prohibited routine clinical applications.53
Reports of very small (<1 cm) carcinomas that lack any evidence of residual adenoma have raised the possibility that some cancers may arise de novo.54 These cancers represent fewer than 5% of all CRCs. Some studies suggest that they are more likely to be of higher grade than ex-adenoma carcinomas, with a higher risk of lymphatic and blood vessel invasion.54 However, other studies do not support that hypothesis. Hornick and colleagues report that small carcinomas without a dysplastic component shared clinical and molecular characteristics with small carcinomas containing a minimal dysplastic component and with conventional larger carcinomas.55 It is also possible that some of these lesions represent rapid progression from a small adenoma to cancer because of the early acquisition of high-grade genetic alterations (e.g., aneuploidy, TP53 mutations).54
Genetic Model of Colorectal Cancer Progression
Human cancers are characterized by an accumulation of a variety of genetic alterations, including mutations that either activate oncogenes or inactivate tumor suppressor genes.56–58 This accumulation of genetic alterations is a critical event in the progression from adenoma to carcinoma and likely begins in aberrant crypt foci and other precursor cells that may not manifest morphologic features of neoplasms.58–61 To accumulate the array of genetic alterations typical of most CRCs, tumor cells must acquire mutations and epigenetic alterations at an increased rate compared with normal crypt epithelial cells.62 Increased acquisition and tolerance of mutations is a hallmark of CRC development, and is referred to as genome instability.63,64 Genes involved in the maintenance of the genome have been likened to “caretakers” of the genome.65 There are at least two general forms of genome instability important to the development of colorectal neoplasia: chromosomal instability (CIN) and microsatellite instability (MSI).66 The third major driving pathway for the development of CRC is the widespread accumulation of epigenetic gene silencing (CpG island methylator phenotype, or CIMP).43,66,67
Chromosomal Instability.
CIN is characterized by a persistently increased rate of gains and losses of chromosomal material; it is present in 70% to 80% of CRCs.68 The acquisition of abnormalities in the number of whole chromosomes results in aneuploidy. In addition to whole-chromosome abnormalities, CRCs have other forms of somatic copy number abnormalities, including abnormalities of whole chromosomal arms as well as focal gains and losses. The underlying genetic basis of aneuploidy in human cancers is poorly understood, although most studies have focused on genes involved in regulation of mitotic spindle assembly and segregation. CIN is a major underlying genetic aberration in the conventional adenoma–carcinoma progression pathway and is therefore the predominant form of instability in left-sided neoplasms.66,69
Microsatellite Instability.
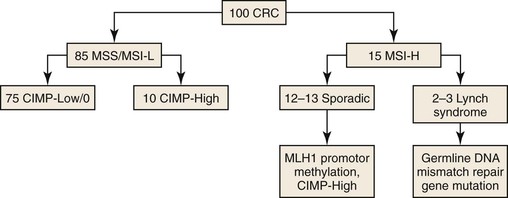
MSI is characterized by widespread alterations in the sizes of repetitive DNA sequences. It is present in approximately 10% to 15% of CRCs.70 MSI is caused by defective DNA mismatch repair (MMR) (Fig. 27.2) (see Hereditary Nonpolyposis Colorectal Cancer). In addition to alterations in the sizes of repetitive DNA sequences, MSI results in markedly increased rates of mutation of coding sequences (somatic hypermutation). In general, CRCs with MSI do not harbor abnormalities in chromosomal number or the focal regions of subchromosomal gains or losses that typify cancers with CIN. In most CRCs with MSI, the underlying defect in MMR function is caused by epigenetic CpG island hypermethylation of the promoter region of the MLH1 gene. This is a characteristic feature of many CRCs arising in the serrated neoplastic pathway, and most of these cancers are high-frequency CIMP (discussed later).44,71 MSI is also the mechanism that underlies the progression of Lynch syndrome cancers, which are caused by inherited defects in DNA MMR (see later discussion). In Lynch syndrome, MSI develops in conventional adenomas and drives rapid progression to cancer.
CpG Island Methylator Phenotype.
CpG island methylator phenotype (CIMP) is the acquisition of widespread hypermethylation of CpG dinucleotides in the promoter regions of genes.45,72,73 Referred to as an epigenetic alteration (because it does not change the DNA sequence), this is a major mechanism of inactivation of tumor suppressor genes such as TP16, CDHI, and MLH1. Widespread CpG island methylation in a single cancer stands in stark contrast to the very limited methylation silencing that occurs in most CRCs and is known as high-frequency CIMP (CIMP-H).67,70,74 CIMP-H is a characteristic feature of CRCs that arise in the serrated pathway, and it is present in 20% to 30% of CRCs, including almost all cancers that also have MLH1 hypermethylation silencing. The underlying genetic basis of the CIMP-H phenotype is poorly understood, but there is evidence that genetic factors and environmental exposures (e.g., smoking, estrogen withdrawal) may be associated with the development of serrated pathway carcinomas.45 A working hypothesis is evolving wherein genetic and epidemiologic factors contribute to abnormal methylation events in serrated polyps of the right colon, which predisposes them to methylation silencing of MLH1, MGMT, and other important genes. Presumably, these events drive progression from dysplasia to adenocarcinoma43,75–77 (Table 27.5). The subsequent carcinomas that develop are referred to as “serrated” adenocarcinomas by some authorities (see later discussion), and they are often found to be MSI-H or CIMP-H, or both, on molecular phenotyping.78,79
Table 27.5
Molecular Pathologic Classification of Colorectal Cancer
| Proportion (%) | Precursor | CIMP Status | Mismatch Repair Status | Microsatellite Instability Status | MLH1 Gene Status | Chromosomal Status |
| 75-80 | Adenoma | CIMP-L/0 | Proficient MMR | MSS/MSI-L | No methylation | Unstable (aneuploid) |
| 40 | Adenoma | CIMP-0 | Proficient MMR | MSS | No methylation | Unstable (aneuploid) |
| 30-35 | Adenoma | CIMP-L | Proficient MMR | MSS | No methylation | Unstable (aneuploid) |
| 5 | Adenoma | CIMP-L | Proficient MMR | MSI-L | No methylation | Unstable (aneuploid) |
| 10 | Serrated adenoma/polyp | CIMP-H | Deficient MMR (sporadic) | MSI-H | Methylated | Stable (diploid) |
| 2-3 | Serrated adenoma/polyp | CIMP-L/0 | Deficient MMR (sporadic) | MSI-H | Methylated | Stable (diploid) |
| 5-10 | Serrated adenoma/polyp | CIMP-H | Proficient MMR | MSS | No methylation | Stable (diploid) |
| 2-3 | Adenoma | CIMP-0 | Deficient MMR (Lynch syndrome) | MSI-H | No methylation | Stable (diploid) |
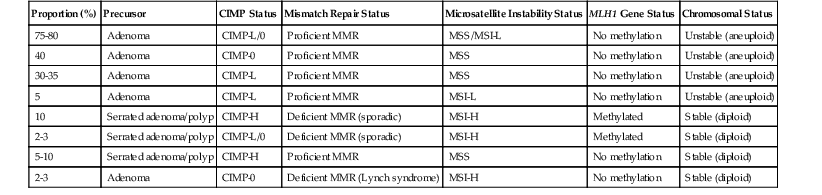
CIMP, CpG island methylator phenotype; -H, high frequency; -L, low frequency; MMR, mismatch repair; MSI, microsatellite instability; MSS, microsatellite stable.
Implications of Progression Pathways for Screening and Prevention.
Screening colonoscopy is more effective for the prevention of left-sided than right-sided CRC, and this difference is thought to be caused by the predominance of the conventional adenoma–carcinoma progression pathway in the left colon compared with the serrated adenoma–carcinoma progression pathway in the right colon.37,50,80 The failure of screening to prevent right-sided colon cancer to the same degree as left-sided cancer appears to be related to the difficulties that exist in the endoscopic identification of SSA/Ps as well as their particular molecular pathogenetic characteristics. Serrated polyps are more difficult to completely excise, which could directly lead to colonoscopic screening failures.51,81,82 Serrated polyps are also at greater risk of undergoing rapid progression in a relatively small lesion, secondary to the acquisition of DNA MMR deficiency and MSI.37 Indeed, studies of interval cancers have found that they are much more likely to exhibit MSI and CIMP-H, both features of CRCs arising in the serrated pathway.83,84
Molecular Pathway Classification of Colorectal Cancer.
A molecular-pathologic classification of CRC has been proposed that is based, in large part, on the presence or absence of CIN (i.e., CIN− versus CIN+; diploid versus aneuploid), the MMR status (microsatellite stable [MSS] versus MSI), and the status of the methylation pathway (CIMP+ versus CIMP−)69,75,85 (see Table 27.5). Although the clinical relevance of the aneuploid and (MMR)-deficient pathways has been delineated (see Prognostic Factors), the possible clinicopathologic associations of the methylator pathway have not been as well defined.
WNT Signaling Abnormalities.
APC was first identified as the gene mutated in individuals with familial adenomatous polyposis.86 In this syndrome, affected individuals inherit one mutant copy of APC that is functionally inactive. In tumorigenesis, the second allelic copy of the APC gene is also inactivated, which fulfills Knudsen’s paradigm for tumor suppressor gene inactivation. APC mutations are also present in 70% to 80% of sporadic CRCs, where mutations occur early during neoplastic development, and even in dysplastic aberrant crypt foci.58,86 The apparent necessity of APC inactivation for the development of early adenomas has resulted in its designation as a “gatekeeper” gene of colorectal neoplasia.65
APC is an important component of the Wingless (WNT) pathway, an evolutionarily conserved signaling cascade that is critical to embryonic development and intestinal epithelial renewal.86,87 APC mutations abrogate its role in binding β-catenin, thereby releasing β-catenin from phosphorylation regulation by glycogen synthase kinase-3β GSK3β and allowing it to accumulate in the nucleus, where it is involved in activating transcription of a number of other downstream targets, such as cyclin D and Myc.86 Recent microarray studies suggest that the transcriptional profile of β-catenin activation resembles the program of intestinal crypt stem cells. APC is also involved in cytoskeletal interactions, and, although the most important potential role in tumorigenesis has not been precisely defined, APC has been directly implicated in cell–cell adhesion, migration, chromosomal segregation (genome stability), and apoptosis.58,86
Further evidence of the importance of WNT signaling abnormalities in colorectal tumorigenesis is evidenced by the presence of mutations in the β-catenin gene (CTNNB1) in some tumors that lack APC mutations.58 In contrast to the inactivating APC mutations, the CTNNB1 mutations target amino acid residues integral to phosphorylation, resulting in oncogenic activation of WNT signaling.58 Finally, mutations have been rarely identified in other WNT signaling members in CRCs with wild-type APC, including AXIN1, AXIN2, and TCF4.58
Other Genetic Alterations.
Activating mutations in KRAS are present in approximately 40% of CRCs and were one of the first genetic alterations described in any type of solid human tumor.59 KRAS mutations typically occur early, in aberrant crypt foci or small adenomas, and result in constitutive activation of the gene. KRAS mutations are a major determinant of response to anti-EGFR therapy (see Targeted Therapy). Mutation and inactivation of the TP53 gene occurs in approximately 50% to 70% of carcinomas, often at the point of development of high-grade dysplasia.88 Among the most widely studied of human tumor suppressor genes, TP53 is important in the DNA damage response and the cell cycle arrest pathway.
Molecular Profiles of Colorectal Cancer.
Technologic advancements in the past 5 years have allowed major strides in the comprehensive analysis of human cancers. Although many studies focus on the results of one profiling technology, the potential advances of these approaches are most impressive when multiple methodologies are applied to individual tumors. The Cancer Genome Atlas has integrated multiple platforms, including exome sequencing, DNA copy number, promoter methylation, and messenger RNA (mRNA) and microRNA expression, as well as subset whole-genome sequencing, to develop comprehensive profiles of CRC.89 These profiles have provided remarkable insights into the molecular aberrations present in CRC, ranging from the grouping of cancers into distinct genome instability classes to the identification of 24 genes that are mutated at significant frequencies (including APC, TP53, SMAD4, PIK3CA, KRAS, ARID1A, SOX9, and FAM123B). In addition, potentially drug-targetable amplifications of ERBB2 and IGF2 have been found to be present in small subsets of cases, raising the possibility of novel therapeutic approaches personalized to these patients. These and other studies suggest that significant advances in biomarkers and personalized therapies may be achievable in the next 5 to 10 years.90,91
Gross Features and Specimen Handling
The principles for handling, evaluating, and processing CRC resection specimens for pathologic examination have been well described and are part of the College of American Pathologists’ protocol for examination of these specimens.92 The fundamental principles of gross examination are similar for both colonic and rectal resection specimens, but key differences should be observed when dealing with rectal specimens, particularly from total mesorectal excision (TME).
With large resection specimens that include multiple subsegments of the colon or rectum, lymph nodes should be designated as either regional or nonregional, definitions of which are available in the American Joint Committee on Cancer’s AJCC Cancer Staging Manual (seventh edition).2 Metastases to nonregional lymph nodes are considered to be stage M1.2
For carcinomas of the colon, the distance of tumor from the proximal and distal margins should be measured in the fresh state if possible. All colonic cancer resections have a radial resection margin; in areas of the colon not completely invested by peritoneum (i.e., some cecal tumors, all ascending and descending colon tumors), the radial resection margin is the posterior bare area, which should be inked and sampled for histologic examination. In other segments of the colon (e.g., transverse and sigmoid colon), the radial margin is represented by the vascular ties on the so-called “mesocolic” or “mesenteric” margin. In addition to radial margins, it is imperative to assess whether there is tumor extending through a serosal surface. Suspicious areas are areas of serosa that are roughened, granular, or hypervascular; such areas should be thoroughly sampled for histologic examination. All lymph nodes must be found; this is best achieved by removing the fat close to the bowel wall and then using inspection and palpation to identify nodes. It is important to examine the fat that remains adhered to the bowel wall, because this is often a location for small, potentially positive nodes (see Special Studies for a discussion of methods to enhance lymph node dissection.)
Distinguishing colonic from rectal cancers can be problematic; tumors in the nonperitonealized portion of the left colon are considered rectal in origin, and all tumors located within 15 cm of the anal verge are also considered rectal. Tumors more proximally located for which the exact location is unclear are designated “rectosigmoid.” The point at which the peritoneum no longer surrounds the bowel completely is considered the true rectosigmoid junction; the teniae coli terminate at the junction of the rectum.
For carcinomas of the rectum when a mesorectal excision procedure has been performed (the standard of practice for all rectal cancers) (Fig. 27.3), the quality and completeness of the mesorectal excision should be assessed before inking and sectioning3 and graded as either complete, nearly complete, or incomplete (Table 27.6). This procedure is best done in the fresh state but can be done in fixed specimens. The mesorectum is the fatty soft tissue envelope containing lymph nodes, blood vessels, and nerves that surrounds the rectum and is enveloped by fascia. The quality of the mesorectal excision procedure and the distance of the tumor from the radial margin are related to the local recurrence and overall prognosis in rectal cancer patients,94–98 and the procedure for gross examination of these specimens is aimed at optimizing these assessments. To assess the quality of the mesorectal surgery, four parameters are evaluated: the bulk of the mesorectum, the presence and depth of any defects in the mesorectum, the presence or absence of coning, and the appearance of the nonperitonealized margin. A complete TME is defined by the presence of an intact mesorectum with only minor irregularities of an otherwise smooth surface, no defects deeper than 5 mm, no coning toward the distal margin, and a smooth circumferential margin on transverse slicing. A nearly complete TME has moderate bulk to the mesorectum, some irregularity to the surface, and moderate coning, with some defects greater than 5 mm but no visible muscularis propria except at the insertion of the levator muscles. An incomplete TME has only a little bulk to the mesorectum, defects that extend to the muscularis propria, a greater degree of coning, and an irregular circumferential margin. The nonperitonealized margin is assessed after slicing of the specimen, but other parameters are assessed on the unopened specimen.
Table 27.6
Parameters Used to Assess the Quality of Mesorectal Excision Surgery for Rectal Carcinoma
| Grade | Mesorectal Surface and Bulk | Defects in Mesorectum | Coning | Radial Margin |
| Complete | Good bulk, smooth surface | No deeper than 5 mm | None | Smooth |
| Nearly complete | Moderate bulk, irregular surface | Deeper than 5 mm but no visible muscularis propria (except where levator muscles insert) | Moderate | Moderately irregular |
| Incomplete | Little bulk, irregular surface | Down to muscularis propria | Moderate to marked | Irregular |

The nonperitonealized (radial) margins lie beneath the peritoneal reflections (low on the anterior aspect but high on the posterior aspect), and it is good practice to report the distance of a rectal tumor to the peritoneal reflections. Some rectal tumors also have a serosal surface that requires careful assessment, as in colonic tumors. Tumors in the upper (proximal) rectum have a serosal covering anteriorly and a radial margin posteriorly, whereas middle to low (distal) rectal tumors have a circumferential radial resection margin.
These specimens should not be opened through the tumor before fixation; rather, they should be left intact so that the specimen can be transversely sectioned when thoroughly fixed. This is done to optimize the assessment of the distance of the tumor to the radial margin. The distance of tumor from the proximal and distal margins should be assessed in the fresh state if possible, and the specimen should be opened along the anterior aspect from top and bottom, leaving the bowel intact at a level just above and just below the tumor. A loose gauze wick, soaked in formalin, is then placed into the unopened ends of the bowel. All rectal cancer specimens should be fixed for at least 48 hours (preferably 72 to 96 hours) in an adequate volume of clean formalin. Once fixed, the unopened bowel is sliced at 3- to 5-mm intervals, and the slices are laid down on the work surface and inspected for (1) appearance of the circumferential radial margin (i.e., smooth, regular, or irregular); (2) extent of tumor; (3) closest distance of tumor to the circumferential radial margin; and (4) any obviously positive nodes and their distance from the circumferential radial margin (Fig. 27.4). Fat away from the tumor is also examined for lymph nodes, taking care not to double-count nodes that are present in multiple slices. There are usually fewer lymph nodes in patients who have received neoadjuvant therapy. In one large study, patients who had undergone neoadjuvant therapy had a mean of 5 fewer nodes, and 63% of these patients had fewer than 12 nodes retrieved; the smaller number of nodes retrieved was not associated with poorer disease-specific survival.99
The macroscopic appearance of CRCs can be categorized into four general types (Fig. 27.5):
There is much overlap among these four patterns of growth, and there is no evidence that gross configuration is a relevant prognostic indicator independent of the histologic subtype of the tumor.100 It is usually not possible to assess accurately the gross appearance of a rectal tumor, given that the specimens are sliced transversely. The importance of properly assessing the radial margin vastly outweighs the importance of determining the macroscopic type of rectal carcinoma present. In all of these tumor types, the cut surface of the tumor is usually relatively homogeneous in appearance but may show areas of necrosis. There may be dilation of the bowel proximal to the tumor (secondary to obstruction) and alteration of the serosal surface if the tumor extends close to the serosa.
Pathologic Features
Criteria for Malignancy and Biopsy Diagnosis
Tumors in which there is invasion confined to the lamina propria and muscularis mucosae (so-called “intramucosal adenocarcinoma”) are almost never associated with lymph node metastases.2 This fact is usually attributed to the relative paucity of lymphatics in the colorectal mucosa. Therefore, colorectal tumors are considered to be malignant only if they have invaded through the muscularis mucosae into the submucosa (stage T1).2 Although this may make practical sense for management of CRC, it has created some controversy about the appropriate nomenclature for neoplasms that do not invade into the submucosa. In the AJCC classification,2 invasion confined to the mucosa (i.e., lamina propria invasion without submucosal invasion, or intramucosal adenocarcinoma) is still classified as stage Tis because of the retention of this arcane term in certain cancer registries. Unfortunately, with this approach, high-grade dysplasia (intraepithelial neoplasia) is also classified as Tis. At a practical, diagnostic level, because of the lack of malignant potential of lamina propria invasion, use of the diagnostic term “intramucosal carcinoma” is not recommended, and “carcinoma” should be used only in the context of “invasive adenocarcinoma,” which refers to tumors that have infiltrated into or beyond the submucosa.
Biopsy specimens of sessile or flat lesions are usually superficial, are often poorly oriented, and may be ulcerated. The most important aspect of pathologic examination is to determine whether invasion is present. Although the presence of single infiltrating cells or small, markedly irregular glands readily establishes a diagnosis of invasion, some better-differentiated cancers may not show this pattern of growth. In the absence of definitive pathologic features of invasion, a diagnosis of invasion relies on identification of desmoplasia, which is usually present with invasive adenocarcinoma. Differentiation between malignancy-associated desmoplasia and the stromal reaction in an ulcerated adenoma can be difficult. The stroma of an ulcerated adenoma is often more cellular, with more abundant microvasculature, inflammatory cells, and plump fibroblasts. If the muscularis mucosae can be identified, it is helpful to note whether invasion penetrates this layer, but in many biopsy fragments, the muscularis mucosae is difficult to identify clearly. In these cases, the presence of desmoplasia is a helpful indicator that the tumor has penetrated into the submucosa, in which case the carcinoma is at least T1. Desmoplasia is rarely, if ever, associated with lamina propria invasion alone. For those cancers that are within a polyp, the requirement for surgical resection as opposed to polypectomy is determined on the basis of completeness of excision and the presence or absence of high-risk factors for lymph node metastases (see Chapter 22).
Classification of Histologic Subtypes
Most CRCs are adenocarcinomas; by definition, they are considered to be of “usual” morphology (Table 27.7). Histologic complexity may be related to the presence of small components of other histologic patterns in an otherwise typical adenocarcinoma. The criteria used to define the various subtypes of colon cancer, such as “mucinous” adenocarcinoma, are described in the following sections. For colon cancer cases in which a second element of differentiation is present but the pattern is insufficient to meet the specific diagnostic criteria, it is recommended that the presence and percentage of all histologic components be commented on in the pathology report (e.g., “Moderately differentiated adenocarcinoma, with 10% extracellular mucinous component”).
Table 27.7
Histologic Classification of Colorectal Carcinoma
| Histologic Type | Approximate Frequency (%) |
| Recognized World Health Organization Type* | |
| Adenocarcinoma | 75-80 |
| Mucinous (colloid) adenocarcinoma | 8-10 |
| Serrated adenocarcinoma | 10 |
| Signet ring cell carcinoma | 2 |
| Medullary carcinoma | 1.0 |
| Adenosquamous carcinoma | <1.0 |
| Squamous cell carcinoma | <1.0 |
| Small cell (neuroendocrine) carcinoma | <1.0 |
| Undifferentiated carcinoma | <1.0 |
| Mixed carcinoid-adenocarcinoma | <1.0 |
| Other | |
| Choriocarcinoma | <0.1 |
| Clear cell carcinoma | <0.1 |
| Microglandular goblet cell carcinoma | <0.1 |
| Carcinomas with melanin production | <0.1 |
| Spindle cell carcinoma | <0.1 |
* See Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. Vol 3. In: Bosman FT, Jaffe ES, Lakhani SR, Ohgaki H, eds. World Health Organization Classification of Tumours. 4th ed. Lyon, France: IARC; 2009.
Adenocarcinoma, Usual Type
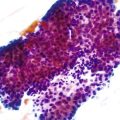
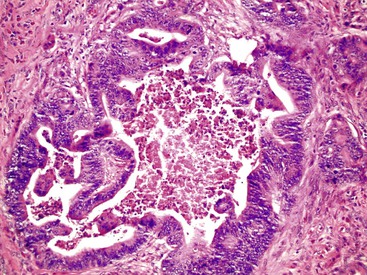
Most adenocarcinomas are moderately to well differentiated or low grade. Typically, there are medium-sized to large glands, with moderate variability in gland size and configuration and a moderate amount of stroma (Fig. 27.6). In well-differentiated tumors, the epithelial cells are usually tall and columnar and become increasingly cuboidal, or polygonal, with decreasing degrees of differentiation. Mitotic figures are usually abundant. Glandular lumina are usually filled with inspissated eosinophilic mucus as well as nuclear and cellular debris, so-called dirty necrosis (Fig. 27.7). When dirty necrosis is present in a metastasis of unknown primary origin, this feature is sometimes used to infer a colorectal primary. In general, there tends to be little difference between the superficial and deeper portions of the tumor, although the leading edge is often associated with gland rupture and more frequent foci of small, irregular, and often infiltrative-appearing glands. Some tumors have a prominent papillary component, particularly at the surface. Desmoplasia can be prominent, as in cancers of the pancreas and biliary tract. In some instances, the appearance of desmoplasia is caused, in part, by extensive tumor necrosis and hyalinization. In addition to glandular cells, a variable number of Paneth cells, neuroendocrine cells, squamous cells, melanocytes, and trophoblasts can be found in usual adenocarcinoma.101 Typically, the presence of these other cell types has no prognostic significance.
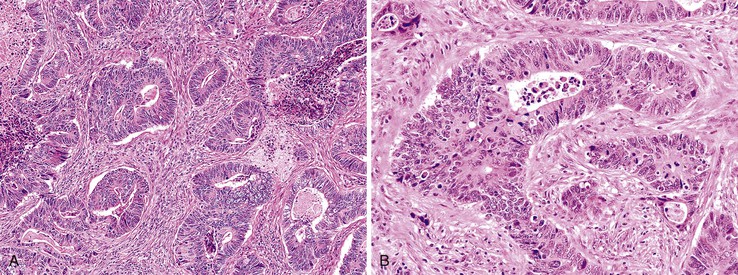
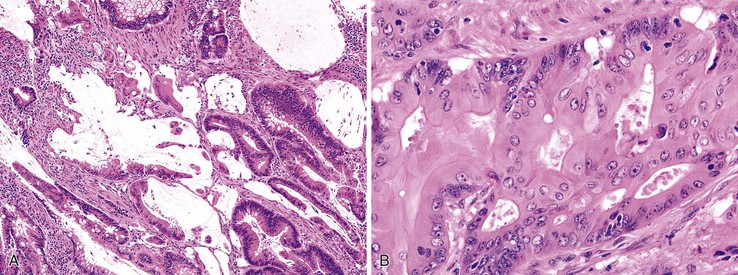
Mucinous Adenocarcinoma
The World Health Organization (WHO) defines mucinous tumors by the amount of extracellular mucin (i.e., tumor composed of >50% mucin). The terms adenocarcinoma with mucinous features or adenocarcinoma with mucinous differentiation are often used to describe tumors that have a significant mucinous component (>10% but <50%). Most mucinous adenocarcinomas have free-floating strips of neoplastic epithelium or individual tumor cells in the mucin (Fig. 27.8). A variable number of signet ring cells also may be seen (Fig. 27.9). If more than 50% of the tumor is composed of signet ring cells, the tumor is best classified as “signet ring cell carcinoma” (or adenocarcinoma, signet ring cell type), even if more than 50% of the tumor is composed of extracellular mucin.
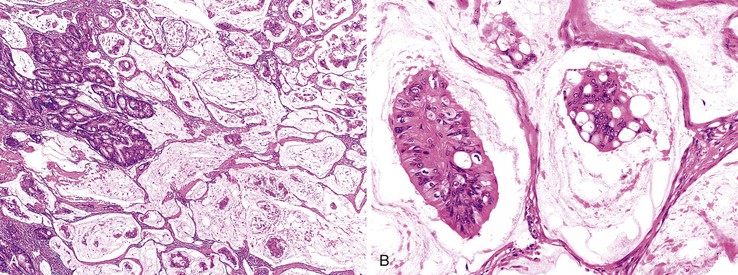
Mucinous adenocarcinomas represent approximately 10% of all colonic cancers.102 They are more common in young individuals and in patients with Lynch syndrome, and they are more likely to be at an advanced stage at presentation. The type of genetic alterations in these tumors suggests that the molecular pathogenesis is different from that of usual adenocarcinomas. Defects in DNA MMR and MSI-H are more common in mucinous adenocarcinomas, but a mucinous phenotype shows a low degree of sensitivity as a marker for this genotype.103 MSI-H mucinous tumors are found more often in younger individuals and are more likely to be exophytic and to have an expanding growth pattern, compared with non–MSI-H mucinous cancers.104 Immunohistochemical staining for TP53 is less frequently positive (30% versus >50% in the usual type of adenocarcinoma), which suggests that mucinous carcinomas are less likely to have a stabilizing point mutation in TP53. Expression of HATH1, a transcription factor that activates MUC2 expression in intestinal epithelium, is maintained in both mucinous and signet ring cell carcinomas but is repressed in nonmucinous cancers, which indicates a possible biologic basis for mucinous neoplasms.105
Mucinous tumors represent a greater proportion of right colon tumors and are more common in females.106 The cut surface is typically soft and gelatinous with little fibrous tissue, which imparts a “colloid” appearance to the tumor. The tumor is often composed of small nodules of mucin.
On microscopic examination, mucinous adenocarcinomas contain glandular structures or individual tumor cells embedded in pools of mucin; the mucin can be highlighted if necessary with periodic acid–Schiff or Alcian blue stains. The margins of the tumor are often dissecting and infiltrative, which probably contributes to the overall poor prognosis of patients with these tumors. In one series of 132 mucinous carcinomas, adjacent precursor adenomas were identified in 31%, a figure similar to that found in usual adenocarcinomas.102
The association between mucinous subtype and survival is controversial. Whereas many studies have shown that these tumors are associated with a relatively poor prognosis, others have not.107–109 Some studies that have examined the behavior of mucinous tumors failed to differentiate mucinous from signet ring cell carcinomas, the latter being more aggressive. A recent meta-analysis showed that mucinous carcinomas do not manifest at a higher stage than usual adenocarcinomas, but there was a 2% to 8% increased hazard of death.106 Compared with usual adenocarcinomas, mucinous tumors are more likely to develop peritoneal implants110 and to invade adjacent viscera101 and less likely to be cured by surgical resection.110 They are also more likely to show lymph node involvement beyond the pericolonic region.101 Of likely greater importance is whether the mucinous tumor is MSI-H or MSI-L/MSS; MSI-H tumors are more likely to behave in a low-grade manner, whereas MSI-L/MSS tumors are more apt to be high-grade in behavior.111
Signet Ring Cell Adenocarcinoma
Signet ring cell adenocarcinoma is defined as a tumor that is composed of at least 50% signet ring cells. For classification purposes, this feature supersedes the presence and amount of extracellular mucin. These tumors represent approximately 0.5% to 1.0% of all CRCs.112–114 They are slightly more common in men (male-to-female ratio, 1.3 : 1) and occur at a younger age (mean, 63.5 years).113 In some studies, more than 50% of signet ring cell adenocarcinomas were detected in individuals younger than 40 years of age.115 Signet ring cell carcinoma is also more common in ulcerative colitis, with approximately one third of tumors occurring in patients with this form of IBD.116
Signet ring cell carcinomas constitute a greater proportion of right-sided colonic tumors.112,113 Synchronous tumors are found in 14% of patients.112 They are usually ulcerating, and approximately two thirds have an infiltrative gross appearance.112,113 A linitis plastica growth pattern occurs in up to 20% of cases.113
Histologically, tumor cells show a characteristic mucin vacuole that pushes the nucleus to the periphery of the cell (Fig. 27.10). A subset of signet ring cells are round and contain centrally located nuclei, without an apparent mucin vacuole. Compared with gastric signet ring cell carcinomas, those in the colorectum are more likely to be associated with abundant extracellular mucin and less commonly result in diffuse infiltration of the tissues. It can be difficult to differentiate colorectal signet ring cell carcinoma from metastatic gastric signet ring carcinoma on morphologic grounds, and immunohistochemistry is not helpful because tumors from both sites usually are negative for MUC1 and thyroid transcription factor 1 (TTF1) and positive for MUC2 and CDX2+.117 Metastatic lung tumors are typically MUC1/TTF1+ and MUC2/CDX2−.
Similar to mucinous carcinomas, signet ring cell carcinomas are more likely to be diagnosed at an advanced stage.118 Approximately 80% of patients have stage III or IV disease at the time of diagnosis.119 Full-thickness penetration of the muscularis propria and peritoneal seeding are more common than in usual adenocarcinomas (36% versus 12%).112–114 Some studies have reported distant metastases in as many as 60% of patients at the time of diagnosis.120,121 As a result, surgical resection is less likely to be curative.115 As with mucinous carcinomas, tumors that are MSI-L/MSS are more aggressive.122 Five-year survival rates of less than 10% are reported.113,114 Peritoneal carcinomatosis is present in virtually all patients who die of their disease; liver metastases are present in fewer than 50% of the cases.112
Undifferentiated Carcinoma
The term undifferentiated carcinoma is essentially restricted to cancers that contain evidence of epithelial differentiation but are without obvious gland formation, although most undifferentiated tumors probably represent extremely poorly differentiated adenocarcinoma. Some authors accept this designation for tumors with a small component (<5%) of gland formation.1
Undifferentiated cancers tend to be bulky and soft because of their high degree of cellularity and relative lack of desmoplasia, and there is often extensive necrosis. There are typically sheets of cells, cords, or trabecular structures, often with an infiltrative growth pattern (Fig. 27.11). The degree of pleomorphism is variable; some tumors show relatively uniform cytologic features, whereas others show marked nuclear variability.
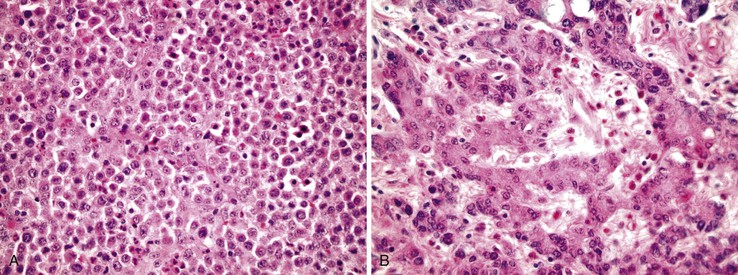
Although pure undifferentiated cancers are rare, many adenocarcinomas contain an undifferentiated component. These types of tumors are best classified as adenocarcinoma and graded on the basis of the overall percentage of glandular elements. Nevertheless, the presence of an undifferentiated component increases the probability that the tumor contains a DNA MMR deficiency, particularly if it is associated with prominent tumor-infiltrating lymphocytes (see Carcinomas with DNA Mismatch Repair Deficiency).
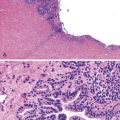
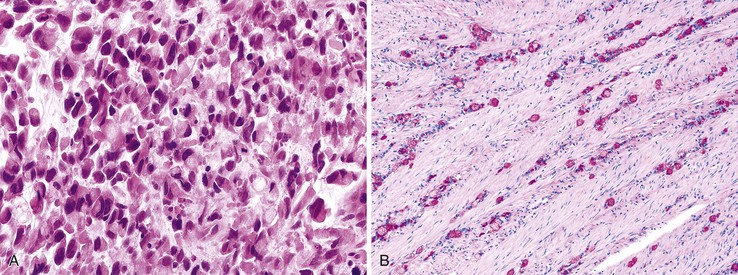
Medullary Carcinoma
Medullary carcinomas are characterized by sheets of polygonal cells with vesicular nuclei, prominent nucleoli, and abundant cytoplasm that are associated with numerous tumor-infiltrating lymphocytes123 (Fig. 27.12). Tumor cells may have an organoid or a trabecular architecture, and focal mucin production may be present. These tumors are more common in women and typically occur in the cecum or proximal colon.124 Medullary carcinoma has also been referred to as “large cell minimally differentiated carcinoma.”125
Most medullary carcinomas are associated with a characteristic genomic profile. These tumors are less likely than usual CRCs to show KRAS and TP53 mutations, and they are more likely to harbor defects in DNA MMR125 (see Carcinomas with DNA Mismatch Repair Deficiency). Even when it is present as a small subcomponent of the tumor, a medullary pattern is often predictive of an underlying DNA MMR deficiency.103 Immunophenotypically, these tumors are usually cytokeratin 20 (CK20) negative, are occasionally CK7 positive, and often show reduced CDX2 expression.125,126 Differentiation from other nonglandular, undifferentiated carcinomas is important, because medullary carcinomas have a more favorable outcome.125,127 Use of immunohistochemistry for calretinin may be helpful in this regard; in one study, calretinin was positive in 73% of medullary carcinomas but only 12% of poorly differentiated adenocarcinomas.126
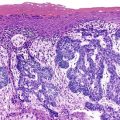
Serrated Adenocarcinoma
The term serrated adenocarcinoma has been used in two settings: to describe cancers that arise from hyperplastic or serrated precursor lesions, including both SSA/Ps and traditional serrated adenomas, and to describe cancers that have a distinctive serrated morphology.75 Consistent with their origin in the serrated pathogenetic pathway, serrated adenocarcinomas are more often proximal than conventional adenocarcinomas. Morphologically, these tumors are characterized by a variety of features; architecturally, there are epithelial serrations, and some degree of mucin production is common. In mucinous areas, tumor cells are often seen in the form of aggregated balls and pseudopapillary rods that project into mucinous areas. Tumor cells have abundant clear or eosinophilic cytoplasm, and the nuclei are vesicular with peripheral condensation of chromatin (Fig. 27.13). Necrosis is uncommon or focal.79,128
Studies of the molecular pathogenesis of serrated carcinomas have revealed significant heterogeneity, although most of these tumors are associated with a high degree of methylation (CIMP).75,79 There are at least two major subtypes of serrated carcinomas: proximal MSI-H cancers that arise from sessile serrated polyps, and distal MSS/MSI-L cancers that arise from traditional serrated adenomas.79 In one study,129 only 16.1% of all serrated carcinomas were MSI-H, but 8.2% of the nonserrated adenocarcinomas were also MSI-H. Therefore, most proximal MSI-H cancers do not show histologically identifiable serrated features even though they are believed to arise, in large part, from serrated precursor lesions. In another study of more than 900 CRCs, 9.1% were classified as serrated adenocarcinomas, and about half of these had an identifiable serrated adenoma precursor.128 In this series, there was a trend toward decreased 5-year survival, particularly for left-sided cancers. Although the presence of a serrated precursor lesion should always be reported, it is probably best to reserve the term serrated carcinoma for tumors that show characteristic features in the malignant component of the tumor, recognizing that this is not considered a distinctive subtype in the current WHO classification and that further clinical validation studies need to be performed.1
Carcinoma with Squamous Metaplasia (Adenoacanthoma)
Adenoacanthomas are rare.130 In one report, they were defined by the presence of adenocarcinoma elements with abundant admixed areas of benign-appearing squamous metaplasia (Fig. 27.14). The natural history and treatment are similar to those of pure adenocarcinomas.
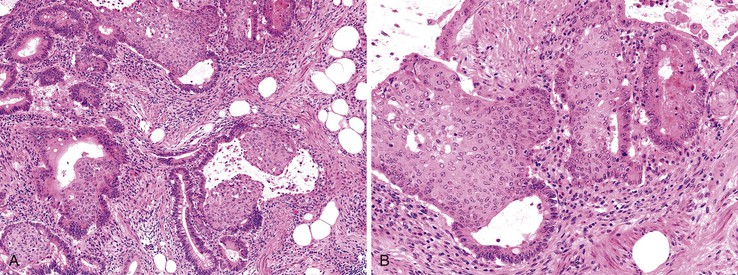
Adenosquamous Carcinoma
Adenosquamous carcinomas are rare neoplasms, accounting for 0.06% of all CRCs in SEER data, although they were reported to be three times more common than pure squamous cell carcinomas in one series.130,131 An association with paraneoplastic hypercalcemia and parathyroid hormone–related protein has been reported with sufficient frequency that a colorectal adenosquamous primary should be considered in the differential diagnosis of this presentation.132,133 This tumor type has also been seen in the setting of chronic ulcerative colitis.132 There is an even distribution between the right and left colon.130 Morphologically, there are malignant squamous and glandular elements, either as separate components or admixed.
These cancers often have a higher stage at presentation than usual adenocarcinomas; in one study, 50% had metastases at the time of diagnosis.130 Although the overall survival rate in stage I/II disease is the same as in comparably staged adenocarcinoma, the rate for stage III/IV disease is significantly lower.130,131,134 The overall 5-year survival rate for all adenosquamous carcinomas is 31%.131
Squamous Cell Carcinoma
Primary squamous cell carcinomas of the colorectum are rare; only 69 cases were reported before 1985, with most series reporting a prevalence of approximately 0.1% among all CRCs.135,136 The etiology and histogenesis are unknown. Most authorities favor an origin from pluripotent stem cells capable of multidirectional differentiation.136 Some have suggested derivation from foci of squamous metaplasia associated with chronic mucosal irritation. In two separate studies, all 31 cases were negative for human papillomavirus (HPV).130,137 One case of HPV-associated dysplasia and invasive squamous cell carcinoma of the rectum was described, suggesting a possible pathogenetic role for this virus in rare cases.138 Most primary squamous cell carcinomas manifest clinically at an advanced pathologic stage. The initial presentation is often at a more advanced patient age.
The pathologic features are similar to those of squamous cell carcinomas in other organs. Diagnosis of primary squamous cell carcinoma requires (1) exclusion of metastasis from other sites (particularly lung); (2) exclusion of an associated squamous-lined fistula tract (which, if present, is likely the site of origin); and (3) differentiation from carcinomas of the anus that extend proximally into the lower rectum.136 The 5-year survival rate for lymph node–negative squamous cell carcinomas is reported to be 85%. However, squamous cell carcinomas have been reported to have a worse prognosis than stage-matched adenocarcinomas.130,136
Sarcomatoid Carcinoma (Spindle Cell Carcinoma, Carcinosarcoma)
Sarcomatoid carcinomas, also termed carcinosarcoma or spindle cell carcinoma, are more common in elderly patients, are often bulky or fleshy, and show abundant hemorrhage. Histopathologically, these tumors reveal a biphasic growth pattern that combines both epithelial and mesenchymal elements. Histologic areas of transition may be observed in some cases.139,140 The spindle cell component may be entirely undifferentiated, or it may show osseous, cartilaginous, or smooth muscle differentiation.141,142
Similar to sarcomatoid carcinomas in other anatomic locations, these tumors usually express keratins and epithelial membrane antigen (EMA) in both the carcinoma and the spindle cell components.141 Carcinoembryonic antigen (CEA) positivity is usually limited to the adenocarcinoma element.143 Focal S100 and myoglobin positivity has also been described in some of these tumors.139 Metastases may show either or both cellular components. Evidence in favor of a common progenitor cell origin stems from confirmation of shared TP53 mutations in the epithelial and sarcomatoid components.142 Further subtyping into sarcomatoid carcinomas (which are keratin positive throughout the tumor) and carcinosarcomas (which are keratin negative in the mesenchymal component) has no apparent clinical or prognostic utility.144 These neoplasms are typically fast growing and aggressive.
Choriocarcinoma
Primary choriocarcinoma of the colorectum is rare.145 These cancers may manifest with massive bleeding from the gastrointestinal tract or from liver metastases.146 Microscopically, the tumor reveals a variable admixture of choriocarcinoma and adenocarcinoma elements. The choriocarcinoma component stains with human chorionic gonadotropin (β-hCG) and α-fetoprotein. On occasion, choriocarcinoma elements predominate in metastases when the primary tumor was only focally β-hCG positive.147 Increased serum levels of β-hCG148 and α-fetoprotein146 have also been reported. These cancers usually show metastasis to the liver at the time of clinical presentation and follow an aggressive clinical course.
Poorly Differentiated Neuroendocrine Carcinoma
Both small cell and, less frequently, large cell poorly differentiated neuroendocrine carcinomas can occur as a primary tumor of the colorectum, where they usually arise within an adenoma.149 These tumors typically manifest in younger patients compared with adenocarcinoma (mean age, 57 years), and they can arise anywhere in the colon or rectum. The morphologic features of these neoplasms are identical to those that occur in the lung. Immunohistochemistry usually demonstrates positivity for chromogranin or synaptophysin (and may be focal), the lesions may also show CDX2 positivity.
Poorly differentiated neuroendocrine carcinomas of the colorectum are aggressive tumors. Approximately 70% of patients have distant metastatic (stage IV) disease at presentation.149,150 The median survival time is 10.4 months, and the 3-year survival rate is 13%.149 A recent study showed that patients with neoplasms composed exclusively of large neuroendocrine cells survived longer than those with other morphologic subtypes.151
Mixed Adenoneuroendocrine Carcinoma
In some cases, distinct separation of pure glandular from pure neuroendocrine tumors is blurred in neoplasms that show evidence of dual differentiation. At one end of the spectrum are adenocarcinomas that contain scattered endocrine cells and neuroendocrine tumors that contain scattered non-neuroendocrine mucinous tumor cells. These features are common and do not affect either tumor behavior or management; they do not warrant classification as distinct entities.
However, tumors that show a significant mixture of adenocarcinoma and neuroendocrine elements (>30% of each component) are referred to as mixed adenoneuroendocrine carcinomas.1 In these composite tumors, the adenocarcinoma and neuroendocrine components are, by definition, mixed intimately. Metastases may show both or only one of the cellular components. There are insufficient studies on the clinical behavior of mixed tumors.151
In contrast to these mixed tumors, neoplasms that exhibit both neuroendocrine and glandular differentiation in individual cells are termed amphicrine neoplasms. On ultrastructural examination, these tumors show dense-core granules and mucin droplets in single cells. One variant in the colon has similar characteristics to appendiceal goblet cell carcinoid tumor.152,153Some contain cells with multiple cellular phenotypes, such as goblet, endocrine, and Paneth cell differentiation (Fig. 27.15). Because these tumors seem to recapitulate the characteristics of normal intestinal crypts, the term crypt cell carcinoma has been proposed.152,153 The malignant potential of these lesions has been well documented, and death is usually the result of intraabdominal rather than disseminated disease.152,153
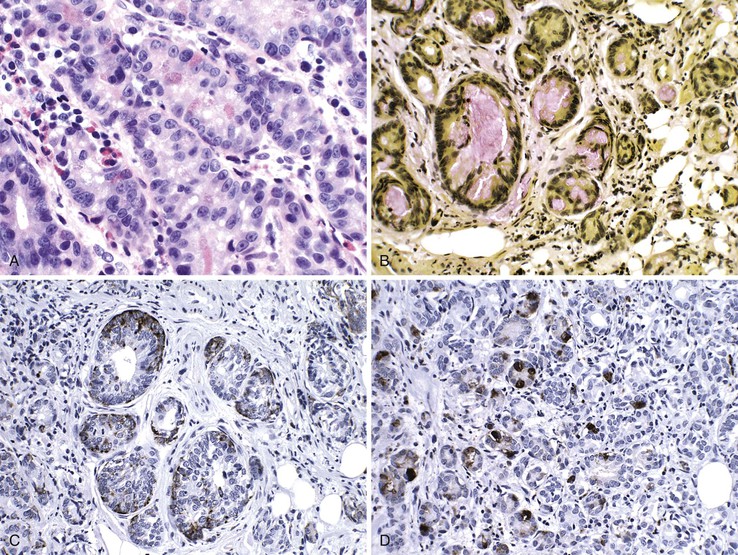
Diffuse Carcinoma
Non–signet ring cell carcinomas in the colorectum may occasionally exhibit a diffuse pattern of growth similar to those that occur in the stomach. In one study, 15 (0.6%) of 2369 CRCs were “diffuse”; only 7 were signet ring cell type, whereas 8 were “lymphangiosis” type.120 The lymphangiosis type of diffuse cancer is an adenocarcinoma, often of moderate differentiation, with extensive lymphatic and vascular spread.120,121 As expected, the prognosis of this type of cancer is extremely poor.120
Other Rare Subtypes of Colon Carcinoma
Melanotic adenocarcinoma of the anorectal junction has been reported as a rare primary tumor, although evidence supports phagocytosis of anal melanocytes rather than true melanocytic differentiation in the pathogenesis of these tumors.154 Clear cell carcinomas have been described in the colon; they have clear cytoplasm but lack cytoplasmic mucin (Fig. 27.16). Strong CEA staining helps distinguish this tumor from a metastatic renal cell carcinoma.155
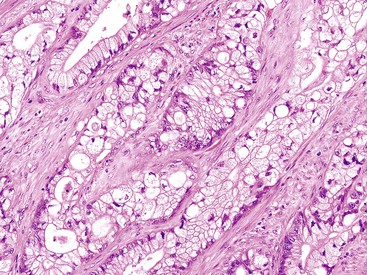
Carcinomas with DNA Mismatch Repair Deficiency
Inactivation of DNA MMR pathways plays an important role in the pathogenesis of 10% to 15% of CRCs. Although some of these cases are the result of hereditary alterations in one or more of the MMR genes (see Hereditary Nonpolyposis Colorectal Cancer [HNPCC] and Lynch syndrome), approximately 80% are sporadic, resulting primarily from promoter hypermethylation and silencing of MLH1 transcription. In addition to the characteristic MSI-H genetic alteration, these cancers reveal a variety of specific clinical and pathologic associations of importance to surgical pathologists (see Lynch Syndrome for discussion of immunohistochemical and molecular diagnostics).70,156
Sporadic MMR-deficient MSI-H CRCs with MLH1 methylation silencing show a marked female predominance (about 4 : 1), increase in prevalence with age, and are particularly common in women older than 70 years.157 Approximately 90% of sporadic MLH1-deficient MSI-H CRCs are located in the right colon, where they constitute approximately 50% of all right-sided cancers in women older than 70 years of age.103 Precursor lesions often reveal characteristics of SSA/Ps in sporadic cases and conventional adenomas in hereditary cases (e.g., HNPCC).158 A number of specific pathologic associations have also been described in MSI-H CRCs, and many of these are similar to HNPCC cancers, which are also caused by DNA MMR deficiency103,159–163 (Table 27.8). MSI-H cancers are also more likely to be multiple, to have a polypoid or exophytic growth pattern, and to show sharply circumscribed, pushing margins and marked grossly visible necrosis. Furthermore, MSI-H tumors are more likely to show mucinous or signet ring cell features, as well as microglandular differentiation. The association between these histologic features and DNA MMR deficiency is high, even when less than 50% of the tumor reveals these features. Therefore, the sensitivity and specificity of these histologic features for the diagnosis of MSI-H cancer vary according to the specific criteria used. The presence of a component of any one of the associated histologic variants (mucinous, signet ring cell, microglandular, or undifferentiated) or of a prominent tumor-infiltrating lymphocytic response provides a relatively sensitive threshold to identify possible MSI-H cancers.111,164
Table 27.8
Pathologic Features of High-Frequency Microsatellite Instability Colorectal Carcinomas
| Pathologic Feature | MSI-H (%) | MSI-L/MSS (%) | Positive Predictive Value (%)* |
| Location in right colon | 90 | 34 | 32 |
| Exophytic/polypoid | 82 | 54 | 21 |
| Signet ring cell component | 13 | 5 | 31 |
| Mucinous type (>50% extracellular mucin) | 15 | 5 | 35 |
| Mucinous component (>10% extracellular mucin) | 22 | 7 | 36 |
| Cribriform pattern | 13 | 28 | 8 |
| Poor differentiation | 38 | 13 | 34 |
| Medullary type | 14 | 0.4 | 71 |
| Medullary component (>10%) | 25 | 3 | 59 |
| Lymphocytosis | 21 | 3 | 55 |
| Pushing margin | 15 | 8 | 25 (NS) |
| Crohn’s-like reaction | 49 | 36 | 19 (NS) |
| “MSI-H” by pathologist | 49 | 11 | 44 |
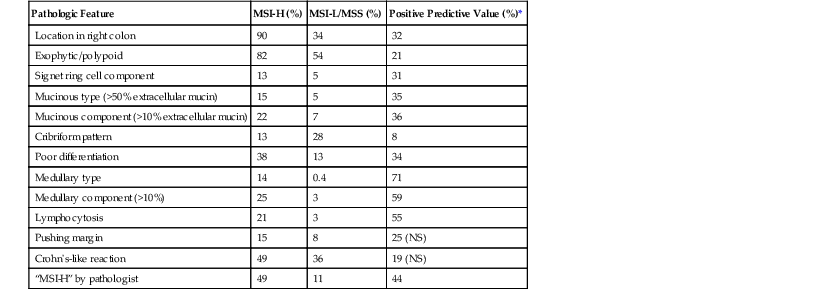
* Assuming a prevalence of MSI-H of 15%.
MSI-H, High-frequency microsatellite instability; MSI-L, low-frequency microsatellite instability; MSS, microsatellite stable; NS, not significant.
From Alexander J, Watanabe T, Wu TT, et al. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527-535; and Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148-156.
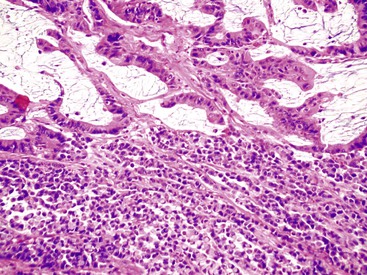
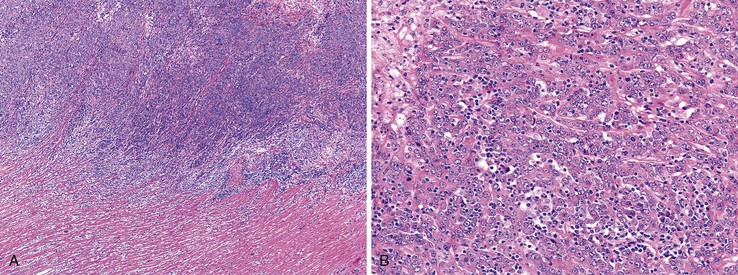
The character of the host lymphoid response is another feature that shows a striking difference between MSI-H and MSS cancers. The presence of tumor-infiltrating lymphocytes (Fig. 27.17) is highly predictive of MSI-H cancer, particularly when present in combination with an undifferentiated tumor (which may or may not be of the classic medullary subtype). In one study, the presence of at least 5 tumor-infiltrating lymphocytes per 10 high-power fields (HPFs) had a sensitivity of 93% and a specificity of 62% for the detection of MSI-H CRC.165 A number of complex algorithms have been proposed in an attempt to achieve a high degree of specificity for the presence of MSI-H in any single colon carcinoma. However, in one study, the pathologist’s “opinion” (based on an overall evaluation of the features described previously and his or her prior experience with known MSI-H cancers) was the best predictive factor.104 Overall, compared with MSI-H HNPCC–associated colon cancer, sporadic MSI-H cancers are more frequently heterogeneous, poorly differentiated, mucinous, proximally located, and, as mentioned earlier, associated with a prominent tumor-infiltrating lymphocyte reaction.166
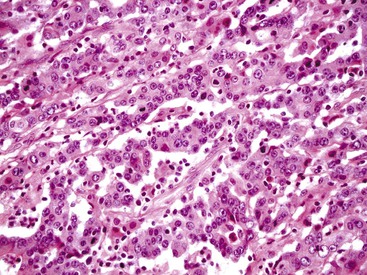
Clinically, patients with MSI-H CRCs are more likely to have an advanced T stage at presentation, but they are less likely to show regional lymph node metastases. In fact, most T4 N0 M0 CRCs are deficient in DNA MMR enzymes.167 When controlled for stage and other standard prognostic parameters, most studies have shown that MSI-H CRCs are associated with better overall survival compared with MSS colon cancers (see Molecular Genetic Markers).167,168
Villous Adenocarcinoma
Recently described, this subtype consists of more than 50% villous architecture. It accounted for 8.6% of all CRCs in one series.169 Fewer than half of the lesions had severe atypia, and differentiation from a benign adenoma with or without prolapse changes was made based on the presence of epithelial islands in desmoplastic stroma, which were present in almost all cases. Clinical follow-up of 35 cases revealed a favorable prognosis.169 Additional studies of diagnostic criteria and outcome are needed before novel management approaches can be considered.
Tumor Grading
Grading is one of the most widely used pathologic variables, but it is also one of the most difficult to define accurately. Furthermore, there is ongoing controversy as to whether tumor grade is an independent prognostic factor,170 although most studies show this to be the case, particularly when a two-tiered system is used.171,172
Grading is based primarily on the proportion of tumor composed of glands, relative to areas that are solid or composed of nests or cords of cells without lumina. Use of a two-tiered system of low- and high-grade is recommended and endorsed by the WHO and AJCC1,2 (Table 27.9). This system replaces the traditional classification of well-differentiated, moderately differentiated, and poorly differentiated tumors. It is more reproducible because well- and moderately differentiated carcinomas behave similarly. In this system, colorectal adenocarcinomas are classified as either low grade (well or moderately differentiated; >50% gland formation) or high grade (poorly differentiated; <50% gland formation). Grading of biopsy specimens often is inaccurate and fails to reflect the final grade of the tumor in the resection specimen.
Table 27.9
Grading of Colorectal Adenocarcinomas
| Grade | Descriptive Nomenclature | Criteria* | AJCC Recommendation |
| GX | Grade cannot be assessed | ||
| G1 | Well differentiated | >95% Gland forming Majority (>75%) of glands are smooth and regular No significant component of high-grade nuclei |
Low grade |
| G2 | Moderately differentiated | 50-95% Gland forming | Low grade |
| G3 | Poorly differentiated | <50% Gland forming | High grade |
| G4 | Undifferentiated | No apparent gland formation† | High grade |

* All criteria must be met.
† Undifferentiated tumors are a distinct subtype.
AJCC, American Joint Committee on Cancer.
From Edge SB, Byrd DR, Compton CC, et al., eds. AJCC Cancer Staging Manual. 7th ed. New York: Springer-Verlag; 2010:156; and Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer: College of American Pathologists consensus statement 1999. Arch Pathol Lab Med. 2000;124:979-994.
Grading of CRCs applies only to adenocarcinomas of the usual type. By definition, signet ring cell carcinomas and small cell carcinomas are high grade. Some also consider all types of mucinous carcinomas to be high grade; grading of mucinous cancers on the basis of degree of gland formation and cytologic features is of questionable practical utility. Tumors that show no or only minimal gland formation are classified as “undifferentiated” and likely represent very poorly differentiated adenocarcinomas in most cases. Most importantly, medullary carcinomas should not be classified as either “poorly differentiated” or “undifferentiated,” because they are associated with a better overall outcome than these other tumors; medullary carcinomas should probably not be graded at all by the usual methods.173 In general, grading should not be performed on the invading edge of carcinomas. The presence of small nests or cords of cells located at the leading edge of the tumor (tumor budding) is an independent indicator of poor prognosis (see Tumor Budding).
Special Studies
Techniques to Facilitate Lymph Node Detection
Several techniques have been developed to increase the yield of lymph nodes found during tissue dissection. The cheapest and simplest method is the use of GEWF solution, a relatively inexpensive mixture of glacial acetic acid, absolute ethanol, distilled water, and formaldehyde, all of which are commonly found in most pathology laboratories. GEWF is a lymph node highlighting solution, not a fat-clearing solution; its use results in lymph nodes’ becoming more readily identified as chalky-white nodules in fat that has been immersed in the solution for at least 24 hours. Most studies that have evaluated the use of GEWF in CRC specimens demonstrated a 50% to 100% increase in nodal yield as well as an increased yield of smaller lymph nodes,174–176 although one study showed only an 11% increase in lymph node yield.177
Fat-clearing techniques involve immersion of the specimen in graded alcohol solutions, followed by xylene to dissolve the fat. Although such methods also increase the yield of lymph nodes,178 they are time-consuming and expensive (due to reagent costs) and involve exposure to noxious solvents.
Sentinel Lymph Nodes
The development of distant metastases in 20% to 30% of patients who have a primary tumor confined to the bowel wall is often cited as evidence of undetectable metastasis at the time of surgical resection and pathologic evaluation. Meticulous pathologic evaluation of sentinel lymph nodes (lymph nodes that have the most direct drainage from the tumor) has been investigated in numerous studies. To identify the sentinel node in patients with CRC, the primary tumor is isolated, and a blue dye is then injected circumferentially around the tumor in the subserosal layer. The first one to four lymph nodes that change color are considered the sentinel lymph nodes. Techniques used to evaluate sentinel lymph nodes include thin slicing, submission in total, multiple tissue levels, and immunohistochemical analysis.
Reports on the sensitivity of sentinel lymph nodes in predicting nodal positivity are varied, with some studies showing increased sensitivity for node detection179,180 and others showing that the sentinel lymph nodes are no more sensitive than other regional lymph nodes for the detection of micrometastases.181–183 A recent meta-analysis of the sentinel lymph node procedure for CRC showed a low sensitivity for sentinel lymph node detection, regardless of T stage, localization, or pathologic technique used.184
Many questions remain about the practical use of the sentinel lymph node procedure in the management of CRC. Much of the problem relates to the fact that the lymph drainage in the mesocolon is anatomically complicated and there is no obvious orderly progression of nodal spread within the mesocolon.185
Data on the clinical significance of micrometastases are scarce, and there are no data to show that chemotherapy for patients with micrometastases is beneficial. It is also not clear whether isolated tumor cells or micrometastases are more likely to be present in a sentinel lymph node than in a nonsentinel lymph node.
Immunophenotype
Routine examination of CRC specimens does not usually require immunohistochemical studies. These studies are more often required for characterization of distinctive subtypes (e.g., small cell carcinoma) or for investigation of primary versus metastatic tumors. The following markers may be helpful in the examination of CRCs:
• Keratins: CRC tumor cells express low-molecular-weight cytokeratins. Most tumors (80% to 100%) are positive for CK20, and as many as 15% are positive for CK7.186 The great majority of CRCs are positive for CK20 and negative for CK7. MSI-H adenocarcinomas often show decreased expression of CK20. A summary of the frequency of specific keratin profiles in colorectal adenocarcinoma is presented in Table 27.10.
Table 27.10
CK7/CK20 Profile of Colorectal Cancer194
| CK7 | CK20 | Prevalence in Colorectal Cancer (%) | Differential |
| Overall Results | |||
| + | 5-15 | ||
| + | 85-100 | ||
| Profiles | |||
| + | + | 5-10 | Pancreas, bile duct, urothelial |
| + | − | 0-5 | Lung, breast, ovary, endometrial |
| − | + | 75-95 | Colorectal, gastric |
| − | − | 0-15 | Prostate, adrenal, hepatocellular, renal, carcinoid |

CK, Cytokeratin.
• Villin: Villin is a microfilament-associated actin-binding protein that shows diffuse cytoplasmic expression with brush border accentuation in more than 90% of colorectal adenocarcinomas. It is also positive in nongastrointestinal tumors that show intestinal differentiation (e.g., lung, bladder).187,188
• CDX2: The nuclear transcription factor CDX2 is expressed in more than 90% of colorectal adenocarcinomas.189,190 Exceptions include medullary carcinomas and some nonmedullary, DNA MMR-deficient carcinomas, which are often negative for CDX2 and CK20 and positive for CK7.125 Expression is related to differentiation, with high-grade tumors positive in approximately 50% of cases.190 There also appears to be an inverse correlation of CDX2 expression with tumor stage.190 CDX2 is expressed in 10% to 30% of other gastrointestinal malignancies, particularly those with intestinal-type differentiation; in 20% to 65% of mucinous ovarian tumors; and in 50% to 100% of urinary bladder adenocarcinomas.187,189,190 CDX2 expression is rare in breast, lung, thyroid, kidney, and endometrial adenocarcinomas, although nonbronchioloalveolar mucinous carcinomas of the lung are frequently positive. Several studies have also postulated a role for CDX2 in neoplastic progression, with loss of expression correlating with more advanced/aggressive disease.
• Mucins: MUC2 is expressed in most CRCs; MUC1 expression may be seen, but expression of MUC5AC is rare.186,191
Differential Diagnosis
Metastasis versus Multiple Synchronous Primary Carcinoma
A small subset of patients with CRC have multiple colonic tumors. These may represent synchronous primary CRCs or metastases from one tumor to other areas of the colorectum. The only definitive diagnostic feature of a primary tumor is the presence of an adjacent precursor lesion (adenoma or serrated polyp). Multiple synchronous carcinomas can be definitively diagnosed only if definite foci of adenomatous or neoplastic serrated epithelium are detected in both tumor locations. However, adenoma adjacent to a colorectal adenocarcinoma occurs in fewer than one third of cases.192 Pathologists should be aware that hyperplastic “transitional” mucosa adjacent to a carcinoma may simulate an adenoma. The presence of surface epithelial maturation and the absence of deep crypt apoptosis and budding glandular architecture are helpful in correctly identifying this reactive change. Metastasis is favored if there is no mucosal involvement or if there is extensive lymphatic involvement. The presence of multiple tumors that reveal similar but unusual morphology favors metastasis from a single primary, whereas the presence of morphologically dissimilar patterns, at least one of which is of an unusual subtype, favors multiple synchronous tumors rather than metastasis. Differences in immunophenotype or molecular marker status may also be helpful in diagnosing multiple synchronous primaries, although the latter is rarely used in practice.
Recurrent or Metastatic Carcinoma versus Metachronous Carcinoma
Patients with CRC are at significantly increased risk for the development of a new cancer. In these patients, it may be difficult to differentiate tumor recurrence from a metachronous cancer. If a precursor lesion is identified in the more recently developed tumor, a diagnosis of a new primary can be established. However, if there is an absence of mucosal involvement or if there is extensive submucosal-mucosal lymphatic spread in association with multiple tumor nodules, particularly when it is also present in the site of the original tumor, a metastasis should be favored. In the remainder of cases, it is often difficult to be definitive diagnosis. In the absence of a precursor lesion, a diagnosis of tumor recurrence is always favored when a tumor develops at a surgical anastomosis line, even if there are no other supporting features.
Colorectal Adenocarcinoma versus Endometrial or Ovarian Adenocarcinoma
The morphologic appearance of some endometrial and endometrioid ovarian carcinomas may show significant overlap with primary CRC. As a result, it may be difficult to firmly establish the correct site of origin of tumors that extensively involve the colorectum and other pelvic organs. The presence of a precursor lesion or the absence of mucosal involvement is helpful in diagnosing a primary colorectal tumor or a noncolorectal tumor, respectively. Some ovarian metastases from a colonic primary may show only unilateral disease and may contain foci suggestive of a primary ovarian tumor of benign nature or low malignant potential.193 Immunohistochemistry may be useful in this differential diagnosis: CRCs are typically CK7−/CK20+, whereas most endometrial and ovarian cancers are CK7+/CK20−.193,194 Furthermore, nuclear expression of WT1 is often positive in ovarian cancers but negative in CRCs.195 CDX2 is positive in approximately 90% of CRCs and is almost always negative in endometrial and nonmucinous ovarian tumors.189,190
Colorectal Adenocarcinoma versus Primary Small-Intestinal Adenocarcinoma
Not uncommonly, CRC may directly invade the small intestine or seed the small intestine by intraabdominal spread, which raises the possibility of a primary small-intestinal carcinoma, particularly when there is small-intestinal mucosal involvement. The presence of a precursor lesion provides definitive evidence of the original site of origin of the tumor. Immunohistochemistry may be helpful, because primary small intestinal adenocarcinomas are typically CK7+ and CK20+, whereas approximately one third of small-intestinal adenocarcinomas are CK7+ and CK20−.196 Villin and CDX2 are positive in approximately 60% of small intestinal carcinomas, compared with 90% of CRCs.
Natural History
The overall survival rates for CRC at 1, 5, and 10 years are 83%, 64% and 58%, respectively.3 As with most solid tumors, survival correlates with the presence of locoregional disease and distant metastasis. For instance, the 5-year survival rate is 90% for patients with localized disease, 69% for those with regional metastases, and 12% for patients with distant metastasis.3However, only 39% of new cases are diagnosed with only localized disease.3 Pathologic staging in colon cancer (AJCC system) is based on depth of invasion into the bowel wall and surrounding structures, spread to regional lymph nodes, and distant metastasis. With progression, tumors may penetrate the peritoneal lining, resulting in intraabdominal spread or invasion into other abdominal and pelvic structures. Spread through lymphatics or blood vessels may occur at any time (or stage) during the evolution of disease. With further progression, cancer may invade the portal vein tributaries, leading to liver metastases, or the vena cava, leading to lung and bone marrow as well as other distant metastases.
Treatment
Surgical Therapy
Patients with colon and rectal cancer are usually managed by surgical resection of the primary tumor, with or without chemotherapy, depending on the stage of the tumor. Surgical resection is undertaken in all patients unless the tumor is deemed locally unresectable or there is a medical contraindication to surgery. In patients with nonobstructing tumors, a segmental colectomy with en bloc removal of regional lymph nodes is performed. Patients with obstructing tumors may undergo a similar one-stage procedure, a resection with diversion, or a diversion and stent procedure followed by resection. There is no minimum number of lymph nodes in a given specimen, because of the many factors that affect lymph node number, but on average the specimens should contain a minimum of 12 lymph nodes to be considered adequate for N stage assessment. Laparascopic-assisted colectomy may be considered if defined criteria are met.197 Rectal cancers that have spread beyond the muscularis propria (stage T3) are often treated with neoadjuvant chemoradiation before resection.198
Surgical resection of liver and pulmonary metastatic disease is also considered if the following criteria are met: (1) the primary tumor was (or will be) resected for cure, (2) complete resection of metastatic disease is feasible, and (3) maintenance of adequate organ function can be preserved. Ablative therapies and re-resection may be considered in selected patients. Reevaluation for conversion of previously unresectable disease to resectable disease is typically performed every 2 months after the initiation of chemotherapy.
Chemotherapy
For almost 4 decades, fluoropyrimidines have been the mainstay of CRC chemotherapy (intravenous 5-fluorouracil [5-FU] plus leucovorin folinic acid, or oral capecitabine [Xeloda]).199 In the past 5 years, two new cytotoxic agents—CPT-11/irinotecan (Campto), a topoisomerase inhibitor, and oxaliplatin (Eloxatin), a third-generation platinum compound—have shown benefit in patients with advanced disease. These chemotherapeutic agents are now widely used in various treatment protocols in the adjuvant and advanced disease settings, typically in combination with 5-FU (for example, the FOLFOX protocol consists of FOLinic acid + 5-Fluorouracil + OXaliplatin); they result in improved response rates and more prolonged progression-free survival.200
Targeted Therapy
The addition of several different targeted therapeutic agents has shown efficacy in patients with resistant metastatic disease. Cetuximab (Erbitux) and panitumumab (Vetibix) are humanized anti–epidermal growth factor receptor (EGFR) antibodies that inhibit downstream signaling of cell growth and proliferation, as well as apoptotic pathways that have been shown to be important in the progression of neoplasia. Both are highly effective in advanced CRCs that do not harbor KRAS mutations.201,202 KRAS mutation analysis is therefore required in the primary tumor or a metastasis before anti-EGFR therapy is initiated (see Molecular Genetic Predictive Markers).198,203 Regorafenib (Stivarga) is a small-molecule multikinase inhibitor that has been shown to be effective in metastatic disease that is resistant to other therapies, including KRAS mutant tumors.204,205 Bevacizumab (Avastin) is a humanized anti–vascular endothelial growth factor (VEGF) antibody that inhibits soluble protein and results in an antiangiogenic effect in tumors. Aflibercept (Zaltrap) is another recently developed VEGF inhibitor that has been approved for use in the United States for treatment of metastatic CRC that has failed other modalities.201,202 The proven benefit of these targeted therapeutic agents is currently restricted to patients with metastatic disease, in whom they may be used alone or in combination with 5-FU and either irinotecan or oxaliplatin.
Investigational Agents
In addition to the aforementioned agents, a number of other chemotherapeutic and biologic compounds are under investigation and development. Although this is more limited in terms of chemotherapeutic agents, there are ongoing or planned trials with new topoisomerase inhibitors and new platinum compounds. In comparison, a wide array of new targeted therapeutic agents are in phase II and III trials, including tyrosine kinase inhibitors, cyclooxygenase-2 inhibitors, matrix metalloproteinase inhibitors, and various forms of immunotherapy (Table 27.11).199,200,206
Table 27.11
Investigational New Targeted Therapeutic Agents in Colorectal Cancer*
| Biologic Activity | Gene | Compound (Drug Class; Company) |
| Angiogenesis | VEGFR-2 | Ramucirumab (monocloncal antibody; ImClone) |
| VEGFR-2/FGFR/PDGF | BIBF1120 (RTKI†; Boerhinger Ingelheim) | |
| Protein kinase C (VEGF pathway) | Enzastaurin (small molecule inhibitor; Eli Lilly) | |
| Tumor growth (EGFR pathway) | EGFR | Necitumumab (monocloncal antibody; BMS/Eli Lilly) |
| EGFR/EGFR-2 | BIBw 2992 (RTKI†; Boerhinger Ingelheim) | |
| Tumor growth (RAS-RAF-MEK-ERK pathway) | RAF | BMS-908662 (small molecule inhibitor; BMS) |
| MEK | Selumetinib (small molecule inhibitor; AstraZeneca) | |
| Tumor growth (IGF-1R pathway) | IGF-1R | Dalotuzuman (monoclonal antibody; Merck) |
| Tumor growth (PI3K-Akt-mTOR pathway) | mTOR | Everolimus (small molecule inhibitor; Novartis) |
| Apoptosis induction | TRAIL (TNFSF10) | Conatumumab (monoclonal antibody; Amgen) |
| Eg5 | LY2523355 (small molecule inhibitor; Eli Lilly) | |
| Immune modulator | TLR9 | MGN1703 (double-stem loop DNA immunostimulator; Mologen) |
| DNA repair | PARP | Veliparib (small molecule inhibitor; Abbott) |
* This table is meant to represent the array of targets and compounds currently being tested in phase II and phase III trials in colorectal cancer and is not meant to be a comprehensive listing of investigational new agents.
† Small-molecule receptor tyrosine kinase inhibitor.
EGFR, Epidermal growth factor receptor; IGF, insulin-like growth factor; VEGF, vascular endothelial growth factor.
Role of Stage and Pathologic Factors in Management
Colon Cancer.
Stage I disease (T1/T2 N0 M0) is managed by surgical resection alone, and these patients are not usually offered adjuvant chemotherapy. Stage I malignant polyps represent a unique management subgroup; localized excision (polypectomy) is considered curative therapy in most cases because of the low risk of lymph node metastases. However, there are two features that predict an appreciable risk of regional lymph node metastases and therefore require segmental resection: poor differentiation and lymphatic or venous invasion (particularly invasion of extramural large veins). The presence of carcinoma within 1 mm of the resection margin is also predictive of residual disease at the polypectomy site. Because proctectomy is associated with higher morbidity, malignant rectal polyps may be managed by polypectomy alone, even in the presence of high-risk features, if the regional lymph nodes are negative by ultrasonography or MRI.
The use of adjuvant therapy in stage II disease is variable and has been the subject of some controversy.203,206–208 In general, chemotherapy is not offered to patients with low-risk stage II disease (T3 N0 M0 and absence of high-risk features), although it may be considered in some patients, particularly those with MMR-proficient tumors (see later discussion).209 Adjuvant therapy is typically offered to patients with high-risk stage II disease, which is defined by the presence of poor differentiation (after exclusion of MSI-H); lymphatic or vascular invasion; bowel obstruction; fewer than 12 lymph nodes examined; perineural invasion; localized perforation; close, indeterminate, or positive margins; or any T4 disease.
In stage III disease, adjuvant chemotherapy is widely used. In this setting, use of 5-FU alone results in a 5% to 6% absolute survival advantage after 5 years of follow-up.207,210,211 More recently, many studies have found that disease-free survival is improved when oxaliplatin is added to a fluoropyrimidine in the adjuvant setting.200 This combination of drugs is now considered the standard choice for patients who do not participate in new cancer trials and in whom they can be tolerated. To date, no proven benefit has been identified for the addition of anti-EGFR or anti-VEGF therapies in controlled clinical trials in the adjuvant setting.200,203
In patients with advanced disease (stage IV, Dukes stage D, distant metastases), the added benefit of chemotherapy is firmly established.199,201,202 At present, most patients are offered a combination of 5-FU, oxaliplatin, and either anti-VEGF therapy (bevacizumab) or an anti-EGFR agent (cetuximab or panitumumab), the choice depending on KRAS mutation status (see later discussion).203