[level-membership-for-pediatrics-category]
Chapter 7 Endocrinology
Long Cases
Congenital adrenal hyperplasia
Background information
Congenital adrenal hyperplasia (CAH) refers to a number of inherited defects in adrenal steroidogenesis, which cause impaired synthesis of cortisol from cholesterol in the adrenal cortex. The most common of these is 21-hydroxylase deficiency (21-OHD), which is caused by a range of mutations in one gene—the CYP21A2 gene on chromosome 6p21.3, which codes for 21-hydroxylase (P450C21). The end result is a lack of cortisol (and usually aldosterone) synthesis by the adrenal cortex. This leads to increased adrenocortical stimulation by hypothalamic corticotropin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH), which induces adrenal glandular hyperplasia—hence the term CAH. CAH is inherited in an autsomal recessive manner. This long case deals with the common form of CAH due to 21-OHD, the preferred term for which, under current nomenclature, is 21-OHD CAH.
Females with classic 21-OHD CAH are born with ambiguous genitalia. The degree of virilisation of the external genitalia is scored by the Prader scale (see the short case on ambiguous genitalia in this chapter). Males with classic salt-wasting 21-OHD CAH appear normal at birth, but then deteriorate with adrenal insufficiency after 1–4 weeks. In these babies, adrenal aldosterone production is insufficient for the distal tubules to reabsorb sodium, leading to salt loss as well as deficiency of cortisol and an excess of androgens. Symptoms can include poor feeding, failure to thrive, vomiting, loss of weight, dehydration, hypotension, hyponatraemia and hyperkalaemic metabolic acidosis, leading to adrenal crisis with vascular collapse and a significant mortality rate. Some degree of aldosterone deficiency occurs in all forms of 21-OHD CAH.
Females with non-classic 21-OHD CAH may present with clitoromegaly, early development of pubic hair, hirsutism, acne, increased growth rate, and gynaecological problems such as oligomenorrhoea, abnormal menses or infertility. Males with non-classic 21-OHD CAH may develop early penile growth, pubic hair, increased growth rate and increased musculature. Deficiency of cytochrome P450 enzyme 21-hydroxylase (CYP21A2) causes 90% of 21-OHD CAH cases. Ten types of mutation in CYP21A2 account for more than 90% of affected cases, although well over 100 have been described, including point mutations, small deletions, small insertions and complex rearrangements of the gene. 21-OHD CAH demonstrates a heterogeneous phenotype, with concordance between phenotype and genotype. The Human Gene Mutation Database, Cardiff (http://www.hgmd.org) lists all known mutations.
Diagnosis
21-OHD CAH can be diagnosed at different ages, as follows.
High-risk pregnancies: prenatal diagnosis of 21-OHD CAH
If the fetus is female, and the proband has two CYP21A2 disease-causing mutations, then molecular testing of fetal DNA is undertaken to identify whether the fetus has inherited both disease-carrying alleles. If the fetus is female and unaffected, dexamethasone is stopped. If the fetus is female and is found by DNA analysis to have classic 21-OHD CAH, dexamethasone is continued to term. Prenatal treatment is solely to prevent virilisation of the genitalia in affected females. It has no effect on any later requirement for hormone replacement therapy. There are no significant side effects of dexamethasone treatment; there is no increased risk of congenital anomalies, and no effect on birth weight, length or head circumference.
Preimplantation genetic diagnosis (PGD) of 21-OHD CAH
This may be offered to families in which the disease-causing mutations have been identified.
Other forms of CAH
There are five forms of CAH. Patients with other forms of CAH (<2% of CAH patients) may present at various ages. Impaired enzyme function at each step of adrenal steroidogenesis leads to a unique set of excess precursors and reduced products. 17-OHD CAH is due to mutation in the gene CYP17A1 on chromosome 10q24.3, which codes for 17-hydroxylase (P450C17). CAH caused by deficiency of 3 beta-hydroxysteroid dehydrogenase (3Beta-HSD) is due to mutation in the gene HSD17B10, on chromosome Xp11.2. HSD17B10 encodes 3-hydroxyacyl-CoA dehydrogenase type II, a mitochondrial protein that catalyses a number of steroids, fatty acids and alcohols. CAH caused by deficiency of steroidogenic acute regulatory protein (StAR) is due to mutation in the STAR gene; the protein encoded by the STAR gene allows the transfer of cholesterol across the mitochondrial membrane, after which it is converted into pregnenolone. For further discussion, see the short case on ambiguous genitalia.
History
Initial diagnosis
2. Immediate pre-diagnosis symptoms:
3. Where, when and how the diagnosis was made, length of hospital stay, education given, any treatment of the mother with steroids while the embryo was in utero, treatment in hospital, treatment at discharge.
Progress of the disease
1. Details of subsequent hospitalisations (frequency, indications, usual length of stay, usual outcome).
2. Complications of the disease; for example, the need for corrective genital surgery, episodes of adrenal crisis, abnormal growth and development, psychosexual developmental aspects (for example, girls with male-type play, physical aggression, low interest in babies or maternal nurturing behaviours; increased incidence of lesbian relationships), inadequately treated hyperandrogenism, hypoglycaemic reactions.
3. Complications of treatment; for example, overtreatment with steroids causing deceleration of linear growth, too much fludrocortisone causing hypertension, degree of control (number of episodes of adrenal crisis).
4. Monitoring of the disease: how often seen in clinic, the usual investigations performed, how often seen by local doctor.
5. Changes in management; for example, the usual increases in steroid dosage on sick days.
6. At what age was the patient administering his or her own steroids?
7. Compliance; for example, previous refusal to take steroids in teenage males.
Current status
1. General health: lethargic or energetic.
2. Current medications: type (e.g. hydrocortisone, fludrocortisone, salt supplement, antiandrogens dose), regimen (how much, when, given by whom, modifications with intercurrent illness), salt craving (not enough fludrocortisone), hypertension (too much fludrocortisone), compliance with treatment.
3. Adrenal insufficiency: how often, what symptoms (e.g. vomiting, lethargy, crying, convulsions, near-syncope, syncope, loss of consciousness), usual precipitants, anticipatory strategies for prevention, response.
4. Hypoglycaemia: any episodes, any suggestion of this (e.g. sweating, pallor, tremulousness, hunger, headache, odd behaviour, lethargy, crying, bad temper, lack of coordination, dizziness, vomiting, convulsions, loss of consciousness).
5. Symptoms attributable to virilisation: acne, increased linear growth, amenorrhoea.
6. Other problems; for example, adolescent self-image, compliance problems.
Social history
1. Impact on child: self-image, reaction of school friends, effects (e.g. virilisation), coping with taking steroids, amount of school missed.
2. Impact on siblings: sibling rivalry, risk of CAH.
3. Impact on parents: family finances, employment, concern regarding future complications, genetic counselling.
4. Social supports: parents groups, access to social worker, government benefits obtained.
5. Coping: who attends the clinic with the patient, the level of education of the child and parents, contingency plans for intercurrent illnesses or severe adrenal crisis causing loss of consciousness, access to local doctor, paediatrician, hospital.
Management
All patients with CAH, regardless of type, require treatment with glucocorticoids. These replace cortisol (which is deficient) and provide negative feedback, suppressing ACTH secretion. This then prevents continued adrenal stimulation, inhibiting excess androgen production (as 17-OHP is not available as a substrate for excess androgen production; this prevents virilisation). Patients with the salt-losing form (for practical purposes, all those with a raised plasma renin activity) also require mineralocorticoid replacement to normalise the sodium balance associated with aldosterone deficiency. Girls with moderate to severe clitoral enlargement and all those with fused labia are offered corrective surgery. The timing of this, and the place of surgery for mild degrees of clitoromegaly, is now a very controversial area, as previous surgical approaches are considered to have led to some loss of clitoral sensation. The overall principles of treatment are given below.
Control of steroid requirements
Prevention of acute complications
Adrenal crises can be averted by anticipatory strategies. All patients with CAH need extra steroid cover for stress: this includes any acute medical condition (such as gastroenteritis, or other viral illnesses), any surgical procedure requiring an anaesthetic and any significant orthopaedic injury (such as a major fracture). Treatment may include hydrocortisone 25–100 mg parenterally (IM or IV), repeated 4–6 hourly until recovery from the acute aspect of the illness: triple the usual dose for 2 days, then double the usual dose for 3 days.
Psychological support
The following basic principles are important:
1. Minimise the number of days off school.
2. Involve the child in management of his or her CAH as appropriate to age and ability.
3. In times of ‘adolescent rebellion’, support and encourage; never resort to threats.
4. Identify and treat negative family responses to CAH, including overindulgence, over-anxiousness, neglect and disinterest, resentment and overcontrolling parents.
5. When the initial diagnosis is made, be aware of depression in patients, siblings and parents, and counsel accordingly. It is important that the parents and child have a full understanding of the pathophysiology of CAH, and all aspects of the psychosexual aspects that can arise should be openly discussed with the parents, and when it is age-appropriate, with the child. There should be no secretive aspects to the family’s handling of this diagnosis, as this could be damaging. There is significant value in sexual counselling for many of these patients and their families.
For the purposes of the long case, the usual problem is that of a non-compliant adolescent. Unfortunately, it is in adolescence—the least receptive time of the patient’s life—that it is crucial to avoid serious complications. For a teenager, immediate peer acceptance, which may involve nights out at parties or hotels, far outweighs the long-term benefits of adequate steroid replacement. Most teenagers with CAH require additional emotional support, which may be achieved through attendance at discussion groups or talking one-on-one with a clinical psychologist.
Types of corticosteroids
The examiners will expect you to be familiar with the various types of steroids and their equivalents. Table 7.1 is a brief guide; refer to the latest MIMS, British National Formulary or equivalent publication for the names of the various preparations available.
| Steroid | Relative glucocorticoid activity | Relative mineralocorticoid activity |
|---|---|---|
| Hydrocortisone (cortisol) | 1 | 1 |
| Cortisone acetate (11-deoxycortisol) | 0.8 | 0.8 |
| Prednisone | 4 | 0.8 |
| Prednisolone | 4 | 0.8 |
| Methylprednisolone (6 alpha-methylprednisolone) | 5 | 0.5 |
| Fludrocortisone (9 alpha-fluorocortisol) | 10 | 125 |
| Betamethasone (9 alpha-fluoro-16 beta-methylprednisolone) | 25 | 0 |
| Dexamethasone (9 alpha-fluoro-16 alpha-methylprednisolone) | 25 | 0 |
21-OHD CAH prenatal diagnosis and intervention
This area may be mentioned in the long-case discussion. Prenatal treatment of CAH attributable to 21-OHD by administration of corticosteroids (dexamethasone) to the mother is most commonly performed in females with a previously affected child. Informed consent must be obtained from the parents before prenatal treatment is contemplated. There are possible maternal adverse effects with CAH, the genital outcome is variable and there may be long-term effects on children, which are presently unknown. Masculinisation of the external genitalia begins at 6–7 weeks’ gestation; if treatment before this suppresses the fetal pituitary–adrenal axis, it could prevent ambiguous genitalia. Of reported cases where prenatal treatment has occurred, it was successful in three quarters of them (one third normal genitalia, two thirds mildly virilised) and unsuccessful in a quarter.
Management of acute adrenocortical insufficiency (adrenal crisis)
Immediate management
1. Assess and secure the airway, breathing and circulation (the ABCs).
2. Insert an IV cannula; take blood as above.
3. Restore the circulating volume with an infusion of normal saline (isotonic, 0.9%) with added glucose to make up to 5% glucose. Start with a 20 mL/kg bolus. Correct hypoglycaemia. Aim to replace fluid and electrolytes over the next 24–48 hours.
4. Give bolus hydrocortisone IV (or IM if IV access is difficult):
5. Hyperkalaemia may need further correction with insulin and glucose.
Diabetes mellitus
Background information
T1DM affects 1 in 1000 children (and 1 in 400 by adolescence). Incidence increases with age. The main genetic factor determining susceptibility to T1DM lies within the major histocompatibility complex (termed IDDM1). There is an association with certain HLA haplotypes: more than 90% of patients with T1DM have HLA-DR3 and/or HLA-DR4 (class II antigens located on the short arm of chromosome 6, at 6p21); 55% have a DR3/DR4 combination, most commonly DR4-DQ8/DR3-DQ2 (1 in 5 in families of diabetics versus 1 in 25 of the general population). DR3/DR4 heterozygosity is seen most frequently in children who develop T1DM under 5 years of age. For children who carry both of the highest-risk HLA haplotypes (DR4-DQ8/DR3-DQ2), the risk of being diagnosed with T1DM by 15 years is 1 in 20; if there is a sibling with T1DM and the same haplotypes, the risk is then 55%. One non-HLA gene is recognised as contributing to around 10% of the family aggregation of T1DM: this is termed IDDM2 and is situated on chromosome 11p5.5. This locus maps to a variable number of tandem nucleotide repeats (VNTR) of the insulin gene. Different sizes of the VNTR are associated with a risk of T1DM, the long form of VNTR being associated with protection from T1DM. A meta-analysis of data combined from most of the genomewide studies of linkage to T1DM, has been carried out by the Type 1 Diabetes Genetics Consortium; this shows that most of the genetic risk for T1DM is conferred by the class II genes encoding HLA-DR and HLA-DQ, as well as one or more additional genes within the HLA region. At least 50 inherited susceptibility loci for T1DM are known. An excellent resource noting all the genes implicated in T1DM, and constantly updated, is T1Dbase (http://www.t1dbase.org); it includes a table of all known T1DM loci, and clear diagrams of chromosomes, showing the position of each locus. Almost every chromosome has at least one locus identified. Apart from the genes in the HLA region, the majority of these loci affect T-cell function, including antigen-driven T-cell activation and cytokine signalling, proliferation or maturation.
Prevention of complications of T1DM
The DCCT Study (Diabetes Control and Complications Trial) involved 1441 volunteers with T1DM, ages 13–39, who had had T1DM for at least 1 but less than 15 years, and no, or minimal, diabetic eye disease; it compared intensive control of blood glucose (keeping HbA1c [glycosylated haemoglobin] as close to 6% as possible) versus standard control of blood glucose. Patients were studied for an average of 6.5 years. The DCCT study showed that intensive blood glucose control decreases the risk of eye disease (retinopathy) by 76%, kidney disease by 50% and nerve disease (neuropathy) by 60%. The trial ended, and was reported, in 1993, but 90% of patients were followed up, and a second study, the EDIC Study (Epidemiology of Diabetes Interventions and Complications), reporting in 2005, assessed both microvacular (eye, kidney and nerve) and macrovascular disease, including incidence and predictors of various forms of cardiovascular disease (including myocardial infarction, cerebrovascular accidents, and requirement for cardiac surgery). The EDIC study showed that intensive blood glucose control decreases the risk of any cardiovascular disease event by 42%, and non-fatal myocardial infarction, cerebrovascular accident or death from any cardiovascular cause by 57%. The EDIC study showed that the benefits noted for the microvascular complications involving the eyes, kidneys and nerves during the DCCT study persisted after that study was completed. This longer-lasting benefit from tight glucose control has been termed ‘metabolic memory’.
Diagnosis
Type 1 diabetes can be diagnosed in children as follows:
• Patients with the ‘classic’ symptoms of T1DM, namely the triad of polydypsia, polyuria and weight loss despite polyphagia, are diagnosed by having a random blood sugar level (BSL; the commonly used term for plasma glucose level) above 11 mmol/L. Some units recommend a level of 14 mmol/L, but in practice the diagnosis is usually clear cut, and the level much higher than either figure.
• Asymptomatic patients require two criteria: a fasting BSL above 7.8 mmol/L, plus a 2-hour postprandial BSL above 11 mmol/L. Alternatively, a formal oral glucose tolerance test (GTT) demonstrating a sustained elevation in BSL above 11 mmol/L at 2 hours is required, as well as a similarly elevated intervening value taken between the time the glucose load (which is 1.75 g/kg, up to 75 g) is given and the 2-hour value. Again, this is fairly theoretical, as doubtful cases requiring a GTT for diagnosis are very rare.
History
Initial diagnosis
1. Initial symptoms: for example, early symptoms preceding the classic triad, lack of weight gain, nocturia, behaviour change, altered school performance, changed vision (blurring), tempo of onset (developing over days, weeks or months pre-diagnosis).
2. Immediate pre-diagnosis symptoms: polydypsia, polyuria, weight loss despite polyphagia.
3. Symptoms associated with ketoacidosis: for example, vomiting, abdominal pain, clouding of consciousness.
4. Where, when and how the diagnosis was made, length of hospital stay, education given, treatment in hospital, treatment at discharge.
Progress of the disease
1. Details of subsequent hospitalisations (frequency, indications, usual length of stay, usual outcome).
2. Complications of the disease: for example, eye problems with cataracts, retinopathy, joint problems with limited joint mobility (LJM), severe hypoglycaemic reactions.
3. Complications of treatment (e.g. fat atrophy or hypertrophy, insulin allergic reactions); degree of control (number of episodes of ketoacidosis, frequency of hypoglycaemic symptoms).
4. Monitoring of the disease: how often seen in clinic, usual investigations performed, how often seen by local doctor.
5. Changes in management: for example, increase in insulin administration from daily to twice daily, twice daily with additional short/ultra-short-acting or basal bolus regimen, use of insulin pump, altered dosages due to occult nocturnal hypoglycaemia, changeover from human insulin to analogues.
6. At what age did the patient begin to administer his or her own insulin?
Current status
1. General health: lethargic or energetic.
2. Insulin type, dose, regimen (which sort, how much, when, given by whom, where, rotation of sites; modifications with raised BSL, sporting activities, intercurrent illness or dining out), compliance with treatment.
3. Diet prescribed (whether portions/exchanges used, glycaemic index, recommended foods), diet actually taken, alcohol intake (adolescents), involvement of dietician, any restrictions (adhered to or not).
4. Hypoglycaemia: how often, what symptoms (e.g. sweating, pallor, tremulousness, hunger, headache, odd behaviour, lethargy, crying, bad temper, lack of coordination, dizziness, vomiting, convulsions, loss of consciousness, early morning headaches after nocturnal ‘hypo’, restless sleep); usual precipitants; anticipatory strategies for prevention of ‘hypos’; response to ‘hypos’ such as taking fast-acting sugars (e.g. glass of orange juice with added sugar, glass of lemonade, jelly beans) followed by a small protein and complex carbohydrate snack (e.g. bread, biscuits); ever any need for intramuscular glucagon? (Note: if no ‘hypos’ have occurred, BSL may have been too high.) Any evidence of ‘hypoglycaemia unawareness’?
5. Control: hypoglycaemia (see above); hyperglycaemia (e.g. any nocturia, polyuria, blurred vision, weight loss, excessive weight gain, disturbance of menstrual periods in postpubertal girls); BSL readings (usual levels, when performed, how often, by whom, response to high level); usual HbAlc levels; urine testing (how often, what indications); amount of school missed in the last few months, vaginal thrush, other infections such as pilonidal sinus, infected ingrown toenail.
6. Other problems: for example, adolescent self-image, compliance issues.
Social history
1. Impact on child: self-image, reaction of school friends, coping with giving insulin, dietary restrictions, exercise, amount of school missed.
2. Impact on siblings: sibling rivalry, risk of diabetes.
3. Impact on parents: family finances, employment, concern regarding future complications, genetic counselling.
4. Social supports: Diabetes Association, access to social worker, government benefits obtained.
5. Coping: who attends the clinic with the patient, level of education of the child and parents, contingency plans for intercurrent illnesses or severe hypoglycaemia causing loss of consciousness, access to local doctor/paediatrician/hospital.
Management
This involves the use of insulin, diet and regular exercise, with the following aims:
1. Control of BSL: maintaining close to normoglycaemia; see below.
2. Prevention of acute complications; for example, hypoglycaemia, ketoacidosis.
3. Ensuring optimum growth and development.
4. Maintaining a normal lifestyle.
5. Adequate education of the patient and parents.
6. Early detection and treatment of associated disease (e.g. Hashimoto’s thyroiditis).
7. Provision of psychological support and counsel.
8. Ensuring adequate access to appropriate social supports.
9. Reducing long-term complications by maintaining good metabolic control.
10. Regular screening for complications and early intervention when they appear (ACE inhibitors for hypertension or proteinuria, etc).
Age-specific aspects of control
The expectations at different ages vary. There are three groups:
1. Infants to preschoolers. The main goal is to avoid hypoglycaemia and preserve cognitive function (in line with the findings of the DCCT). The acceptable range for BSLs at this age is between 6 and 15 mmol/L, and the HbA1c between 8.0 and 9.5 gm%.
2. School-age to puberty. Again, avoidance of hypoglycaemia is a top priority. Acceptable ranges: BSL 4–10 mmol/L; HbA1c 8.0 gm%.
3. Adolescence. Acceptable ranges: BSL 4–8 mmol/L; HbA1c as low as possible. It may be that postpubertal control is more important than prepubertal; this is unclear.
Insulin therapy
Average dosage requirements are as follows:
1. ‘Honeymoon’ period: 0.5 units per kilogram per day (or less).
2. Preadolescent: 1.0 unit per kilogram per day.
3. Adolescent: 1.0–2.0 units per kilogram per day (increase with pubertal growth spurt and reduce later when growth has finished).
Candidates should be familiar with the various insulin regimens. These include:
1. Daily (longer-acting only, or mixed short and longer).
3. Twice daily with additional short or ultrashort.
4. Basal bolus (three pre-meal short or ultra-short, plus longer in evening; only used in motivated adolescents).
5. Premixed (biphasic) insulin (only used for non-compliance, or inability to mix insulin).
Types of insulin/insulin analogue
Rapid-acting (or ultra-short-acting) insulin analogues (insulin lispro, insulin aspart, insulin glulisine)
Other points about insulin
1. Insulin given intramuscularly has a higher peak and shorter duration than when given subcutaneously.
2. Insulin absorption varies with the site of the injection: it is best when injected into the abdominal wall, followed by the upper limbs and then the lower limbs.
3. Insulin should not be injected into an area that is going to be exercised, so if playing tennis, inject into the abdomen, but if doing sit-ups, inject into the thigh.
4. Lipohypertrophy can occur when the same site is used repeatedly for injection. This can be avoided by ensuring rotation of injection sites.
Specific problem areas
Hypoglycaemic episodes
Symptoms of a hypoglycaemic episode (e.g. tremor, hunger, sweating, pallor, confusion, headache) without access to a glucometer, or a BSL reading below 3 mmol/L, should be treated immediately. Fast-acting sugars should be given (e.g. half a glass of lemonade, four jelly beans, or two sugar cubes: each of these approximates half a portion) if consciousness is not impaired.
Alternative modes of insulin delivery
Insulin pumps
An insulin pump is a small computerised device, the size of a pager, which is programmed to supply a basal infusion of insulin, subcutaneously, with increases given at meal times. Pumps have become increasingly sophisticated, and increasingly adopted as a preferred mode of therapy amongst motivated individuals, be they adolescent patients or very motivated parents. They work particularly well in the older, very motivated adolescent who is very comfortable with ‘technology’. Pumps are expensive ($5000–8000), though private health funds do cover them. Blood glucose testing is required 4–6 times per day, and then the pump has to be programmed by the user/parent, based on those measurements, while considering food intake and exercise patterns. When used optimally, compared to standard therapy, the use of pumps is associated with better blood glucose control, better HbA1c levels and improved BMI, and reduction in the risk of long-term complications. Insulin delivery more accurately simulates the way a pancreas would deliver insulin, and pumps allow more flexibility with mealtimes, sleeping patterns, ease of adjustments when unwell and are even associated with improved school performance. Potential disadvantages include not liking the thought of being attached to a piece of machinery/technology constantly, more blood testing, superficial skin infection (or even abscesses) at the infusion site, other site problems (lipohypertrophy, skin reactions to tape, air in lines, kinks in lines) and weight gain (too easy to bolus for unhealthy snacks). A notable disadvantage of pumps is the fourfold risk of diabetic ketoacidosis (DKA); this can occur because only ultra-short-acting insulin is administered via pumps, so if the insulin runs out overnight, within a few hours the blood sugar may be rising very rapidly. For this reason, there must always be insulin and syringes available at any time, so that the patient can revert to these should the need arise. The average age of a child using a pump is around 10 years, in Australia. Children under 12 may have technical difficulties operating these pumps themselves, and may depend on their parents’ competence with the technology. Pumps can be removed for short periods (up to 2 hours) to have a shower, go swimming or play sport, but essentially they are attached 24 hours a day. A pump is only as good as its operator.
Changing over to a pump from syringes and needles involves, initially, changing the total daily insulin dosage by 30%, and setting the basal rate (overall) at 50% of the total daily insulin dosage. A meal bolus is worked out based on an estimated insulin/carbohydrate ratio, the ‘500 rule’, which states that: 500/total daily insulin dose = grams of carbohydrate covered by 1 unit of insulin. A correction bolus is worked out based on estimated insulin sensitivity, the ‘100 rule’, which states that: 100/total daily insulin dose = glucose drop in mml/L, for 1 unit of insulin. Patients should carry relevant guidance information for the usual variances that they are likely to encounter, such as ‘short-term adjustment guideline’ cards, which set out responses to trend arrows, low targets and high BSLs. Exceptions to the above starting parameters include initial poor control (if HbA1c is above 9%, then do not decrease the starting dose by as much as 30%), initial high insulin requirement (if receiving 2 units of insulin per kilogram per day, then start lower, decrease by 50%) and toddlers, where the dosage of insulin may be so small that the insulin needs to be diluted first, before being loaded into the pump.
When just getting started with pump therapy, it is unwise to increase the total insulin by more than 10% in any one given 24-hour period; however, if hypoglycaemia should occur, it is acceptable to reduce the dosage by more than just 10%. Modifications in the longer term require ascertaining which of several bolus rates needs to be adjusted initially; this must be worked through systematically, testing the basal rate first, then testing the correction bolus, then testing the meal bolus—this area is too complicated to discuss briefly here. It is worthwhile candidates spending time with their hospital’s diabetic educators to work through such problems. An excellent resource has been produced by Diabetes Australia: a booklet entitled I’m Considering an Insulin Pump; Information for People with Type 1 Diabetes can be accessed at http://www.diabetesvic.org.au.
Exercise
The only difficulties are the additional considerations about diet and insulin (which are dealt with easily), and that certain groups of patients should not engage in strenuous exercise, namely those with BSL readings above 20 mmol/L, significant ketonuria, significant retinopathy and hypertension.
Monitoring and control
Glycosylated albumin (fructosamine)
Like HbAlc, this gives an indication of recent diabetic control, but over the preceding 2–4 weeks.
Routine follow-up
The other important clinical point is regular eye examination by an ophthalmologist, annually if postpubertal. Recommendations vary for prepubertal children. Routine investigations should include the following.
Initially
1. Screening for coeliac disease (occurs in 3–5% of T1DM): IgA tTG (tissue transglutaminase) antibodies (sensitivity 95%, specificity 90%); IgA EmA (endomysial) antibodies (sensitivity 90%, specificity approaches 100%); in children under 2, antigliadin IgG antibodies (children under 2 have a limited ability to produce IgA antibodies); if IgA deficient, IgG antigliadin antibodies; if screening is positive, small bowel biopsy.
2. Screening for thyroid disease: thyroid function tests, including TSH (15% of patients with T1DM develop Hashimoto’s thyroiditis, and a third of these develop hypothyroidism); if TSH is abnormal, antithyroid antibodies should be tested.
3. Screening for lipid disorders (ideally after overnight fasting): total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides.
Yearly
1. If prepubertal and diabetic for 5 years: check for retinopathy. Retinal photographs.
2. If pubertal and diabetic for 2 years: check for microalbuminuria. The initial screen is the albumin/creatinine ratio. The gold standard is timed overnight urine collection (minimum 8 hours). If microalbuminuria is present, monitor blood pressure. If diabetic for 5 years, check urea, electrolytes, creatinine.
3. If poor control (HbA1c > 9%), check for neuropathy and examine the feet. This should include testing vibration sensation by tuning fork, testing sensation to light touch and testing ankle reflexes.
Complications
Microvascular complications
Retinopathy
This occurs to some degree in 90% or more of patients within 15 years of the onset of their T1DM. The most common lesions seen are ‘background’ lesions, which include microaneurysms, retinal haemorrhages (named according to appearance: dot, blot and flame), hard exudates, cottonwool spots (retinal nerve fibre infarcts) and venous calibre changes (loops, beading). Background retinopathy can be seen in up to one quarter of adolescent patients. More advanced lesions of ‘proliferative’ retinopathy occur in 40% of T1DM patients after 20 years of disease, but are uncommon in adolescence. These changes include signs of neovascularisation, fibrous proliferation and haemorrhage into the vitreous (which can cause sudden visual loss or retinal detachment secondary to fibrosis and traction) and can lead to glaucoma (obstruction to aqueous humour from vascular overgrowth).
Neuropathy
There are several forms of this:
1. Symmetric distal neuropathy, which predominantly affects sensory neurons, causing paraesthesia or pain, but can also involve the motor system.
2. Autonomic neuropathy, which can lead to postural hypotension, arrhythmias (due to denervation hypersensitivity to catecholamines), gastroparesis and impaired response to hypoglycaemia, and may be associated with painless myocardial infarction.
3. Carpal tunnel syndrome, due to the neuropathy affecting the median nerve (often the ulnar nerve is involved as well).
4. Mononeuropathy, which particularly affects cranial nerves.
All of these are infrequent in the paediatric age range. However, nerve conduction studies demonstrating subclinical peripheral nervous system involvement and evidence of sensory neuropathy are not uncommon in adolescent diabetics. The underlying pathology is thought to be a combination of polyol pathway abnormality and microangiopathy affecting the vasa vasorum, leading to axonal loss and segmental demyelination.
Requirement for psychological support
There are three important principles in parenting adolescents with a chronic illness: firstly, staying involved, which is paramount; secondly, expressing understanding while applying limits; and thirdly working as a team. Teenagers want to be understood, but want to be different from their parents. Parents have to learn to accept some things with which they do not agree; one can accept something without agreeing with it (e.g. not wanting to take insulin at school), accept that it is annoying (‘Yeah, it sucks. You must hate this—you must wish you could forget about it.’) but, without agreeing to comply with the teenager’s request, recognise that the issue must be solved (‘How about we figure out a way to sort this out?’). Parents should make it as easy as possible to live with them, recognising that they, the parents, and their teens both will get frustrated but will continually agree that the necessary management aspects for diabetes must be attended to.
Transition from paediatric to adult care
A well-structured transition programme is important. It has been recognised that around one third of patients with T1DM have, in the past, been lost to specialist follow-up during their (planned) transfer from paediatric to adult care. To address this, in Queensland, the Mater Children’s Hospital and Queensland Health together have developed a comprehensive set of Best Practice Guidelines for health professionals, a state-wide model termed the SWEET Diabetes Transition Program. It provides clearly set out steps to guide the process, at key ages (12–13, 15, 16–17, 18+). It aims to actively involve young people in the transition process, allowing flexibility of timing, assigning a transition case worker, the choice of an adult diabetic care provider, and significant preparation and coordination. It is an excellent resource that candidates should utilise, and it can be accessed at http://www.sweet.org.au.
Acute management of diabetic ketoacidosis (DKA)
The principles of treatment can be divided into five main areas, the ‘diabetic pentathlon’:
Some complications
Cerebral oedema
Prevention strategies include slow correction of fluid and glucose abnormalities, and nursing children with the head elevated. Active treatment includes 20% mannitol, 0.5 g/kg IV statim, repeated every 15 minutes, decreasing rate of fluid administration, and intubation and hyperventilation.
Short Cases
Disorders of Sexual Development (ambiguous genitalia)
There are five groups of babies:
1. 46,XX DSD—genetic females with ambiguous or male phenotype.
2. 46,XY DSD—genetic males with ambiguous or female phenotype.
3. Ovotesticular DSD—usually 46,XX, genitalia usually ambiguous, gonads contain both ovarian and testicular components.
4. Sex chromosome aneuploidy DSD—for example, 45,X/46,XY mosaicism, with one streak gonad and one dysgenetic testis (also known as mixed gonadal dysgenesis).
5. Other—dysmorphic syndromes, cloacal anomalies, bladder exstrophy etc.
The most common scenario of these is female with ambiguous genitalia due to CAH.
Until 7 weeks’ gestation, the internal genital tracts are bipotential in both XX and XY embryos. In both, the genital ducts are the wolffian (mesonephric) duct and the müllerian (paramesonephric) duct. In a normal male, the wolffian duct transforms into the epididymis, vas deferens and seminal vesicles. In a normal female, the müllerian duct transforms into the uterus, fallopian tubes and upper portion of the vagina.
Sexual differentiation of the brain occurs between 15 and 25 weeks’ gestation.
Examination for ambiguous genitalia
Differential diagnosis
Virilised female
1. Congenital adrenal hyperplasia (CAH)—deficiency of:
Testicular failure (undervirilised males)
1. Enzymatic—incomplete deficiency of:
Diabetes
Examination
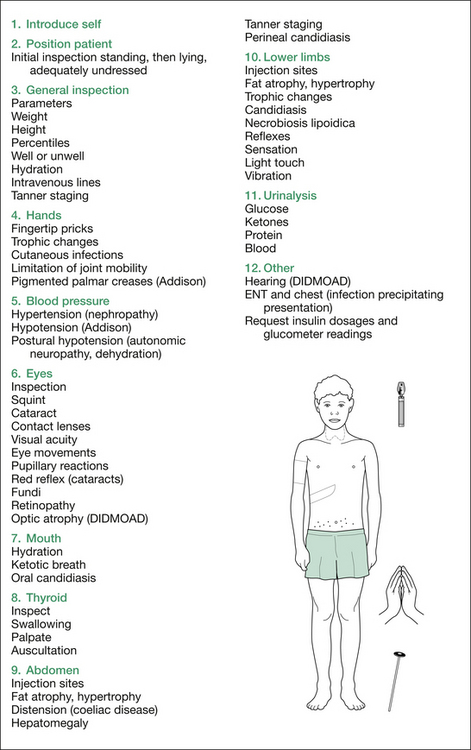
Next, take, or request, the blood pressure (hypertension with nephropathy, postural hypotension with autonomic neuropathy or dehydration in ketoacidosis, hypotension with Addison’s disease).
1. Background lesions include microaneurysms, retinal haemorrhages, hard exudates, cottonwool spots (small retinal infarcts) and venous beading.
2. Proliferative lesions include new vessel networks (arising from peripheral vessels or from vessels overlying the optic disc), vitreous haemorrhage and retinal detachment.
3. Maculopathy, which is rare, includes oedema and hard exudate deposition. Also look for optic atrophy (DIDMOAD).
Figure 7.1 summarises the examination for diabetes.
Short stature
Observation
Firstly, listen carefully to the patient’s age in the introduction. Then, stand back and look for any evidence of an obvious diagnosis (e.g. Turner or Noonan syndromes), any dysmorphism and any disproportion (skeletal dysplasias, rickets), and visually assess the pubertal status. The pubertal status is particularly important. Whether puberty is delayed, normal or precocious will determine the differential diagnoses to be considered as the examination progresses: for example, delayed puberty is consistent with constitutional delayed puberty, pituitary disorders and chronic diseases; normal puberty is consistent with familial short stature; and advanced pubertal staging is consistent with various causes of precocious puberty (see the short case on precocious puberty). Comment on your findings.
Measurements
Interpretation of the US:LS ratio and arm span
• If the US:LS ratio is increased, it suggests short lower limbs (skeletal dysplasias, hypothyroidism).
• If the US:LS ratio is decreased, it suggests a short trunk (scoliosis, spondylodysplasia, osteogenesis imperfecta [OI]) or short neck (Klippel–Feil sequence).
• Next, measure the child’s arm span, and compare this to the total height. The normal arm span minus height values: −3 cm from birth to 7 years, 0 cm from 8 to 12 years, +1 cm (girls) and +4 cm (boys) at 14 years.
• A short arm span can occur with skeletal dysplasias, and an apparently long arm span with a short neck, trunk or legs.
• If the span is less than the height with a high US:LS, this may indicate short limbs and a normal trunk, or normal limbs and a long trunk.
• If the span is greater than the height with a low US:LS, this indicates a short trunk and normal limbs.
• If the span is less than the height with a low or normal US:LS ratio, this indicates short trunk and limbs.
By now, you may well have a good indication of the type of short stature with which you are dealing.
Manoeuvres
Inspect from front
A set of manoeuvres can be performed that very rapidly screens for a number of syndromes that may be relevant in assessing short children. With each manoeuvre, stand opposite the child and demonstrate, so that he or she will mirror your movements.
Ask the child to make a fist and look for a shortened fourth metacarpal (pseudohypoparathyroidism).
Examination
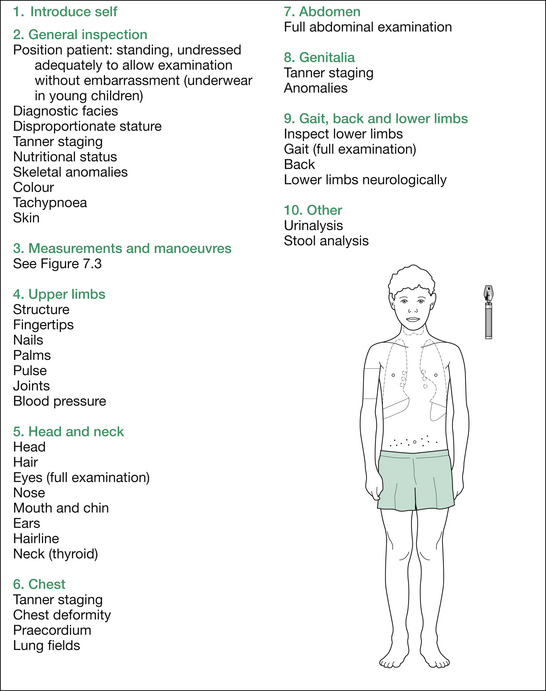
The formal physical examination flows well if commenced at the hands, working up to the head, and then downwards: essentially a ‘head-to-toe’ pattern. Table 7.5 (‘Additional information’) at the end of the section on short stature outlines the findings sought at each point.
| Hands and feet together |
| To detect: |
At the completion of the comprehensive physical examination, request results of urinalysis (e.g. type 1 diabetes mellitus [T1DM], chronic kidney disease [CKD]) and stool analysis (malabsorption from cystic fibrosis [CF], coeliac disease, inflammatory bowel disease [IBD]). Finally, summarise your findings succinctly and give a brief differential diagnosis, placing the most likely diagnosis first (see Figure 7.2).
Investigations
At this stage, you may be asked what investigations you would perform. Depending on the findings, of course, the answers vary. Generally, a bone age is most useful and, in girls, a chromosomal analysis is always warranted to exclude Turner syndrome. Other investigations often ordered include thyroid function tests (hypothyroidism), electrolytes, urea and creatinine (CKD), tissue transglutaminase antibodies (coeliac disease) and growth hormone (GH) provocation testing (clonidine, arginine or glucagon), the latter only being performed after other preliminary testing is done (see Table 7.4), and poor height velocity is demonstrated. Low IGF-1 and IGFBP-3 levels are useful in predicting GH deficiency, or GH resistance, although normal levels do not exclude GH deficiency (see Table 7.4).
Table 7.5 Additional information: a more comprehensive listing of possible physical findings in children with short stature
| General inspection |
| Diagnostic facies |
BSL = blood sugar level; CAH = congenital adrenal hyperplasia; CCF = congestive cardiac failure; CF = cystic fibrosis; CHD = congenital heart disease; CKD = chronic kidney disease; CLD = chronic liver disease; FAS = fetal alcohol syndrome; IBD = inflammatory bowel disease; ICP = intracranial pressure; JIA = juvenile idiopathic arthritis; LS = lower segment; NF = neurofibromatosis; OI = osteogenesis imperfecta; RTA = renal tubular acidosis; SCA = sickle cell anaemia; T1DM = type 1 diabetes mellitus; TORCH = intrauterine infections with toxoplasmosis, other (e.g. HIV, syphilis), rubella, cytomegalovirus, herpes (both simplex and varicella); US = upper segment; VSD = ventricular septal defect.
Table 7.4 Some useful investigations in the child with short stature
| Investigation | Indications/relevance |
|---|---|
| Blood | |
| Full blood examination and film | Chronic disease, anaemia |
| Erythrocyte sedimentation rate | Inflammatory bowel disease |
| Electrolytes, urea, creatinine | Chronic kidney disease |
| Fasting blood glucose | Diabetes |
| Calcium, phosphate, SAP | Rickets, hypophosphatasia |
| Liver function tests | CLD, nutritional deficiency |
| Tissue transglutaminase antibodies | Coeliac disease |
| Pancreatic isoamylase | Shwachman syndrome |
| Thyroid function tests, TSH | Hypothyroidism |
| Karyotype | Syndromes, e.g. Turner, Down |
| Somatomedin C (also called insulin-like growth factor, IGF1) | GH deficiency, coeliac, Crohn, malnutrition, hypothyroidism, T1DM (poorly controlled) |
| Insulin-like growth factor binding protein 3 (IGFBP-3) | GH deficiency |
| GH stimulation tests (arginine, clonidine, glucagon, GHRH) | GH deficiency |
| LH, FSH, prolactin, oestradiol, testosterone | Hypogonadism |
| Dexamethasone suppression test (combined high and low dose) | Cushing’s syndrome |
| Sweat | |
| Sweat conductivity | Cystic fibrosis |
| Imaging | |
| Bone age | Maturational delay, precocious puberty, hypothyroidism, hypopituitarism |
| Skeletal survey | Skeletal dysplasias |
| Skull X-ray | Craniopharyngioma |
| MRI of brain | Intracranial tumour |
CLD = chronic liver disease; FSH = follicle-stimulating hormone; GH = growth hormone; GHRH = growth hormone releasing hormone; LH = luteinising hormone; SAP = serum alkaline phosphatase; TSH = thyroid-stimulating hormone.
Table 7.4 lists some relatively simple investigations that are often useful in the child with short stature. More common diagnoses appearing in the examination setting include constitutional delay in growth and puberty [maturational delay] and Russell–Silver syndrome.
Aetiologies
The mnemonic IS NICE lists various headings/causes for aetiologies of short stature:
I. Idiopathic (constitutional delay in growth and puberty [maturational delay]; familial short stature)/Intrauterine (SGA, TORCH, fetal alcohol syndrome [FAS])
S. Skeletal causes (dysplasias, OI)/Spinal defects (scoliosis, kyphosis)/Syndromes (Russell–Silver, Kallman)/Septo-optic dysplasia (SOD)
 Nutritional (e.g. malabsorption)/Nurturing (deprivation)
Nutritional (e.g. malabsorption)/Nurturing (deprivation)
I. Iatrogenic (steroids, radiation)
C. Chronic diseases (CKD, congenital heart disease, CF, IBD)/Chromosomal (Turner, Down)/Craniopharyngioma (or other central tumour)
E. Endocrine (e.g. GH deficiency, GH insensitivity, hypopituitarism, hypothyroidism, Cushing, T1DM, pseudohypoparathyroidism)
Alternative introduction to short stature—endocrine
Occasionally, the lead-in for short stature has been: ‘This child is short. Would you please examine the endocrine system’. For those who have learned a comprehensive approach such as that previously outlined, this can cause some concern. This should not be the case, as all that is required is some abbreviation of the previous system, with elimination of the irrelevant parts. Remember that assessment for Turner syndrome is part of an endocrine assessment, as is evaluation for Kallmann syndrome, septo-optic dysplasia, craniopharyngioma or other central tumour (i.e. do not forget to test the visual acuity and visual fields, examine the optic fundi and perform a gait examination) and rickets.
Tall stature
Measurements
Next, measure the patient yourself. Stand the patient against a wall, position the head and heels appropriately and record the height. Measure the lower segment (LS)—that is, the distance from the pubic symphysis to the ground—then calculate the upper segment (US), which is the height minus the LS. Work out the US:LS ratio. Normal values at various ages are listed in Table 7.2.
| Age | Ratio |
|---|---|
| Birth | 1.7 |
| 3 years | 1.3 |
| 8 years or more | 1.0 |
Next, measure the arm span and compare this to the total height. The normal arm span minus height values at various ages are given in Table 7.3. A long arm span occurs in Marfanoid or eunuchoid patients, or patients with other diagnoses complicated by a shortened trunk, caused by scoliosis (e.g. Sotos) or kyphosis (e.g. pituitary gigantism). If the arm span to height ratio is > 1.05, then this indicates long limbs (e.g. Marfan syndrome or eunuchoid body habitus).
Table 7.3 Normal arm span minus height values
| Age | Value |
|---|---|
| Birth to 7 years | −3 cm |
| 8–12 years | 0 |
| 14 years | +1 cm in girls |
Next, measure the head circumference and request the weight.
Request percentile charts, progressive measurements (if not given) and calculate the height velocity (be aware of the normal range and nature of percentile charts for this measurement). Request the birth parameters (B–W and Sotos can have large birth weights) and the parents’ heights, percentiles and onsets of puberty (ask about age of menarche for women and age when men started shaving).
Manoeuvres
For a list of manoeuvres, see Table 7.6.
Table 7.7 Additional information: details of possible findings in the child with tall stature
| Introduction |
| Impression of mental state: intellectual impairment (homocystinuria, Sotos, Beckwith–Wiedemann [B–W], Klinefelter) |
| General inspection |
| Body habitus |
CAH = congenital adrenal hyperplasia; CVA = cerebrovascular accident: MEN = multiple endocrine neoplasia; NF-1 = neurofibromatosis type 1.
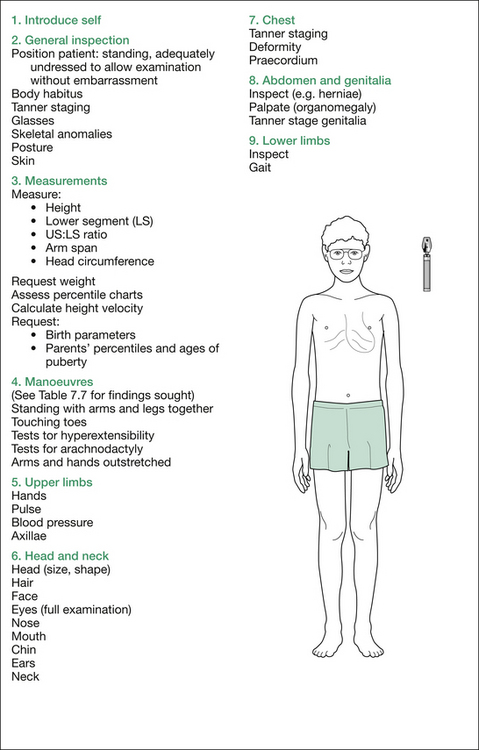
Remember, as you proceed, to explain to the examiners what you are doing and why, so that the significance of each manoeuvre will be appreciated. The formal physical examination proceeds from the hands, working up to the head and then downwards; that is, head to toe, as outlined in Figure 7.3. A comprehensive listing of possible findings is given in Table 7.7 at the end of this section.
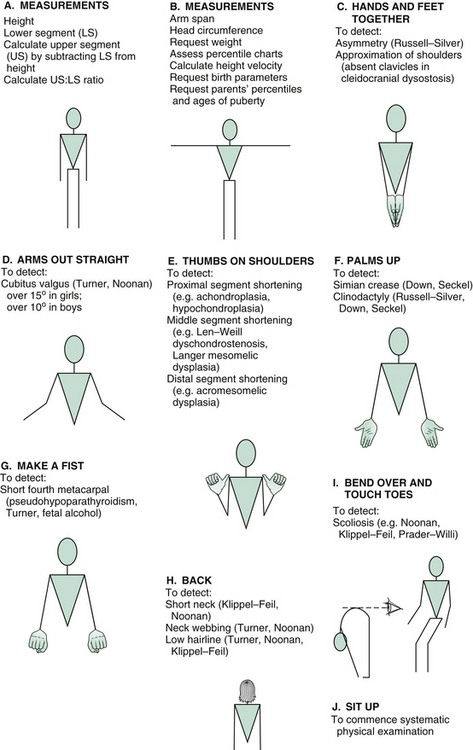
Table 7.8 Additional information: details of possible findings on obesity examination
| Introduction |
| Impression of mental state |
| General inspection |
| Growth parameters |
| Height |
| Head circumference: enlarged (intracranial tumour, hydrocephalus with spina bifida) |
| Percentile charts |
| Calculate height velocity |
| Request |
| Distribution of obesity: central (Cushing) |
| Skin |
| Truncal striae |
| Tanner staging |
| Respiratory rate |
| Head |
| Shape: prominent forehead and bitemporal narrowing (PW) |
| Size: large (brain tumour, hydrocephalus with spina bifida) |
| Hair |
| Face |
| Eyes |
| Retinitis pigmentosa (B–B, Alström) |
| Papilloedema (pituitary tumour, benign intracranial hypertension in Cushing) |
| Hypertensive retinopathy (Cushing) |
| Nose |
| Mouth and chin |
| Cleft lip (repaired) |
| Cleft palate (repaired) |
| Single central incisor |
| Neck |
| Goitre (hypothyroidism) |
| Obliteration of supraclavicular hollow, i.e. supraclavicular fat pads (Cushing) |
| Elevated JVP (RVF in Pickwickian) |
| Abdomen |
| Striae (Cushing) |
| Hepatomegaly (RVF with Pickwickian syndrome) |
| Adrenal mass (Cushing syndrome due to adrenal tumour) |
| Genitalia |
| Lower limbs |
| Inspection |
| Measure: limb lengths (for shortening, as above) |
| Palpate: ankle oedema (RVF in Pickwickian) |
| Manoeuvres |
| Stand child with legs together and re-inspect for: |
| Hold arms up against resistance (proximal myopathy in Cushing) |
| Squat (proximal myopathy); can ‘race’ child with repeated squats; normal child should win |
| Walk, looking for limp with: |
| Trendelenberg’s test: positive with avascular necrosis of femoral head or SCFE |
| Lay patient down again for completion of lower limb examination |
| Lower limbs completion |
| Hip examination: limitation of internal rotation or abduction (SCFE) |
| Ankle jerks: delayed relaxation (hypothyroidism) |
B–B = Bardet–Biedl syndrome; GH = growth hormone; JVP = jugular venous pressure; PHPT = pseudohypoparathyroidism; PW = Prader–Willi syndrome; RVF = right ventricular failure; SCFE = slipped capital femoral epiphysis.
Investigations
At this stage, you may be asked what investigations would clarify the diagnosis. This, of course, depends on the findings and may include: a slit-lamp ophthalmological assessment if Marfan syndrome or homocystinuria are possibilities; urine homocystine and blood homocystine and methionine levels if homocystinuria is likely; an electrocardiogram, chest X-ray and echocardiogram if Marfan syndrome seems likely; plasma somatomedin C concentration and a cerebral CT or MRI scan if a pituitary cause needs to be excluded; and skeletal X-rays if McC–A syndrome is likely (multiple areas of bony fibrous dysplasia) or to confirm and document the degree of scoliosis or kyphosis in patients likely to have Marfan syndrome or homocystinuria (Figure 7.4).
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 7 Endocrinology
Long Cases
Congenital adrenal hyperplasia
Background information
Congenital adrenal hyperplasia (CAH) refers to a number of inherited defects in adrenal steroidogenesis, which cause impaired synthesis of cortisol from cholesterol in the adrenal cortex. The most common of these is 21-hydroxylase deficiency (21-OHD), which is caused by a range of mutations in one gene—the CYP21A2 gene on chromosome 6p21.3, which codes for 21-hydroxylase (P450C21). The end result is a lack of cortisol (and usually aldosterone) synthesis by the adrenal cortex. This leads to increased adrenocortical stimulation by hypothalamic corticotropin-releasing hormone (CRH) and pituitary adrenocorticotropic hormone (ACTH), which induces adrenal glandular hyperplasia—hence the term CAH. CAH is inherited in an autsomal recessive manner. This long case deals with the common form of CAH due to 21-OHD, the preferred term for which, under current nomenclature, is 21-OHD CAH.
Females with classic 21-OHD CAH are born with ambiguous genitalia. The degree of virilisation of the external genitalia is scored by the Prader scale (see the short case on ambiguous genitalia in this chapter). Males with classic salt-wasting 21-OHD CAH appear normal at birth, but then deteriorate with adrenal insufficiency after 1–4 weeks. In these babies, adrenal aldosterone production is insufficient for the distal tubules to reabsorb sodium, leading to salt loss as well as deficiency of cortisol and an excess of androgens. Symptoms can include poor feeding, failure to thrive, vomiting, loss of weight, dehydration, hypotension, hyponatraemia and hyperkalaemic metabolic acidosis, leading to adrenal crisis with vascular collapse and a significant mortality rate. Some degree of aldosterone deficiency occurs in all forms of 21-OHD CAH.
Females with non-classic 21-OHD CAH may present with clitoromegaly, early development of pubic hair, hirsutism, acne, increased growth rate, and gynaecological problems such as oligomenorrhoea, abnormal menses or infertility. Males with non-classic 21-OHD CAH may develop early penile growth, pubic hair, increased growth rate and increased musculature. Deficiency of cytochrome P450 enzyme 21-hydroxylase (CYP21A2) causes 90% of 21-OHD CAH cases. Ten types of mutation in CYP21A2 account for more than 90% of affected cases, although well over 100 have been described, including point mutations, small deletions, small insertions and complex rearrangements of the gene. 21-OHD CAH demonstrates a heterogeneous phenotype, with concordance between phenotype and genotype. The Human Gene Mutation Database, Cardiff (http://www.hgmd.org) lists all known mutations.
Diagnosis
21-OHD CAH can be diagnosed at different ages, as follows.
High-risk pregnancies: prenatal diagnosis of 21-OHD CAH
If the fetus is female, and the proband has two CYP21A2 disease-causing mutations, then molecular testing of fetal DNA is undertaken to identify whether the fetus has inherited both disease-carrying alleles. If the fetus is female and unaffected, dexamethasone is stopped. If the fetus is female and is found by DNA analysis to have classic 21-OHD CAH, dexamethasone is continued to term. Prenatal treatment is solely to prevent virilisation of the genitalia in affected females. It has no effect on any later requirement for hormone replacement therapy. There are no significant side effects of dexamethasone treatment; there is no increased risk of congenital anomalies, and no effect on birth weight, length or head circumference.
Preimplantation genetic diagnosis (PGD) of 21-OHD CAH
This may be offered to families in which the disease-causing mutations have been identified.
Other forms of CAH
There are five forms of CAH. Patients with other forms of CAH (<2% of CAH patients) may present at various ages. Impaired enzyme function at each step of adrenal steroidogenesis leads to a unique set of excess precursors and reduced products. 17-OHD CAH is due to mutation in the gene CYP17A1 on chromosome 10q24.3, which codes for 17-hydroxylase (P450C17). CAH caused by deficiency of 3 beta-hydroxysteroid dehydrogenase (3Beta-HSD) is due to mutation in the gene HSD17B10, on chromosome Xp11.2. HSD17B10 encodes 3-hydroxyacyl-CoA dehydrogenase type II, a mitochondrial protein that catalyses a number of steroids, fatty acids and alcohols. CAH caused by deficiency of steroidogenic acute regulatory protein (StAR) is due to mutation in the STAR gene; the protein encoded by the STAR gene allows the transfer of cholesterol across the mitochondrial membrane, after which it is converted into pregnenolone. For further discussion, see the short case on ambiguous genitalia.
History
Initial diagnosis
2. Immediate pre-diagnosis symptoms:
3. Where, when and how the diagnosis was made, length of hospital stay, education given, any treatment of the mother with steroids while the embryo was in utero, treatment in hospital, treatment at discharge.
Progress of the disease
1. Details of subsequent hospitalisations (frequency, indications, usual length of stay, usual outcome).
2. Complications of the disease; for example, the need for corrective genital surgery, episodes of adrenal crisis, abnormal growth and development, psychosexual developmental aspects (for example, girls with male-type play, physical aggression, low interest in babies or maternal nurturing behaviours; increased incidence of lesbian relationships), inadequately treated hyperandrogenism, hypoglycaemic reactions.
3. Complications of treatment; for example, overtreatment with steroids causing deceleration of linear growth, too much fludrocortisone causing hypertension, degree of control (number of episodes of adrenal crisis).
4. Monitoring of the disease: how often seen in clinic, the usual investigations performed, how often seen by local doctor.
5. Changes in management; for example, the usual increases in steroid dosage on sick days.
6. At what age was the patient administering his or her own steroids?
7. Compliance; for example, previous refusal to take steroids in teenage males.
Current status
1. General health: lethargic or energetic.
2. Current medications: type (e.g. hydrocortisone, fludrocortisone, salt supplement, antiandrogens dose), regimen (how much, when, given by whom, modifications with intercurrent illness), salt craving (not enough fludrocortisone), hypertension (too much fludrocortisone), compliance with treatment.
3. Adrenal insufficiency: how often, what symptoms (e.g. vomiting, lethargy, crying, convulsions, near-syncope, syncope, loss of consciousness), usual precipitants, anticipatory strategies for prevention, response.
4. Hypoglycaemia: any episodes, any suggestion of this (e.g. sweating, pallor, tremulousness, hunger, headache, odd behaviour, lethargy, crying, bad temper, lack of coordination, dizziness, vomiting, convulsions, loss of consciousness).
5. Symptoms attributable to virilisation: acne, increased linear growth, amenorrhoea.
6. Other problems; for example, adolescent self-image, compliance problems.
Social history
1. Impact on child: self-image, reaction of school friends, effects (e.g. virilisation), coping with taking steroids, amount of school missed.
2. Impact on siblings: sibling rivalry, risk of CAH.
3. Impact on parents: family finances, employment, concern regarding future complications, genetic counselling.
4. Social supports: parents groups, access to social worker, government benefits obtained.
5. Coping: who attends the clinic with the patient, the level of education of the child and parents, contingency plans for intercurrent illnesses or severe adrenal crisis causing loss of consciousness, access to local doctor, paediatrician, hospital.
Management
All patients with CAH, regardless of type, require treatment with glucocorticoids. These replace cortisol (which is deficient) and provide negative feedback, suppressing ACTH secretion. This then prevents continued adrenal stimulation, inhibiting excess androgen production (as 17-OHP is not available as a substrate for excess androgen production; this prevents virilisation). Patients with the salt-losing form (for practical purposes, all those with a raised plasma renin activity) also require mineralocorticoid replacement to normalise the sodium balance associated with aldosterone deficiency. Girls with moderate to severe clitoral enlargement and all those with fused labia are offered corrective surgery. The timing of this, and the place of surgery for mild degrees of clitoromegaly, is now a very controversial area, as previous surgical approaches are considered to have led to some loss of clitoral sensation. The overall principles of treatment are given below.
Control of steroid requirements
Prevention of acute complications
Adrenal crises can be averted by anticipatory strategies. All patients with CAH need extra steroid cover for stress: this includes any acute medical condition (such as gastroenteritis, or other viral illnesses), any surgical procedure requiring an anaesthetic and any significant orthopaedic injury (such as a major fracture). Treatment may include hydrocortisone 25–100 mg parenterally (IM or IV), repeated 4–6 hourly until recovery from the acute aspect of the illness: triple the usual dose for 2 days, then double the usual dose for 3 days.
Psychological support
The following basic principles are important:
1. Minimise the number of days off school.
2. Involve the child in management of his or her CAH as appropriate to age and ability.
3. In times of ‘adolescent rebellion’, support and encourage; never resort to threats.
4. Identify and treat negative family responses to CAH, including overindulgence, over-anxiousness, neglect and disinterest, resentment and overcontrolling parents.
5. When the initial diagnosis is made, be aware of depression in patients, siblings and parents, and counsel accordingly. It is important that the parents and child have a full understanding of the pathophysiology of CAH, and all aspects of the psychosexual aspects that can arise should be openly discussed with the parents, and when it is age-appropriate, with the child. There should be no secretive aspects to the family’s handling of this diagnosis, as this could be damaging. There is significant value in sexual counselling for many of these patients and their families.
For the purposes of the long case, the usual problem is that of a non-compliant adolescent. Unfortunately, it is in adolescence—the least receptive time of the patient’s life—that it is crucial to avoid serious complications. For a teenager, immediate peer acceptance, which may involve nights out at parties or hotels, far outweighs the long-term benefits of adequate steroid replacement. Most teenagers with CAH require additional emotional support, which may be achieved through attendance at discussion groups or talking one-on-one with a clinical psychologist.
Types of corticosteroids
The examiners will expect you to be familiar with the various types of steroids and their equivalents. Table 7.1 is a brief guide; refer to the latest MIMS, British National Formulary or equivalent publication for the names of the various preparations available.
| Steroid | Relative glucocorticoid activity | Relative mineralocorticoid activity |
|---|---|---|
| Hydrocortisone (cortisol) | 1 | 1 |
| Cortisone acetate (11-deoxycortisol) | 0.8 | 0.8 |
| Prednisone | 4 | 0.8 |
| Prednisolone | 4 | 0.8 |
| Methylprednisolone (6 alpha-methylprednisolone) | 5 | 0.5 |
| Fludrocortisone (9 alpha-fluorocortisol) | 10 | 125 |
| Betamethasone (9 alpha-fluoro-16 beta-methylprednisolone) | 25 | 0 |
| Dexamethasone (9 alpha-fluoro-16 alpha-methylprednisolone) | 25 | 0 |
21-OHD CAH prenatal diagnosis and intervention
This area may be mentioned in the long-case discussion. Prenatal treatment of CAH attributable to 21-OHD by administration of corticosteroids (dexamethasone) to the mother is most commonly performed in females with a previously affected child. Informed consent must be obtained from the parents before prenatal treatment is contemplated. There are possible maternal adverse effects with CAH, the genital outcome is variable and there may be long-term effects on children, which are presently unknown. Masculinisation of the external genitalia begins at 6–7 weeks’ gestation; if treatment before this suppresses the fetal pituitary–adrenal axis, it could prevent ambiguous genitalia. Of reported cases where prenatal treatment has occurred, it was successful in three quarters of them (one third normal genitalia, two thirds mildly virilised) and unsuccessful in a quarter.
Management of acute adrenocortical insufficiency (adrenal crisis)
Immediate management
1. Assess and secure the airway, breathing and circulation (the ABCs).
2. Insert an IV cannula; take blood as above.
3. Restore the circulating volume with an infusion of normal saline (isotonic, 0.9%) with added glucose to make up to 5% glucose. Start with a 20 mL/kg bolus. Correct hypoglycaemia. Aim to replace fluid and electrolytes over the next 24–48 hours.
4. Give bolus hydrocortisone IV (or IM if IV access is difficult):
5. Hyperkalaemia may need further correction with insulin and glucose.
Diabetes mellitus
Background information
T1DM affects 1 in 1000 children (and 1 in 400 by adolescence). Incidence increases with age. The main genetic factor determining susceptibility to T1DM lies within the major histocompatibility complex (termed IDDM1). There is an association with certain HLA haplotypes: more than 90% of patients with T1DM have HLA-DR3 and/or HLA-DR4 (class II antigens located on the short arm of chromosome 6, at 6p21); 55% have a DR3/DR4 combination, most commonly DR4-DQ8/DR3-DQ2 (1 in 5 in families of diabetics versus 1 in 25 of the general population). DR3/DR4 heterozygosity is seen most frequently in children who develop T1DM under 5 years of age. For children who carry both of the highest-risk HLA haplotypes (DR4-DQ8/DR3-DQ2), the risk of being diagnosed with T1DM by 15 years is 1 in 20; if there is a sibling with T1DM and the same haplotypes, the risk is then 55%. One non-HLA gene is recognised as contributing to around 10% of the family aggregation of T1DM: this is termed IDDM2 and is situated on chromosome 11p5.5. This locus maps to a variable number of tandem nucleotide repeats (VNTR) of the insulin gene. Different sizes of the VNTR are associated with a risk of T1DM, the long form of VNTR being associated with protection from T1DM. A meta-analysis of data combined from most of the genomewide studies of linkage to T1DM, has been carried out by the Type 1 Diabetes Genetics Consortium; this shows that most of the genetic risk for T1DM is conferred by the class II genes encoding HLA-DR and HLA-DQ, as well as one or more additional genes within the HLA region. At least 50 inherited susceptibility loci for T1DM are known. An excellent resource noting all the genes implicated in T1DM, and constantly updated, is T1Dbase (http://www.t1dbase.org); it includes a table of all known T1DM loci, and clear diagrams of chromosomes, showing the position of each locus. Almost every chromosome has at least one locus identified. Apart from the genes in the HLA region, the majority of these loci affect T-cell function, including antigen-driven T-cell activation and cytokine signalling, proliferation or maturation.
Prevention of complications of T1DM
The DCCT Study (Diabetes Control and Complications Trial) involved 1441 volunteers with T1DM, ages 13–39, who had had T1DM for at least 1 but less than 15 years, and no, or minimal, diabetic eye disease; it compared intensive control of blood glucose (keeping HbA1c [glycosylated haemoglobin] as close to 6% as possible) versus standard control of blood glucose. Patients were studied for an average of 6.5 years. The DCCT study showed that intensive blood glucose control decreases the risk of eye disease (retinopathy) by 76%, kidney disease by 50% and nerve disease (neuropathy) by 60%. The trial ended, and was reported, in 1993, but 90% of patients were followed up, and a second study, the EDIC Study (Epidemiology of Diabetes Interventions and Complications), reporting in 2005, assessed both microvacular (eye, kidney and nerve) and macrovascular disease, including incidence and predictors of various forms of cardiovascular disease (including myocardial infarction, cerebrovascular accidents, and requirement for cardiac surgery). The EDIC study showed that intensive blood glucose control decreases the risk of any cardiovascular disease event by 42%, and non-fatal myocardial infarction, cerebrovascular accident or death from any cardiovascular cause by 57%. The EDIC study showed that the benefits noted for the microvascular complications involving the eyes, kidneys and nerves during the DCCT study persisted after that study was completed. This longer-lasting benefit from tight glucose control has been termed ‘metabolic memory’.
Diagnosis
Type 1 diabetes can be diagnosed in children as follows:
• Patients with the ‘classic’ symptoms of T1DM, namely the triad of polydypsia, polyuria and weight loss despite polyphagia, are diagnosed by having a random blood sugar level (BSL; the commonly used term for plasma glucose level) above 11 mmol/L. Some units recommend a level of 14 mmol/L, but in practice the diagnosis is usually clear cut, and the level much higher than either figure.
• Asymptomatic patients require two criteria: a fasting BSL above 7.8 mmol/L, plus a 2-hour postprandial BSL above 11 mmol/L. Alternatively, a formal oral glucose tolerance test (GTT) demonstrating a sustained elevation in BSL above 11 mmol/L at 2 hours is required, as well as a similarly elevated intervening value taken between the time the glucose load (which is 1.75 g/kg, up to 75 g) is given and the 2-hour value. Again, this is fairly theoretical, as doubtful cases requiring a GTT for diagnosis are very rare.
History
Initial diagnosis
1. Initial symptoms: for example, early symptoms preceding the classic triad, lack of weight gain, nocturia, behaviour change, altered school performance, changed vision (blurring), tempo of onset (developing over days, weeks or months pre-diagnosis).
2. Immediate pre-diagnosis symptoms: polydypsia, polyuria, weight loss despite polyphagia.
3. Symptoms associated with ketoacidosis: for example, vomiting, abdominal pain, clouding of consciousness.
4. Where, when and how the diagnosis was made, length of hospital stay, education given, treatment in hospital, treatment at discharge.
Progress of the disease
1. Details of subsequent hospitalisations (frequency, indications, usual length of stay, usual outcome).
2. Complications of the disease: for example, eye problems with cataracts, retinopathy, joint problems with limited joint mobility (LJM), severe hypoglycaemic reactions.
3. Complications of treatment (e.g. fat atrophy or hypertrophy, insulin allergic reactions); degree of control (number of episodes of ketoacidosis, frequency of hypoglycaemic symptoms).
4. Monitoring of the disease: how often seen in clinic, usual investigations performed, how often seen by local doctor.
5. Changes in management: for example, increase in insulin administration from daily to twice daily, twice daily with additional short/ultra-short-acting or basal bolus regimen, use of insulin pump, altered dosages due to occult nocturnal hypoglycaemia, changeover from human insulin to analogues.
6. At what age did the patient begin to administer his or her own insulin?
Current status
1. General health: lethargic or energetic.
2. Insulin type, dose, regimen (which sort, how much, when, given by whom, where, rotation of sites; modifications with raised BSL, sporting activities, intercurrent illness or dining out), compliance with treatment.
3. Diet prescribed (whether portions/exchanges used, glycaemic index, recommended foods), diet actually taken, alcohol intake (adolescents), involvement of dietician, any restrictions (adhered to or not).
4. Hypoglycaemia: how often, what symptoms (e.g. sweating, pallor, tremulousness, hunger, headache, odd behaviour, lethargy, crying, bad temper, lack of coordination, dizziness, vomiting, convulsions, loss of consciousness, early morning headaches after nocturnal ‘hypo’, restless sleep); usual precipitants; anticipatory strategies for prevention of ‘hypos’; response to ‘hypos’ such as taking fast-acting sugars (e.g. glass of orange juice with added sugar, glass of lemonade, jelly beans) followed by a small protein and complex carbohydrate snack (e.g. bread, biscuits); ever any need for intramuscular glucagon? (Note: if no ‘hypos’ have occurred, BSL may have been too high.) Any evidence of ‘hypoglycaemia unawareness’?
5. Control: hypoglycaemia (see above); hyperglycaemia (e.g. any nocturia, polyuria, blurred vision, weight loss, excessive weight gain, disturbance of menstrual periods in postpubertal girls); BSL readings (usual levels, when performed, how often, by whom, response to high level); usual HbAlc levels; urine testing (how often, what indications); amount of school missed in the last few months, vaginal thrush, other infections such as pilonidal sinus, infected ingrown toenail.
6. Other problems: for example, adolescent self-image, compliance issues.
Social history
1. Impact on child: self-image, reaction of school friends, coping with giving insulin, dietary restrictions, exercise, amount of school missed.
2. Impact on siblings: sibling rivalry, risk of diabetes.
3. Impact on parents: family finances, employment, concern regarding future complications, genetic counselling.
4. Social supports: Diabetes Association, access to social worker, government benefits obtained.
5. Coping: who attends the clinic with the patient, level of education of the child and parents, contingency plans for intercurrent illnesses or severe hypoglycaemia causing loss of consciousness, access to local doctor/paediatrician/hospital.
Management
This involves the use of insulin, diet and regular exercise, with the following aims:
1. Control of BSL: maintaining close to normoglycaemia; see below.
2. Prevention of acute complications; for example, hypoglycaemia, ketoacidosis.
3. Ensuring optimum growth and development.
4. Maintaining a normal lifestyle.
5. Adequate education of the patient and parents.
6. Early detection and treatment of associated disease (e.g. Hashimoto’s thyroiditis).
7. Provision of psychological support and counsel.
8. Ensuring adequate access to appropriate social supports.
9. Reducing long-term complications by maintaining good metabolic control.
10. Regular screening for complications and early intervention when they appear (ACE inhibitors for hypertension or proteinuria, etc).
Age-specific aspects of control
The expectations at different ages vary. There are three groups:
1. Infants to preschoolers. The main goal is to avoid hypoglycaemia and preserve cognitive function (in line with the findings of the DCCT). The acceptable range for BSLs at this age is between 6 and 15 mmol/L, and the HbA1c between 8.0 and 9.5 gm%.
2. School-age to puberty. Again, avoidance of hypoglycaemia is a top priority. Acceptable ranges: BSL 4–10 mmol/L; HbA1c 8.0 gm%.
3. Adolescence. Acceptable ranges: BSL 4–8 mmol/L; HbA1c as low as possible. It may be that postpubertal control is more important than prepubertal; this is unclear.
Insulin therapy
Average dosage requirements are as follows:
1. ‘Honeymoon’ period: 0.5 units per kilogram per day (or less).
2. Preadolescent: 1.0 unit per kilogram per day.
3. Adolescent: 1.0–2.0 units per kilogram per day (increase with pubertal growth spurt and reduce later when growth has finished).
Candidates should be familiar with the various insulin regimens. These include:
1. Daily (longer-acting only, or mixed short and longer).
3. Twice daily with additional short or ultrashort.
4. Basal bolus (three pre-meal short or ultra-short, plus longer in evening; only used in motivated adolescents).
5. Premixed (biphasic) insulin (only used for non-compliance, or inability to mix insulin).
