[level-membership-for-pediatrics-category]
Chapter 457 Enzymatic Defects
457.1 Pyruvate Kinase Deficiency
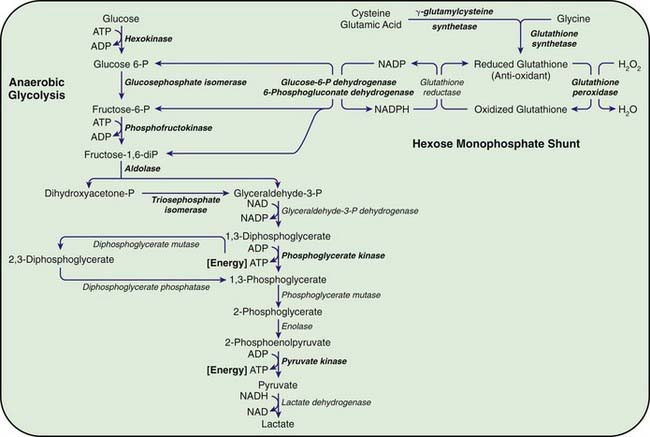
Congenital hemolytic anemia occurs in persons homozygous or compound heterozygous for autosomal recessive genes that cause either a marked reduction in red blood cell (RBC) pyruvate kinase (PK) or production of an abnormal enzyme with decreased activity. Generation of adenosine triphosphate (ATP) within RBCs is impaired, and low levels of ATP, pyruvate, and the oxidized form of nicotinamide adenine dinucleotide (NAD+) are found (Fig. 457-1). The concentration of 2,3-diphosphoglycerate is increased; this isomer is beneficial in facilitating oxygen release from hemoglobin but detrimental in inhibiting hexokinase and enzymes of the hexose monophosphate shunt. In addition, an unexplained decrease occurs in the sum of the adenine (ATP, adenosine diphosphate, and adenosine monophosphate) and pyridine (NAD+ and the reduced form of NAD) nucleotides, further impairing glycolysis. As a consequence of decreased ATP, RBCs cannot maintain their potassium and water content; the cells become rigid, and their life span is considerably reduced.
457.2 Other Glycolytic Enzyme Deficiencies
Chronic nonspherocytic hemolytic anemias of varying severity have been associated with deficiencies of other enzymes in the glycolytic pathway, including hexokinase, glucose phosphate isomerase, and aldolase, which are inherited as autosomal recessive disorders. Phosphofructokinase deficiency, which occurs primarily in Ashkenazi Jews in the USA, results in hemolysis associated with a myopathy classified as glycogen storage disease type VII (Chapter 81.1). Clinically, hemolytic anemia is complicated by muscle weakness, exercise intolerance, cramps, and possibly myoglobinuria. Enzyme assays for phosphofructokinase yield low values for RBCs and muscle.
Deficiencies of Enzymes of the Hexose Monophosphate Pathway
The most important function of the hexose monophosphate pathway is to maintain glutathione in its reduced state (GSH) as protection against the oxidation of RBCs (see Fig. 457-1). Approximately 10% of the glucose taken up by RBCs passes through this pathway to provide the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary for the conversion of oxidized glutathione to GSH. Maintenance of GSH is essential for the physiologic inactivation of oxidant compounds, such as hydrogen peroxide, that are generated within RBCs. If glutathione, or any compound or enzyme necessary for maintaining it in the reduced state, is decreased, the SH groups of the RBC membrane are oxidized and the hemoglobin becomes denatured and may precipitate into RBC inclusions called Heinz bodies. Once Heinz bodies have formed, an acute hemolytic process results from damage to the RBC membrane by the precipitated hemoglobin, the oxidant agent, and the action of the spleen. The damaged RBCs then are rapidly removed from the circulation.
457.3 Glucose-6-Phosphate Dehydrogenase Deficiency and Related Deficiencies
George B. Segel and Lisa R. Hackney
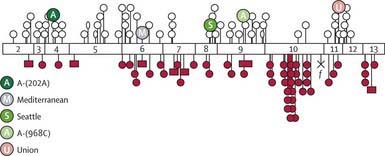
The deficiency is caused by inheritance of any of a large number of abnormal alleles of the gene responsible for the synthesis of the G6PD protein. About 140 mutations have been described in the gene responsible for the synthesis of the G6PD protein. Many of these mutations are single base changes leading to amino acid substitutions and destabilization of the G6PD enzyme. The gene for G6PD has been cloned and sequenced. A web-accessible database catalogs G6PD mutations (www.bioinf.org.uk/g6pd). Some of the mutations that cause episodic vs chronic hemolysis are shown in Figure 457-2. Milder disease is associated with mutations near the amino terminus of the G6PD molecule, and chronic nonspherocytic hemolytic anemia is associated with mutations clustered near the carboxyl terminus. The normal enzyme found in most populations is designated G6PD B+. A normal variant, designated G6PD A+, is common in Americans of African descent.
Episodic or Induced Hemolytic Anemia
Etiology
G6PD catalyzes the conversion of glucose 6-phosphate to 6-phosphogluconic acid. This reaction produces NADPH, which maintains glutathione in the reduced, functional state (see Fig. 457-1). Reduced glutathione provides protection against oxidant threats from certain drugs and infections that would otherwise cause precipitation of hemoglobin (Heinz bodies) or damage the RBC membrane.
Clinical Manifestations
Most individuals with G6PD deficiency are asymptomatic, with no clinical manifestations of illness unless triggered by infection, drugs, or ingestion of fava beans. Typically, hemolysis ensues in about 24-48 hr after a patient has ingested a substance with oxidant properties. In severe cases, hemoglobinuria and jaundice result, and the hemoglobin concentration may fall precipitously. Drugs that elicit hemolysis in these individuals include aspirin, sulfonamides, rasburicase, and antimalarials, such as primaquine (Table 457-1). The degree of hemolysis varies with the inciting agent, amount ingested, and severity of the enzyme deficiency. In some individuals, ingestion of fava beans also produces an acute, severe hemolytic syndrome, known as favism. Fava beans contain divicine, isouramil, and convicine, which ultimately lead to production of hydrogen peroxide and other reactive oxygen products. Favism is thought to be more frequently associated with the G6PD B− variant.
Table 457-1 AGENTS PRECIPITATING HEMOLYSIS IN GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
MEDICATIONS
Antibacterials
Antimalarials
Others
CHEMICALS
ILLNESS
From Asselin BL, Segel GB: In Rakel R, editor: Conn’s current therapy, Philadelphia, 1994, WB Saunders, p 341.
Laboratory Findings
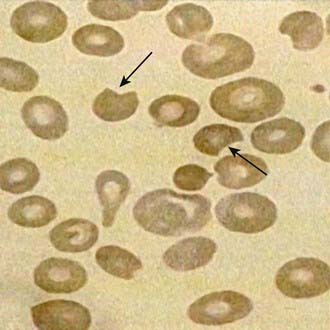
The onset of acute hemolysis usually results in a precipitous fall in hemoglobin and hematocrit. If the episode is severe, the hemoglobin binding proteins, such as haptoglobin, are saturated, and free hemoglobin may appear in the plasma and subsequently in the urine. Unstained or supravital preparations of RBCs reveal precipitated hemoglobin, known as Heinz bodies. The RBC inclusions are not visible on the Wright-stained blood film. Cells that contain these inclusions are seen only within the first 3-4 days of illness because they are rapidly cleared from the blood. Also, the blood film may contain red cells with what appears to be a bite taken from their periphery and polychromasia (evidence of bluish, larger RBCs), representing reticulocytosis (Fig. 457-3).
Chronic Hemolytic Anemias Associated with Deficiency of G6pd or Related Factors
Chronic nonspherocytic hemolytic anemia has been associated with profound deficiency of G6PD caused by enzyme variants, particularly those defective in quantity, activity, or stability. The gene defects leading to chronic hemolysis are located primarily in the region of the NADP binding site near the carboxyl terminus of the protein (see Fig. 457-2). These include the Loma Linda, Tomah, Iowa, Beverly Hills, Nashville, Riverside, Santiago de Cuba, and Andalus variants. Persons with G6PD B− enzyme deficiency occasionally have chronic hemolysis, and the hemolytic process may worsen after ingestion of oxidant drugs. Splenectomy is of little value in these types of chronic hemolysis.
Other enzyme defects may impair the regeneration of GSH as an oxidant “sump” (see Fig. 457-1). Mild, chronic nonspherocytic anemia has been reported in association with decreased RBC GSH, resulting from γ-glutamylcysteine or glutathione synthetase deficiencies. Deficiency of 6-phosphogluconate dehydrogenase (6PDG) has been associated primarily with drug-induced hemolysis, and hemolysis with hyperbilirubinemia has been related to a deficiency of glutathione peroxidase in newborn infants.
Ayi K, Min-Oo G, Serghides L, et al. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008;358:1805-1810.
Beutler E. Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 1994;331:169-173.
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64-74.
Fujii H, Miwa S. Other erythrocyte enzyme deficiencies associated with non-haematological symptoms: phosphoglycerate kinase and phosphofructokinase deficiency. Baillieres Best Pract Res Clin Haematol. 2000;13:141-148.
Kaplan M, Beutler E, Vreman HJ, et al. Neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient heterozygotes. Pediatrics. 1999;104:68-74.
Kaplan M, Muraca M, Hammerman C, et al. Bilirubin conjugation, reflected by conjugated bilirubin fractions, in glucose-6-phosphate dehydrogenase-deficient neonates: a determining factor in the pathogenesis of hyperbilirubinemia. Pediatrics. 1998;102:E37.
Martinov MV, Plotnikov AG, Vitvitsky VM, et al. Deficiencies of glycolytic enzymes as a possible cause of hemolytic anemia. Biochim Biophys Acta. 2000;1474:75-87.
McMullin MF. The molecular basis of disorders of red cell enzymes. J Clin Pathol. 1999;52:241-244.
Min-Oo G, Gros P. Erythrocyte variants and the nature of their malaria protective effect. Cell Microbiol. 2005;7:753-763.
Valentin C, Pissard S, Martin J, et al. Triose phosphate isomerase deficiency in 3 French families: two novel null alleles, a frameshift mutation (TPI Alfortville) and an alteration in the initiation codon (TPI Paris). Blood. 2000;96:1130-1135.
Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: from genetics. Baillieres Best Pract Res Clin Haematol. 2000;13:57-81.
Zanella A, Fermo E, Bianchi P, et al. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130:11-25.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 457 Enzymatic Defects
457.1 Pyruvate Kinase Deficiency
Congenital hemolytic anemia occurs in persons homozygous or compound heterozygous for autosomal recessive genes that cause either a marked reduction in red blood cell (RBC) pyruvate kinase (PK) or production of an abnormal enzyme with decreased activity. Generation of adenosine triphosphate (ATP) within RBCs is impaired, and low levels of ATP, pyruvate, and the oxidized form of nicotinamide adenine dinucleotide (NAD+) are found (Fig. 457-1). The concentration of 2,3-diphosphoglycerate is increased; this isomer is beneficial in facilitating oxygen release from hemoglobin but detrimental in inhibiting hexokinase and enzymes of the hexose monophosphate shunt. In addition, an unexplained decrease occurs in the sum of the adenine (ATP, adenosine diphosphate, and adenosine monophosphate) and pyridine (NAD+ and the reduced form of NAD) nucleotides, further impairing glycolysis. As a consequence of decreased ATP, RBCs cannot maintain their potassium and water content; the cells become rigid, and their life span is considerably reduced.
457.2 Other Glycolytic Enzyme Deficiencies
Chronic nonspherocytic hemolytic anemias of varying severity have been associated with deficiencies of other enzymes in the glycolytic pathway, including hexokinase, glucose phosphate isomerase, and aldolase, which are inherited as autosomal recessive disorders. Phosphofructokinase deficiency, which occurs primarily in Ashkenazi Jews in the USA, results in hemolysis associated with a myopathy classified as glycogen storage disease type VII (Chapter 81.1). Clinically, hemolytic anemia is complicated by muscle weakness, exercise intolerance, cramps, and possibly myoglobinuria. Enzyme assays for phosphofructokinase yield low values for RBCs and muscle.
Deficiencies of Enzymes of the Hexose Monophosphate Pathway
The most important function of the hexose monophosphate pathway is to maintain glutathione in its reduced state (GSH) as protection against the oxidation of RBCs (see Fig. 457-1). Approximately 10% of the glucose taken up by RBCs passes through this pathway to provide the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary for the conversion of oxidized glutathione to GSH. Maintenance of GSH is essential for the physiologic inactivation of oxidant compounds, such as hydrogen peroxide, that are generated within RBCs. If glutathione, or any compound or enzyme necessary for maintaining it in the reduced state, is decreased, the SH groups of the RBC membrane are oxidized and the hemoglobin becomes denatured and may precipitate into RBC inclusions called Heinz bodies. Once Heinz bodies have formed, an acute hemolytic process results from damage to the RBC membrane by the precipitated hemoglobin, the oxidant agent, and the action of the spleen. The damaged RBCs then are rapidly removed from the circulation.
[/not-level-membership-for-pediatrics-category]
