Chapter 11 Epilepsies Due to Monogenic Disorders of Metabolism
This chapter describes a number of inherited metabolic disorders in which epilepsy is a prominent symptom (Table 11-1). These specific disorders were selected among the large group of metabolic diseases for one or more of the following reasons: (1) relatively high prevalence, (2) presentation in adulthood, (3) knowledge of the underlying genetic defect, and (4) availability of a treatment. Disorders that are very rare, that present exclusively in infancy or childhood, in which epilepsy is uncommon, and for which there is no known genetic defect and no specific treatment will not be discussed.

Neuronal Ceroid Lipofuscinoses
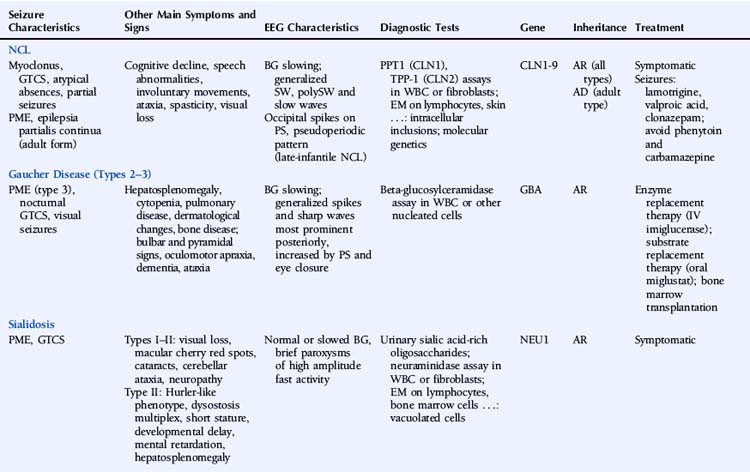
Epilepsy is a feature of all types of NCL and is usually the first symptom of disease in the late-infantile form.1 Seizures can manifest as myoclonic jerks, generalized tonic-clonic seizures (primary or secondarily), atypical absences, or partial seizures. Adult NCL may present as a progressive myoclonic epilepsy2 or as epilepsia partialis continua.3 Northern epilepsy is characterized by childhood-onset generalized tonic-clonic or complex partial seizures decreasing after puberty and not accompanied by myoclonus.4
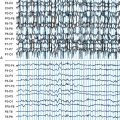
The EEG is characterized by slowing of background activity and the presence of generalized spike-wave and polyspike-wave complexes and bursts of slow waves.5 High-voltage, polyphasic spikes in the occipital region with photic stimulation at 1 to 2 Hz, as well as a pseudoperiodic pattern, have been described in the late-infantile form.6,7
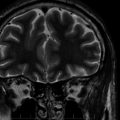
Brain magnetic resonance imaging (MRI) demonstrates cerebral and cerebellar atrophy, T2-hyperintensity of the lobar white matter, thinning of the cerebral cortex, and thalamic T2-hypointensity.8
The diagnostic testing strategy in the NCLs depends mainly on the age of onset. Diagnosis is based on enzymatic assays, electron microscopy, and molecular genetic testing. Two types of enzymatic deficiency have been identified in the NCLs. Palmitoyl-protein thioesterase 1 (PPT1) activity is absent in NCLs caused by mutations in the CLN1/PPT1 gene, and tripeptidyl-peptidase 1 (TPP-1) activity is usually absent in NCLs caused by mutations of the CLN2/TPP1 gene. Enzymatic assays can be performed on leukocytes or fibroblasts. Enzymatic deficiencies in the other NCLs are unknown. Electron microscopy of lymphocytes and tissue biopsies (usually skin) shows typical intracellular inclusions consisting of autofluorescent lipopigment storage material. Eight genes—CLN1/PPT1, CLN2/TPP1, CLN3, CLN5, CLN6, CLN7/MFSD8, CLN8, and CLN10/CTSD—are known to be associated with NCL.9 Numerous different mutations have been identified, but most genes carry a small number of common mutations. The genes at the CLN4 ad CLN9 loci have not been identified. The NCLs are characterized by extensive phenotypic and genetic heterogeneity.
Therapy for the NCLs is limited to symptomatic treatment at present. Promising future treatments include enzyme replacement therapy, gene therapy, and stem cell therapy.10 Lamotrigine has been reported to be effective and well tolerated in the infantile and juvenile forms.11–12 In juvenile NCL, valproic acid is a valuable alternative, and clonazepam may be useful as add-on therapy.13 Carbamazepine and phenytoin should be avoided, as they may increase seizure activity.14,15
Gaucher Disease
Gaucher disease (GD) is an autosomal recessive lysosomal disorder caused by a deficiency of the enzyme beta-glucocerebrosidase (also called acid beta-glucosidase or acid beta-glucosylceramidase) and accumulation of glucosylceramide (GL1) and other glycolipids. Three major clinical subtypes (1, 2, and 3) and two other subtypes (perinatal lethal and cardiovascular) are recognized based on clinical presentation. Type 1 (nonneuronopathic form) usually has no primary central nervous system symptoms. Types 2 (acute neuronopathic form) and 3 (subacute neuronopathic form) are characterized by the presence of primary neurologic disease. They are classically distinguished by age of onset and rate of disease progression, but it is now increasingly recognized that neuronopathic GD represents a phenotypic continuum, ranging from severely affected infants to asymptomatic adults. GD prevalence estimates vary between 1/57,000 and 1/86,000.16,17
GD is a multisystemic disorder characterized by varying degrees of hematological, skeletal, pulmonary, and neurological involvement. Hepatosplenomegaly usually precedes neurological manifestations. Neurological symptoms may include bulbar and pyramidal signs, oculomotor apraxia, dementia, and ataxia. Epilepsy is mainly a symptom of type 3 GD and usually presents as progressive myoclonic epilepsy (also called type 3a GD).18 The myoclonus may be spontaneous, stimulus sensitive, or induced by action. Other reported seizure types include generalized tonic-clonic seizures during sleep and visual seizures.19,20
EEG shows gradual background slowing and generalized spikes and sharp waves, most prominent over the posterior regions and increased by photic stimulation and eye closure.20,21 Brain MRI may show mild cerebral atrophy.
The GBA gene is the only gene known to be associated with GD. At least 200 GBA mutations have been identified.22,23 Four common mutations account for the majority of cases. Molecular genetic testing in a proband is not necessary to confirm the diagnosis, but may be considered for genetic counseling purposes, primarily for carrier detection among at-risk relatives. Genotype-phenotype correlation in GD is poor. Although the genotypic spectrum in patients presenting with progressive myoclonic epilepsy is different from that in other patients with type 3 GD, there appears to be no specific shared genotype.24
Affected individuals should be monitored regularly, including medical history, physical and neurological examination, blood tests—especially hemoglobin concentration and platelet count—assessment of spleen and liver volumes, screening for pulmonary hypertension, and skeletal involvement. Enzyme replacement therapy with imiglucerase, an intravenous recombinant glucosylceramidase enzyme preparation, can reverse systemic involvement. The effectiveness of enzyme replacement therapy for the treatment of neurologic disease remains to be established, although a few reports have suggested some benefit.25–27 Onset of progressive myoclonic seizures while on enzyme replacement therapy appears to indicate a poor prognosis.28 Substrate reduction therapy with the oral agent miglustat is another treatment option in individuals with mild to moderate GD for whom enzyme replacement therapy is not a therapeutic option. A case report described neurologic improvement in a patient with type 3 GD and myoclonic epilepsy on combined enzyme replacement and substrate reduction therapy.29 Individuals with chronic neurologic GD and progressive disease despite enzyme replacement therapy may be candidates for bone marrow transplantation.
Sialidosis
Type I sialidosis (also known as the “normosomatic” type or cherry red spot–myoclonus syndrome) usually presents in the second or third decade and is characterized by progressive visual loss, bilateral macular cherry red spots, cataracts, progressive generalized myoclonus, generalized tonic-clonic seizures, and cerebellar ataxia. The myoclonus is often induced by action. Peripheral neuropathy has also been reported.30 Intellect is usually preserved. Type II sialidosis (also known as the “dysmorphic” type) is the more severe, early onset form and is additionally associated with a Hurler-like phenotype, dysostosis multiplex, short stature, developmental delay, mental retardation, and hepatosplenomegaly.
Reports of EEG observations in sialidosis are rare. Brief paroxysms of high amplitude fast activity on a normal or slightly slowed background were described in one study.31 Jerk-locked back-averaging shows a consistent temporal relationship between the EEG spikes and myoclonic jerks.32 Brain MRI shows progressive cerebral and pontocerebellar atrophy.33
The sialidoses are caused by mutations in the NEU1 gene. Its product, neuraminidase or lysosomal sialidase, has a dual physiologic function: it participates in intralysosomal catabolism of sialated glycoconjugates and is involved in cellular immune response. More than 40 different mutations have been characterized.34–36 In general, there is a close correlation between the residual enzyme activity and the clinical disease severity.37
Treatment is symptomatic. At present, there is no disease-specific treatment available, but potential treatment strategies such as gene therapy and enzyme replacement therapy are under study.38 Treatment of seizures and myoclonus is aspecific. One report described successful treatment of myoclonus with 5-hydroxytryptophan as add-on therapy.39
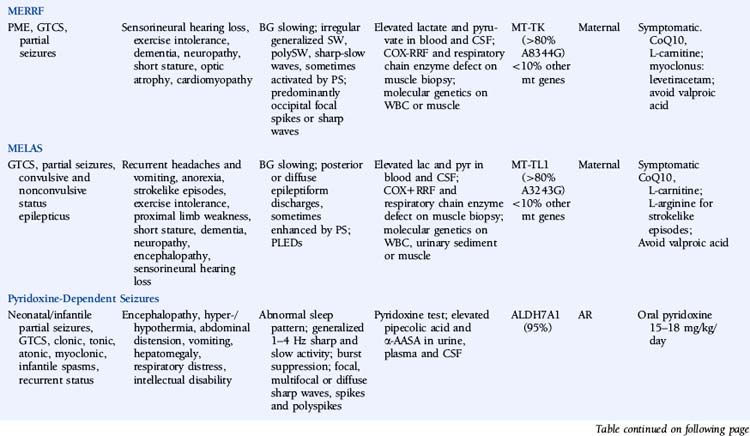
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF)
The disease is characterized by myoclonus, which is often the first symptom, followed by epilepsy and ataxia. Seizures are usually generalized myoclonic or tonic-clonic, but partial seizures have been reported in atypical cases.40 Other common manifestations include sensorineural hearing loss, exercise intolerance, dementia, peripheral neuropathy, short stature, optic atrophy, and cardiomyopathy. Pigmentary retinopathy, ophthalmoparesis, pyramidal signs, and multiple lipomas are occasionally observed. The disease manifestations are very heterogeneous, and overlap syndromes between MERRF and mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) have been reported.41
Lactate and pyruvate levels in blood and cerebrospinal fluid (CSF) are commonly elevated at rest and increase excessively after moderate activity. EEG findings include slowing of background activity and bursts of atypical, irregular generalized spike-wave complexes, polyspike-wave complexes or sharp-slow wave complexes, sometimes activated by photic stimulation.40,42,43 These discharges are often, but not always, related to generalized myoclonic jerks. Focal spikes or sharp waves are also seen, most commonly over the occipital regions. Brain MRI often shows brain atrophy and basal ganglia calcification. Muscle biopsy shows typical cytochrome oxidase (COX) negative ragged-red fibers. Biochemical studies in muscle usually show defects in respiratory chain enzyme activity, especially COX deficiency, but may occasionally be normal. The diagnosis is confirmed by molecular genetic testing, demonstrating mutations in the mitochondrial DNA (mtDNA) gene MT-TK, encoding tRNALys. Over 80% of affected individuals with typical findings carry the A8344G mutation.44 Three additional mutations (T8356C, G8363A, and G8361A) account for about 10% of affected individuals. The mutations appear to exert their pathogenic effects through impairment of protein synthesis.45 The remaining 10% of affected individuals may have other mutations in MT-TK or mutations in a number of other mitochondrial genes. Mutations are usually present in all tissues and can thus be detected in mtDNA from blood leukocytes. However, the occurrence of heteroplasmy (differences in cellular mutational load) can result in variations of tissue distribution of mutated mtDNA. Hence, in some cases the mutation may be undetectable in leukocytes and may only be detected in other tissues, most reliably in skeletal muscle. For the same reason, accurate prediction of phenotype in oligosymptomatic individuals or at-risk family members based on test results is not possible.
There is no specific treatment for the disease. Empirical treatment with coenzyme Q10 (50 to 100 mg 3x/day) and L-carnitine (1000 mg 3×/day), aiming to improve mitochondrial function, is often used.46 No controlled studies have compared the efficacy of various AEDs in MERRF. Valproic acid should be used with caution because of the increased risk of hepatotoxicity.47 A number of reports have described substantial improvement of myoclonus with levetiracetam.48,49 Potential future treatments for MERRF include selective inhibition of mutant mtDNA replication by peptide nucleic acids and import of nuclear-encoded tRNALys into mitochondria.50,51
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes (MELAS)
MELAS is another disorder caused by mutations in mtDNA and thus is transmitted by maternal inheritance. Onset is typically in childhood, though infantile and adult onset has been reported. The most common presenting symptoms are seizures, recurrent headaches, anorexia, and recurrent vomiting. Seizures can be generalized or partial and are often associated with strokelike episodes of transient hemiparesis or cortical blindness. Generalized convulsive and complex partial status epilepticus, as well as epilepsia partialis continua, have been reported.52–54 Other common symptoms and signs include exercise intolerance, proximal limb weakness, short stature, encephalopathy, dementia, sensorineural hearing loss, and peripheral neuropathy. Less-common symptoms include myoclonus, ataxia, episodic coma, optic atrophy, cardiomyopathy, pigmentary retinopathy, ophthalmoplegia, diabetes mellitus, hirsutism, gastrointestinal dysmotility, and nephropathy. MELAS should be suspected based on the following features: (1) strokelike episodes before age 40; (2) encephalopathy with seizures and/or dementia; and (3) lactic acidosis and/or ragged-red fibers on muscle biopsy, or both. The diagnosis may be confirmed if at least two of the following are also present: normal early psychomotor development, recurrent headache, or recurrent vomiting.55 There is a wide variability in clinical presentation, and overlap syndromes between MELAS and other mitochondrial disorders have been reported.
Lactate and pyruvate levels in blood and CSF are commonly elevated at rest and increase excessively after moderate activity. EEG commonly shows slowing of alpha-rhythm and posterior or diffuse epileptiform discharges, sometimes enhanced by photic stimulation.40,56–58 Periodic lateralized epileptiform discharges (PLEDs) with alternating focus have been reported during episodes of complex partial status epilepticus.52,59 Basal ganglia calcifications are commonly seen on computed tomography (CT). Brain MRI during strokelike episodes shows cortico-subcortical T2- and FLAIR-hyperintense lesions in the posterior regions that slowly spread in the weeks following initial symptoms.60–62 Diffusion-weighted MRI (DWI) demonstrates increased apparent diffusion coefficient (ADC) values in these lesions, distinguishing them from classic ischemic strokes.61 Muscle biopsy typically shows COX-positive ragged red fibers and an abundance of mitochondria. Biochemical analysis of respiratory chain enzymes in muscle usually shows multiple partial defects, but it can also be normal. The diagnosis is confirmed by molecular genetic testing, demonstrating mutations in the mtDNA gene MT-TL1, encoding tRNALeu. tRNALeu is essential for mitochondrial protein synthesis, specifically for the incorporation of leucine into nascent proteins. The A3243G mutation is present in over 80% of individuals with typical clinical findings.63 The remainder of cases are caused by other mutations in MT-TL1 or mutations in other mtDNA genes, most commonly MT-ND5. Mutations are usually present in all tissues and can thus be detected in leukocytes. However, the percentage of mutated mtDNA decreases in blood with age in patients harboring the A3243G mutation,64 and in some cases the pathogenic mutation may be undetectable in leukocytes because of heteroplasmy. In such cases, urinary sediment has proven the most useful among accessible tissues for detecting the A3243G mutation.65,66 A muscle biopsy is recommended in the rare instance in which the MT-TL1 A3243G mutation cannot be detected by standard techniques in leukocytes or urinary sediment from an individual with classic MELAS. Accurate prediction of phenotype in oligosymptomatic individuals or at-risk family members based on genetic test results is not possible.
No specific treatment for MELAS exists. Coenzyme Q10 (50 to 100 mg 3×/day) and L-carnitine (1000 mg 3×/day) may be of some benefit. L-arginine has been reported to improve strokelike symptoms when given in the acute phase and to decrease frequency and severity of strokelike episodes when given between episodes.67,68 Beneficial effects of oral succinate were reported in one individual.69 Seizures usually respond to traditional AEDs, though valproic acid may aggravate seizures.70,71
Pyridoxine-Dependent Seizures
The hallmark of the disease consists of intractable seizures not controlled with AEDs but responding to pyridoxine. Seizures classically present soon after birth, but onset up to the age of 3 years has been reported.72–74 Seizures may be partial, generalized tonic-clonic, clonic, tonic, atonic, myoclonic, or infantile spasms. Prolonged seizures and recurrent episodes of status epilepticus are typical. Periods of encephalopathy frequently precede seizures. Systemic manifestations may also occur, including hyper- or hypothermia, abdominal distension, vomiting, hepatomegaly, respiratory distress with hypoxemia, and metabolic acidosis, all of which resolve with pyridoxine treatment.75 Intellectual disability is common. Several atypical cases have been reported in the literature, including seizures that initially respond to AEDs and then become intractable,76 seizures that only respond to pyridoxine after several months,77 and seizure-free intervals of up to several months after discontinuation of pyridoxine.74,78
EEG background activity may be normal or abnormal, but normal sleep patterns are absent. Other EEG abnormalities include generalized bursts of 1 to 4 Hz sharp and slow activity, burst suppression patterns, and focal, multifocal, or diffuse sharp waves, spikes, and polyspikes.75,79,80 The interictal EEG may remain normal in patients with later onset. Brain imaging shows a variety of abnormalities.81,82 The most typical structural abnormality is hypoplasia of the posterior part of the corpus callosum. Other findings include cerebellar hypoplasia with megacisterna magna and hydrocephalus. Untreated seizures can be associated with intraventricular and/or subarachnoid hemorrhage and white matter changes, which may be partially reversible.83,84
The diagnosis can be established on a clinical basis by administering pyridoxine 100 mg intravenously while monitoring the EEG. In individuals with pyridoxine-dependent seizures, clinical seizures generally cease over several minutes. If a clinical response is not demonstrated after 10 minutes, the dose should be repeated. In some patients, doses exceeding 500 mg have been required.85,86 A corresponding change should be observed in the EEG, though in some cases the response may be delayed by several hours. Close systemic monitoring is essential, as neurologic and cardiorespiratory depression following this trial has been reported in some cases.77,84,87 In older children or during the interictal phase, pyridoxine can be administered orally at 15 to 30 mg/kg/day. In individuals with pyridoxine-dependent seizures, clinical seizures should cease within a week.
Elevated levels of pipecolic acid and α-aminoadipic semialdehyde (α-AASA) in urine, plasma, and CSF can be used as biomarkers for the disease.88,89
Mutations in the gene ALDH7A1, encoding antiquitin (ATQ1), are responsible for the disease in 95% of typical cases with elevated urinary α-AASA.88,89 ATQ-1 is an aldehyde dehydrogenase. The mutations result in absent or strongly reduced α-AASA dehydrogenase enzyme activity, leading to increased levels of Δ1-piperideine-6-carboxylate (P6C). P6C, in turn, condenses with and inactivates pyridoxal 5′-phosphate (PLP), a cofactor of glutamic acid decarboxylase (GAD). This probably results in abnormal metabolism of neurotransmitters. At least one family not linked to the 5q31 locus that harbors the ALDH7A1 gene has been reported, indicating genetic heterogeneity.90
The main prognostic factor is age at onset: patients presenting after 1 month of age do considerably better than those with earlier onsets.75 The question of whether early initiation of treatment improves outcome is currently a matter of debate.86,91 The recommended daily dose of oral pyridoxine is 15 to 18 mg/kg, with a maximum of 500 mg/day.75 Once seizures are controlled, all AEDs can be withdrawn, and seizure control will be maintained on pyridoxine monotherapy. Because the disease manifestations may exacerbate during intercurrent illnesses such as gastroenteritis or respiratory infection, it is recommended to double the daily dose of pyridoxine for several days until the acute illness resolves. Therapy should be maintained life-long. For couples who have a child with the disorder, it has been recommended that mothers take 50 to 100 mg of pyridoxine per day during the last half of subsequent pregnancies to reduce the severity of intellectual impairment in a possibly affected fetus.80,91
Glucose Transporter Type 1 (Glut1) Deficiency Syndrome
Glut1-deficiency syndrome is an autosomal dominant disorder usually presenting as an infantile-onset epileptic encephalopathy. Children are normal at birth and start presenting refractory seizures between ages 1 to 18 months.92 Other symptoms include deceleration of head growth, psychomotor retardation, ataxia, and spasticity. Apneic episodes, behavioral arrests, and abnormal episodic eye movements simulating opsoclonus may precede the onset of seizures by several months.93 Seizures can be generalized tonic and/or clonic, atypical absences, partial, myoclonic, or astatic.92 More rarely seizures may be absent.94,95 Other paroxysmal events including intermittent ataxia, confusion, lethargy, or somnolence; alternating hemiparesis, abnormalities of movement or posture such as myoclonus and dystonia, total body paralysis, sleep disturbances, and recurrent headaches have also been described.95 It is unclear whether these events represent epileptic or nonepileptic phenomena. Dysarthria and predominantly expressive dysphasia are common. Most patients exhibit abnormal movements, ranging from motor restlessness to dystonia. Neurologic symptoms generally fluctuate and may be triggered by fasting or fatigue. Rare cases diagnosed in adulthood have been reported.96
EEG abnormalities include generalized 2- to 4-Hz spike-wave discharges, generalized slowing or attenuation, focal epileptiform discharges, and focal slowing or attenuation. A significant proportion of cases has normal interictal EEGs.92,97,98 A reduction of epileptiform discharges during postprandial EEG recording has been reported in some cases.98,99 Brain imaging usually shows no structural abnormalities.95,100 Cerebral fluorodeoxyglucose-positron emission tomography (FDG-PET) reveals a global decrease in glucose uptake with relative preservation of basal ganglia metabolism.100
The biochemical hallmark of the disease is hypoglycorrhachia with normal blood glucose levels, measured following a 4-hour fast. The absolute CSF glucose concentration seldom, if ever, exceeds 40 mg/dL. The ratio of CSF glucose concentration to blood glucose concentration is about 0.33. CSF lactate concentration is low to normal.95 The diagnosis is supported by the presence of reduced 3-0-methyl-D-glucose (3-OMG) uptake in erythrocytes, although normal values have been reported.101,102
Glut1-deficiency syndrome is caused by mutations in the gene SLC2A1 (solute carrier family 2, facilitated glucose transporter member 1).103 Mutations are detected in about 80% of affected individuals. Whole-gene deletions have also been reported.103–105 Most mutations occur de novo. Glut1 is the major glucose transporter in the mammalian blood–brain barrier, responsible for glucose entry into the brain. It is selectively expressed in brain endothelial cells, astroglia, and erythrocytes.106
The seizures are usually refractory to conventional AEDs. The ketogenic diet is highly effective in controlling the seizures and is well tolerated in most cases; however, neurobehavioral and cognitive deficits persist.107 Seizures sometimes recur despite the diet.108,109 Barbiturates, diazepam, and chloral hydrate are known to inhibit transport of glucose and thus should be avoided.109,110 Methylxanthines (e.g., caffeine) and ethanol also inhibit glucose transport by Glut1 and may worsen symptoms of Glut1-deficiency syndrome.111,112
1. Nardocci N, Verga ML, Binelli S, Zorzi G, Angelini L, Bugiani O. Neuronal ceroid-lipofuscinosis: a clinical and morphological study of 19 patients. Am J Med Genet. 1995;57:137-141.
2. Sadzot B, Reznik M, Arrese-Estrada JE, Franck G. Familial Kufs’ disease presenting as a progressive myoclonic epilepsy. J Neurol. 2000;247:447-454.
3. Gambardella A, Pasquinelli G, Cittadella R, et al. Kufs’ disease presenting as late-onset epilepsia partialis continua. Neurology. 1998;51:1180-1182.
4. Ranta S, Lehesjoki AE. Northern epilepsy, a new member of the NCL family. Neurol Sci. 2000;21:S43-S47.
5. Sinha S, Satishchandra P, Santosh V, Gayatri N, Shankar SK. Neuronal ceroid lipofuscinosis: a clinicopathological study. Seizure. 2004;13:235-240.
6. Taratuto AL, Saccoliti M, Sevlever G, et al. Childhood neuronal ceroid-lipofuscinoses in Argentina. Am J Med Genet. 1995;57:144-149.
7. Veneselli E, Biancheri R, Perrone MV, Buoni S, Fois A. Neuronal ceroid lipofuscinoses: clinical and EEG findings in a large study of Italian cases. Neurol Sci. 2000;21:S75-81.
8. D’Incerti L. MRI in neuronal ceroid lipofuscinosis. Neurol Sci. 2000;21:S71-73.
10. Hobert JA, Dawson G. Neuronal ceroid lipofuscinoses therapeutic strategies: past, present and future. Biochim Biophys Acta. 2006;1762:945-953.
11. Aberg L, Heiskala H, Vanhanen SL, et al. Lamotrigine therapy in infantile neuronal ceroid lipofuscinosis (INCL). Neuropediatrics. 1997;28:77-79.
12. Aberg L, Kirveskari E, Santavuori P. Lamotrigine therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia. 1999;40:796-799.
13. Aberg LE, Backman M, Kirveskari E, Santavuori P. Epilepsy and antiepileptic drug therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia. 2000;41:1296-1302.
14. Philippart M. Diagnosis and treatment of typical and atypical forms of lipopigment storage disorders. Am J Med Genet Suppl. 1988;5:291-298.
15. Philippart M, Messa C, Chugani HT. Spielmeyer-Vogt (Batten, Spielmeyer-Sjögren) disease. Distinctive patterns of cerebral glucose utilization. Brain. 1994;117(Pt 5):1085-1092.
16. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249-254.
17. Poorthuis BJ, Wevers RA, Kleijer WJ, et al. The frequency of lysosomal storage diseases in the Netherlands. Hum Genet. 1999;105:151-156.
18. Patterson MC, Horowitz M, Abel RB, et al. Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher’s disease. Neurology. 1993;43:1993-1997.
19. Filocamo M, Mazzotti R, Stroppiano M, Grossi S, Dravet C, Guerrini R. Early visual seizures and progressive myoclonus epilepsy in neuronopathic Gaucher disease due to a rare compound heterozygosity (N188S/S107L). Epilepsia. 2004;45:1154-1157.
20. Tuzun E, Baykan B, Gurses C, Gokyigit A. Long-term follow-up of electroencephalographic and clinical findings of a case with Gaucher’s disease type 3a. Seizure. 2000;9:469-472.
21. Nishimura R, Omos-Lau N, Ajmone-Marsan C, Barranger JA. Electroencephalographic findings in Gaucher disease. Neurology. 1980;30:152-159.
22. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005;129:178-188.
23. Tsuji S, Choudary PV, Martin BM, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316:570-575.
24. Park JK, Orvisky E, Tayebi N, et al. Myoclonic epilepsy in Gaucher disease: genotype-phenotype insights from a rare patient subgroup. Pediatr Res. 2003;53:387-395.
25. Altarescu G, Hill S, Wiggs E, et al. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher’s disease. J Pediatr. 2001;138:539-547.
26. Erikson A, Forsberg H, Nilsson M, Astrom M, Mansson JE. Ten years’ experience of enzyme infusion therapy of Norrbottnian (type 3) Gaucher disease. Acta Paediatr. 2006;95:312-317.
27. Vellodi A, Bembi B, de Villemeur TB, et al. Management of neuronopathic Gaucher disease: a European consensus. J Inherit Metab Dis. 2001;24:319-327.
28. Frei KP, Schiffmann R. Myoclonus in Gaucher disease. Adv Neurol. 2002;89:41-48.
29. Capablo JL, Franco R, de Cabezon AS, Alfonso P, Pocovi M, Giraldo P. Neurologic improvement in a Type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia. 2007;48:1406-1408.
30. Steinman L, Tharp BR, Dorfman LJ, et al. Peripheral neuropathy in the cherry-red spot-myoclonus syndrome (sialidosis type I). Ann Neurol. 1980;7:450-456.
31. Panzica F, Canafoglia L, Franceschetti S, et al. Movement-activated myoclonus in genetically defined progressive myoclonic epilepsies: EEG-EMG relationship estimated using autoregressive models. Clin Neurophysiol. 2003;114:1041-1052.
32. Tobimatsu S, Fukui R, Shibasaki H, Kato M, Kuroiwa Y. Electrophysiological studies of myoclonus in sialidosis type 2. Electroencephalogr Clin Neurophysiol. 1985;60:16-22.
33. Palmeri S, Villanova M, Malandrini A, et al. Type I sialidosis: a clinical, biochemical and neuroradiological study. Eur Neurol. 2000;43:88-94.
34. Loren DJ, Campos Y, d’Azzo A, et al. Sialidosis presenting as severe nonimmune fetal hydrops is associated with two novel mutations in lysosomal alpha-neuraminidase. J Perinatol. 2005;25:491-494.
35. Pattison S, Pankarican M, Rupar CA, Graham FL, Igdoura SA. Five novel mutations in the lysosomal sialidase gene (NEU1) in type II sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum Mutat. 2004;23:32-39.
36. Seyrantepe V, Poupetova H, Froissart R, Zabot MT, Maire I, Pshezhetsky AV. Molecular pathology of NEU1 gene in sialidosis. Hum Mutat. 2003;22:343-352.
37. Bonten EJ, Arts WF, Beck M, et al. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum Mol Genet. 2000;9:2715-2725.
38. Oheda Y, Kotani M, Murata M, et al. Elimination of abnormal sialylglycoproteins in fibroblasts with sialidosis and galactosialidosis by normal gene transfer and enzyme replacement. Glycobiology. 2006;16:271-280.
39. Gascon G, Wallenberg B, Daif AK, Ozand P. Successful treatment of cherry red spot-myoclonus syndrome with 5-hydroxytryptophan. Ann Neurol. 1988;24:453-455.
40. Canafoglia L, Franceschetti S, Antozzi C, et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. 2001;56:1340-1346.
41. Zeviani M, Muntoni F, Savarese N, et al. A MERRF/MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA (Lys) gene. Eur J Hum Genet. 1993;1:80-87.
42. Serra G, Piccinnu R, Tondi M, Muntoni F, Zeviani M, Mastropaolo C. Clinical and EEG findings in eleven patients affected by mitochondrial encephalomyopathy with MERRF-MELAS overlap. Brain Dev. 1996;18:185-191.
43. So N, Berkovic S, Andermann F, Kuzniecky R, Gendron D, Quesney LF. Myoclonus epilepsy and ragged-red fibres (MERRF). 2. Electrophysiological studies and comparison with other progressive myoclonus epilepsies. Brain. 1989;112(Pt 5):1261-1276.
44. Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931-937.
45. Masucci JP, Davidson M, Koga Y, Schon EA, King MP. In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNA(Lys) gene: two genotypes produce similar phenotypes. Mol Cell Biol. 1995;15:2872-2881.
46. DiMauro S, Hirano M, Schon EA. Mitochondrial encephalomyopathies: therapeutic approaches. Neurol Sci. 2000;21:S901-908.
47. Krahenbuhl S, Brandner S, Kleinle S, Liechti S, Straumann D. Mitochondrial diseases represent a risk factor for valproate-induced fulminant liver failure. Liver. 2000;20:346-348.
48. Crest C, Dupont S, Leguern E, Adam C, Baulac M. Levetiracetam in progressive myoclonic epilepsy: an exploratory study in 9 patients. Neurology. 2004;62:640-643.
49. Mancuso M, Galli R, Pizzanelli C, Filosto M, Siciliano G, Murri L. Antimyoclonic effect of levetiracetam in MERRF syndrome. J Neurol Sci. 2006;243:97-99.
50. Kolesnikova OA, Entelis NS, Jacquin-Becker C, et al. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum Mol Genet. 2004;13:2519-2534.
51. Taylor RW, Chinnery PF, Turnbull DM, Lightowlers RN. Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nat Genet. 1997;15:212-215.
52. Leff AP, McNabb AW, Hanna MG, Clarke CR, Larner AJ. Complex partial status epilepticus in late-onset MELAS. Epilepsia. 1998;39:438-441.
53. Montagna P, Gallassi R, Medori R, et al. MELAS syndrome: characteristic migrainous and epileptic features and maternal transmission. Neurology. 1988;38:751-754.
54. Veggiotti P, Colamaria V, Dalla Bernardina B, Martelli A, Mangione D, Lanzi G. Epilepsia partialis continua in a case of MELAS: clinical and neurophysiological study. Neurophysiol Clin. 1995;25:158-166.
55. Hirano M, Ricci E, Koenigsberger MR, et al. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2:125-135.
56. Fujimoto S, Mizuno K, Shibata H, et al. Serial electroencephalographic findings in patients with MELAS. Pediatr Neurol. 1999;20:43-48.
57. Majamaa-Voltti KA, Winqvist S, Remes AM, et al. A 3-year clinical follow-up of adult patients with 3243A>G in mitochondrial DNA. Neurology. 2006;66:1470-1475.
58. Tulinius MH, Hagne I. EEG findings in children and adolescents with mitochondrial encephalomyopathies: a study of 25 cases. Brain Dev. 1991;13:167-173.
59. Corda D, Rosati G, Deiana GA, Sechi G. “Erratic” complex partial status epilepticus as a presenting feature of MELAS. Epilepsy Behav. 2006;8:655-658.
60. Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61:1238-1244.
61. Kolb SJ, Costello F, Lee AG, et al. Distinguishing ischemic stroke from the stroke-like lesions of MELAS using apparent diffusion coefficient mapping. J Neurol Sci. 2003;216:11-15.
62. Yonemura K, Hasegawa Y, Kimura K, Minematsu K, Yamaguchi T. Diffusion-weighted MR imaging in a case of mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. AJNR Am J Neuroradiol. 2001;22:269-272.
63. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651-653.
64. Pyle A, Taylor RW, Durham SE, et al. Depletion of mitochondrial DNA in leucocytes harbouring the 3243A-G mtDNA mutation. J Med Genet. 2007;44:69-74.
65. McDonnell MT, Schaefer AM, Blakely EL, et al. Noninvasive diagnosis of the 3243AG mitochondrial DNA mutation using urinary epithelial cells. Eur J Hum Genet. 2004;12:778-781.
66. Shanske S, Pancrudo J, Kaufmann P, et al. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A. 2004;130:134-137.
67. Koga Y, Akita Y, Nishioka J, et al. MELAS and L-arginine therapy. Mitochondrion. 2007;7:133-139.
68. Kubota M, Sakakihara Y, Mori M, Yamagata T, Momoi-Yoshida M. Beneficial effect of L-arginine for stroke-like episode in MELAS. Brain Dev. 2004;26:481-483. discussion 480
69. Oguro H, Iijima K, Takahashi K, et al. Successful treatment with succinate in a patient with MELAS. Intern Med. 2004;43:427-431.
70. Lam CW, Lau CH, Williams JC, Chan YW, Wong LJ. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) triggered by valproate therapy. Eur J Pediatr. 1997;156:562-564.
71. Lin CM, Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab Brain Dis. 2007;22:105-109.
72. Bachman DS. Late-onset pyridoxine-dependency convulsions. Ann Neurol. 1983;14:692-693.
73. Coker SB. Postneonatal vitamin B6-dependent epilepsy. Pediatrics. 1992;90:221-223.
74. Goutieres F, Aicardi J. Atypical presentations of pyridoxine-dependent seizures: a treatable cause of intractable epilepsy in infants. Ann Neurol. 1985;17:117-120.
75. Baxter P. Pyridoxine-dependent and pyridoxine-responsive seizures. Dev Med Child Neurol. 2001;43:416-420.
76. Gospe SMJr, Olin KL, Keen CL. Reduced GABA synthesis in pyridoxine-dependent seizures. Lancet. 1994;343:1133-1134.
77. Bass NE, Wyllie E, Cohen B, Joseph SA. Pyridoxine-dependent epilepsy: the need for repeated pyridoxine trials and the risk of severe electrocerebral suppression with intravenous pyridoxine infusion. J Child Neurol. 1996;11:422-424.
78. Bankier A, Turner M, Hopkins IJ. Pyridoxine dependent seizures—a wider clinical spectrum. Arch Dis Child. 1983;58:415-418.
79. Mikati MA, Trevathan E, Krishnamoorthy KS, Lombroso CT. Pyridoxine-dependent epilepsy: EEG investigations and long-term follow-up. Electroencephalogr Clin Neurophysiol. 1991;78:215-221.
80. Nabbout R, Soufflet C, Plouin P, Dulac O. Pyridoxine dependent epilepsy: a suggestive electroclinical pattern. Arch Dis Child Fetal Neonatal Ed. 1999;81:F125-F129.
81. Baxter P, Griffiths P, Kelly T, Gardner-Medwin D. Pyridoxine-dependent seizures: demographic, clinical, MRI and psychometric features, and effect of dose on intelligence quotient. Dev Med Child Neurol. 1996;38:998-1006.
82. Gospe SMJr, Hecht ST. Longitudinal MRI findings in pyridoxine-dependent seizures. Neurology. 1998;51:74-78.
83. Jardim LB, Pires RF, Martins CE, et al. Pyridoxine-dependent seizures associated with white matter abnormalities. Neuropediatrics. 1994;25:259-261.
84. Tanaka R, Okumura M, Arima J, Yamakura S, Momoi T. Pyridoxine-dependent seizures: report of a case with atypical clinical features and abnormal MRI scans. J Child Neurol. 1992;7:24-28.
85. Clarke TA, Saunders BS, Feldman B. Pyridoxine-dependent seizures requiring high doses of pyridoxine for control. Am J Dis Child. 1979;133:963-965.
86. Haenggeli CA, Girardin E, Paunier L. Pyridoxine-dependent seizures, clinical and therapeutic aspects. Eur J Pediatr. 1991;150:452-455.
87. Kroll JS. Pyridoxine for neonatal seizures: an unexpected danger. Dev Med Child Neurol. 1985;27:377-379.
88. Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307-309.
89. Plecko B, Paul K, Paschke E, et al. Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat. 2007;28:19-26.
90. Bennett CL, Huynh HM, Chance PF, Glass IA, Gospe SMJr. Genetic heterogeneity for autosomal recessive pyridoxine-dependent seizures. Neurogenetics. 2005;6:143-149.
91. Baxter P, Aicardi J. Neonatal seizures after pyridoxine use. Lancet. 1999;354:2082-2083.
92. Leary LD, Wang D, Nordli DRJr, Engelstad K, De Vivo DC. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia. 2003;44:701-707.
93. De Vivo DC, Leary L, Wang D. Glucose transporter 1 deficiency syndrome and other glycolytic defects. J Child Neurol. 2002;17(Suppl 3):3S15. 3S1523, discussion 13S24-15
94. Overweg-Plandsoen WC, Groener JE, Wang D, et al. GLUT-1 deficiency without epilepsy—an exceptional case. J Inherit Metab Dis. 2003;26:559-563.
95. Wang D, Pascual JM, Yang H, et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005;57:111-118.
96. Klepper J, Willemsen M, Verrips A, et al. Autosomal dominant transmission of GLUT1 deficiency. Hum Mol Genet. 2001;10:63-68.
97. Boles RG, Seashore MR, Mitchell WG, Kollros PR, Mofidi S, Novotny EJ. Glucose transporter type 1 deficiency: a study of two cases with video-EEG. Eur J Pediatr. 1999;158:978-983.
98. von Moers A, Brockmann K, Wang D, et al. EEG features of glut-1 deficiency syndrome. Epilepsia. 2002;43:941-945.
99. Ito Y, Gertsen E, Oguni H, et al. Clinical presentation, EEG studies, and novel mutations in two cases of GLUT1 deficiency syndrome in Japan. Brain Dev. 2005;27:311-317.
100. Pascual JM, Van Heertum RL, Wang D, Engelstad K, De Vivo DC. Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol. 2002;52:458-464.
101. Fujii T, Ho YY, Wang D, et al. Three Japanese patients with glucose transporter type 1 deficiency syndrome. Brain Dev. 2007;29:92-97.
102. Klepper J, Garcia-Alvarez M, O’Driscoll KR, et al. Erythrocyte 3-O-methyl-D-glucose uptake assay for diagnosis of glucose-transporter-protein syndrome. J Clin Lab Anal. 1999;13:116-121.
103. Seidner G, Alvarez MG, Yeh JI, et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998;18:188-191.
104. Vermeer S, Koolen DA, Visser G, et al. A novel microdeletion in 1(p34.2p34.3), involving the SLC2A1 (GLUT1) gene, and severe delayed development. Dev Med Child Neurol. 2007;49:380-384.
105. Wang D, Kranz-Eble P, De Vivo DC. Mutational analysis of GLUT1 (SLC2A1) in Glut-1 deficiency syndrome. Hum Mutat. 2000;16:224-231.
106. Pardridge WM, Boado RJ, Farrell CR. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J Biol Chem. 1990;265:18035-18040.
107. Klepper J, Leiendecker B, Bredahl R, et al. Introduction of a ketogenic diet in young infants. J Inherit Metab Dis. 2002;25:449-460.
108. Klepper J, Scheffer H, Leiendecker B, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005;36:302-308.
109. Klepper J, Fischbarg J, Vera JC, Wang D, De Vivo DC. GLUT1-deficiency: barbiturates potentiate haploinsufficiency in vitro. Pediatr Res. 1999;46:677-683.
110. Klepper J, Florcken A, Fischbarg J, Voit T. Effects of anticonvulsants on GLUT1-mediated glucose transport in GLUT1 deficiency syndrome in vitro. Eur J Pediatr. 2003;162:84-89.
111. Ho YY, Yang H, Klepper J, Fischbarg J, Wang D, De Vivo DC. Glucose transporter type 1 deficiency syndrome (Glut1DS): methylxanthines potentiate GLUT1 haploinsufficiency in vitro. Pediatr Res. 2001;50:254-260.
112. Krauss SW, Diamond I, Gordon AS. Selective inhibition by ethanol of the type 1 facilitative glucose transporter (GLUT1). Mol Pharmacol. 1994;45:1281-1286.