[level-membership-for-pathology-category]
19 ENDOCRINE SYSTEM
THYROID GLAND
Histologic organization of the thyroid gland
Each lobe of the thyroid gland consists of numerous follicles. The thyroid follicle, or acinus, is the structural and functional unit of the gland. It consists of a single layer of cuboidal epithelial cells, the follicular epithelium (Figures 19-1 and 19-2), enclosing a central lumen containing a colloid substance rich in thyroglobulin, an iodinated glycoprotein, yielding a periodic acid–Schiff (PAS)–positive reaction.
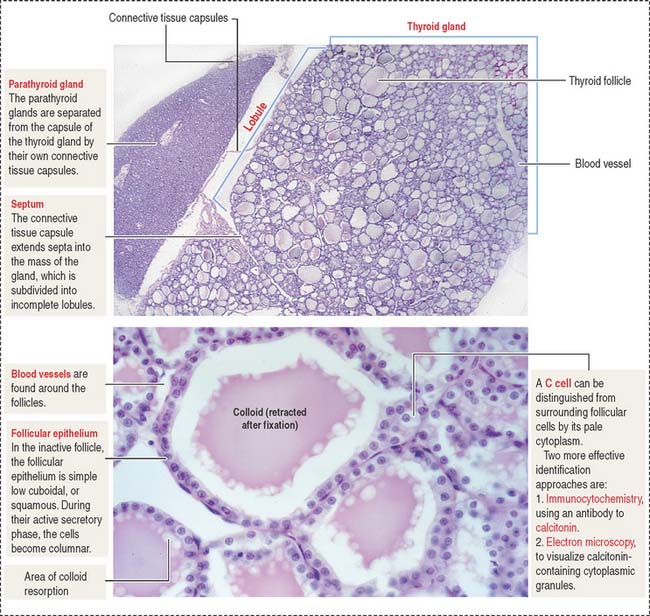
When the thyroid gland is active, the follicular epithelium is columnar, and colloid droplets may be seen within the cells as well as large apical pseudopodia and microvilli (see Figure 19-2).
Function of the thyroid gland
The synthesis and secretion of thyroid hormones involve two phases (Figure 19-3): (1) an exocrine phase and (2) an endocrine phase.
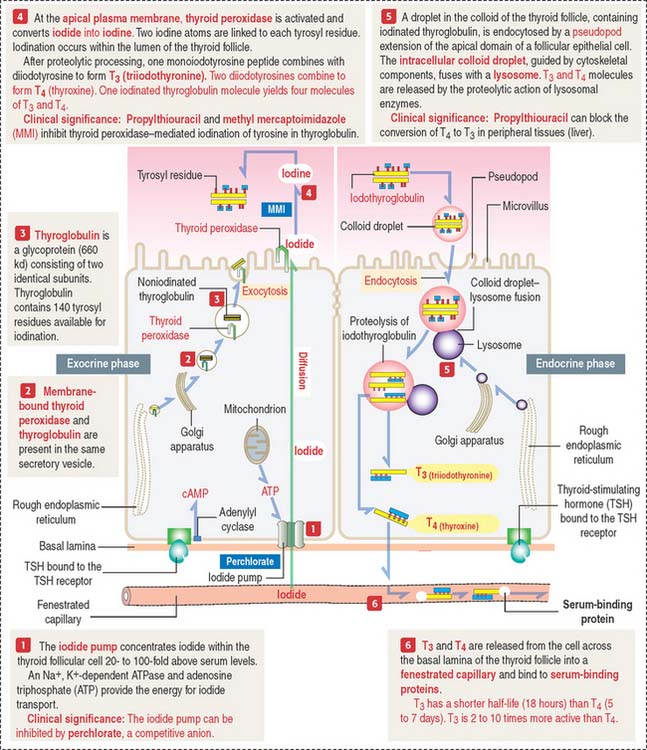
Both phases are regulated by TSH by a mechanism that includes receptor binding and cyclic adenosine monophosphate (cAMP) production, as discussed in Chapter 3, Cell Signaling.
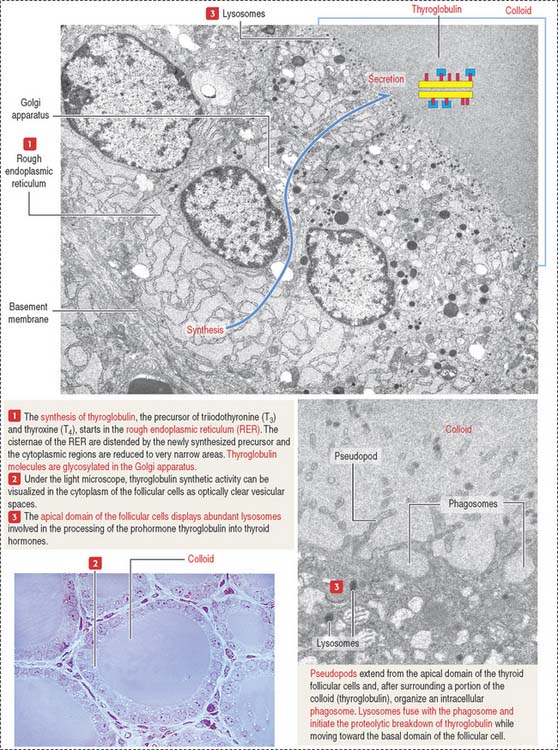
The exocrine phase (see Figure 19-3) consists of (1) the uptake of inorganic iodide from the blood, (2) the synthesis of thyroglobulin, and (3) the incorporation of iodine into tyrosyl residues of thyroglobulin by thyroid peroxidase.
The rough endoplasmic reticulum and Golgi apparatus are sites involved in the synthesis and glycosylation of thyroglobulin, a 660-kd glycoprotein composed of two identical subunits. Thyroglobulin is packed in secretory vesicles and released by exocytosis into the colloidal lumen. Thyroglobulin contains about 140 tyrosine residues available for iodination.
Thyroid peroxidase is activated during exocytosis. Activated thyroid peroxidase oxidizes iodide to iodine within the colloid; the iodine is then transferred to acceptor tyrosyl residues of thyroglobulin. Thyroid peroxidase activity and the iodination process can be inhibited by propylthiouracil and methyl mercaptoimidazole (MMI). These antithyroid drugs are used to inhibit the production of thyroid hormones by hyperactive glands.
The endocrine phase starts with the TSH-stimulated endocytosis of iodinated thyroglobulin into the follicular cell (see Figure 19-3):
Clinical significance: Hyperthyroidism (Graves’ disease) and hypothyroidism
Graves’ disease is an autoimmune disease in which the thyroid gland is hyperfunctional (Figure 19-4). Autoantibodies (called thyroid-stimulating immunoglobulins or TSIs), produced by plasma cells derived from sensitized T cells against TSH receptors present at the basal surface of thyroid follicular cells, bind to the receptor and mimic the effect of TSH, stimulating cAMP production.
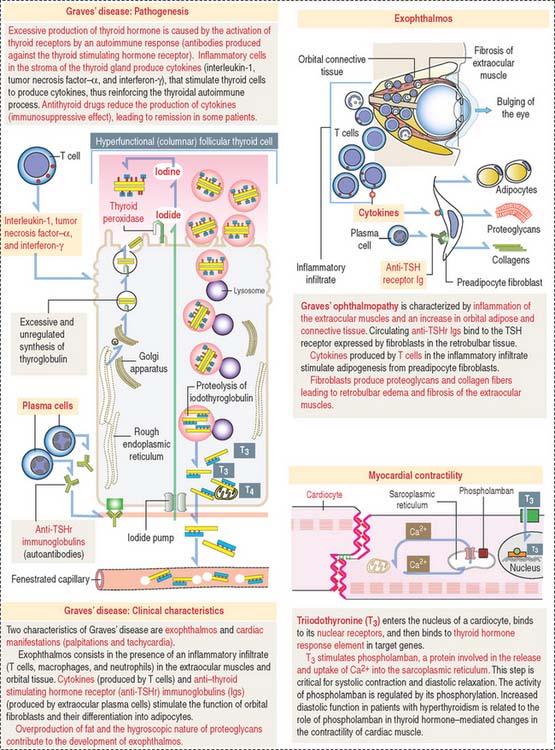
As a result, thyroid follicular cells become columnar and secrete large amounts of thyroid hormones in the blood circulation in an unregulated fashion. Enlargement of the thyroid gland (goiter), bulging of the eyes (exophthalmos; see Figure 19-4), tachycardia, warm skin, and fine finger tremors are typical clinical features.
Hashimoto’s disease is an autoimmune disease associated with hypothyroidism.
It is caused by autoantibodies targeted to thyroid peroxidase and thyroglobulin. Antibodies to thyroid peroxidase are known as antimicrosomal antibodies. A progressive destruction of the thyroid follicles leads to a decrease in the function of the thyroid gland.
PARATHYROID GLANDS
Histologic organization of the parathyroid glands
The parenchyma of the parathyroid glands consists of two cell populations supplied by sinusoidal capillaries (Figure 19-5): (1) the more numerous chief or principal cell, and (2) the oxyphil or acidophilic cell. Cells are arranged in cordlike or follicular-like clusters.
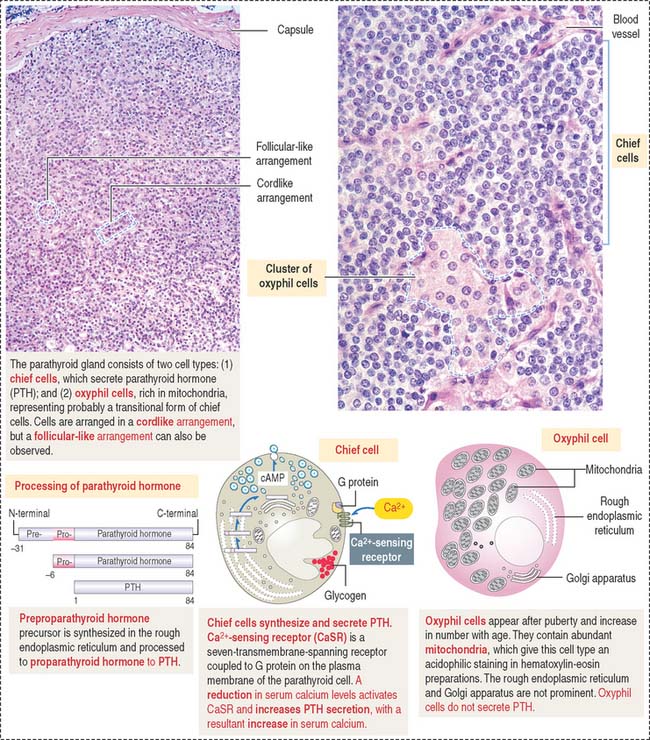
Function of the parathyroid hormone
Parathyroid hormone regulates the Ca2+ and PO43- balance in blood by acting on two main sites:
Parathyroid hormone binds to a cell surface receptor of the osteoblast to regulate the synthesis of three proteins essential for the differentiation and function of osteoclasts (Figure 19-6; see also the discussion of osteoclasts in Chapter 4, Connective Tissue):
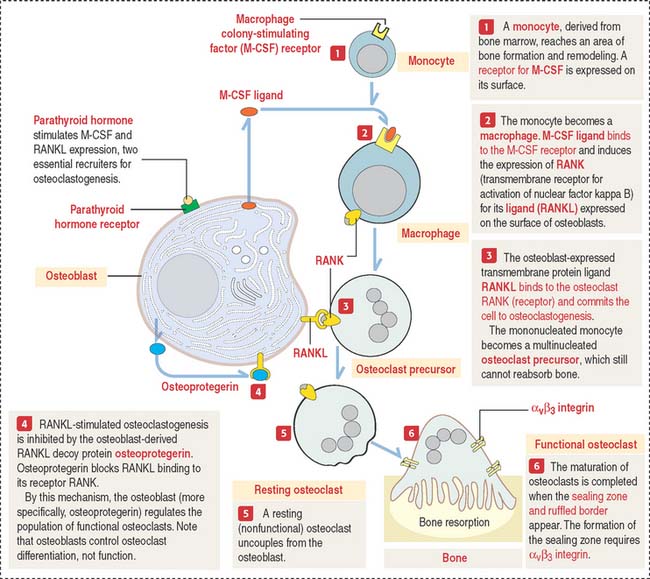
You should be aware that RANKL not only regulates osteoclastogenesis but also is expressed by dendritic cells, T and B cells, components of the immune system. This is an important consideration in anti-RANKL treatment of some forms of osteoporosis, as discussed under Bone in Chapter 4, Connective Tissue.
Clinical significance: Hyperparathyroidism, hypoparathyroidism, and CaSR mutations
C CELLS (THYROID FOLLICLE)
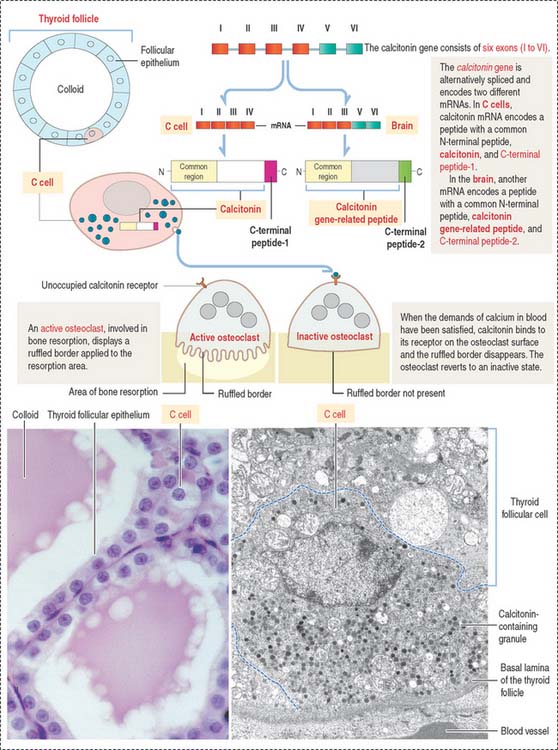
C cells derive from neural crest cells and are associated with thyroid follicles. C cells (1) represent about 0.1% of the mass of thyroid tissue, (2) may be present within (or outside) the thyroid follicle but are not in contact with the colloid, and (3) produce calcitonin, encoded by a gene located on the short arm of chromosome 11 (Figure 19-7).
VITAMIN D
Vitamin D2 is formed in the skin by the conversion of 7-dehydrocholesterol to cholecalciferol following exposure to ultraviolet light (Figure 19-8). Cholecalciferol is then absorbed into the blood circulation and transported to the liver where it is converted to 25-hydroxycholecalciferol by the addition of a hydroxyl group to the side chain.
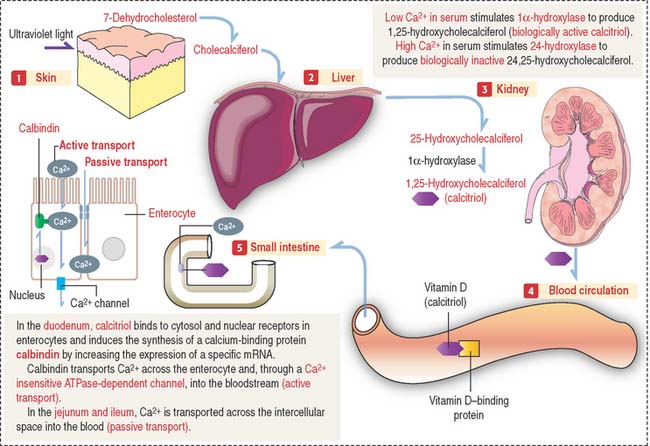
In the nephron, two events can occur:
The main function of vitamin D is to stimulate calcium absorption by the intestinal mucosa. Calcium is absorbed by (1) transcellular absorption (active mechanism) in the duodenum, an active process that involves the import of calcium by enterocytes through voltage-insensitive channels, its transport across the cell—assisted by the carrier protein calbindin—and its release from the cell by a calcium-ATPase—mediated mechanism; and (2) paracellular absorption (passive mechanism) in the jejunum and ileum, through tight junctions into the intercellular spaces, and into blood. A small fraction (about 10%) of calcium absorption takes place in the large intestine by active and passive mechanisms.
ADRENAL GLAND
Functions of the fetal adrenal cortex
During the early stage of gestation, the adrenal cortex synthesizes dehydroepiandrosterone, a precursor of the synthesis of estrogen by the placenta. A lack of 3β-hydroxysteroid dehydrogenase activity prevents the synthesis of progesterone, glucocorticoids, and androstenedione. The interaction between the fetal adrenal cortex and the placenta is known as the fetoplacental unit (see Chapter 23, Fertilization, Placentation, and Lactation).
Histologic organization of the adrenal cortex
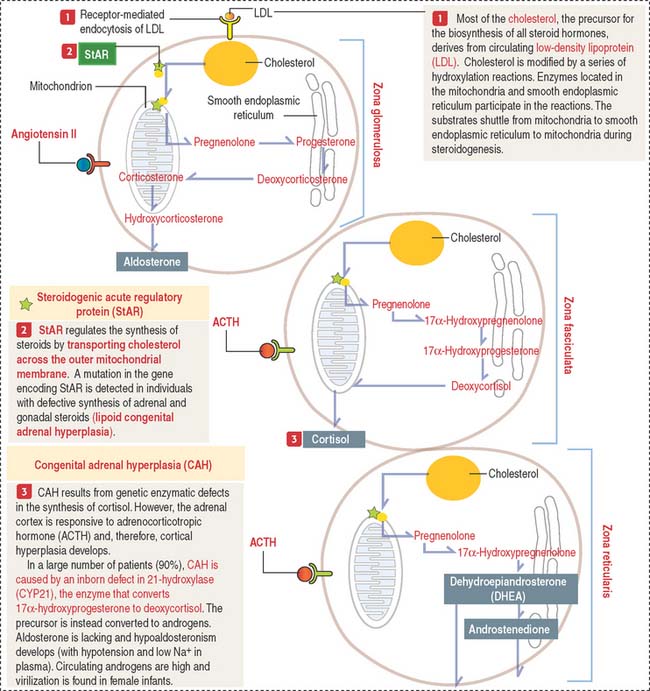
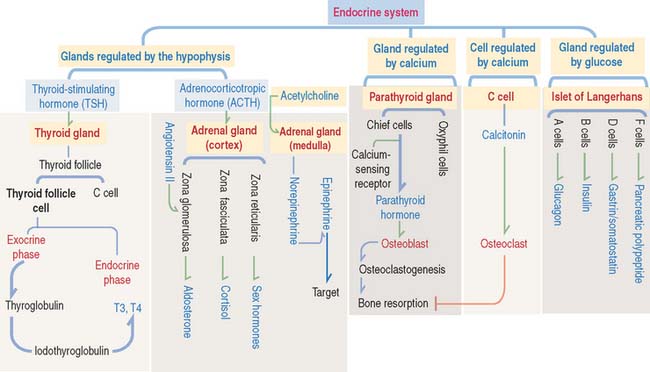
The adrenal cortex consists of three concentric zones (Figures 19-9 and 19-10). (1) The outermost layer of the cortex is the zona glomerulosa. (2) The middle layer of the cortex is the zona fasciculata. (3) The innermost layer of the cortex is the zona reticularis.
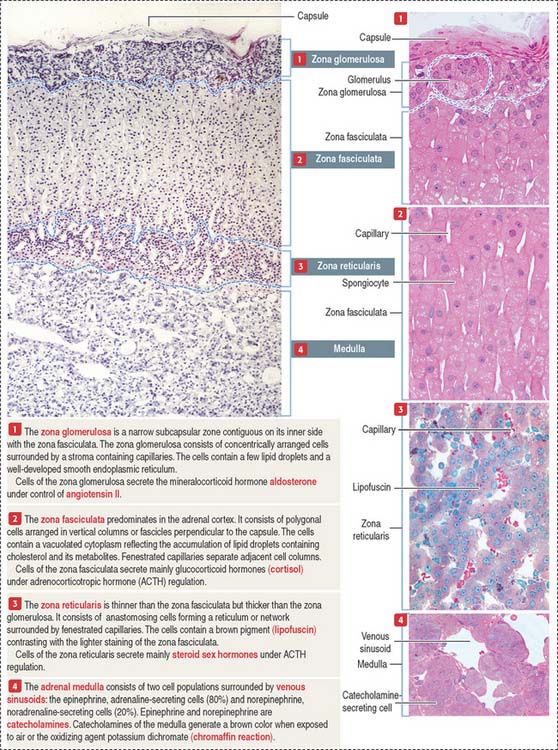
Cells of the zona glomerulosa produce the mineralocorticoid aldosterone (Figures 19-11 and 19-12). Although the zona fasciculata is often associated with glucocorticoid production—mainly cortisol—and the zona reticularis with androgen production, the functional distinctions between the two layers are not precise and they appear as a functional unit. In addition, these two layers are stimulated by corticotropin (ACTH), whereas the zona glomerulosa is primarily angiotensin II–dependent. Angiotensin II stimulates both the growth of the zona glomerulosa and the synthesis of aldosterone (see Figure 19-12).
Angiotensin II is an octapeptide derived from the conversion of the angiotensin I decapeptide in the pulmonary circulation by angiotensin-converting enzyme (see Chapter 14, Urinary System). Aldosterone has a half-life of 20 to 30 minutes and acts directly on the distal convoluted tubule and collecting tubule, where it increases Na+ reabsorption and excretion of K+.
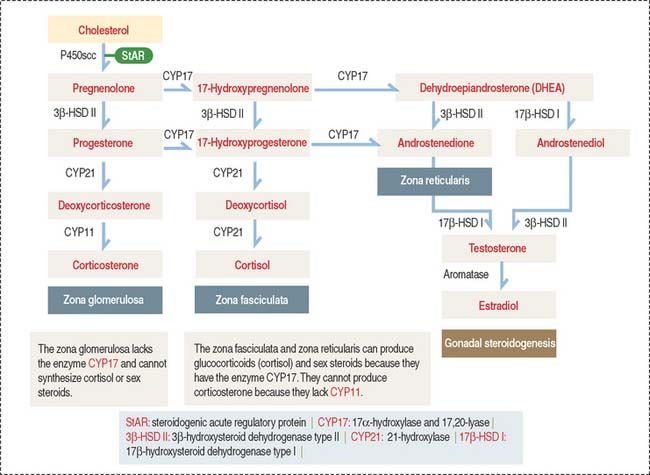
The zona glomerulosa (Latin glomus, ball) has the following characteristics (see Figure 19-9): (1) it lies under the capsule; (2) it represents 10% to 15% of the cortex; (3) its cells aggregate into a glomerulus-like arrangement and have a moderate amount of lipid droplets in the cytoplasm; and (4) it lacks the enzyme 17α-hydroxylase and, therefore, cannot produce cortisol or sex steroids.
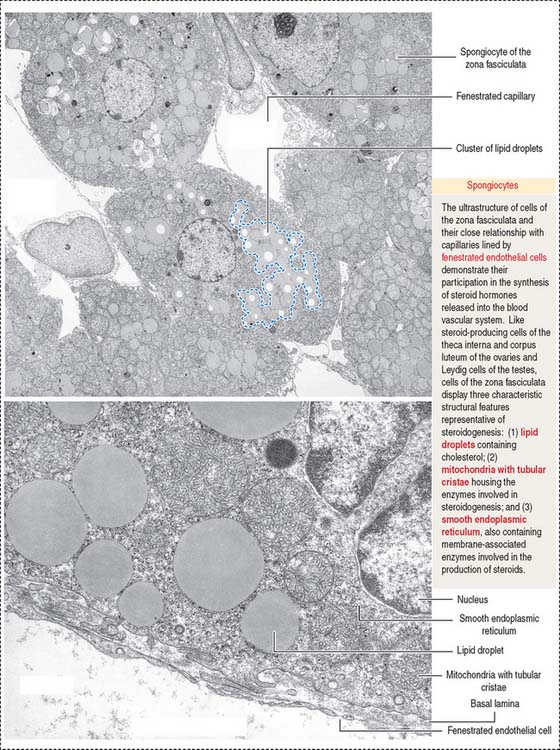
The zona fasciculata (Latin fascis, bundle) makes up 75% of the cortex. It is formed by cuboid cells, with the structural features of steroid-producing cells (see Figure 19-10), arranged in longitudinal cords separated by cortical fenestrated capillaries, or sinusoids (see Figure 19-11).
The cytoplasm of zona fasciculata cells shows three components that characterize their steroidogenic function: (1) the steroid hormone precursor cholesterol stored in abundant lipid droplets (see Figure 19-11); when lipids are extracted during histologic preparation or are unstained by the standard hematoxylin-eosin (H&E) procedures, the cells of the zona fasciculata display a foamy appearance and are called spongiocytes; (2) mitochondria with tubular cristae containing steroidogenic enzymes; and (3) well-developed smooth endoplasmic reticulum, also with enzymes involved in the synthesis of steroid hormones (see Figure 19-11).
Cells of the zona fasciculata and zona reticularis cannot produce aldosterone but contain 17α –hydroxylase necessary for the production of glucocorticoids—cortisol—and cortisol—and the enzyme 17, 20-hydroxylase, required for the production of sex hormones.
Cortisol has two major effects: (1) A metabolic effect: Cortisol’s effects are opposite to those of insulin. In the liver, cortisol stimulates gluconeogenesis to increase the concentration of glucose in blood. (2) An anti-inflammatory effect: Cortisol suppresses tissue responses to injury and decreases cellular and humoral immunity.
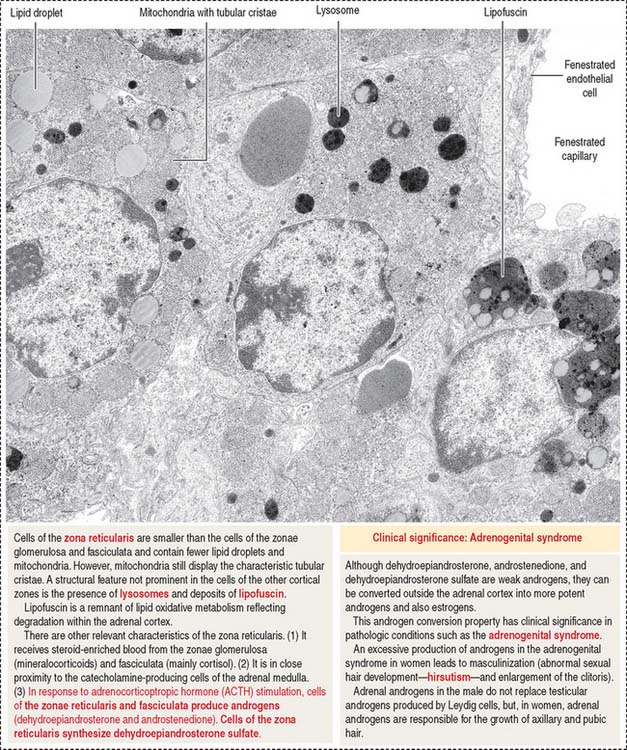
The cells of this zone are acidophilic, due to abundant lysosomes, large lipofuscin granules, and fewer lipid droplets (see Figure 19-11). Although cells of the zona fasciculata can synthesize androgens, the primary site of adrenal sex hormone production is the zona reticularis. Dehydroepiandrosterone (DHEA) and androstenedione are the predominant androgens produced by the cortex of the adrenal gland (see Figures 19-12 and 19-13). DHEA sulfate is synthesized in the zona reticularis.
Adrenal medulla
Chromaffin cells (Figure 19-14) are modified sympathetic postganglionic neurons—without postganglionic processes—derived from the neural crest and forming epithelioid cords surrounded by fenestrated capillaries. The cytoplasm of chromaffin cells contains membrane-bound dense granules consisting in part of matrix proteins, called chromogranins, and one class of catecholamine, either epinephrine or norepinephrine (adrenaline or noradrenaline). Some granules contain both epinephrine and norepinephrine. Minimal secretion of dopamine also occurs, but the role of adrenal dopamine is not known.
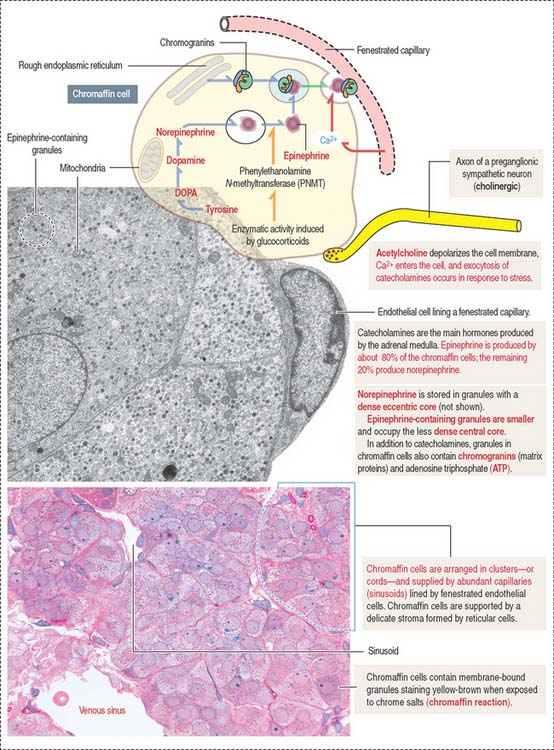
Two different chromaffin cell types are present. About 80% of the cells produce epinephrine and 20% synthesize norepinephrine. These two cell populations can be distinguished at the electron microscope level by the morphology of the membrane-bound granules. Norepinephrine is stored in granules with a dense eccentric core. Epinephrine-containing granules are smaller and occupy the less dense central core. Note an important difference with cells of the adrenal cortex: cells from the adrenal cortex do not store their steroid hormones in granules.
Catecholamines are synthesized from tyrosine to DOPA (3,4-dihydroxyphenylalanine) in the presence of tyrosine hydroxylase (see Figure 19-14). DOPA is converted to dopamine by DOPA decarboxylase. Dopamine is transported into existing granules and converted inside them by dopamine β-hydroxylase to norepinephrine. The membrane of the granules contains the enzymes required for catecholamine synthesis and ATP-driven pumps for the transport of substrates.
Blood supply to the adrenal gland
Similar to all endocrine organs, the adrenal glands are highly vascularized. Arterial blood derives from three different sources (Figure 19-15): (1) the inferior phrenic artery, which gives rise to the superior adrenal artery; (2) the aorta, from which the middle adrenal artery branches out; and (3) the renal artery, which gives rise to the inferior adrenal artery.
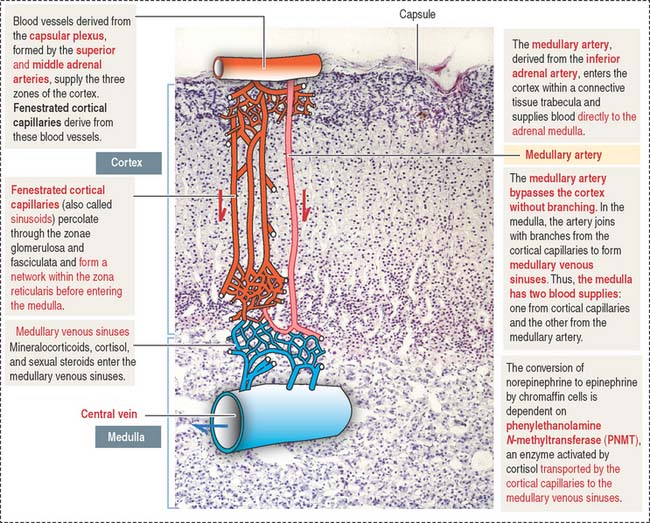
There are no veins or lymphatics in the adrenal cortex. The adrenal cortex and medulla are drained by the central vein, present in the adrenal medulla.
Clinical significance: Abnormal secretory activity of the adrenal cortex
An acute destruction of the adrenal gland by meningococcal septicemia in infants is the cause of Waterhouse-Friderichsen syndrome. A chronic destruction of the adrenal cortex by an autoimmune process or tuberculosis results in the classic Addison’s disease. In Addison’s disease, ACTH secretion increases because of the cortisol deficiency. ACTH can cause an increase in skin pigmentation, in particular in the skin folds and gums. The loss of mineralocorticoids leads to hypotension and circulatory shock. A loss of cortisol decreases vasopressive responses to catecholamines and leads to an eventual drop in peripheral resistance, thereby contributing to hypotension. A deficiency in cortisol causes muscle weakness (asthenia).
ENDOCRINE PANCREAS
Islets of Langerhans
The pancreas has two portions (Figures 19-16 and 19-17):
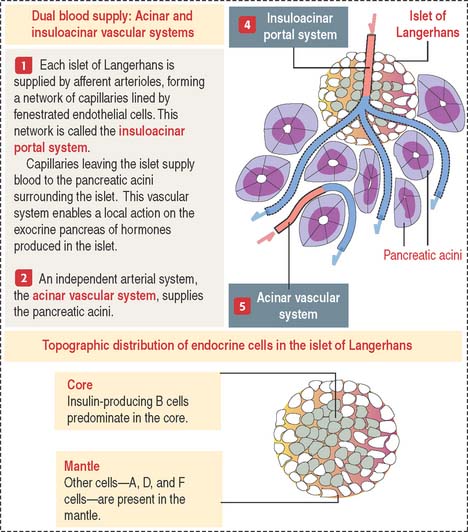
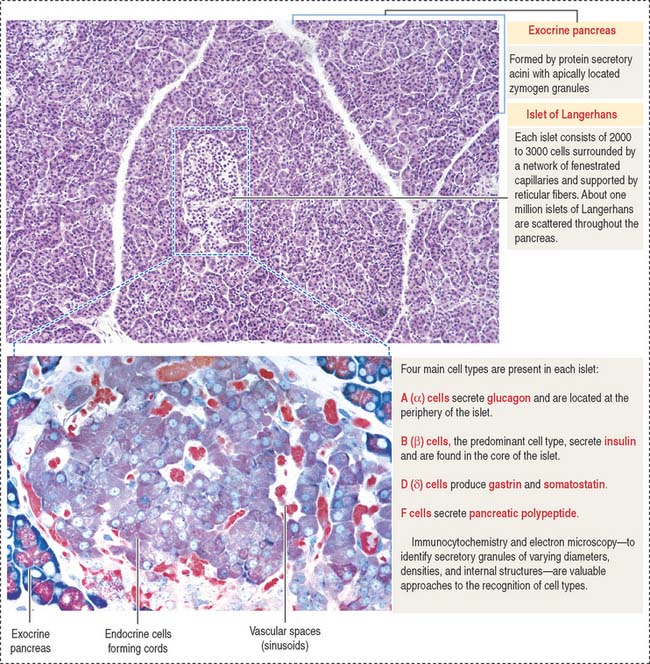
Each islet of Langerhans is formed by two components:
An independent vascular system, the acinar vascular system, supplies blood directly to the exocrine pancreatic acini.
B cells (β cells) produce insulin, a 6-kd polypeptide consisting of two chains (Figure 19-18): (1) chain A, with 21 amino acids; and (2) chain B, with 30 amino acids. Chains A and B are linked by disulfide bonds.
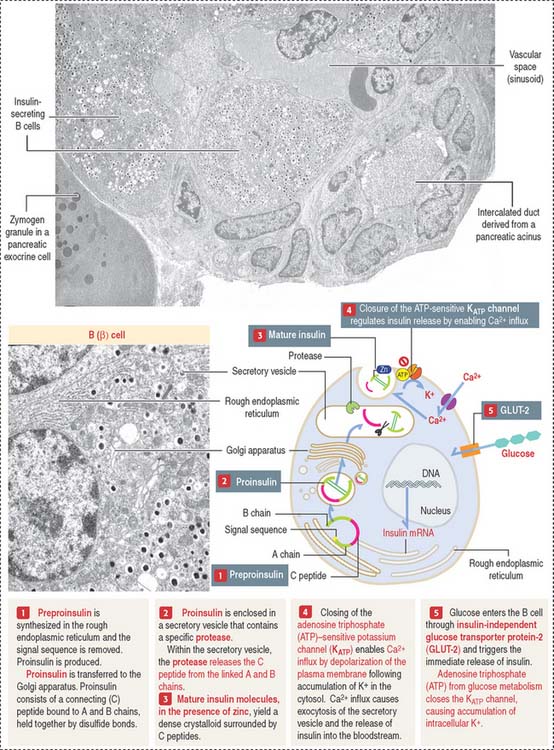
If glucose levels remain high, new synthesis of insulin occurs. GLUT-2 is also present in hepatocytes. Insulin is required for increasing the transport of glucose in cells (predominantly in hepatocytes, skeletal and cardiac muscle, fibroblasts, and adipocytes). This is accomplished by (1) the transmembrane transport of glucose and amino acids, (2) the formation of glycogen in hepatocytes and skeletal and cardiac muscle cells, and (3) the conversion of glucose to triglycérides in adipose cells (Figure 19-19).
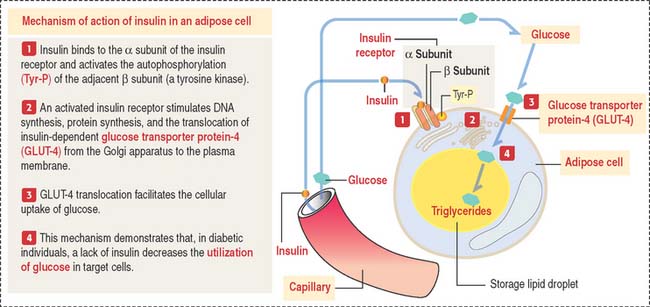
Note the functional difference between GLUT-2 and GLUT-4: (1) GLUT-2 is insulin-independent and serves to transport glucose to insular B cells and hepatocytes; (2) GLUT-4 is insulin-dependent and serves to remove glucose from blood.
Neither C peptide nor zinc is present in glucagon-containing secretory granules.
D (δ) cells produce gastrin (see discussion of enteroendocrine cells in Chapter 15, Upper Digestive Segment) and somatostatin. Somatostatin is a 14 amino-acid peptide identical to somatostatin produced in the hypothalamus. Somatostatin inhibits the release of insulin and glucagon in a paracrine manner.
Cell types in the islets of Langerhans can be identified by (1) immunocytochemistry, using antibodies specific for each cell product; (2) electron microscopy, to distinguish the size and structure of the secretory granules; and (3) the cell distribution in the islet. B cells are centrally located (core distribution) and surrounded by the other cell types (mantle distribution; see Figure 19-16).
Clinical significance: ATP-sensitive K+ channel and insulin secretion
KATP channel modulates the influx of Ca2+ through voltage-gated Ca2+ channels. In the normal resting state, the KATP channel is open and the voltage-gated Ca2+ channel remains closed. No insulin is secreted. When glucose is taken up by B cells by GLUT-2, the KATP channel closes utilizing ATP derived from glucose metabolism. K+ accumulates in the cell, the Ca2+ channel opens by membrane depolarization, and the influx of Ca2+ triggers insulin exocytosis (see Figure 19-18).
Clinical significance: Insulin and diabetes
Hyperglycemia can be the result of the following (Figure 19-20):
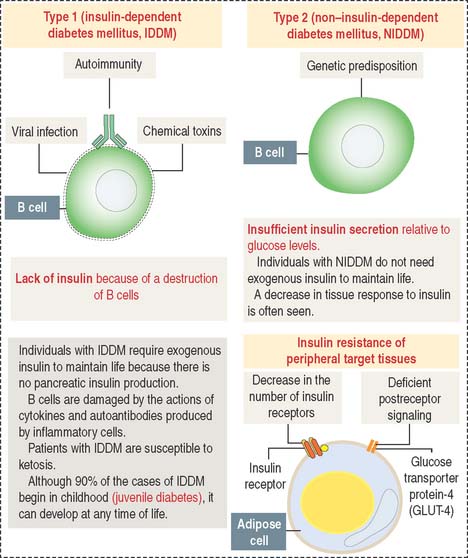
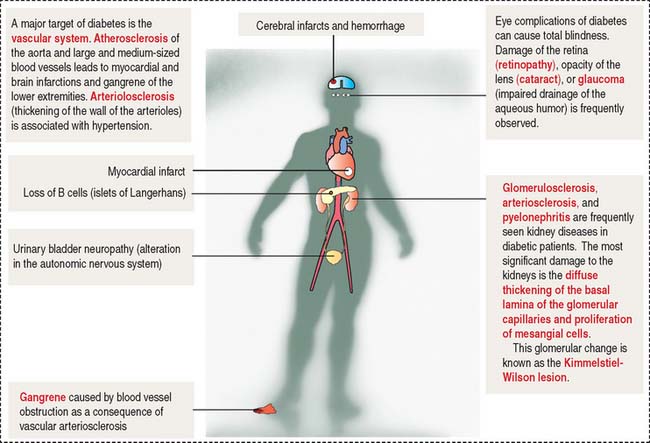
The symptoms and consequences of type 1 and type 2 diabetes are generally similar. Hyperglycemia, polyuria (increased frequency of micturition and urine volume), and polydipsia (sensation of thirst and increased fluid intake) are the three characteristic symptoms. The clinical forms of diabetes mellitus are summarized in Figure 19-20. The late complications of diabetes mellitus are summarized in Figure 19-21.
Essential concepts
Hyperparathyroidism is caused by an adenoma (benign tumor) of the parathyroid gland. Excessive secretion of parathyroid hormone causes hypercalcemia, phosphaturia, and hypercalciuria. Complications include the formation of renal stones and bone cysts caused by excessive removal of mineralized bone. Inactivating mutations of CaSR cause familial benign hypercalcemia. Activating mutations of CaSR result in idiopathic hypoparathyroidism.
In children, a deficiency of vitamin D causes rickets. In adults, it causes osteomalacia.
Mutations in the sulfonyurea receptor (Sur 1) gene and the inward rectifying K+ channel (Kir6.2) gene, components of the KATP channel, are seen in patients with neonatal diabetes mellitus.
Diabetes is characterized by hyperglycemia, polyuria, and polydipsia.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
19 ENDOCRINE SYSTEM
THYROID GLAND
Histologic organization of the thyroid gland
Each lobe of the thyroid gland consists of numerous follicles. The thyroid follicle, or acinus, is the structural and functional unit of the gland. It consists of a single layer of cuboidal epithelial cells, the follicular epithelium (Figures 19-1 and 19-2), enclosing a central lumen containing a colloid substance rich in thyroglobulin, an iodinated glycoprotein, yielding a periodic acid–Schiff (PAS)–positive reaction.
When the thyroid gland is active, the follicular epithelium is columnar, and colloid droplets may be seen within the cells as well as large apical pseudopodia and microvilli (see Figure 19-2).
Function of the thyroid gland
The synthesis and secretion of thyroid hormones involve two phases (Figure 19-3): (1) an exocrine phase and (2) an endocrine phase.
Both phases are regulated by TSH by a mechanism that includes receptor binding and cyclic adenosine monophosphate (cAMP) production, as discussed in Chapter 3, Cell Signaling.
The exocrine phase (see Figure 19-3) consists of (1) the uptake of inorganic iodide from the blood, (2) the synthesis of thyroglobulin, and (3) the incorporation of iodine into tyrosyl residues of thyroglobulin by thyroid peroxidase.
The rough endoplasmic reticulum and Golgi apparatus are sites involved in the synthesis and glycosylation of thyroglobulin, a 660-kd glycoprotein composed of two identical subunits. Thyroglobulin is packed in secretory vesicles and released by exocytosis into the colloidal lumen. Thyroglobulin contains about 140 tyrosine residues available for iodination.
Thyroid peroxidase is activated during exocytosis. Activated thyroid peroxidase oxidizes iodide to iodine within the colloid; the iodine is then transferred to acceptor tyrosyl residues of thyroglobulin. Thyroid peroxidase activity and the iodination process can be inhibited by propylthiouracil and methyl mercaptoimidazole (MMI). These antithyroid drugs are used to inhibit the production of thyroid hormones by hyperactive glands.
The endocrine phase starts with the TSH-stimulated endocytosis of iodinated thyroglobulin into the follicular cell (see Figure 19-3):
Clinical significance: Hyperthyroidism (Graves’ disease) and hypothyroidism
Graves’ disease is an autoimmune disease in which the thyroid gland is hyperfunctional (Figure 19-4). Autoantibodies (called thyroid-stimulating immunoglobulins or TSIs), produced by plasma cells derived from sensitized T cells against TSH receptors present at the basal surface of thyroid follicular cells, bind to the receptor and mimic the effect of TSH, stimulating cAMP production.
As a result, thyroid follicular cells become columnar and secrete large amounts of thyroid hormones in the blood circulation in an unregulated fashion. Enlargement of the thyroid gland (goiter), bulging of the eyes (exophthalmos; see Figure 19-4), tachycardia, warm skin, and fine finger tremors are typical clinical features.
Hashimoto’s disease is an autoimmune disease associated with hypothyroidism.
It is caused by autoantibodies targeted to thyroid peroxidase and thyroglobulin. Antibodies to thyroid peroxidase are known as antimicrosomal antibodies. A progressive destruction of the thyroid follicles leads to a decrease in the function of the thyroid gland.
PARATHYROID GLANDS
Histologic organization of the parathyroid glands
The parenchyma of the parathyroid glands consists of two cell populations supplied by sinusoidal capillaries (Figure 19-5): (1) the more numerous chief or principal cell, and (2) the oxyphil or acidophilic cell. Cells are arranged in cordlike or follicular-like clusters.
Function of the parathyroid hormone
Parathyroid hormone regulates the Ca2+ and PO43- balance in blood by acting on two main sites:
Parathyroid hormone binds to a cell surface receptor of the osteoblast to regulate the synthesis of three proteins essential for the differentiation and function of osteoclasts (Figure 19-6; see also the discussion of osteoclasts in Chapter 4, Connective Tissue):
You should be aware that RANKL not only regulates osteoclastogenesis but also is expressed by dendritic cells, T and B cells, components of the immune system. This is an important consideration in anti-RANKL treatment of some forms of osteoporosis, as discussed under Bone in Chapter 4, Connective Tissue.
Clinical significance: Hyperparathyroidism, hypoparathyroidism, and CaSR mutations
C CELLS (THYROID FOLLICLE)
C cells derive from neural crest cells and are associated with thyroid follicles. C cells (1) represent about 0.1% of the mass of thyroid tissue, (2) may be present within (or outside) the thyroid follicle but are not in contact with the colloid, and (3) produce calcitonin, encoded by a gene located on the short arm of chromosome 11 (Figure 19-7).
VITAMIN D
Vitamin D2 is formed in the skin by the conversion of 7-dehydrocholesterol to cholecalciferol following exposure to ultraviolet light (Figure 19-8). Cholecalciferol is then absorbed into the blood circulation and transported to the liver where it is converted to 25-hydroxycholecalciferol by the addition of a hydroxyl group to the side chain.
In the nephron, two events can occur:
The main function of vitamin D is to stimulate calcium absorption by the intestinal mucosa. Calcium is absorbed by (1) transcellular absorption (active mechanism) in the duodenum, an active process that involves the import of calcium by enterocytes through voltage-insensitive channels, its transport across the cell—assisted by the carrier protein calbindin—and its release from the cell by a calcium-ATPase—mediated mechanism; and (2) paracellular absorption (passive mechanism)
[/not-level-membership-for-pathology-category]
