168 Endocrine and Metabolic Crises in the Pediatric Intensive Care Unit
Increasing numbers of endocrine and metabolic conditions are being recognized, and the number of children in treatment programs for them is increasing. Although improved screening programs and therapy may decrease the number of children requiring critical care for these conditions, it is likely they will be recognized in increasing numbers of critically ill children for the foreseeable future. There is also increasing awareness of the significance of metabolic changes such has hypo- and hyperglycemia in the pediatric intensive care unit (PICU).1 General principles of PICU management apply to patients with endocrine and metabolic crises (Table 168-1).2,3 Crises may cause damage with long-term sequelae for the child and family; however, they also present unique diagnostic opportunities. The intensivist has a particular responsibility to:
TABLE 168-1 Principles of Management of Metabolic and Endocrine Crises
| Principle | Specifics of Conditions |
|---|---|
| Airway management | Many patients have depressed level of consciousness, and airway management is essential to prevent complications. |
| Breathing support | Acidotic patients may make huge respiratory effort; ventilatory support may help decrease the metabolic demands on these patients. Although administration of sodium bicarbonate may help to settle some of the acidosis-related symptoms such as hyperventilation, bicarbonate may aggravate some problems seen in conjunction with urea cycle defects. Give bicarbonate only if the plasma bicarbonate <10 mmol/L, and then only half correct deficits. |
| Circulatory support | Ensure adequate circulating volume; this may be a particular issue if there has been excessive fluid loss from vomiting or diarrhea. |
| Disability | Control seizures using anticonvulsant agents. Administer pyridoxine if possibility of pyridoxine dependency. |
| Dialysis to remove toxins where necessary | Hemodialysis is the most efficient means of removing toxins such as ammonia and leucine. Hemofiltration is less efficient but may be more applicable in critically ill children. Peritoneal dialysis is slower but has the advantage of ease of initiation.1 In some conditions, it may be possible to remove toxins by stimulating alternative pathways of metabolism. |
| Ensure that glucose is maintained in the normal range | A normal glucose level should be maintained at all times. Excessive administration of glucose in the mitochondrial energy chain problem may exacerbate lactic acidosis. Also, attempt to provide an adequate energy supply (may use medium-chain fatty acids where appropriate). Minimize energy demands on patient. |
| Fluids | In general, provide 1.5× normal fluid maintenance requirements to accelerate excretion of water-soluble toxins. In the context of encephalopathy (MSUD or urea cycle defects), be careful to avoid overhydration, which may contribute to development of cerebral edema. |
| Feeds | If there is accumulation of a product, this needs to be eliminated from the diet (e.g., fructose, galactose). Start with protein-free diet, but do not continue beyond 2 days, because the catabolic state also creates problems. If diagnosis not identified, need gradual reintroduction of feeds and nutrition. If there is deficiency of any nutrient (e.g., carnitine, which may have a primary or a secondary deficiency), supplement that nutrient. Ensure there is an adequate energy source along a metabolic route that is functional. Provide specific vitamin therapy where indicated. |
| Family support and information | The diagnosis of an inborn error of metabolism has major implications for families, and considerable support is required.2 |
| Treat infection | Infections are an important component of pediatric ICU presentation of inborn errors of metabolism. Some conditions such as galactosemia are related to specific infections such as Escherichia coli. Other conditions are related to pyogenic infections because of neutropenia. Children who are in a poor nutritional or metabolic state are more susceptible to infection. Intercurrent infections may be the precipitating factor for metabolic decompensation. |
| Investigations | A wide variety of investigations are relevant to inborn errors of metabolism. Biochemical testing on a range of body fluids and on tissues is fundamental to accurate diagnosis of the problem. Biochemical tests may range from simple screening tests to more complex tests on tissue culture. Imaging techniques such as CT, MRI, magnetic resonance spectroscopy, and echocardiography may be relevant. Functional tests such as EEG, ECG, and EMG may be useful in diagnosis. Increasingly, genetic diagnosis is available if children have recognized genetic mutations. |
| Monitor response to therapy | Clinical monitoring is essential. Biochemical monitoring of the appropriate metabolites is essential to ensure that metabolic control is established. |
CT, computed tomography; ECG, electrocardiogram; EEG, electroencephalogram; EMG, electromyogram; ICU, intensive care unit; MRI, magnetic resonance imaging.
A particular problem of endocrine and metabolic crises is that laboratory investigation of specific conditions may take time while patients require urgent therapeutic intervention. Because it may not always be possible to follow algorithms of investigation, a reasonable approach is to collect all relevant specimens immediately,6 store them appropriately, and liaise with laboratory services to use the specimens in a logical and cost-effective manner to confirm the diagnosis.
 Endocrine Crises
Endocrine Crises
Abnormalities of Glucose Control
Abnormalities of glucose control, including diabetic ketoacidosis, are the most common endocrine crises encountered in the PICU. Hypoglycemia and hyperglycemia are associated with increased mortality1,7 in sick children and may be part of a wide variety of disease processes. Measurement of blood glucose is part of the initial biochemical evaluation of any sick child, particularly if a depressed level of consciousness or shock is present. When an abnormal glucose level has been identified, it must be addressed and levels be remeasured at appropriate intervals until the problem has been resolved. The situation is further complicated by technical issues in the measurement of blood glucose,8,9 with differences between blood and plasma glucose level (glucose concentration in plasma is approximately 11% higher than whole blood because of the higher water content in plasma, but this may be affected by anemia or polycythemia); differences between arterial, venous, and capillary glucose levels (which may also vary depending on clinical context),8 and potentially significant differences between measurement techniques.9 A particular concern is that in general, inaccuracies increase at lower glucose levels.9 Generally, central laboratory devices are taken as the standard, although there is increasing utilization (and convenience) of point-of-care devices.
Hypoglycemia
Hypoglycemia may be associated with devastating damage to the brain and requires immediate attention. In general, a diagnosis of hypoglycemia depends on the presence of symptoms and a low blood glucose level, and resolution of symptoms on correction of the low glucose level. Unfortunately, symptoms of hypoglycemia are relatively nonspecific, ranging from lethargy, poor feeding, hypotonia, and “jitteriness” to convulsions, apneic episodes, cardiovascular collapse, and sudden infant death syndrome (SIDS). Hypoglycemia may be hidden in the complex of critical illness, particularly if patients are deeply sedated and paralyzed. In addition, some diabetic patients have reduced awareness of hypoglycemia.10 Thus, regular monitoring of blood glucose is an important component of the management of any critically ill child.
Hypoglycemia immediately following birth may be common, but there are considerable controversies in the definition of hypoglycemia in this period.11–13 Table 168-2 lists an approach to hypoglycemia immediately following birth.
TABLE 168-2 At-Risk Infants for Whom Routine Monitoring of Blood Glucose Is Recommended
| Associated with Changes in Maternal Metabolism |
From Cornblath M, Hawdon JM, Williams AF et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105:1141-5.
Symptomatic hypoglycemia occurs more frequently during the neonatal period than in any other period of childhood. Infants at particular risk include infants with poor hepatic glycogen stores (e.g., preterm or small-for-gestational-age infants); poor glucose intake (e.g., preterm or ill infants); and hyperinsulinism, either primary or secondary to high intrauterine glucose levels (e.g., infants of diabetic mothers).14 Hypoglycemia also may be a feature of perinatal illness including asphyxia, polycythemia, hypothermia, septicemia, and respiratory distress syndrome. Much less common causes include growth hormone15 or adrenal insufficiency,16 inborn errors of metabolism, and glucagon insufficiency. Drugs administered to the mother during pregnancy, including oral hypoglycemic agents, also must be considered.
In childhood, hypoglycemia may result from inadequate glucose intake (prolonged starvation, malabsorption); defects in glycogenolysis (glycogen storage disorders) or gluconeogenesis (fructose-1,6-diphosphatase deficiency, ethanol intoxication, Jamaican vomiting sickness, etc.), fatty acid oxidation disorders and defects in ketogenesis, deficiency of gluconeogenic hormones (e.g., adrenalin, corticosteroids, glucagons, growth hormone, thyroid hormone), excessive insulin secretion (hyperinsulinism), and a variety of specific disorders including abnormalities of amino acid metabolism.17
As soon as hypoglycemia is noted, specimens should be collected immediately for appropriate tests (Table 168-3). Treatment of hypoglycemia with intravenous (IV) glucose should be initiated promptly. An initial bolus dose of 0.5 g/kg of glucose (may need 0.5-2 g/kg in neonates) should be given as a 10% or 25% (in older children) dextrose solution, followed by an ongoing infusion of glucose at a rate of 4 to 8 mg/kg/min. The concentration of the ongoing infusion depends on the fluid requirements of the child and the availability of central venous access (for higher concentrations). Glucagon may be given at a dose of 0.1 to 0.3 mg/kg (IV or intramuscularly [IM]) but is unlikely to be effective in patients with low glycogen stores, glycogen storage disorders, or hepatic dysfunction. Hydrocortisone at a dose of 5 mg/kg every 12 hours may be useful in some patients. Diazoxide and IV octreotide decrease insulin release and may be useful in the management of hyperinsulinemia.
TABLE 168-3 Investigation of Hypoglycemia
| Blood glucose | Measurement of glucose using blood from capillary specimens and using test strips may be unreliable (particularly in poorly perfused patients or patients with high hematocrit); where possible, low glucose levels should be confirmed using laboratory assays on venous or arterial blood. |
| Actual glucose intake | Hypoglycemia in the presence of normal glucose intake or after brief fast suggests hyperinsulinism. Hypoglycemia after hours of fasting is associated with fatty acid oxidation defects and endocrine insufficiency. |
| Non–glucose-reducing substances in the urine | Particularly in neonates and probably not relevant in older children. If present in the urine, consider galactosemia, hereditary fructose intolerance, or tyrosinemia. |
| Serum and urinary ketones | Low ketones suggest hyperinsulinism or fatty acid oxidation problem. |
| Serum free fatty acids | Free fatty acids are low in hyperinsulinism but high in fatty acid oxidation defect. |
| Serum insulin (and C peptide), cortisol, glucagon, growth hormone, and thyroid levels | Normal serum insulin in the presence of hypoglycemia is evidence of hyperinsulinism. C peptide may be necessary to ascertain whether exogenous insulin was administered. Release of C peptide may not be as pulsatile as that of insulin. |
| Serum ammonia | To recognize hyperinsulinism/hyperammonemia syndrome |
| Urinary organic acids and serum amino acids | To diagnose fatty acid oxidation defects (urinary organic acids). Aminoacidopathies such as MSUD, propionic acidemia, isovaleric acidemia, methylmalonic acidemia, and tyrosinemia may also present with hypoglycemia. |
| Total and free carnitine with acylcarnitine profile | To recognize primary and secondary deficiency of carnitine and fatty acid oxidation defects |
Neonates
In the neonatal period, glucose is not the only energy source from oxidative metabolism in the brain, and alternative energy sources such as ketones may be used.11 In fact, breast-fed babies routinely have lower glucose levels and higher ketone levels than formula-fed infants. Recent reviews have highlighted that “there is inadequate information in the literature to define any one value of glucose below which irreparable hypoglycemic injury to the central nervous system occurs, at any one time or for any defined period of time, in a population of infants or in any given infant.”18 However, there is evidence that hypoglycemic injury is more likely to occur at very low levels of glucose (20-25 mg/dL [1.1-1.4 mmol/L]) and if hypoglycemia is prolonged, is the consequence of hyperinsulinemia (when alternative energy sources for the brain may be very limited), and in the presence of other potential injuries.18,19
A suggested approach to hypoglycemia in the neonatal period is shown in Figure 168-1. Although the threshold for treatment in the asymptomatic neonate is 25 to 30 mg/dL (1.1-1.4 mmol/L), the recommended levels during treatment are above 45 mg/dL (2.5 mmol/L).
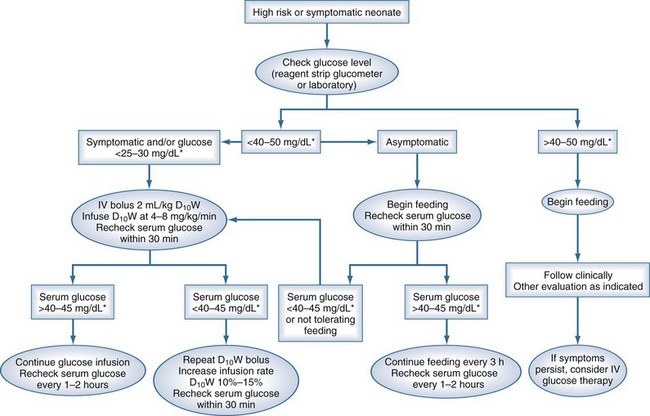
Figure 168-1 A suggested approach to management of the neonate with low glucose.
(From Rozance PJ, Hay WW. Hypoglycemia in newborn infants: features associated with adverse outcomes. Biol Neonate 2006;90:74-86.)
Although the exact definition of hypoglycemia in children is controversial, a minimal level of 2.6 mmol/L or greater should be maintained to ensure normal neural function.12,20,21 It probably is safer to maintain a level of greater than 3.5 mmol/L. Because there are multiple causes for hypoglycemia, and symptoms may not be due to the hypoglycemia alone, it is essential to identify the cause.
Hypoglycemia is associated with severe illness. A wide range of illnesses including infections,22 cyanotic and acyanotic congenital heart disease, and cardiomyopathy/myocarditis have been associated with hypoglycemia. Hepatic failure from infection, toxin ingestion, or drug reactions may be associated with severe hypoglycemia, and Reye syndrome classically presents with hypoglycemia. Toxins such as salicylates and ethanol also may cause hypoglycemia. Hypoglycemia has been linked with increased mortality from malaria,23–26 gastroenteritis,27 and acute bacterial meningitis28 among other conditions. Hypoglycemia also has been described as a complication of therapy for leukemia with mercaptopurine and methotrexate.29,30 Although severe illness or sepsis may be an adequate explanation for hypoglycemia, a diagnosis of sepsis should not exclude the possibility of an endocrine or metabolic crisis.
Hyperinsulinemic Hypoglycemia
Hyperinsulinism is the most common cause of persistent or recurrent hypoglycemia in infancy.31–33 Hyperinsulinism may be secondary to risk factors in the perinatal period (associated with high maternal glucose levels,34 rhesus incompatibility, intrauterine growth retardation,35 and perinatal asphyxia36) but may also be congenital37,38 or associated with Beckwith-Weidemann syndrome and some other developmental syndromes.33
Although most patients with hyperinsulinemic hypoglycemia present in the neonatal period, first presentation may be during infancy and occasionally during childhood,39 when the condition may be more likely to respond to medical therapy. Neonates with hypoglycemia may have the macrosomia typical of infants of diabetic mothers, but hyperinsulinemic hypoglycemia may occur in apparently normal infants of normal or low birth weight. Hypertrophic cardiomyopathy and hepatomegaly may be seen31 in affected infants. The characteristic features of hyperinsulinism include hypoglycemia with glucose requirements of greater than 6 to 8 mg/kg/min to maintain normoglycemia, absence of ketonemia and ketonuria, low plasma free fatty acids and branch chain amino acids, detectable insulin at the time of hypoglycemia, and response to glucagon administration.31 The combination of hypoglycemia with low free fatty acids and absence of ketonemia is responsible for the potentially devastating effects of this condition on the brain, as it is deprived of both normal and alternate substrates.32
Hyperinsulinemic hypoglycemia with hyperammonemia (previously called leucine-sensitive hypoglycemia) is well described40,41 and is attributed to mutations in the gene for glutamate dehydrogenase. Patients generally respond well to therapy with diazoxide, and consumption of extra carbohydrate before protein meals may help ameliorate symptoms. Special low-leucine milks are available.
Congenital hyperinsulinemic hypoglycemia is caused by abnormalities in genes controlling the secretion of insulin by the beta cells of the pancreas, with abnormalities described in seven genes.32
Clinically, hyperinsulinemic hypoglycemic patients may be categorized by their response to diazoxide (5-20 mg/kg/d in 2-3 divided doses), with most responding. Exceptions include those with congenital hyperinsulinemia related to focal hyperinsulinemia and those with diffuse hyperinsulinemia related to inactivating mutations in ABCC8 and KCNJ11. Unfortunately, diazoxide may predispose to fluid retention, and use must be carefully monitored. Chlorothiazide (7-10 mg/kg/day in 2 divided doses) may be added (particularly in neonates).32 Nifedipine (0.25-2.5 mg/kg/d in 3 divided doses) may also be useful in some patients.42
A suggested approach to ongoing diagnosis and management is outlined in Figure 168-2, showing a marked change from previous practice. In those patients who are responsive to diazoxide, that will remain the basis of therapy. In those with no response to diazoxide, genetic testing (for homozygous or compound heterozygous mutations in ABCC8 and KCNJ11), followed by fluorine-18 (18F)-dopa positron emission tomography (PET) scanning for those with potentially focal pancreatic lesions will enable identification of those who may benefit from resection of the pancreas. Pancreatic islets cells take up L-3, 4-dihydroxyphenylalanine (L-dopa), where it is converted to dopamine by dopa-decarboxylase. Uptake of the positron-emitting tracer 18F-dopa PET is increased in beta cells with a high rate of insulin synthesis and secretion provides visualization of the focal lesion.43–46 Patients with focal lesions should respond to partial pancreatectomy, which may be done laparoscopically.47,48 Diffuse disease that is unresponsive to diazoxide therapy will require a near-total pancreatectomy and may be associated with a high incidence of both endocrine and exocrine problems.49
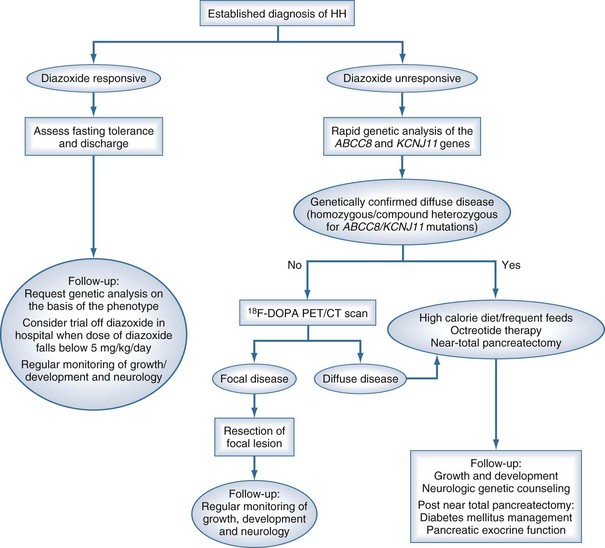
Figure 168-2 Flow chart outlining the management cascade of neonates with hyperinsulinemic hypoglycemia (HH). Clinically, HH can be classified into diazoxide-responsive and diazoxide-unresponsive disease. A fluorine-18 L-3, 4-dihydroxyphenylalanine positron emission tomography (18F-dopa PET) scan is currently only indicated in neonates who are unresponsive to diazoxide and do not have genetically confirmed diffuse disease.
(From Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child 2009;94:450–7.)
Close long-term follow up will be required in all these patients, and there may be significant neurologic and psychological problems to be dealt with.49
Ketotic Hypoglycemia
Although ketotic hypoglycemia (“accelerated starvation”) is probably the most common cause of hypoglycemia in previously healthy children,50 it is unlikely to present in the PICU. This condition usually affects children aged 6 months to 8 years, and the clinical features include ketosis, severe nausea, and hypoglycemia, usually occurring in the morning after a moderate fast. Treatment consists of ensuring that there is an adequate and regular intake of glucose, particularly during intercurrent infections. Urinary ketones may act as a warning signal, because the ketosis usually precedes the onset of hypoglycemia by several hours.
Adrenal Insufficiency
Adrenal insufficiency after high-dose inhaled corticosteroid therapy has presented with hypoglycemia51–54 and should be considered if there is a past history of inhaled steroid use (particularly fluticasone).53,55 Adrenal insufficiency also may occur after adrenal bleeds (e.g., after meningococcal septicemia or difficult delivery), as part of adrenal disease (e.g., congenital adrenal hyperplasia or hypoplasia) in which ambiguous genitalia may (or may not) be a pointer in females, or as part of hypopituitarism (e.g., congenital, after craniopharyngioma, or after cranial irradiation56). Some patients with primary adrenal insufficiency may present with hypoglycemia, particularly during acute illnesses.16,57 Adrenoleukodystrophy should be considered as part of the etiologic diagnosis in any male patient with Addison’s disease (pigmentation may be a clue) and should be tested for by measurement of very-long-chain fatty acids.58,57
There has been considerable interest in adrenocorticoid deficiency in children with critical illness and particularly acute severe sepsis (see Chapter 131).59–63 Currently, supplementary steroids are recommended for children with acute severe sepsis and catecholamine resistant shock,64,65 but there are not clear definitions for either adrenal insufficiency or catecholamine resistance, nor are there firm recommendations for the dose of adrenal replacement therapy.
Adrenocorticoid deficiency also has been shown in preterm infants.66 A randomized controlled study of “stress” dose hydrocortisone therapy in hypotensive very low-birthweight infants showed that steroids were effective in treating refractory hypotension.67
Congenital adrenal hyperplasia is associated rarely with hypoglycemia. Female patients are usually diagnosed early in life as a result of virilization, whereas male patients tend to present later. Patients with the salt-losing form of congenital adrenal hyperplasia present with hyponatremic dehydration and shock, usually associated with hyperkalemia. Because patients with salt-wasting 21-hydroxylase deficiency also may have catecholamine deficiency, shock may be a significant feature. Diagnosis is based on the clinical picture, typical electrolyte pattern, hypoaldosteronism, and hyperreninemia.68 Long-term treatment consists of hydrocortisone (to suppress excess secretion of corticotropin-releasing hormone and corticotropin), 10 to 20 mg/m2 of body surface area per day in three divided doses, although larger doses may be required during adrenal crises, together with mineralocorticoid replacement (0.1-0.2 mg of fludrocortisone daily) and sodium chloride supplementation. Little is known about the dose of hydrocortisone required during critical illness, although Charmandari et al.69 showed that when 6-hourly bolus doses of 15 mg/m2 of hydrocortisone are given, high immediate serum levels are achieved, followed by rapid decline to undetectable levels by 4 hours after administration. These authors postulated that continuous infusion of hydrocortisone may be more appropriate in critical illness.
Growth Hormone Deficiency
In the neonatal period, growth hormone deficiency presents with hypoglycemia (possibly with seizures), prolonged jaundice, and in boys, micropenis and undescended testes. Growth failure becomes apparent only toward the end of the first year of life. In later childhood, growth failure is a more common presentation, and hypoglycemia rarely occurs56 unless associated with adrenocorticotropic hormone deficiency.
Hyperglycemia Other Than Diabetes Mellitus
Hyperglycemia is relatively common in the PICU.70 In a retrospective study of 948 nondiabetic patients admitted to the PICU, there was a high prevalence of hyperglycemia, with 70.4% of patients having a glucose value above 120 mg/dL, 44.5% above 150 mg/dL, and 22.3% above 200 mg/dL within 10 days of admission. A 2.5-fold increased risk of dying was seen if the maximum glucose obtained within 24 hours of admission was over 150 mg/dL and a 5.68-fold increased risk if the maximum glucose obtained within 10 days of admission to the PICU was over 120 mg/dL.71 However, that study was retrospective and not corrected for severity of illness. In addition, ascertainment bias was present,72 so the study could not really provide insight in terms of causality or the potential impact of therapy.73
Hyperglycemia is common in a wide variety of conditions including bronchiolitis,74 sepsis, hemolytic uremic syndrome,75 tetanus,76 and toxin ingestion (e.g., theophylline poisoning77). Other studies have confirmed that high glucose levels are not only common in the PICU population but are associated with increased mortality and/or morbidity in a wide variety of conditions.78–81
An initial report from an adult surgical unit (predominantly cardiac)82 provided evidence that “tight” control of glucose levels was associated with a significant improvement in patient outcomes. The same group studied a cohort of patients in a medical ICU, and there was no difference in mortality between groups.83 There have been numerous studies in a variety of adult critical care populations since that time, with positive effects of tight glucose control noted on cholestasis,84 renal function,85 neurologic and neuromuscular complications,86,87 and endothelial function.88 Unfortunately, there have also been increased reports of iatrogenic hypoglycemia, and a recent meta-analysis of studies in adults89 concluded that tight glucose control was not associated with an improvement in hospital mortality and was associated with an increased incidence of hypoglycemia. Subsequently, a large randomized controlled trial (RCT) of adult patients compared “tight” (81-108 mg/dL or 4.5-6.0 mmol/L) with “conventional” glucose control (target of ≤180 mg/dL or ≤10.0 mmol/L). The 90-day mortality was higher in the group on tight glucose control, and subgroup analysis showed that the outcomes favored conventional control in all groups except trauma patients and patients on steroids.
Pediatricians have been more cautious in their approach to control of hyperglycemia, but a number of protocols for PICU management of hyperglycemia have been implemented and reported.90,91 An RCT of protocols for tight glucose control in the PICU showed no difference between a paper-based protocol and a computerized decision support tool.92
In a study of glycemic control in 177 postoperative cardiac patients,93 there was no difference in glucose levels on day 1 between survivors and nonsurvivors, but the 5-day mean peak glucose levels were significantly higher in nonsurvivors. Insulin usage was higher in the nonsurvivors, and nonsurvivors had more hypoglycemic events. The authors speculated that targeting a more permissive glucose level of 90-140 mg/dL (5-7.7 mmol/L) might be associated with both improved outcomes and reduced risk of hypoglycemia. In a retrospective review of 100 postoperative cardiac patients, there was high incidence of hyperglycemia (and an association with higher severity of illness), and implementation of a pediatric glycemic control protocol had a low incidence of hypoglycemia.79
A prospective RCT of 700 critically ill children (317 infants and 383 children) admitted to PICU94 randomized patients to targeted blood glucose levels (throughout PICU stay) of 2.8 to 4.4 mmol/L in infants and 3.9 to 5.6 mmol/L in children, with insulin infusion throughout PICU stay (intensive group [n=349]) or to insulin infusion only to prevent blood glucose from exceeding 11.9 mmol/L (conventional group [n=351]). Mean blood glucose concentrations were lower in the intensive group than in the conventional group, and hypoglycemia (glucose ≤2.2 mmol/L) occurred in 87 (25%) patients in the intensive group (P < 0.0001) versus 5 (1%) patients in the conventional group. Severe hypoglycemia (blood glucose less than 117 mmol/L) occurred in 17 (5%) of the intensive group versus 3 (1%) of the conventional group (P=0.001). Duration of PICU stay was reduced in the intensively treated group (5.51 days [95% CI, 4.65-6.37] versus 6.15 days [95% CI, 5.25-7.05]; P=0.017). The number of patients with stay in PICU longer than the median was 132 (38%) in the intensive group versus 165 (47%) in the conventional group (P=0.013). Nine (3%) patients died in the intensively treated group versus 20 (6%) in the conventional group (P = 0.038). There is ongoing debate about appropriate targets for glucose, optimization of protocols, balance of nutrient and glucose intake versus insulin therapy, and the like.
In the context of major burns there is also some evidence that insulin therapy to maintain lower blood glucose levels may be associated with improvements in metabolism.95
A recent review96 concluded:
Pediatricians have been reluctant to implement tight glucose control in PICU because of concerns about the deleterious effects of hypoglycemia.97,98
A recent review of hyperglycemia in the preterm infant99 suggested the following pragmatic approach to management: confirm hyperglycemia with laboratory test; treat any underlying problem such as sepsis, stress, etc.; calculate glucose infusion rates, and if above 12 mg/kg/min, reduce infusion rate; treat with insulin if glucose is over 10 mmol/L (or other symptoms such as polyuria), but start cautiously with very low doses; finally, if hyperglycemia persists, consider other diagnoses such as diabetes.
Diabetes Mellitus
Children with diabetes mellitus have a higher mortality than healthy children,100,101 with standardized mortality ratios of 2.15102 to 4.2,103 although some deaths are not directly related to diabetes. The highest mortality is in children aged 1 to 4 years, in whom the standardized mortality ratios may be 9.2104 to 13.7.105 Most deaths attributable to diabetes mellitus occur as a consequence of diabetic ketoacidosis (DKA) or hyperglycemia, with the remainder attributable to hypoglycemia.104 Diabetic ketoacidosis is relatively common at the time of first presentation, particularly in younger children,106 in whom diagnosis may be delayed. Although the incidence of type 1 diabetes mellitus has been increasing in many parts of the world, the hospitalization rate for DKA in established and new cases of type 1 diabetes mellitus has not increased in Canada and Europe since the 1990s107,108 because of earlier diagnosis and safer ambulatory management with the help of a multidisciplinary team.
The mortality rate in the developed world for DKA ranges from 0.15% to 0.31%109 but may be far higher in other settings.110 The most common cause of death among patients with DKA is cerebral edema. Other causes of death in DKA include electrolyte disturbances, hypoglycemia, pulmonary edema, rhabdomyolysis, infections (including mucormycosis), and thrombosis. The management of DKA in childhood has been extensively reviewed elsewhere,109,111,112 with current recommendations.
Cerebral Edema in Diabetic Ketoacidosis
In affluent countries, symptomatic cerebral edema occurs in 0.5% to 1% of pediatric DKA episodes,113 with risks being higher in young children and previously undiagnosed diabetics. Mortality is high (21%-24%), and 15% to 26% of survivors will have permanent morbidity (including pituitary insufficiency).113
The exact mechanisms of cerebral edema in DKA are not clear,114 although some imaging studies suggest that cerebral edema may be related to vasogenic factors rather than osmotic factors.115 Cerebral hyperemia has also been demonstrated as part of abnormal autoregulation.116,117 Factors that have been associated with the development of cerebral edema include the administration of bicarbonate, a higher plasma urea, arterial partial pressure of carbon dioxide (PCO2),118,119 and a smaller increase in plasma sodium concentration during therapy.120 However, a recent systematic review121 of the literature concluded that there was no clear evidence that treatment was related to the development of cerebral edema. Cerebral ischemia and reperfusion injury have also been considered.122 Cerebral edema may be present before therapy for DKA in 5% of cases, although most cases develop 4 to 12 hours after initiation of therapy.118,123 The clinical signs of cerebral edema in DKA are variable and include headache, deterioration in level of consciousness, inappropriate slowing of pulse rate, and increased blood pressure. However, children with no clinical signs of cerebral edema have been documented to have brain swelling,124 and a significant proportion of children have disrupted memory function following episodes of DKA.125
Adverse outcomes have been associated with greater neurologic depression at the time of diagnosis, high initial serum urea nitrogen,118,126 and intubation with hyperventilation to a PCO2 less than 22 mm Hg.126,127
Although the biochemical derangements of hyperglycemia, metabolic acidosis with ketosis, and electrolyte abnormalities are the most obvious problems in DKA, significant derangements in other systems have been documented, including plasma tryptophan levels,128 thiamine levels,129 cytokine130 and lymphocyte responses,130 and coagulation abnormalities.130 There is little doubt that DKA is associated with a thrombotic state131 and an increased incidence of cerebrovascular accidents, and care should be taken about the use of femoral central venous access because this may have a higher than usual complication rate in these patients.132 A reported case of myocardial infarction related to DKA133 may be a complication of the thrombotic state. Although myocardial function is generally normal in DKA, myocarditis134 has been noted in occasional case reports, whereas pulmonary edema may be more common than previously recognized.135 Prolongation of the QTc interval may be common in DKA (it correlates with ketosis), and careful cardiac monitoring is essential.136
Principles of Management
Management of DKA should be coordinated by an experienced diabetes team. The biochemical criteria for the diagnosis of DKA include a serum glucose concentration above 11 mmol/L (~200 mg/dL), ketonemia and ketonuria, and acidosis with venous pH below 7.3, or serum bicarbonate level below 15 mEq/L.112 The severity of DKA is defined by the level of acidosis, with mild having venous pH less than 7.3 (or bicarbonate <15 mmol/L); moderate, pH less than 7.2 (or bicarbonate <10 mmol/L); and severe, pH less than 7.1 (or bicarbonate <5 mmol/L).112 Children with severe DKA should be managed in a specialized diabetic unit or in the PICU.
Baseline Assessment
An admission weight should be obtained if at all possible, and future therapy should be based on this weight. Blood samples should be taken for the following investigations: serum or plasma glucose, electrolytes (including bicarbonate or total carbon dioxide), blood urea nitrogen, creatinine, osmolality, venous (or arterial in critically ill patient) pH, PCO2, calcium, phosphorus, and magnesium concentrations (if possible), HbA1c, hemoglobin and hematocrit or complete blood count. Measurement of blood β-hydroxybutyrate concentration, if available, is useful to confirm ketoacidosis and may be used to monitor the response to treatment.137–140 Urine specimens should be analyzed for ketones. Electrocardiograms may be useful if delays are expected in getting potassium results.
Fluid Management
The objectives of fluid and electrolyte replacement therapy are restoration of circulating volume, replacement of sodium and body fluid deficit, improved renal function with enhanced clearance of glucose and ketones from the blood, and minimization of risk of cerebral edema.112 There is a wide range in the amount and rate of fluid and electrolyte loss in patients presenting with DKA (depending on the rate of onset and duration of symptoms, the severity of vomiting or diarrhea or both, and the fluid ingested by the patient).112 There is a wide range of intravascular status ranging from normovolemia to severe hypovolemia (uncommon). Clinical assessment of dehydration is notoriously inaccurate, and there is an unpredictable rate of ongoing fluid loss related to the osmotic diuresis. In the (unusual) presence of hypovolemic shock, it is reasonable to infuse 0.9% saline using aliquots of 5 to 10 mL/kg until an acceptable blood pressure is obtained.141 Typically, 10 to 20 mL/kg needs to be infused over 1 to 2 hours.109 Ringer’s lactate may be a reasonable alternative, because administration of large volumes of 0.9% saline has been associated with the development of hyperchloremic acidosis. There is no evidence to support the use of colloid solutions.
Thereafter the acceptable principles are that hypovolemia, rapid changes in plasma osmolality, and large volumes of sodium uptake should be avoided. Fluid therapy should be calculated to achieve rehydration over 48 hours.109,112,113 Careful monitoring of fluid balance is essential to ensure that patients are neither losing excessive fluid (via osmotic diuresis) nor gaining excessive fluid. Fluid with a tonicity less than that of 0.45% saline should not be used, and a positive balance of around 6 mmol of sodium chloride per kilogram over 24 hours should be regarded as the upper limit.141 The rate of fluid infusion rarely exceeds 1.5 to 2 times the usual daily requirement.
Despite the fact that almost all patients with DKA are potassium depleted, serum potassium levels frequently are increased at presentation. With initiation of insulin therapy and correction of acidosis, there is rapid intracellular movement of potassium, and careful monitoring of potassium levels is essential. As soon as potassium levels are less than 5.5 mEq/L, 30 to 40 mEq/L of potassium should be added to the fluid infusions, and 0.5 to 1 mEq/kg/h of potassium may be required to correct potassium deficits. Potassium may be given as chloride or phosphate. Although severe hypophosphatemia is relatively common,142 and symptomatic hypophosphatemia has been reported,143 there is no evidence that phosphate administration is routinely necessary in the management of DKA, and the clinical effects of severe hypophosphatemia rarely are seen in DKA. Theoretically, phosphate administration may reduce insulin resistance and depletion of adenosine triphosphate and have positive effects on 2,3-diphosphoglycerate.144 Administration of potassium phosphate helps decrease the chloride load given to patients with DKA. Potassium phosphate may be used safely,145 provided that calcium levels are monitored carefully.146,147 Glucose must be added to the infusion of fluids when the glucose levels are 14 to 17 mmol/L to avoid hypoglycemia.
Bicarbonate
The use of bicarbonate in DKA is extremely limited. Many studies have shown no clinical benefit from its administration.148–150 More recently, bicarbonate administration has been associated with the development of cerebral edema. It should not be given routinely, not in bolus form, and possibly only in patients who have a pH of less than 6.9 despite appropriate correction of intravascular volume and ongoing adequate insulin therapy.
Treat Underlying Cause
In previously undiagnosed patients, the cause of DKA is insulin deficiency. Even in previously diagnosed patients, most episodes of DKA probably are related to insulin omission or treatment error, although children 3 years old or younger are more likely to have a bacterial infection.151 If infection is suspected as the precipitating cause of DKA, aggressive therapy with antibiotics and drainage of any pus should be instituted. Routine prophylactic antibiotic therapy is not indicated in DKA.
Monitoring
Although some patients are hypovolemic on presentation, there is little evidence that invasive hemodynamic monitoring is necessary. Careful monitoring of sodium levels is essential because smaller changes in serum sodium with therapy have been associated with development of cerebral edema.118 Hyperlipidemia may decrease the aqueous phase of serum and artificially reduce sodium levels; this can be corrected using the following formula152:
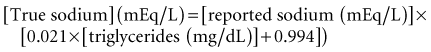
The osmotic load of glucose also decreases serum sodium levels, with a decrease in sodium concentration of approximately 1.6 to 1.8 mEq/L per 100 mg/dL increase in glucose153 (alternatively, Corrected sodium = measured Na + 2([plasma glucose − 5.6]/5.6) (mmol/L). The expectation is that with decreasing levels of hyperglycemia and hyperlipidemia, sodium levels should increase. This increase may be offset, however, by urinary losses of sodium secondary to osmotic diuresis. Careful and frequent monitoring of potassium and glucose levels is essential. If phosphate is being administered, calcium levels should be monitored. Regular acid-base monitoring is required.
Monitoring of end-tidal PCO2154 or transcutaneous PCO2155 could be used as a noninvasive method for continuous monitoring of response to therapy for DKA. The only proviso (as pointed out by the authors and in an accompanying editorial156) is that any changes in respiratory drive or efficiency of the respiratory system may mask changes in acid-base that otherwise might be reflected by capnometry.
Investigations for Possible Cerebral Edema in Diabetic Ketoacidosis
Although cerebral edema is the most common cause of depressed level of consciousness in DKA, there are other causes that are amenable to alternative therapy, including cerebral venous thrombosis157 and acute hydrocephalus.158 Other abnormalities such as brain infarction159 and extrapontine myelinolysis160 have been shown. Computed tomography (CT) of patients with a depressed level of consciousness may be recommended to exclude other treatable pathology. Because the risks are relatively low, however, excluding other pathology must be balanced against the risks associated with moving ill patients to the radiology suite.
Mannitol has been used for the management of cerebral edema161 (0.25-1 g/kg over 20 minutes), although there are no controlled studies. Hypertonic saline (5-10 mL/kg of 3% saline) may be an alternative to mannitol.162 Hyperventilation after intubation for cerebral edema may be associated with worse outcomes.126
Thyroid Insufficiency
Neonates exposed to large amounts of iodine in iodine-containing antiseptics may develop transient hypothyroidism163–167 (also called the Wolff-Chaikoff effect) as a result of transcutaneous absorption of iodine. This condition also has been shown in infants undergoing cardiac catheterization and cardiac surgery.168 Care should be taken to limit the exposure of infants to iodine-containing agents. Triiodothyronine supplementation may be considered in children who have been exposed to significant amounts of iodine before or during a critical illness.
The sick euthyroid syndrome has been well documented in the PICU, particularly in patients undergoing cardiac surgery. The subject has been reviewed elsewhere.169 Although there may be benefit to some children from triiodothyronine supplementation after cardiac surgery,170–172 there is no established role for triiodothyronine supplementation after cardiac surgery.173
Children with Down syndrome have a high incidence of hypothyroidism.174,175 Attention should be paid to the possible need for triiodothyronine supplementation in critically ill children with Down syndrome.
 Metabolic Crises
Metabolic Crises
Epidemiology
Population data on inborn errors of metabolism suggest that there is a minimal incidence of 35 to 40 per 100,000 live births176,177 in countries such as Canada or Italy, while the incidence may be as high as 150 per 100,000 live births in other countries.178 In addition, some conditions have a particularly high incidence in particular population groups (e.g., maple syrup urine disease has an incidence of 568 per 100,000 births in the Mennonite community in Pennsylvania). Inborn errors of metabolism have a diverse presentation and are part of the differential diagnosis of many children admitted to the PICU with acute illness. Until more recently, only conditions such as phenylketonuria and galactosemia had been identified at birth using screening programs. With increasing availability of technology such as tandem mass spectrometry, screening of other inborn errors of metabolism (including fatty acid oxidation abnormalities and aminoacidopathies) has been introduced in some parts of the world,179–182 and this potentially may decrease the number of children presenting with acute metabolic decompensation.
There is evidence that SIDS may be related to inborn errors of metabolism in at least 1% of cases,183,184 and inborn errors of metabolism must be considered as part of the differential diagnosis of any infant who presents to the PICU or neonatal ICU after a near-SIDS episode. A family history of SIDS also should raise the possibility of inborn errors of metabolism in siblings presenting to the PICU with acute illness.
Although there are a bewildering number of inborn errors of metabolism, many are amenable to therapy, and screening may be performed using relatively simple tests. Patients with incurable conditions may derive considerable relief of suffering from diagnosis and appropriate therapy. Even when a condition is not amenable to therapy, it is important to make a diagnosis to facilitate counseling for the family involved and prevent unnecessary suffering in future children. Long-term management of most inborn errors of metabolism requires a team approach including metabolic experts, dietitians, geneticists, biochemists, and social workers to elucidate the exact nature of the problem, provide appropriate therapy and therapeutic plans, and give genetic and family counseling. Although many screening tests for inborn errors of metabolism can be done in most diagnostic laboratories, the specialized tests required to identify the exact nature of an inborn error of metabolism can be done at relatively few laboratories. Despite the complexity of inborn errors of metabolism, there are principles germane to the management of all children who are admitted to a PICU, and these should apply (see Table 168-1).
When to Consider an Inborn Error of Metabolism in the Pediatric Intensive Care Unit
Inborn errors of metabolism may be classified into diagnostically useful groups185: (1) disorders that give rise to intoxication (e.g., organic acidemias and urea cycle defects); (2) disorders involving energy metabolism (e.g., fatty acid oxidation defects and respiratory chain defects); (3) disorders involving complex molecules in which symptoms are permanent, progressive, and independent of intercurrent events (e.g., peroxisomal disorders, lysosomal disorders, and congenital defects of glycosylation); and (4) those disorders that present with seizures (particularly in the neonatal period). The conditions most likely to present acutely in the PICU are conditions involving intoxication and energy metabolism. There is overlap, however, between all of these groups in terms of clinical presentation. There also may be considerable variation in the clinical presentation of conditions that have the same underlying genetic abnormality; this may apply even within families.
Although the clinical features of an inborn error of metabolism may be related primarily to the accumulation of a toxic metabolite, the condition may be complicated by the relative deficiency of another compound or increased stress put on other metabolic pathways by the primary problem.186 Management may involve limiting the intake of potentially toxic substances, increasing the removal of toxic substances, supplementation of deficient substances, and supplementation of other metabolic pathways that are being stressed.
Inborn errors of metabolism should be considered as part of the differential diagnosis of any child or infant who presents with a severe illness, particularly during the neonatal period.185 Acute symptoms that are particularly associated with inborn errors of metabolism include encephalopathy (acute or acute on chronic), intractable seizures, hepatic failure, cardiomyopathy, metabolic acidosis, and hypoglycemia (Table 168-4). Family history of SIDS or of previous childhood deaths may suggest an inborn error of metabolism. Particular attention should be paid to the identification of specific risk factors for the differential diagnoses, including drug exposure, prolonged rupture of membranes, and perinatal asphyxial episodes.
TABLE 168-4 Factors That Should Alert the Intensivist to the Possibility of an Inborn Error of Metabolism
| History | |
| General | Population group with high incidence of inborn errors of metabolism |
| Consanguinity of parents | |
| Previous history of apparent SIDS or childhood deaths in the family | |
| Presence of dysmorphic features associated with inborn error of metabolism | |
| During pregnancy | Previous history multiple spontaneous abortions |
| Hyperemesis may be associated with fat oxidation disorders,240 as may frank hepatic symptoms such as acute fatty liver of pregnancy or the more severe HELLP syndrome (hemolysis, liver enzymes, low platelets). | |
| In neonatal period | Deterioration after apparently being normal at birth, particularly if Apgar scores and early neonatal period were normal |
| Earliest signs of inborn error of metabolism in the neonatal period may include lethargy and poor feeding, which may progress rapidly to obvious depressed level of consciousness. | |
| Depressed level of consciousness without obvious explanation | |
| Vomiting is an unusual clinical feature of illness in neonates and is strongly associated with inborn errors of metabolism. | |
| Strange odors | |
| In childhood | Previous history of being “sickly” with episodes of intermittent vomiting |
| Previous hospital admissions (even for apparent respiratory symptoms as this may be acidosis) | |
| Unusual dietary preferences by the child | |
| Onset of virtually any organ dysfunction (liver, heart, renal, etc.) may be related to inborn error. | |
| Examination | |
| General in neonatal period | Dysmorphic features that may be associated with inborn errors of metabolism |
| Strange odors | |
| Neurologic signs in inborn errors of metabolism tend to include increased tone and abnormal movements, in contrast to the features of sepsis, which usually is associated with decreased tone. | |
| In childhood | Acute or intermittent ataxia is a common feature of inborn errors of metabolism in children. |
SIDS, sudden infant death syndrome.
Clinical Presentations of Inborn Errors of Metabolism
Intractable Seizures
Seizures (in isolation) are an uncommon presentation of inborn errors of metabolism and, with the exception of the pyridoxine-dependent seizures, tend to be associated with other clinical and metabolic abnormalities. In neonates or some infants presenting with intractable seizures, (particularly if associated with grimacing and abnormal eye movements), pyridoxine-dependent seizures (PDS),187 pyridoxine phosphate oxidase deficiency (PNPO) hypophosphatasia, and folinic acid–responsive seizures188 should be considered. The clinical diagnosis of both PDS and PNPO depends on demonstration that seizure control is dependent on continuous pharmacologic doses of pyridoxine or pyridoxal-5′-phosphate, respectively. However, it has recently been shown that detection of elevated levels of α-amino-adipic semialdehyde in blood, urine, or cerebrospinal fluid (CSF), along with the demonstration of mutations in the ALDH7A1 (antiquitin) gene, confirm a diagnosis of PDE189; whereas an abnormal pattern of CSF catecholamine and indole amine metabolite levels, together with elevated CSF and plasma glycine and threonine concentrations and urinary vanillactic acid excretion are characteristic but not uniformly present in PNPO.190,191 It has also been shown that patients with folinic acid–dependent seizures have the same mutation in the antiquitin gene.188 There is a considerable range in clinical presentation, and pyridoxine-dependent seizures probably should be considered in any infant up to age 18 months presenting with seizures.
Patients with defects in transport of glucose across the blood-brain barrier associated with mutations in the GLUT1 gene may present with seizures. The only clue is the presence of low CSF glucose in the presence of normal blood glucose. Patients may improve on a ketogenic diet.192
Biotinidase deficiency (an autosomal recessively inherited disorder of biotin recycling—estimated incidence of biotinidase deficiency is about 1 in 60,000193) can be ameliorated or prevented by administering pharmacologic doses of the vitamin biotin (5-20 mg daily independent of age). A large proportion of cases present with seizures and hypotonia, associated with failure to thrive, and rash or alopecia. Some 50% of cases have ataxia, developmental delay, and eye problems (conjunctivitis and optic atrophy), with more than 75% developing hearing loss. There is a considerable variation in clinical presentation, even within affected families,194 with features ranging from mild episodes of seizure and ataxia to severe metabolic failure and death. Onset of symptoms may occur at any time from the neonatal period through to adulthood. Untreated individuals may have ketoacidosis, lactic acidosis, and/or hyperammonemia, with a wide range of other metabolic anomalies.194 Diagnosis can be made from analysis of organic acids in urine, whereas an enzyme assay can be done on blood. Guidelines for testing have recently been published.194
Intractable tonic/clonic seizures also may be a feature of molybdenum cofactor deficiency.195,196 This condition presents in early infancy with seizures, encephalopathy in the absence of metabolic acidosis, hypoglycemia or hyperammonemia, and failure to thrive. Imaging of the brain initially shows cerebral edema, which may progress to cerebral atrophy. There are typical imaging findings.197 Clinical features, CT findings, and neuropathology may be similar to that seen in severe hypoxic-ischemic brain injury.198,199 Lens dislocation may be a clinical feature.200 Uric acid levels are low, whereas urinary amino acid analysis shows increased S-sulfocysteine. Sulfite may be demonstrated on fresh urine specimens. Electrospray tandem mass spectrometry of urine or urine-soaked filter paper may facilitate rapid diagnosis.201 Seizures may be part of the clinical presentation of many other disorders, including seizures with lactic acidosis (Leigh disease; mitochondrial encephalopathy lactic acidosis and strokelike episodes [MELAS]; mitochondrial encephalopathy with ragged red fibers [MERRF]), GM2 gangliosidosis, and peroxisomal disorders. Other clinical features predominate in these conditions and should direct investigation.
Encephalopathy
The inborn errors of metabolism that present with acute encephalopathy vary with age. In the neonatal period, the common inborn errors of metabolism include urea cycle defects (with hyperammonemia), maple syrup urine disease, nonketotic hyperglycinemia, and organic acidopathies.202 All of these conditions, with the exception of nonketotic hyperglycinemia, also may present during childhood. During childhood, the common inborn errors of metabolism presenting with acute encephalopathy include fatty acid oxidation defects and maple syrup urine disease.
Investigation
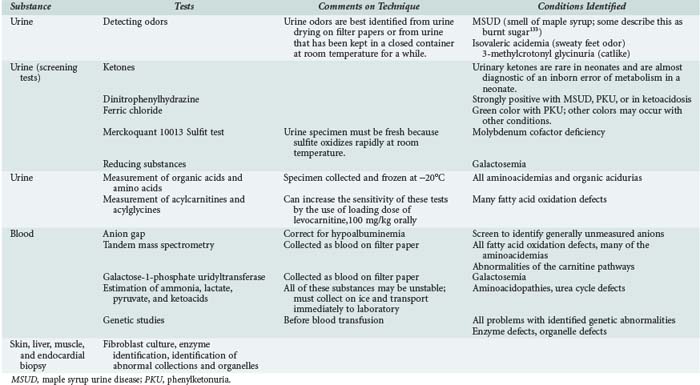
The specimens that normally would be collected for diagnosis of sepsis should be collected, including blood culture, hemoglobin, white blood cell count (including differential), and platelets. Serum electrolytes should be checked, including sodium, potassium, calcium, phosphate, and magnesium. Liver function tests are essential because acute hepatic failure may cause acute encephalopathy, and the liver may be affected by inborn errors of metabolism. Specimens for testing for inborn errors of metabolism must be collected at the time of presentation, because this may provide the best opportunity for diagnosis (Table 168-5).
Blood Gas Analysis
Arterial blood gas analysis should be performed, with particular attention to the presence of metabolic acidosis and calculation of the anion gap (this should be corrected for the presence of hypoalbuminemia).203–205
Specific Inborn Errors of Metabolism
Maple Syrup Urine Disease
The urine may smell like maple syrup, but the smell is also similar to that of burned sugar.202 The urine smell may be difficult to detect in the first few days of life, then may be detected on diapers that have been allowed to dry.206 Urine tests for ketones are usually strongly positive, and dinitrophenylhydrazine is usually positive, although both tests may be negative before 3 days of age.206 Tandem mass spectrometry is the quickest and most efficient screening test in neonates. Leucine levels can be checked rapidly on whole-blood filter paper specimens, or quantitative amino acid analysis should be done on plasma or serum. Principles of management have been to remove leucine using dialysis and to reduce the production of leucine by dietary manipulation. Hemodialysis has been shown to decrease leucine levels rapidly,207 particularly if used in conjunction with dietary therapy. Previously, exchange transfusion, peritoneal dialysis, and hemofiltration were reported to decrease leucine levels. Morton and colleagues206 have used a protocol consisting of total caloric intake of 120 to 140 kcal/kg/d, with lipid forming 40% to 50% of calories; 3 to 4 g/kg/d of protein as essential and nonessential amino acids, with 80 to 120 mg/kg/d each of isoleucine and valine and 250 mg/kg/d each of glutamine and alanine, with tyrosine, histidine, and threonine supplemented to normalize plasma amino acid ratios; careful attention to sodium balance to ensure that serum sodium is kept at greater than 140 mEq/L; and hyperosmolar therapy if cerebral edema develops. This protocol produces decreases in leucine equal to that seen after dialysis. Recent studies suggest that norleucine may have a place in reducing brain injury in patients with MSUD.208
Isovaleric Aciduria, Methylmalonic Aciduria, and Propionic Acidemia
Strokelike episodes are a feature of isovaleric aciduria, methylmalonic aciduria, and propionic acidemia in later life, although there may be a wide range of neurologic presentations including hypotonia and developmental delay. Extrapyramidal signs related to infarction of the basal ganglia may be a feature of methylmalonic aciduria and propionic acidemia. Neutropenia, thrombocytopenia, and anemia are common in the neonatal presentation, whereas neutropenia also may be a feature of a later presentation. Sepsis may be a significant component of clinical exacerbations, particularly in propionic acidemia. Pancreatitis has been reported to be associated with these disorders.209 Cardiomyopathy also may develop, particularly during metabolic decompensation.210 Isovaleric aciduria, propionic acidemia, and methylmalonic aciduria are diagnosed by the organic acid profiles, and tandem mass spectroscopy may be useful by looking at the acylcarnitine profiles.
Nonketotic Hyperglycinemia
Nonketotic hyperglycinemia presents in early infancy with severe encephalopathy in the absence of acidosis, ketosis, hypoglycemia, hyperammonemia, or any other clinical abnormalities. Although the outcome is almost invariably poor, there have been more recent descriptions of transient neonatal hyperglycinemia.211 There also is an association of abnormality of the corpus callosum with nonketotic hyperglycinemia.212 Diagnosis is confirmed by the presence of high CSF glycine. The enzyme defect can be confirmed on a liver biopsy specimen.213,214 Sodium benzoate may be helpful in therapy, possibly in combination with imipramine.215
Hypoglycemia and Inborn Errors of Metabolism
Investigation and Management
Urinary Ketones
If urinary ketones are positive, the clinician should assess for urinary and plasma organic acids and quantitative amino acids. High urinary ketones in the presence of hepatomegaly suggest glycogen storage disease type 1, fructose-1,6-diphosphatase (FDPase) deficiency, or β-ketothiolase deficiency.216 In the last-mentioned condition, lactate levels are normal, whereas they are increased in glycogen storage disease type 1 and FDPase deficiency. In the absence of hepatomegaly, high ketones suggest ketotic hypoglycemia or deficiencies of growth hormone or glucocorticoids.
Specific Conditions Associated with Hypoglycemia
Hyperammonemia
Investigation
Ammonia is potentially toxic, and therapy must be instituted urgently to remove ammonia. It is crucial to collect appropriate diagnostic specimens at the time of presentation because it may be difficult to establish a diagnosis when dialysis and other therapy have been instituted. The following tests enable an approach to diagnosis.218
Management
Principles of management for hyperammonemia consist of the following:
Metabolic Acidosis
Acid also may be produced by bacterial overgrowth in the bowel and absorbed, as occurs in D-lactic acidosis.219 D-Lactic acid is not detected by routine blood tests for lactic acid, which employ a lactic dehydrogenase, but is detected by urinary assays for organic acids. These patients present with acidosis with increased anion gap.
Lactic Acidosis
The clinical course of pyruvate dehydrogenase deficiency may be extremely variable, and diagnosis is confirmed by studies of enzyme activity in cultured fibroblasts. The lactic acidosis in pyruvate dehydrogenase deficiency can be ameliorated by a ketogenic diet,220 although many factors must be considered before embarking on this diet, including its protein content, particularly if there is associated renal failure, and the long-term problems of ketogenic diets.221 Dichloroacetic acid may be helpful in some cases.222 Many cases have been reported in which thiamine was associated with clinical improvement, although high levels may be required.223
Lactic acidosis occurs in all of the conditions affecting the metabolism of pyruvate through the tricarboxylic acid cycle. Abnormalities include pyruvate dehydrogenase deficiency and mitochondrial energy cycle defects. The mitochondrial energy cycle problems frequently are associated with persistent lactic acidosis, myopathy, failure to thrive, psychomotor retardation, and seizures. Other symptoms that may be present in mitochondrial energy conditions in children include antenatal problems,224 cardiomyopathy225–227 and cardiac arrhythmias,225 sensorineural hearing loss,228 stroke and abnormalities of central respiratory drive,229 and diabetes mellitus.230
Acquired defects in mitochondrial function have been associated with severe lactic acidosis in adults and children on antiretroviral therapy.231 Lactic acidosis also may be a secondary phenomenon of defects of organic acid metabolism, including 3-hydroxy-3-methylglutaryl-CoA lyase deficiency, propionic acidemia, and methylmalonic acidemia.
Cardiomyopathy
A few conditions may present with cardiac problems apparently in isolation. In the differential diagnosis of myocarditis/cardiomyopathy, many conditions need to be excluded, including carnitine deficiency, trifunctional protein defects, or isolated long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. In the latter two conditions, urinary organic acid analysis at the time of the acute illness shows the presence of medium-chain and long-chain dicarboxylic acids. At least one form of very-long-chain acyl-CoA dehydrogenase deficiency can present as an acute cardiomyopathy. For all these conditions, measurement of acylcarnitines using tandem mass spectrometry allows diagnosis. Diagnosis is confirmed using enzyme activity in cultured fibroblasts. At least one case report232 shows that substantial clinical improvement can be achieved by elimination of long-chain fatty acids from the diet (replacing with medium-chain fatty acids). Many of the disorders of the mitochondrial energy chain have poor myocardial function as a component of their multiple symptoms, but echocardiography may be needed to show more subtle features of poor contractility.
Hepatopathology
In one study of infants presenting to a transplant service in acute hepatic failure, inborn errors of metabolism were responsible for the hepatic failure in 42.5% of the patients. Of these patients, 35% had hepatorenal tyrosinemia, whereas 50% had mitochondrial abnormalities. Hereditary fructose intolerance and galactosemia together were present in less than 9% of patients.233
Hepatorenal tyrosinemia may present in the neonatal period as acute hepatic failure. It is difficult to distinguish from acute viral hepatitis, because plasma amino acid levels may be similar in both situations. Alpha-fetoprotein levels may be substantially elevated in hepatorenal tyrosinemia and may be a distinguishing feature. The coagulopathy tends to be relatively severe in hepatorenal tyrosinemia, and coagulopathy may be the only presenting feature of hepatorenal tyrosinemia.234 Patients tend to have moderate to severe anemia. The response to treatment with 2-(2-nitro-4-triflu-oromethylbenzoyl)-1,3-cyclohexandion (NTBC) may be dramatic.235–238
Galactosemia is characterized by the development of hypoglycemia in the neonatal period in association with jaundice (initially unconjugated, but subsequently conjugated), marked increase in transaminase levels, some abnormality of coagulation, and moderate hypoalbuminemia. Severe cerebral edema occasionally may be a dominant feature. Management has been reviewed elsewhere.239 There is a close association with Escherichia coli septicemia, and any infant presenting with E. coli septicemia should be investigated for galactosemia. Galactosuria clears rapidly if feeds are stopped. A screening test is available on blood collected on filter paper (semiquantitative measure of galactose-1-phosphate uridyltransferase). The diagnosis can be confirmed on a quantitative measurement of galactose-1-phosphate uridyltransferase. Wilson’s disease may present as acute hepatitis, but rarely before age 5 years.
Key Points
Boles RG, Buck EA, Blitzer MG, et al. Retrospective biochemical screening of fatty acid oxidation disorders in post-mortem livers of 418 cases of sudden death in the first year of life. J Pediatr. 1998;132:924-933.
Dunger DB, Sperling MA, Acerini CL, et al. ESPE/LWPES consensus statement on DKA in children and adolescents. Arch Dis Child. 2004;89:188-194.
Durand P, Debray D, Mandel R, et al. Acute liver failure in infancy: A 14-year experience of a pediatric liver transplantation center. J Pediatr. 2001;139:871-876.
Marcin JP, Glaser N, Barnett P, et al. Factors associated with adverse outcomes in children with DKA-related cerebral edema. J Pediatr. 2002;141:793-797.
Morton DH, Strauss KA, Robinson DL, et al. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109:999-1008.
1 Wintergerst KA, Buckingham B, Gandrud L, Wong BJ, Kache S, Wilson DM. Association of hypoglycemia, hyperglycemia, and glucose variability with morbidity and death in the pediatric intensive care unit. Pediatrics. 2006;118(1):173-179.
2 Daschner M, Schaefer F. Emergency dialysis in neonatal metabolic crises. Adv Ren Replace Ther. 2002;9(1):63-69.
3 Cederbaum JA, LeMons C, Rosen M, Ahrens M, Vonachen S, Cederbaum SD. Psychosocial issues and coping strategies in families affected by urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S72-S80.
4 Packman W, Henderson SL, Mehta I, et al. Psychosocial issues in families affected by maple syrup urine disease. J Genet Couns. 2007;16(6):799-809.
5 Enns GM, Packman W. The adolescent with an inborn error of metabolism: medical issues and transition to adulthood. Adolesc Med. 2002;13(2):315-329. vii
6 Kappy MS, Bajaj L. Recognition and treatment of endocrine/metabolic emergencies in children: part I. Adv Pediatr. 2002;49:245-272.
7 Osier FH, Berkley JA, Ross A, Sanderson F, Mohammed S, Newton CR. Abnormal blood glucose concentrations on admission to a rural Kenyan district hospital: prevalence and outcome. Arch Dis Child. 2003;88(7):621-625.
8 Tonyushkina K, Nichols JH. Glucose meters: a review of technical challenges to obtaining accurate results. J Diabetes Sci Technol. 2009;3(4):971-980.
9 Rice MJ, Pitkin AD, Coursin DB. Review article: glucose measurement in the operating room: more complicated than it seems. Anesth Analg. 2010;110(4):1056-1065.
10 Ly TT, Gallego PH, Davis EA, Jones TW. Impaired awareness of hypoglycemia in a population-based sample of children and adolescents with type 1 diabetes. Diabetes Care. 2009;32(10):1802-1806.
11 Hay WWJr, Raju TN, Higgins RD, Kalhan SC, Devaskar SU. Knowledge gaps and research needs for understanding and treating neonatal hypoglycemia: workshop report from Eunice Kennedy Shriver National Institute of Child Health and Human Development. J Pediatr. 2009;155(5):612-617.
12 Cornblath M, Hawdon JM, Williams AF, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics. 2000;105(5):1141-1145.
13 Cornblath M, Ichord R. Hypoglycemia in the neonate. Semin Perinatol. 2000;24(2):136-149.
14 Maayan-Metzger A, Lubin D, Kuint J. Hypoglycemia rates in the first days of life among term infants born to diabetic mothers. Neonatology. 2009;96(2):80-85.
15 Binder G, Weidenkeller M, Blumenstock G, Langkamp M, Weber K, Franz AR. Rational approach to the diagnosis of severe growth hormone deficiency in the newborn. J Clin Endocrinol Metab. 2010;95(5):2219-2226.
16 Keil MF, Bosmans C, Van Ryzin C, Merke DP. Hypoglycemia during acute illness in children with classic congenital adrenal hyperplasia. J Pediatr Nurs. 2010;25(1):18-24.
17 Hoe FM. Hypoglycemia in infants and children. Adv Pediatr. 2008;55:367-384.
18 Rozance PJ, Hay WW. Hypoglycemia in newborn infants: Features associated with adverse outcomes. Biol Neonate. 2006;90(2):74-86.
19 Salhab WA, Wyckoff MH, Laptook AR, Perlman JM. Initial hypoglycemia and neonatal brain injury in term infants with severe fetal acidemia. Pediatrics. 2004;114(2):361-366.
20 Koh TH, Aynsley-Green A, Tarbit M, Eyre JA. Neural dysfunction during hypoglycaemia. Arch Dis Child. 1988;63(11):1353-1358.
21 Cornblath M, Schwartz R, Aynsley-Green A, Lloyd JK. Hypoglycemia in infancy: the need for a rational definition. A Ciba Foundation discussion meeting. Pediatrics. 1990;85(5):834-837.
22 Jan IS, Tsai TH, Chen JM, et al. Hypoglycemia associated with bacteremic pneumococcal infections. Int J Infect Dis. 2009;13(5):570-576.
23 Maitland K, Levin M, English M, et al. Severe P. falciparum malaria in Kenyan children: evidence for hypovolaemia. QJM. 2003;96(6):427-434.
24 Jaffar S, Van Hensbroek MB, Palmer A, Schneider G, Greenwood B. Predictors of a fatal outcome following childhood cerebral malaria. Am J Trop Med Hyg. 1997;57(1):20-24.
25 Marsh K, Forster D, Waruiru C, et al. Indicators of life-threatening malaria in African children. N Engl J Med. 1995;332(21):1399-1404.
26 Willcox ML, Forster M, Dicko MI, Graz B, Mayon-White R, Barennes H. Blood glucose and prognosis in children with presumed severe malaria: is there a threshold for ‘hypoglycaemia’? Trop Med Int Health. 2010;15(2):232-240.
27 Bennish ML, Azad AK, Rahman O, Phillips RE. Hypoglycemia during diarrhea in childhood. Prevalence, pathophysiology, and outcome. N Engl J Med. 1990;322(19):1357-1363.
28 Mwangi I, Berkley J, Lowe B, Peshu N, Marsh K, Newton CR. Acute bacterial meningitis in children admitted to a rural Kenyan hospital: increasing antibiotic resistance and outcome. Pediatr Infect Dis J. 2002;21(11):1042-1048.
29 Halonen P, Salo MK, Schmiegelow K, Makipernaa A. Investigation of the mechanisms of therapy-related hypoglycaemia in children with acute lymphoblastic leukaemia. Acta Paediatr. 2003;92(1):37-42.
30 Ziino O, Russo D, Orlando MA, Benigno V, Locatelli F, Arico M. Symptomatic hypoglycemia in children receiving oral purine analogues for treatment of childhood acute lymphoblastic leukemia. Med Pediatr Oncol. 2002;39(1):32-34.
31 Aynsley-Green A, Hussain K, Hall J, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82(2):F98-F107.
32 Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia. Arch Dis Child. 2009;94(6):450-457.
33 Kapoor RR, James C, Hussain K. Hyperinsulinism in developmental syndromes. Endocr Dev. 2009;14:95-113.
34 Straussman S, Levitsky LL. Neonatal hypoglycemia. Curr Opin Endocrinol Diabetes Obes. 2010;17(1):20-24.
35 Fafoula O, Alkhayyat H, Hussain K. Prolonged hyperinsulinaemic hypoglycaemia in newborns with intrauterine growth retardation. Arch Dis Child Fetal Neonatal Ed. 2006;91(6):F467.
36 Collins JE, Leonard JV. Hyperinsulinism in asphyxiated and small-for-dates infants with hypoglycaemia. Lancet. 1984;2(8398):311-313.
37 Arnoux JB, de Lonlay P, Ribeiro MJ, et al. Congenital hyperinsulinism. Early Hum Dev. 2010;86(5):287-294.
38 de Lonlay P, Giurgea I, Robert JJ, et al. Hyperinsulinemic hypoglycemia in children. Ann Endocrinol (Paris). 2004;65(1):96-98.
39 Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003;149(1):43-51.
40 El-Gharbawy AH. Hyperinsulinism/Hyperammonemia Syndrome: a synopsis. Mol Genet Metab. 2005;84(2):101-103.
41 De Lonlay P, Benelli C, Fouque F, et al. Hyperinsulinism and hyperammonemia syndrome: report of twelve unrelated patients. Pediatr Res. 2001;50(3):353-357.
42 Bas F, Darendeliler F, Demirkol D, Bundak R, Saka N, Gunoz H. Successful therapy with calcium channel blocker (nifedipine) in persistent neonatal hyperinsulinemic hypoglycemia of infancy. J Pediatr Endocrinol Metab. 1999;12(6):873-878.
43 Mohnike K, Blankenstein O, Minn H, Mohnike W, Fuchtner F, Otonkoski T. [18F]-dopa positron emission tomography for preoperative localization in congenital hyperinsulinism. Horm Res. 2008;70(2):65-72.
44 Mohnike K, Blankenstein O, Christesen HT, et al. Proposal for a standardized protocol for 18F-dopa PET (PET/CT) in congenital hyperinsulinism. Horm Res. 2006;66(1):40-42.
45 Hardy OT, Hernandez-Pampaloni M, Saffer JR, et al. Accuracy of [18F]fluorodopa positron emission tomography for diagnosing and localizing focal congenital hyperinsulinism. J Clin Endocrinol Metab. 2007;92(12):4706-4711.
46 Hardy OT, Hernandez-Pampaloni M, Saffer JR, et al. Diagnosis and localization of focal congenital hyperinsulinism by 18F-fluorodopa PET scan. J Pediatr. 2007;150(2):140-145.
47 Bax KN, van der Zee DC. The laparoscopic approach toward hyperinsulinism in children. Semin Pediatr Surg. 2007;16(4):245-251.
48 De Vroede M, Bax NM, Brusgaard K, Dunne MJ, Groenendaal F. Laparoscopic diagnosis and cure of hyperinsulinism in two cases of focal adenomatous hyperplasia in infancy. Pediatrics. 2004;114(4):e520-e522.
49 Greene SA, Aynsley-Green A, Soltesz G, Baum JD. Management of secondary diabetes mellitus after total pancreatectomy in infancy. Arch Dis Child. 1984;59(4):356-359.
50 Daly LP, Osterhoudt KC, Weinzimer SA. Presenting features of idiopathic ketotic hypoglycemia. J Emerg Med. 2003;25(1):39-43.
51 Macdessi JS, Randell TL, Donaghue KC, Ambler GR, van Asperen PP, Mellis CM. Adrenal crises in children treated with high-dose inhaled corticosteroids for asthma. Med J Aust. 2003;178(5):214-216.
52 Todd GR, Acerini CL, Ross-Russell R, Zahra S, Warner JT, McCance D. Survey of adrenal crisis associated with inhaled corticosteroids in the United Kingdom. Arch Dis Child. 2002;87(6):457-461.
53 Todd GR, Acerini CL, Buck JJ, et al. Acute adrenal crisis in asthmatics treated with high-dose fluticasone propionate. Eur Respir J. 2002;19(6):1207-1209.
54 Drake AJ, Howells RJ, Shield JP, Prendiville A, Ward PS, Crowne EC. Symptomatic adrenal insufficiency presenting with hypoglycaemia in children with asthma receiving high dose inhaled fluticasone propionate. BMJ. 2002;324(7345):1081-1082.
55 Randell TL, Donaghue KC, Ambler GR, Cowell CT, Fitzgerald DA, van Asperen PP. Safety of the newer inhaled corticosteroids in childhood asthma. Paediatr Drugs. 2003;5(7):481-504.
56 Geffner ME. Hypopituitarism in childhood. Cancer Control. 2002;9(3):212-222.
57 Artavia-Loria E, Chaussain JL, Bougneres PF, Job JC. Frequency of hypoglycemia in children with adrenal insufficiency. Acta Endocrinol Suppl (Copenh). 1986;279:275-278.
58 Ronghe MD, Barton J, Jardine PE, et al. The importance of testing for adrenoleucodystrophy in males with idiopathic Addison’s disease. Arch Dis Child. 2002;86(3):185-189.
59 Hatherill M, Tibby SM, Hilliard T, Turner C, Murdoch IA. Adrenal insufficiency in septic shock. Arch Dis Child. 1999;80(1):51-55.
60 Menon K, Clarson C. Adrenal function in pediatric critical illness. Pediatr Crit Care Med. 2002;3(2):112-116.
61 Menon K, Ward RE, Lawson ML, et al. A prospective multicenter study of adrenal function in critically ill children. Am J Respir Crit Care Med. 2010;182:246-251.
62 Agus M. One step forward: an advance in understanding of adrenal insufficiency in the pediatric critically ill. Crit Care Med. 2005;33(4):911-912.
63 Langer M, Modi BP, Agus M. Adrenal insufficiency in the critically ill neonate and child. Curr Opin Pediatr. 2006;18(4):448-453.
64 Brierley J, Choong K, Cornell T, et al. 2007 American College of Critical Care Medicine clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock*. Crit Care Med. 2008 Nov 28. Epub ahead of print
65 Brierley J, Carcillo JA, Choong K, et al. Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med. 2009;37(2):666-688.
66 Ng PC, Lee CH, Lam CW, et al. Transient adrenocortical insufficiency of prematurity and systemic hypotension in very low birthweight infants. Arch Dis Child Fetal Neonatal Ed. 2004;89(2):F119-F126.
67 Ng PC, Lee CH, Bnur FL, et al. A double-blind, randomized, controlled study of a “stress dose” of hydrocortisone for rescue treatment of refractory hypotension in preterm infants. Pediatrics. 2006;117(2):367-375.
68 Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349(8):776-788.
69 Charmandari E, Lichtarowicz-Krynska EJ, Hindmarsh PC, Johnston A, Aynsley-Green A, Brook CG. Congenital adrenal hyperplasia: management during critical illness. Arch Dis Child. 2001;85(1):26-28.
70 Faustino EV, Apkon M. Persistent hyperglycemia in critically ill children. J Pediatr. 2005;146(1):30-34.
71 Izquierdo R. Hyperglycemia and mortality. J Pediatr. 2005;146(1):5-7.
72 Bowlby D, Rapaport R, Hojsak J. Hyperglycemia in critically ill children. J Pediatr. 2006;148(6):847. author reply 847-8
73 Kong MY, Alten J, Tofil N. Is hyperglycemia really harmful? A critical appraisal of “Persistent hyperglycemia in critically ill children” by Faustino and Apkon (J Pediatr 2005; 146:30-34). Pediatr Crit Care Med. 2007;8(5):482-485.
74 Branco RG, Tasker RC. Glycemic level in mechanically ventilated children with bronchiolitis. Pediatr Crit Care Med. 2007;8(6):546-550.
75 Crawford BA, Roy LP, Knight JF. Hyperglycaemia complicating haemolytic uraemic syndrome. J Paediatr Child Health. 1990;26(4):225-226.
76 O’Keefe SJ, Wesley A, Jialal I, Epstein S. The metabolic response and problems with nutritional support in acute tetanus. Metabolism. 1984;33(5):482-487.
77 Polak M, Rolon MA, Chouchana A, Czernichow P. Theophylline intoxication mimicking diabetic ketoacidosis in a child. Diabetes Metab. 1999;25(6):513-515.
78 Cochran A, Scaife ER, Hansen KW, Downey EC. Hyperglycemia and outcomes from pediatric traumatic brain injury. J Trauma. 2003;55(6):1035-1038.
79 Preissig CM, Rigby MR, Maher KO. Glycemic control for postoperative pediatric cardiac patients. Pediatr Cardiol. 2009;30(8):1098-1104.
80 Falcao G, Ulate K, Kouzekanani K, Bielefeld MR, Morales JM, Rotta AT. Impact of postoperative hyperglycemia following surgical repair of congenital cardiac defects. Pediatr Cardiol. 2008;29(3):628-636.
81 Day KM, Haub N, Betts H, Inwald DP. Hyperglycemia is associated with morbidity in critically ill children with meningococcal sepsis. Pediatr Crit Care Med. 2008;9(6):636-640.
82 van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345(19):1359-1367.
83 Van den Berghe G, Wilmer A, Hermans G, et al. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354(5):449-461.
84 Mesotten D, Wauters J, Van den Berghe G, Wouters PJ, Milants I, Wilmer A. The effect of strict blood glucose control on biliary sludge and cholestasis in critically ill patients. J Clin Endocrinol Metab. 2009;94(7):2345-2352.
85 Schetz M, Vanhorebeek I, Wouters PJ, Wilmer A, Van den Berghe G. Tight blood glucose control is renoprotective in critically ill patients. J Am Soc Nephrol. 2008;19(3):571-578.
86 Hermans G, Wilmer A, Meersseman W, et al. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Respir Crit Care Med. 2007;175(5):480-489.
87 Van den Berghe G, Schoonheydt K, Becx P, Bruyninckx F, Wouters PJ. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64(8):1348-1353.
88 Langouche L, Vanhorebeek I, Vlasselaers D, et al. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest. 2005;115(8):2277-2286.
89 Wiener RS, Wiener DC, Larson RJ. Benefits and risks of tight glucose control in critically ill adults: a meta-analysis. JAMA. 2008;300(8):933-944.
90 Preissig CM, Hansen I, Roerig PL, Rigby MR. A protocolized approach to identify and manage hyperglycemia in a pediatric critical care unit. Pediatr Crit Care Med. 2008;9(6):581-588.
91 Verhoeven JJ, Brand JB, van de Polder MM, Joosten KF. Management of hyperglycemia in the pediatric intensive care unit; implementation of a glucose control protocol. Pediatr Crit Care Med. 2009;10(6):648-652.
92 Faraon-Pogaceanu C, Banasiak KJ, Hirshberg EL, Faustino EV. Comparison of the effectiveness and safety of two insulin infusion protocols in the management of hyperglycemia in critically ill children. Pediatr Crit Care Med. 2010;11:741-749.
93 Ulate KP, Lima Falcao GC, Bielefeld MR, Morales JM, Rotta AT. Strict glycemic targets need not be so strict: a more permissive glycemic range for critically ill children. Pediatrics. 2008;122(4):e898-e904.
94 Vlasselaers D, Milants I, Desmet L, et al. Intensive insulin therapy for patients in paediatric intensive care: a prospective, randomised controlled study. Lancet. 2009;373(9663):547-556.
95 Fram RY, Cree MG, Wolfe RR, et al. Intensive insulin therapy improves insulin sensitivity and mitochondrial function in severely burned children. Crit Care Med. 2010;38(6):1475-1483.
96 Gunst J, Van den Berghe G. Blood glucose control in the intensive care unit: benefits and risks. Semin Dial. 2010;23(2):157-162.
97 Vlasselaers D. Blood glucose control in the intensive care unit: discrepancy between belief and practice. Crit Care. 2010;14(3):145.
98 Preissig CM, Rigby MR. A disparity between physician attitudes and practice regarding hyperglycemia in pediatric intensive care units in the United States: a survey on actual practice habits. Crit Care. 2010;14(1):R11.
99 Ogilvy-Stuart AL, Beardsall K. Management of hyperglycaemia in the preterm infant. Arch Dis Child Fetal Neonatal Ed. 2010;95(2):F126-F131.
100 Bruno G, Cerutti F, Merletti F, et al. Short-term mortality risk in children and young adults with type 1 diabetes: the population-based Registry of the Province of Turin, Italy. Nutr Metab Cardiovasc Dis. 2009;19(5):340-344.
101 Patterson CC, Dahlquist G, Harjutsalo V, et al. Early mortality in EURODIAB population-based cohorts of type 1 diabetes diagnosed in childhood since 1989. Diabetologia. 2007;50(12):2439-2442.
102 Dahlquist G, Kallen B. Mortality in childhood-onset type 1 diabetes: a population-based study. Diabetes Care. 2005;28(10):2384-2387.
103 Feltbower RG, Bodansky HJ, Patterson CC, et al. Acute complications and drug misuse are important causes of death for children and young adults with type 1 diabetes: results from the Yorkshire Register of diabetes in children and young adults. Diabetes Care. 2008;31(5):922-926.
104 Edge JA, Ford-Adams ME, Dunger DB. Causes of death in children with insulin dependent diabetes 1990-96. Arch Dis Child. 1999;81(4):318-323.
105 Warner DP, McKinney PA, Law GR, Bodansky HJ. Mortality and diabetes from a population based register in Yorkshire 1978-93. Arch Dis Child. 1998;78(5):435-438.
106 Orlowski JP, Cramer CL, Fiallos MR. Diabetic ketoacidosis in the pediatric ICU. Pediatr Clin North Am. 2008;55(3):577-587. x
107 Hirasing RA, Reeser HM, de Groot RR, Ruwaard D, van Buuren S, Verloove-Vanhorick SP. Trends in hospital admissions among children aged 0-19 years with type I diabetes in The Netherlands. Diabetes Care. 1996;19(5):431-434.
108 Curtis JR, To T, Muirhead S, Cummings E, Daneman D. Recent trends in hospitalization for diabetic ketoacidosis in Ontario children. Diabetes Care. 2002;25(9):1591-1596.
109 Dunger DB, Sperling MA, Acerini CL, et al. European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics. 2004;113(2):e133-e140.
110 Jayashree M, Singhi S. Diabetic ketoacidosis: predictors of outcome in a pediatric intensive care unit of a developing country. Pediatr Crit Care Med. 2004;5(5):427-433.
111 Cooke DW, Plotnick L. Management of diabetic ketoacidosis in children and adolescents. Pediatr Rev. 2008;29(12):431-435. quiz 436
112 Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis in children and adolescents with diabetes. Pediatr Diabetes. 2009;10(Suppl 12):118-133.
113 Wolfsdorf J, Glaser N, Sperling MA, American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: A consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150-1159.
114 Fiordalisi I, Novotny WE, Holbert D, Finberg L, Harris GD, Critical Care Management Group. An 18-yr prospective study of pediatric diabetic ketoacidosis: an approach to minimizing the risk of brain herniation during treatment. Pediatr Diabetes. 2007;8(3):142-149.
115 Glaser NS, Marcin JP, Wootton-Gorges SL, et al. Correlation of clinical and biochemical findings with diabetic ketoacidosis-related cerebral edema in children using magnetic resonance diffusion-weighted imaging. J Pediatr. 2008;153(4):541-546.
116 Roberts JS, Vavilala MS, Schenkman KA, Shaw D, Martin LD, Lam AM. Cerebral hyperemia and impaired cerebral autoregulation associated with diabetic ketoacidosis in critically ill children. Crit Care Med. 2006;34(8):2217-2223.
117 Ackerman A. Cerebral edema in pediatric diabetic ketoacidosis: Can six patients make a difference? Crit Care Med. 2006;34(8):2258-2259.
118 Glaser N, Barnett P, McCaslin I, et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344(4):264-269.
119 Tasker RC, Lutman D, Peters MJ. Hyperventilation in severe diabetic ketoacidosis. Pediatr Crit Care Med. 2005;6(4):405-411.
120 Glaser N. Cerebral edema in children with diabetic ketoacidosis. Curr Diabetes Rep. 2001;1(1):41-46.
121 Brown TB. Cerebral oedema in childhood diabetic ketoacidosis: is treatment a factor? Emerg Med J. 2004;21(2):141-144.
122 Glaser N. Cerebral injury and cerebral edema in children with diabetic ketoacidosis: could cerebral ischemia and reperfusion injury be involved? Pediatr Diabetes. 2009;10(8):534-541.
123 Edge JA, Hawkins MM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85(1):16-22.
124 Glaser NS, Wootton-Gorges SL, Buonocore MH, et al. Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr Diabetes. 2006;7(2):75-80.
125 Ghetti S, Lee JK, Sims CE, Demaster DM, Glaser NS. Diabetic ketoacidosis and memory dysfunction in children with type 1 diabetes. J Pediatr. 2010;156(1):109-114.
126 Marcin JP, Glaser N, Barnett P, et al. Factors associated with adverse outcomes in children with diabetic ketoacidosis-related cerebral edema. J Pediatr. 2002;141(6):793-797.
127 Marcin JP, Glaser N, Kuppermann N. Ventilation in pediatric diabetic ketoacidosis–not too much, but not too little. Pediatr Crit Care Med. 2005;6(4):489-490.
128 Carl GF, Hoffman WH, Blankenship PR, Litaker MS, Hoffman MG, Mabe PA. Diabetic ketoacidosis depletes plasma tryptophan. Endocr Res. 2002;28(1-2):91-102.
129 Clark JA, Burny I, Sarnaik AP, Audhya TK. Acute thiamine deficiency in diabetic ketoacidosis: Diagnosis and management. Pediatr Crit Care Med. 2006;7(6):595-599.
130 Hoffman WH, Burek CL, Waller JL, Fisher LE, Khichi M, Mellick LB. Cytokine response to diabetic ketoacidosis and its treatment. Clin Immunol. 2003;108(3):175-181.
131 Carl GF, Hoffman WH, Passmore GG, et al. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res. 2003;29(1):73-82.
132 Gutierrez JA, Bagatell R, Samson MP, Theodorou AA, Berg RA. Femoral central venous catheter-associated deep venous thrombosis in children with diabetic ketoacidosis. Crit Care Med. 2003;31(1):80-83.
133 Batra AS, Acherman RJ, Wong P, Silka MJ. Acute myocardial infarction in a 12-year-old as a complication of hyperosmolar diabetic ketoacidosis. Pediatr Crit Care Med. 2002;3(2):194-196.
134 Mokuno T, Sawai Y, Oda N, et al. A case of myocarditis associated with IDDM. Diabetes Care. 1996;19(4):374-378.
135 Hoffman WH, Locksmith JP, Burton EM, et al. Interstitial pulmonary edema in children and adolescents with diabetic ketoacidosis. J Diabetes Complications. 1998;12(6):314-320.
136 Kuppermann N, Park J, Glatter K, Marcin JP, Glaser NS. Prolonged QT interval corrected for heart rate during diabetic ketoacidosis in children. Arch Pediatr Adolesc Med. 2008;162(6):544-549.
137 Rewers A, McFann K, Chase HP. Bedside monitoring of blood beta-hydroxybutyrate levels in the management of diabetic ketoacidosis in children. Diabetes Technol Ther. 2006;8(6):671-676.
138 Ham MR, Okada P, White PC. Bedside ketone determination in diabetic children with hyperglycemia and ketosis in the acute care setting. Pediatr Diabetes. 2004;5(1):39-43.
139 Vanelli M, Chiari G, Capuano C, Iovane B, Bernardini A, Giacalone T. The direct measurement of 3-beta-hydroxy butyrate enhances the management of diabetic ketoacidosis in children and reduces time and costs of treatment. Diabetes Nutr Metab. 2003;16(5-6):312-316.
140 Prisco F, Picardi A, Iafusco D, et al. Blood ketone bodies in patients with recent-onset type 1 diabetes (a multicenter study). Pediatr Diabetes. 2006;7(4):223-228.
141 Carlotti AP, Bohn D, Halperin ML. Importance of timing of risk factors for cerebral oedema during therapy for diabetic ketoacidosis. Arch Dis Child. 2003;88(2):170-173.
142 Camp MA, Allon M. Severe hypophosphatemia in hospitalized patients. Miner Electrolyte Metab. 1990;16(6):365-368.
143 de Oliveira Iglesias SB, Pons Leite H, de Carvalho WB. Hypophosphatemia-induced seizure in a child with diabetic ketoacidosis. Pediatr Emerg Care. 2009;25(12):859-861.
144 Clerbaux T, Reynaert M, Willems E, Frans A. Effect of phosphate on oxygen-hemoglobin affinity, diphosphoglycerate and blood gases during recovery from diabetic ketoacidosis. Intensive Care Med. 1989;15(8):495-498.
145 Becker DJ, Brown DR, Steranka BH, Drash AL. Phosphate replacement during treatment of diabetic ketosis. Effects on calcium and phosphorus homeostasis. Am J Dis Child. 1983;137(3):241-246.
146 Winter RJ, Harris CJ, Phillips LS, Green OC. Diabetic ketoacidosis. Induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67(5):897-900.
147 Zipf WB, Bacon GE, Spencer ML, Kelch RP, Hopwood NJ, Hawker CD. Hypocalcemia, hypomagnesemia, and transient hypoparathyroidism during therapy with potassium phosphate in diabetic ketoacidosis. Diabetes Care. 1979;2(3):265-268.
148 Viallon A, Zeni F, Lafond P, et al. Does bicarbonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med. 1999;27(12):2690-2693.
149 Lever E, Jaspan JB. Sodium bicarbonate therapy in severe diabetic ketoacidosis. Am J Med. 1983;75(2):263-268.
150 Green SM, Rothrock SG, Ho JD, et al. Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998;31(1):41-48.
151 Flood RG, Chiang VW. Rate and prediction of infection in children with diabetic ketoacidosis. Am J Emerg Med. 2001;19(4):270-273.
152 Goldman MH, Kashani M. Spurious hyponatremia in diabetic ketoacidosis with massive lipid elevations. J Med Soc N J. 1982;79(7):591-592.
153 Oh G, Anderson S, Tancredi D, Kuppermann N, Glaser N. Hyponatremia in pediatric diabetic ketoacidosis: reevaluating the correction factor for hyperglycemia. Arch Pediatr Adolesc Med. 2009;163(8):771-772.
154 Garcia E, Abramo TJ, Okada P, Guzman DD, Reisch JS, Wiebe RA. Capnometry for noninvasive continuous monitoring of metabolic status in pediatric diabetic ketoacidosis. Crit Care Med. 2003;31(10):2539-2543.
155 McBride ME, Berkenbosch JW, Tobias JD. Transcutaneous carbon dioxide monitoring during diabetic ketoacidosis in children and adolescents. Paediatr Anaesth. 2004;14(2):167-171.
156 Tobias JD. Noninvasive monitoring of the response to therapy during diabetic ketoacidosis: is end-tidal CO2 useful? Crit Care Med. 2003;31(10):2562-2563.
157 Keane S, Gallagher A, Ackroyd S, McShane MA, Edge JA. Cerebral venous thrombosis during diabetic ketoacidosis. Arch Dis Child. 2002;86(3):204-205.
158 Eskandar EN, Weller SJ, Frim DM. Hydrocephalus requiring urgent external ventricular drainage in a patient with diabetic ketoacidosis and cerebral edema: case report. Neurosurgery. 1997;40(4):836-838. discussion 838-9
159 Roe TF, Crawford TO, Huff KR, Costin G, Kaufman FR, Nelson MDJr. Brain infarction in children with diabetic ketoacidosis. J Diabetes Complications. 1996;10(2):100-108.
160 Bonkowsky JL, Filloux FM. Extrapontine myelinolysis in a pediatric case of diabetic ketoacidosis and cerebral edema. J Child Neurol. 2003;18(2):144-147.
161 Roberts MD, Slover RH, Chase HP. Diabetic ketoacidosis with intracerebral complications. Pediatr Diabetes. 2001;2(3):109-114.
162 Kamat P, Vats A, Gross M, Checchia PA. Use of hypertonic saline for the treatment of altered mental status associated with diabetic ketoacidosis. Pediatr Crit Care Med. 2003;4(2):239-242.
163 Theodoropoulos T, Braverman LE, Vagenakis AG. Iodide-induced hypothyroidism: a potential hazard during perinatal life. Science. 1979;205(4405):502-503.
164 Pyati SP, Ramamurthy RS, Krauss MT, Pildes RS. Absorption of iodine in the neonate following topical use of povidone iodine. J Pediatr. 1977;91(5):825-828.
165 Smerdely P, Lim A, Boyages SC, et al. Topical iodine-containing antiseptics and neonatal hypothyroidism in very-low-birthweight infants. Lancet. 1989;2(8664):661-664.
166 l’Allemand D, Gruters A, Heidemann P, Schurnbrand P. Iodine-induced alterations of thyroid function in newborn infants after prenatal and perinatal exposure to povidone iodine. J Pediatr. 1983;102(6):935-938.
167 l’Allemand D, Gruters A, Beyer P, Weber B. Iodine in contrast agents and skin disinfectants is the major cause for hypothyroidism in premature infants during intensive care. Horm Res. 1987;28(1):42-49.
168 Linder N, Sela B, German B, et al. Iodine and hypothyroidism in neonates with congenital heart disease. Arch Dis Child Fetal Neonatal Ed. 1997;77(3):F239-F240.
169 Ross OC, Petros A. The sick euthyroid syndrome in paediatric cardiac surgery patients. Intensive Care Med. 2001;27(7):1124-1132.
170 Mainwaring RD, Capparelli E, Schell K, Acosta M, Nelson JC. Pharmacokinetic evaluation of triiodothyronine supplementation in children after modified Fontan procedure. Circulation. 2000;101(12):1423-1429.
171 Portman MA, Fearneyhough C, Ning XH, Duncan BW, Rosenthal GL, Lupinetti FM. Triiodothyronine repletion in infants during cardiopulmonary bypass for congenital heart disease. J Thorac Cardiovasc Surg. 2000;120(3):604-608.
172 Bettendorf M, Schmidt KG, Grulich-Henn J, Ulmer HE, Heinrich UE. Tri-iodothyronine treatment in children after cardiac surgery: a double-blind, randomised, placebo-controlled study. Lancet. 2000;356(9229):529-534.
173 Dimmick S, Badawi N, Randell T. Thyroid hormone supplementation for the prevention of morbidity and mortality in infants undergoing cardiac surgery. Cochrane Database Syst Rev 2004;(3)(3):CD004220.
174 Gruneiro de Papendieck L, Chiesa A, Bastida MG, Alonso G, Finkielstain G, Heinrich JJ. Thyroid dysfunction and high thyroid stimulating hormone levels in children with Down’s syndrome. J Pediatr Endocrinol Metab. 2002;15(9):1543-1548.
175 Tuysuz B, Beker DB. Thyroid dysfunction in children with Down’s syndrome. Acta Paediatr. 2001;90(12):1389-1393.
176 Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105(1):e10.
177 Dionisi-Vici C, Rizzo C, Burlina AB, et al. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140(3):321-327.
178 Moammar H, Cheriyan G, Mathew R, Al-Sannaa N. Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983-2008. Ann Saudi Med. 2010;30(4):271-277.
179 Jones PM, Bennett MJ. The changing face of newborn screening: diagnosis of inborn errors of metabolism by tandem mass spectrometry. Clin Chim Acta. 2002;324(1-2):121-128.
180 Lee NC, Tang NL, Chien YH, et al. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010;100(1):46-50.
181 Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304-2312.
182 Vockley J. Newborn screening: After the thrill is gone. Mol Genet Metab. 2007;92(1-2):6-12.
183 Centers for Disease Control and Prevention (CDC). Contribution of selected metabolic diseases to early childhood deaths–Virginia, 1996-2001. MMWR Morb Mortal Wkly Rep. 2003;52(29):677-679.
184 Dott M, Chace D, Fierro M, et al. Metabolic disorders detectable by tandem mass spectrometry and unexpected early childhood mortality: a population-based study. Am J Med Genet A. 2006;140(8):837-842.
185 Champion MP. An approach to the diagnosis of inherited metabolic disease. Arch Dis Child Educ Pract Ed. 2010;95(2):40-46.
186 Anadiotis G, Ierardi-Curto L, Kaplan PB, Berry GT. Ornithine transcarbamylase deficiency and pancreatitis. J Pediatr. 2001;138(1):123-124.
187 Hunt ADJr, Stokes JJr, McCrory WW, Stroud HH. Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics. 1954;13(2):140-145.
188 Gallagher RC, Van Hove JL, Scharer G, et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65(5):550-556.
189 Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12(3):307-309.
190 Hoffmann GF, Schmitt B, Windfuhr M, et al. Pyridoxal 5′-phosphate may be curative in early-onset epileptic encephalopathy. J Inherit Metab Dis. 2007;30(1):96-99.
191 Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077-1086.
192 Klepper J, Leiendecker B. GLUT1 deficiency syndrome–2007 update. Dev Med Child Neurol. 2007;49(9):707-716.
193 Wolf B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab. 2010;100(1):6-13.
194 Cowan TM, Blitzer MG, Wolf B, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet Med. 2010;12(7):464-470.
195 Endres W, Shin YS, Gunther R, Ibel H, Duran M, Wadman SK. Report on a new patient with combined deficiencies of sulphite oxidase and xanthine dehydrogenase due to molybdenum cofactor deficiency. Eur J Pediatr. 1988;148(3):246-249.
196 Slot HM, Overweg-Plandsoen WC, Bakker HD, et al. Molybdenum-cofactor deficiency: an easily missed cause of neonatal convulsions. Neuropediatrics. 1993;24(3):139-142.
197 Appignani BA, Kaye EM, Wolpert SM. CT and MR appearance of the brain in two children with molybdenum cofactor deficiency. AJNR Am J Neuroradiol. 1996;17(2):317-320.
198 Rupar CA, Gillett J, Gordon BA, et al. Isolated sulfite oxidase deficiency. Neuropediatrics. 1996;27(6):299-304.
199 Topcu M, Coskun T, Haliloglu G, Saatci I. Molybdenum cofactor deficiency: report of three cases presenting as hypoxic-ischemic encephalopathy. J Child Neurol. 2001;16(4):264-270.
200 Beemer FA, Duran M, Wadman SK, Cats BP. Absence of hepatic molybdenum cofactor. An inborn error of metabolism associated with lens dislocation. Ophthalmic Paediatr Genet. 1985;5(3):191-195.
201 Ito T, van Kuilenburg AB, Bootsma AH, et al. Rapid screening of high-risk patients for disorders of purine and pyrimidine metabolism using HPLC-electrospray tandem mass spectrometry of liquid urine or urine-soaked filter paper strips. Clin Chem. 2000;46(4):445-452.
202 Clarke JTR. A clinical guide to inherited metabolic diseases, 3rd ed. Cambridge University Press; 2006.
203 Fencl V, Jabor A, Kazda A, Figge J. Diagnosis of metabolic acid-base disturbances in critically ill patients. Am J Respir Crit Care Med. 2000;162(6):2246-2251.
204 Durward A, Mayer A, Skellett S, et al. Hypoalbuminaemia in critically ill children: incidence, prognosis, and influence on the anion gap. Arch Dis Child. 2003;88(5):419-422.
205 Hatherill M, Waggie Z, Purves L, Reynolds L, Argent A. Correction of the anion gap for albumin in order to detect occult tissue anions in shock. Arch Dis Child. 2002;87(6):526-529.
206 Morton DH, Strauss KA, Robinson DL, Puffenberger EG, Kelley RI. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002;109(6):999-1008.
207 Puliyanda DP, Harmon WE, Peterschmitt MJ, Irons M, Somers MJ. Utility of hemodialysis in maple syrup urine disease. Pediatr Nephrol. 2002;17(4):239-242.
208 Zinnanti WJ, Lazovic J, Griffin K, et al. Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain. 2009;132(Pt 4):903-918.
209 Kahler SG, Sherwood WG, Woolf D, et al. Pancreatitis in patients with organic acidemias. J Pediatr. 1994;124(2):239-243.
210 Massoud AF, Leonard JV. Cardiomyopathy in propionic acidaemia. Eur J Pediatr. 1993;152(5):441-445.
211 Aliefendioglu D, Tana Aslan A, Coskun T, Dursun A, Cakmak FN, Kesimer M. Transient nonketotic hyperglycinemia: two case reports and literature review. Pediatr Neurol. 2003;28(2):151-155.
212 Paupe A, Bidat L, Sonigo P, Lenclen R, Molho M, Ville Y. Prenatal diagnosis of hypoplasia of the corpus callosum in association with non-ketotic hyperglycinemia. Ultrasound Obstet Gynecol. 2002;20(6):616-619.
213 Applegarth DA, Toone JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab. 2001;74(1-2):139-146.
214 Applegarth DA, Toone JR. Glycine encephalopathy (nonketotic hyperglycinaemia) : review and update. J Inherit Metab Dis. 2004;27(3):417-422.
215 Wiltshire EJ, Poplawski NK, Harrison JR, Fletcher JM. Treatment of late-onset nonketotic hyperglycinaemia: effectiveness of imipramine and benzoate. J Inherit Metab Dis. 2000;23(1):15-21.
216 Pretorius CJ, Loy Son GG, Bonnici F, Harley EH. Two siblings with episodic ketoacidosis and decreased activity of succinyl-CoA:3-ketoacid CoA-transferase in cultured fibroblasts. J Inherit Metab Dis. 1996;19(3):296-300.
217 Antozzi C, Zeviani M. Cardiomyopathies in disorders of oxidative metabolism. Cardiovasc Res. 1997;35(2):184-199.
218 Brusilow SW. Arginine, an indispensable amino acid for patients with inborn errors of urea synthesis. J Clin Invest. 1984;74(6):2144-2148.
219 Bongaerts G, Bakkeren J, Severijnen R, et al. Lactobacilli and acidosis in children with short small bowel. J Pediatr Gastroenterol Nutr. 2000;30(3):288-293.
220 Wexler ID, Hemalatha SG, McConnell J, et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology. 1997;49(6):1655-1661.
221 Weber TA, Antognetti MR, Stacpoole PW. Caveats when considering ketogenic diets for the treatment of pyruvate dehydrogenase complex deficiency. J Pediatr. 2001;138(3):390-395.
222 Stacpoole PW, Barnes CL, Hurbanis MD, Cannon SL, Kerr DS. Treatment of congenital lactic acidosis with dichloroacetate. Arch Dis Child. 1997;77(6):535-541.
223 Naito E, Ito M, Yokota I, Saijo T, Matsuda J, Kuroda Y. Thiamine-responsive lactic acidaemia: role of pyruvate dehydrogenase complex. Eur J Pediatr. 1998;157(8):648-652.
224 von Kleist-Retzow JC, Cormier-Daire V, Viot G, et al. Antenatal manifestations of mitochondrial respiratory chain deficiency. J Pediatr. 2003;143(2):208-212.
225 Marin-Garcia J, Goldenthal MJ. Fatty acid metabolism in cardiac failure: biochemical, genetic and cellular analysis. Cardiovasc Res. 2002;54(3):516-527.
226 Marin-Garcia J, Goldenthal MJ. Understanding the impact of mitochondrial defects in cardiovascular disease: a review. J Card Fail. 2002;8(5):347-361.
227 Marin-Garcia J, Goldenthal MJ, Filiano JJ. Cardiomyopathy associated with neurologic disorders and mitochondrial phenotype. J Child Neurol. 2002;17(10):759-765.
228 Choong K, Clarke JT, Cutz E, Pollit RJ, Olpin SE. Lethal cardiac tachyarrhythmia in a patient with neonatal carnitine-acylcarnitine translocase deficiency. Pediatr Dev Pathol. 2001;4(6):573-579.
229 Howard RS, Russell S, Losseff N, et al. Management of mitochondrial disease on an intensive care unit. QJM. 1995;88(3):197-207.
230 Choo-Kang AT, Lynn S, Taylor GA, et al. Defining the importance of mitochondrial gene defects in maternally inherited diabetes by sequencing the entire mitochondrial genome. Diabetes. 2002;51(7):2317-2320.
231 Rey C, Prieto S, Medina A, Perez C, Concha A, Menendez S. Fatal lactic acidosis during antiretroviral therapy. Pediatr Crit Care Med. 2003;4(4):485-487.
232 Touma EH, Rashed MS, Vianey-Saban C, et al. A severe genotype with favourable outcome in very long chain acyl-CoA dehydrogenase deficiency. Arch Dis Child. 2001;84(1):58-60.
233 Durand P, Debray D, Mandel R, et al. Acute liver failure in infancy: a 14-year experience of a pediatric liver transplantation center. J Pediatr. 2001;139(6):871-876.
234 Croffie JM, Gupta SK, Chong SK, Fitzgerald JF. Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics. 1999;103(3):675-678.
235 Barkaoui E, Debray D, Habes D, Ogier H, Bernard O. Favorable outcome of treatment with NTBC of acute liver insufficiency disclosing hereditary tyrosinemia type I. Arch Pediatr. 1999;6(5):540-544.
236 Holme E, Lindstedt S. Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione). J Inherit Metab Dis. 1998;21(5):507-517.
237 Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4(4):805-814.
238 Ros Viladoms J, Vilaseca Busca MA, Lambruschini Ferri N, Mas Comas A, Gonzalez Pascual E, Holme E. Evolution of a case of tyrosinemia type I treated with NTBC. An Esp Pediatr. 2001;54(3):305-309.
239 Walter JH, Collins JE, Leonard JV. Recommendations for the management of galactosaemia. UK Galactosaemia Steering Group. Arch Dis Child. 1999;80(1):93-96.
240 Walter JH. Inborn errors of metabolism and pregnancy. J Inherit Metab Dis. 2000;23(3):229-236.
