Chapter 26 Epidemiology and Pathophysiology of Mesenteric Vascular Disease
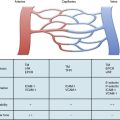
Severe acute intestinal ischemia results from sudden symptomatic reduction in intestinal blood flow of sufficient magnitude to potentially result in intestinal infarction.1 Acute ischemia of the small bowel and/or right colon may result from mesenteric arterial occlusion (embolus or thrombosis), mesenteric venous occlusion, and nonocclusive processes, especially vasospasm (Fig. 26-1). Isolated dissections of the superior mesenteric artery (SMA), either in association with cystic medial degeneration of the arteries or, more commonly, as progression of an existing dissection in the descending thoracic aorta into the SMA and celiac artery, may also result in acute intestinal ischemia.
Acute Arterial Occlusive Mesenteric Ischemia
Acute Mesenteric Arterial Embolism
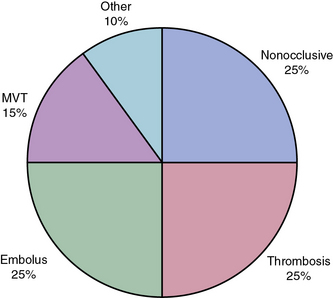
Roughly 25% of all cases of acute mesenteric ischemia are due to emboli to the SMA, 25% of cases are due to thrombosis of preexisting atherosclerotic lesions, and the remaining 50% are due to a variety of other etiologies.2 Mesenteric emboli can originate from left atrial or ventricular mural thrombi or from cardiac valvular lesions.1 These thrombi are most often associated with cardiac dysrhythmias such as atrial fibrillation, global myocardial dysfunction with poor ejection fraction, or discrete hypokinetic regions produced by previous myocardial infarction (MI).3 About 15% of emboli lodge at the origin of the SMA, but the majority lodge 3 to 10 cm distally in the tapered segment of the SMA just past the origin of the middle colic artery.2 More than 20% of emboli to the SMA are associated with concurrent emboli to another arterial bed.4 Intestinal ischemia due to embolic arterial occlusion can be compounded by reactive mesenteric vasospasm, further reducing collateral flow and exacerbating the ischemic insult.2,5
Acute Mesenteric Arterial Thrombosis
Thrombosis of the SMA or the celiac artery is usually associated with preexisting critical stenoses. Many of these patients have histories consistent with chronic mesenteric ischemia (CMI), including postprandial pain, weight loss, “food fear,” and early satiety.1,6 Superior mesenteric artery thrombosis can be regarded as a complication of untreated chronic intestinal ischemia.1 The SMA plaque likely progresses slowly to a critical stenosis over years until thrombosis occurs.
Unlike embolic occlusions, thrombosis of the SMA generally occurs flush with the aortic origin of the vessel. Aortic dissection involving the visceral vessels, though rare, can cause acute mesenteric ischemia. The intimal flap of the dissection can extend into, compress, or exclude the mesenteric orifice.5 Acute mesenteric ischemia is also an uncommon (< 1%) but serious complication of cardiac surgery, with a reported mortality rate of greater than 50% in most series.5,7 Presumably, the nonpulsatile perfusion delivered by most extracorporeal circuits allows severely stenotic visceral vessels to occlude during cardiopulmonary bypass. Identified risk factors for this complication include prolonged cross-clamp times, use of intraaortic balloon counterpulsation, low cardiac output syndromes, blood transfusion, triple-vessel disease, coronary artery disease (CAD), and peripheral artery disease (PAD).7
Pathophysiology of Occlusive Acute Mesenteric Ischemia
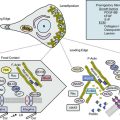
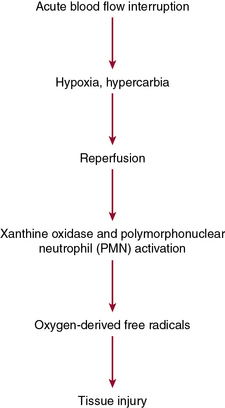
Acute mesenteric ischemia, whether the underlying cause is embolic or thrombotic, may eventually lead to intestinal infarction (Fig. 26-2). Hypoxia and hypercarbia that occur during flow interruption, and reperfusion injury once intestinal blood flow is restored, all contribute to tissue loss8 (Fig. 26-3). Reperfusion injury is believed to be principally mediated by activation of the enzyme xanthine oxidase and recruitment and activation of circulating neutrophils (PMNs).9 The mechanism of injury likely involves production of oxygen-derived free radicals by xanthine oxidase that then causes profound local tissue injury through lipid peroxidation, membrane disruption, and increased microvascular permeability.5,9 The ischemic endothelium recruits PMNs in an autocrine and paracrine manner by secreting chemotactic cytokines (tumor necrosis factor [TNF]-α, interleukin [IL]-1, platelet-derived growth factor [PDGF]) that perpetuate further damage to the reperfused tissue. Once activated, PMNs degranulate, releasing myeloperoxidase, collagenases, and elastases that further injure already ischemic and vulnerable tissue.5,10 Activation of this endogenous inflammatory cascade is not restricted to the injured organ and may also have deleterious systemic effects, with cardiac, pulmonary, and other organ system dysfunction.5
Natural History of Acute Mesenteric Arterial Occlusive Disease
The mortality rate for occlusive acute mesenteric ischemia exceeds 70% in most series.1,2,5,11,12 Occlusive acute intestinal ischemia resulting from SMA embolism has a more favorable prognosis than that resulting from SMA thrombosis.1 Survival following acute intestinal ischemia due to SMA thrombosis is rare. The more favorable prognosis associated with embolism is attributable to the fact that most emboli lodge distally in the SMA (beyond the origin of the middle colic artery), thus allowing perfusion of the proximal intestine via middle colic and jejunal artery branches.1 Thrombotic occlusion of the SMA usually occurs proximal to the middle colic artery, and therefore completely interrupts mid-gut arterial perfusion in patients with poorly developed celiac artery or inferior mesenteric artery (IMA) collateral flow.1,3,6
Nonocclusive Mesenteric Ischemia
Epidemiology
Nonocclusive mesenteric ischemia (NOMI) accounts for 25% of all episodes of acute intestinal ischemia.1,5,13,14 In NOMI, microscopic arterial blood flow is inadequate to supply perfusion to the bowel. The result is intestinal ischemia and infarction in the presence of a patent macroscopic vasculature.15 Previous reports15–17 have identified multiple risk factors for development of NOMI (Box 26-1). Mesenteric arterial vasospasm may occur following elective revascularization procedures for chronic SMA occlusion. In such cases, vasoconstriction of small and medium-sized vessels is precipitated by early enteral feeding.18 Without prompt intervention, NOMI may progress from localized intestinal ischemia to transmural infarction, peritonitis, and death.19,20 Mortality is high regardless of treatment, owing to the underlying medical conditions that precipitate NOMI and frequent delays in diagnosis.5,13–15
 Box 26-1
Box 26-1Pathophysiology
Nonocclusive mesenteric ischemia was first recognized in autopsies of patients with small-intestinal gangrene in the absence of arterial or venous occlusion.21,22 Investigation of the regulatory mechanisms of mesenteric circulation has demonstrated that the pathophysiology of NOMI is multifactorial. Virtually all patients with NOMI have a severe coexisting illness, commonly severe cardiac failure.23 It is postulated that hypoperfusion from cardiac failure, resulting in peripheral hypoxemia and splanchnic vasoconstriction, precipitates intestinal ischemia.13,14 Mesenteric vasoconstriction, intestinal hypoxia, and ischemia-reperfusion injury all contribute to development of NOMI.
Mesenteric vasoconstriction, the hallmark of NOMI, represents an exaggerated homeostatic mechanism induced by excessive sympathetic activity during cardiogenic shock or hypovolemia. The body attempts to maintain cardiac and cerebral perfusion at the expense of splanchnic and peripheral circulations. Experimental evidence suggests that the mediators of this response are endothelin-1 (ET-1), nitric oxide (NO), vasopressin, and angiotensin (Ang).24,25 Endothelin-1 is a potent vasoconstrictor secreted from endothelial cells (ECs); in concert with other vasoactive peptides, it regulates myogenic cells in the vascular wall. Nitric oxide can have paradoxical effects on vascular tone, depending on local concentration.26 At low concentrations it acts as a vasodilator, whereas at higher concentrations it acts as an oxygen-derived free radical, impairing mitochondrial energy production.
The splanchnic autoregulatory system is affected by local arteriolar smooth muscle relaxation and vasodilation, as well as increased cellular oxygen extraction.27 Adequate oxygen delivery may be maintained despite declining perfusion pressures until a critical threshold is reached. In experimental models, maximal extraction is reached at a pressure of 40 mmHg, but beyond this point, oxygen consumption declines, and ischemia ensues.28 Neri et al. recently demonstrated that postoperative cardiac surgery patients who develop NOMI had persistent deficits between oxygen delivery (DO2) and consumption (VO2) because of poor circulatory reserve. In contrast, postoperative cardiac patients who did not develop NOMI were able to normalize their DO2:VO2 ratio by optimizing their cardiac output.14 In the presence of impaired perfusion, blood flow is not evenly distributed in the bowel wall. The mucosa retains its perfusion at the expense of the serosal layers through mucosal production of NO, prostaglandins (PGs), and stimulation of dopamine-I receptors.27 Histological damage is first observed at the villous tip and progresses to the deeper muscularis, submucosa, and mucosa within a few hours.
Once set in motion, mesenteric vasospasm may persist despite correction of the precipitating event.13,14 The etiology of persistent vasoconstriction once adequate blood flow is restored is unknown, but it may respond to direct intraarterial papaverine infusion or other vasodilators, including iloprost.29 This phenomenon of protracted vasoconstriction, however, plays an important role in development and maintenance of occlusive and nonocclusive intestinal ischemia, and may also complicate mesenteric revascularization.18
Use of vasoconstrictor agents and digitalis has been associated with the majority of cases of NOMI. Vasoactive agents, including α-adrenergic drugs and vasopressin, produce splanchnic vasoconstriction directly, whereas digoxin preparations alter mesenteric vasoreactivity by stimulating arterial and venous smooth muscle cell (SMC) contraction.27,29 This may enhance mesenteric arteriolar vasoconstriction in the setting of acute venous hypertension.29
Restoration of blood flow to the ischemic intestine may be complicated by reperfusion injury. During critical ischemia, adenosine triphosphate (ATP) levels are depleted, causing distortion of ATP-dependent cell membrane systems. This results in loss of cellular homeostasis, with cellular swelling and electrolyte imbalances.27 Reduction in ATP levels also generates large amounts of adenosine, a precursor of hypoxanthine. Within the swollen cells, calcium accumulates and triggers hydrolysis of the enzyme xanthine dehydrogenase into xanthine oxidase, which reacts with intracellular hypoxanthine to produce uric acid and toxic oxygen free radicals.5,27 These free radicals exert direct damage to cellular membranes, causing capillary leak syndrome, and incite endogenous inflammatory cascades that cause widespread tissue injury. The deleterious effects of free radicals are usually limited by endogenous scavengers such as glutathione, catalase, superoxide dismutase, and NO.27 However, in cases of prolonged ischemia, the capacity of this scavenger system to eliminate reactive oxygen species (ROS) is exceeded, and continued damage occurs. The degree of reperfusion injury is thus related to the frequency and duration of the ischemic episodes. Clark and Gewertz demonstrated that two short 15-minute periods of low flow followed by reperfusion resulted in a more severe histological injury than a single 30-minute period of ischemia.30,31
In NOMI, a similar scenario exists: hypoperfusion may be partial and occasionally repetitive. It is believed that episodic reperfusion creates a local environment replete with primed neutrophils within the ischemic bed that are capable of degranulating and releasing superoxide. This concept is substantiated by recent experimental evidence that reperfusion injury may be attenuated by reperfusion with leukodepleted blood or by blockade of EC surface receptors for leukocyte adherence.32 In addition, several compounds including N-acetylcysteine and vitamin E have been shown in animal models to reduce tissue damage caused by reactive oxygen species.33 Application of these novel approaches in human NOMI awaits further study.
Mesenteric Venous Thrombosis
Mesenteric venous thrombosis (MVT) refers to thrombosis of the veins draining the intestine (inferior mesenteric, superior mesenteric, splenic, and portal veins). The obstruction in venous return leads to edema, distention, and in some cases, infarction of the affected segments.1,5
Epidemiology
Mesenteric venous thrombosis is a comparatively rare form of mesenteric ischemia. The presentation may vary from asymptomatic to fulminant with intestinal infarction and hemodynamic collapse. Mesenteric venous thrombosis was first described by Elliot in 1895 as “thrombosis of the portomesenteric venous system.”34 In 1935, Warren and Eberhard characterized MVT as a distinct clinical entity. These authors reported a 34% mortality rate following intestinal resection for venous thrombosis.35
Mesenteric venous thrombosis currently constitutes no more than 5% to 15% of all cases of acute mesenteric ischemia.36 Only 372 patients with MVT were reported from 1911 to 1984,37 and MVT accounted for only 6.2% of 1167 patients treated for mesenteric ischemia at the Mayo Clinic from 1972 to 1993.38 Ottinger and Austen found that MVT represented 0.006% of hospital admissions.39 It is estimated that intestinal infarction due to MVT is encountered in less than 1 in 1000 laparotomies for acute abdomen.40
Pathophysiology
Mesenteric venous thrombosis can be classified as primary or secondary. Primary MVT is defined as spontaneous idiopathic thrombosis of mesenteric veins not associated with any other disease or etiological factor.1,5,36 The number of patients in this group has decreased substantially in the past decade because of increased recognition of inherited thrombotic disorders and hypercoagulable states.
Patients in whom an etiological factor can be identified are said to have secondary MVT. Currently, however, a causative factor can be identified in only 35% of patients with MVT.36,38 Known causes of secondary MVT are shown in Box 26-2. Oral contraceptive use accounts for 9% to 18% of episodes of MVT in young women.41 Protein C and protein S deficiency, antithrombin III deficiency, dysfibrinogenemia, abnormal plasminogen, polycythemia vera, thrombocytosis, sickle cell disease (SCD), and factor V Leiden mutation have all been associated with MVT.1,5,36,41,42 Localized secondary MVT has also been reported, most commonly associated with volvulus, intussusception, or mechanical bowel strangulation.
 Box 26-2
Box 26-2Location of the thrombus may be predicted by etiology. Thrombosis due to an intraabdominal cause such as inflammatory conditions or surgery starts in the larger vessels at the site of compression and propagates distally to involve the smaller venous arcades and arcuate channels.41 In contrast, thrombosis due to an underlying hypercoagulable state usually begins in the small vessels and later involves the larger vessels.36,41 Occlusion of the venae rectae and the intramural vessels interferes with adequate venous drainage, with eventual hemorrhagic infarction of the involved bowel segment. The transition from normal to ischemic bowel is usually gradual, unlike that seen with acute embolic or thrombotic occlusion.
Natural History
The natural history of MVT varies based on the etiology. In most cases, it usually does not result in gangrenous bowel.1,5,41,42 Similarly, symptomatic manifestations are diverse. Patients may present with a benign abdominal examination and few symptoms or with profound hemodynamic collapse. Most patients have abdominal pain. Although it can be sudden in onset, most frequently it begins insidiously and worsens over time. Approximately 50% of patients have pain from 5 to 30 days before seeking medical attention, and 27% report abdominal pain for more than 1 month.43 In a recent review, only 16% of patients had severe peritonitis, and 33% required bowel resection.42 Despite improved diagnostic modalities and more aggressive treatment regimens, symptomatic acute MVT is an indicator of poor prognosis, with an approximate 10% to 20% 30-day mortality rate and a 3-year survival rate of 35%.42,44 Patients with evidence of chronic thrombosis fare somewhat better because collateral venous channels form and thereby augment intestinal venous drainage.
Chronic Mesenteric Ischemia
Symptomatic chronic mesenteric arterial insufficiency is a well-described but infrequently encountered clinical problem. Although the earliest report of chronic intestinal ischemia was by Councilman in 1894,45 in 1918, Goodman credited Baccelli as the first individual to correctly associate postprandial pain with CMI. Eighteen years later, Dunphy suggested that the abdominal pain associated with chronic mesenteric arterial occlusion was a possible precursor of later intestinal infarction.46 Later, Shaw and Maynard performed the first successful endarterectomy for CMI.47 Morris et al. described the technique of retrograde aortovisceral bypass in 1962.48
Epidemiology
Chronic mesenteric ischemia results from atherosclerosis in 90% of cases.1,5,49–51 Nonatherosclerotic causes of CMI are listed in Box 26-3. Nonatherosclerotic etiologies have been described in young adults and children as young as 30 months of age.49–51 In general, Risk factors for atherosclerotic-associated CMI are similar to those of other atherosclerotic conditions, including a positive family history, sedentary lifestyle, hypertension, hypercholesterolemia, and smoking.1,5,46–51 In contrast to other atherosclerotic vascular diseases, approximately 60% of patients with CMI are female, and nearly 50% of patients have a history of earlier cardiovascular surgery.51 Symptomatic CMI generally manifests at a mean age of about 58 years.1,5,46,52–56 More than one third of patients have hypertension, coronary artery disease, and/or cerebrovascular disease.1,5,53–56 Nearly 20% have evidence of chronic renal insufficiency, and 10% have diabetes.49,56
 Box 26-3 Nonatherosclerotic Conditions Associated with Chronic Mesenteric Ischemia
Box 26-3 Nonatherosclerotic Conditions Associated with Chronic Mesenteric Ischemia
Although there is a high prevalence of mesenteric atherosclerosis, the clinical syndrome of symptomatic mesenteric ischemia is uncommon.1,54,55 An autopsy series of unselected patients found significant stenoses in approximately 50% of celiac arteries, 30% of SMAs, and 30% of IMAs.52 In a more recent series of 120 consecutive autopsies, however, rates of significant stenoses in the celiac, SMA, and IMA were not quite as high, with 22%, 16%, and 10% incidence, respectively.55 The prevalence of potentially flow-limiting stenosis within the mesenteric vessels increases with age, with up to 67% of those older than 80 years of age having more than 50% stenosis in some mesenteric artery.55 Aortograms performed for aortic aneurysmal or occlusive disease demonstrate significant stenosis of the celiac artery in 33% of cases and SMA lesions in nearly 20%.54–56
Pathophysiology and Natural History
Chronic mesenteric ischemia occurs when the blood supply is insufficient to meet the metabolic demands of the bowel, resulting from increased motility, secretion, and absorption after meals.57 The infrequent occurrence of symptomatic disease may be explained in part by the extensive mesenteric collateral circulation, which includes both viscerovisceral (celiac artery-SMA-IMA), and parietovisceral (hypogastric-IMA) blood flow.1,5,55–57 The slow development of a chronic high-grade stenosis or occlusion of one or more of the major mesenteric vessels may thus be fully compensated by collateral blood flow. In addition, recent evidence suggests that preexisting significant stenoses in even remote arterial beds may provide protective effects through the mechanism of ischemic preconditioning.58–60
It has been proposed that the pathophysiology of symptomatic CMI involves a regional vascular steal phenomenon.61 Investigators have used tonometric assessment of splanchnic blood flow in dogs with 50% stenoses of both the celiac artery and SMA to show that food intake reduced intestinal perfusion by 50%. This reduction was associated with a significant decrease in intestinal intramural pH that was attributed to steal from the intestinal to the gastric circulation stimulated by a food bolus within the stomach.61
Rarely, single-vessel disease of the SMA may produce symptoms characteristic of CMI. The vast majority of patients who present with symptomatic CMI, however, have arteriographic evidence of multivessel visceral artery disease.1,5,49–52,55,56
A variety of pain syndromes characterize patients with CMI. In general, the symptoms consist of upper abdominal cramping or aching pain beginning 20 to 30 minutes after eating. At first the pain may be of short duration, but later it may become more persistent and last for 3 to 4 hours after eating. As the disease progresses, the amount of food that precipitates abdominal pain may decrease. Patients avoid eating to prevent the resulting abdominal pain. Most patients with CMI suffer weight loss secondary to diminished nutritional intake; malabsorption is not the primary mechanism of weight loss.57
No form of bowel activity is “classic” for CMI. Patients may have diarrhea (which can potentially exacerbate their nutritional depletion), constipation, or normal bowel habits. Without intervention, such patients develop severe protein-calorie malnutrition, and CMI can progress to visceral infarction.49,50,55–57
Most fatal cases of CMI occur in patients with a prolonged history of chronic abdominal complaints.1,49–51,55–57 Such cases are frequently characterized by months of abdominal complaints and multiple negative endoscopies, CT scans, and other diagnostic tests. In retrospect, the diagnosis is usually obvious. A high index of suspicion and prompt intervention are clearly indicated in cases of unexplained abdominal pain and weight loss. Early diagnosis may prevent acute thrombosis of stenotic vessels and the often fatal complication of intestinal infarction.1,49–51,55–57
1 Moneta G. Acute mesenteric ischemia. In: Rutherford R.B., ed. Diagnosis of intestinal ischemia. Philadelphia: WB Saunders; 2000:2508.
2 McKinsey J., Gewertz B.L. Acute mesenteric ischemia. Surg Clin North Am. 1997;77:307.
3 Park W.M., Gloviczki P., Cherry K.J. Contemporary management of acute mesenteric ischemia: factors associated with survival. J Vasc Surg. 2002;35:445.
4 Kaleya R.N., Sammartano R.J., Boley S.J. Aggressive approach to acute mesenteric ischemia. Surg Clin North Am. 1992;72:157.
5 Schwartz L.B., McKinsey J.F., Gewertz B.L. Visceral ischemic syndromes. In: Moore W.S., ed. Vascular surgery: a comprehensive review. ed 6. Philadelphia: WB Saunders; 2002:572.
6 Sreenarasimhaiah J. Diagnosis and management of intestinal ischemic disorders. BMJ. 2003;326:1372.
7 Ghosh S., Roberts N., Firmin R.K., et al. Risk factors for intestinal ischemia in cardiac surgical patients. Eur J Cardiothorac Surg. 2002;21:411.
8 Wyers M.C. Acute mesenteric ischemia: diagnostic approach and surgical treatment. Semin Vasc Surg. 2010;23:9–20.
9 Kozuch P.L., Brandt L.J. Review article: diagnosis and management of mesenteric ischaemia with an emphasis on pharmacotherapy. Aliment Pharmacol Ther. 2005;21:201–215.
10 Korthius R.J., Anderson D.C., Granger D.N. Role of neutrophil-endothelial cell adhesion in inflammatory disorders. J Crit Care. 1994;9:47.
11 Lock G. Acute mesenteric ischemia: classification, evaluation and therapy. Acta Gastroenterol Belg. 2002;65:220.
12 Edwards M.S., Cherr G.S., Craven T.E., et al. Acute occlusive mesenteric ischemia: surgical management and outcomes. Ann Vasc Surg. 2003;17:72.
13 Trompeter M., Brazda T., Remy C.T., et al. Non-occlusive mesenteric ischemia: etiology, diagnosis, and interventional therapy. Eur Radiol. 2001;12:1179.
14 Neri E., Sassi C., Massetti M., et al. Nonocclusive mesenteric ischemia in patients with acute aortic dissection. J Vasc Surg. 2002;36:738.
15 Howard T.J., Plaskon L.A., Wiebke E.A., et al. Nonocclusive mesenteric ischemia remains a diagnostic dilemma. Am J Surg. 1996;171:405.
16 Lock G., Scholmerich J. Non-occlusive mesenteric ischemia. Hepatogastroenterology. 1995;42:234.
17 Wilcox M.G., Howard T.J., Plakson L.A., et al. Current theories of pathogenesis and treatment of nonocclusive mesenteric ischemia. Dig Dis Sci. 1995;40:709.
18 Gewertz B.L., Zarins C.K. Postoperative vasospasm after antegrade mesenteric revascularization: a report of three cases. J Vasc Surg. 1991;14:382.
19 Patel A., Kaleya R.N., Sammartano R.J. Pathophysiology of mesenteric ischemia. Surg Clin North Am. 1992;72:31.
20 Williams L.F., Anastasia L.F., Hasiotis C.A., et al. Non-occlusive mesenteric infarction. Am J Surg. 1967;114:376.
21 Case records of the Massachusetts General Hospital (Case 35082). N Engl J Med. 1949;240:308.
22 Haglund U., Lundgren O. Nonocclusive acute intestinal vascular failure. Br J Surg. 1979;66:155.
23 Trompeter M., Brazda T., Remy C.T., et al. Non-occlusive mesenteric ischemia: etiology, diagnosis, and interventional therapy. Eur Radiol. 2002;12:1179–1187.
24 Mitsuyoshi A., Obama K., Shinkura N., et al. Survival in nonocclusive mesenteric ischemia: early diagnosis by multidetector row computed tomography and early treatment with continuous intravenous high-dose prostaglandin E1. Ann Surg. 2007;246:229–235.
25 Luckner G., Jochberger S., Mayr V.D., et al. Vasopressin as adjunct vasopressor for vasodilatory shock due to non-occlusive mesenteric ischemia. Anaesthesist. 2006;55:283–286.
26 Lamarque D., Whittle B.J. Increase in gastric intramucosal hydrogen ion concentration following endotoxin challenge in the rat and the actions of nitric oxide synthase inhibitors. Clin Exp Pharmacol Physiol. 2001;28:164.
27 Kolkman J.J., Mensink P.B. Non-occlusive mesenteric ischemia: a common disorder in gastroenterology and intensive care. Best Pract Res Clin Gastroenterol. 2003;17:457.
28 Mesh C.L., Gewertz B.L. The effect of hemodilution on blood flow regulation in normal and postischemic intestine. J Surg Res. 1990;48:183.
29 Char D., Hines G. Chronic mesenteric ischemia: diagnosis and treatment. Heart Dis. 2001;3:231.
30 Kim E.H., Gewertz B.L. Chronic digitalis administration alters mesenteric vascular reactivity. J Vasc Surg. 1987;5:382.
31 Clark E.T., Gewertz B.L. Intermittent ischemia potentiates intestinal reperfusion injury. J Vasc Surg. 1991;13:606.
32 Toledo-Pereyra L.H. Leukocyte depletion, ischemic injury and organ preservation. J Surg Res. 2011;169:188–189.
33 Dimakakos P.B., Kotsis T., Kondi-Pafiti A., et al. Oxygen free radicals in abdominal aortic surgery: an experimental study. J Cardiovasc Surg (Torino). 2002;43:77.
34 Elliot J.W. The operative relief of gangrene of intestine due to occlusion of the mesenteric vessels. Ann Surg. 1895;21:9.
35 Warren S., Eberhard T.P. Mesenteric venous thrombosis. Surg Gynecol Obstet. 1935;61:102.
36 Rhee R.Y., Gloviczki P. Mesenteric venous thrombosis. Surg Clin North Am. 1997;77:327.
37 Abdu R., Zakhour B.J., Dallis D.J. Mesenteric venous thrombosis—1911–1984. Surgery. 1987;101:383.
38 Rhee R.Y., Glovickzi P., Medonca C.T., et al. Mesenteric venous thrombosis: still a lethal disease in the 1990s. J Vasc Surg. 1994;20:688.
39 Ottinger L.W., Austen W.G. A study of 136 patients with mesenteric infarction. Surg Gynecol Obstet. 1967;124:251.
40 Kazmers A. Intestinal ischemia caused by venous thrombosis. In Vascular Surgery. Philadelphia: WB Saunders; 1995. p 526
41 Kumar S., Sarr M.G., Kamath P.S. Mesenteric venous thrombosis. N Engl J Med. 2001;345:686.
42 Harward T.R., Green D., Bergan J.J., et al. Mesenteric venous thrombosis. J Vasc Surg. 1989;9:328.
43 Morasch M.D., Ebaugh J.L., Chiou A.C., et al. Mesenteric venous thrombosis: a changing clinical entity. J Vasc Surg. 2001;34:680.
44 Harnik I.G., Brandt L.J. Mesenteric venous thrombosis. Vasc Med. 2010;15:407–418.
45 Councilman W.T. Three cases of occlusion of the superior mesenteric artery. Boston Med Surg J. 1894;130:410.
46 Taylor L.M., Porter J.M. Treatment of chronic intestinal ischemia. Semin Vasc Surg. 1990;3:186.
47 Shaw R.S., Maynard E.P.III. Acute and chronic thrombosis of the mesenteric arteries associated with malabsorption: a report of two cases successfully treated with thromboendarterectomy. N Engl J Med. 1958;258:874.
48 Morris G.C., Crawford E.S., Cooley D.A., et al. Revascularization of the celiac and superior mesenteric arteries. Arch Surg. 1962;84:113.
49 White C.J. Chronic mesenteric ischemia: diagnosis and management. Prog Cardiovasc Dis. 2011;54:36–40.
50 Sanders B.M., Dalsing M.C. Mesenteric ischemia affects young adults with predisposition. Ann Vasc Surg. 2003;17:270.
51 Zeller T., Rastan A., Sixt S. Chronic atherosclerotic mesenteric ischemia. Vasc Med. 2010;15:333–338.
52 Crawford E.S., Morris G.C., Myhre H.O., et al. Celiac axis, superior mesenteric artery, and inferior mesenteric artery occlusion: surgical considerations. Surgery. 1977;82:856.
53 Shanley C.J., Ozaki C.K., Zelenock G.B. Bypass grafting for chronic mesenteric ischemia. Surg Clin North Am. 1997;77:381.
54 Cleveland T.J., Nawaz S., Gaines P.A. Mesenteric arterial ischemia: diagnosis and therapeutic options. Vasc Med. 2002;7:311.
55 Chang J.B., Stein T. Mesenteric ischemia: acute and chronic. Ann Vasc Surg. 2003;17:323.
56 Chang J.B., Stein T.A. Mesenteric ischemia. Asian J Surg. 2003;26:55.
57 van Bockel J.H., Geelkerken R.H., Wasser M.N. Chronic splanchnic ischemia. Best Pract Res Clin Gastroenterol. 2001;15:99.
58 Heusch G., Schulz R. Remote preconditioning. J Mol Cell Cardiol. 2002;34:1279.
59 Cinel I., Avlan D., Cinel L., et al. Ischemic preconditioning reduces intestinal apoptosis in rats. Shock. 2003;19:588.
60 Asoyek S., Cinel I., Avlan D., et al. Intestinal ischemic preconditioning protects the intestine and reduces bacterial translocation. Shock. 2002;18:476.
61 Poole J.W., Sammartano R.J., Boley S.J. Hemodynamic basis of the pain of chronic mesenteric ischemia. Am J Surg. 1987;153:171.