CHAPTER 176 Encephalocele
Encephalocele is herniation of cerebral tissue, meninges, and cerebrospinal fluid (CSF) outside the confines of the skull. The condition is also termed cephalocele to encompass any combination of these intracranial elements.1 Encephalocele has been recognized since antiquity; it was first depicted in ancient sculptures and later in medieval artwork to represent demonic creatures.2,3 Early written descriptions came from the Dutch physician Petrus Forestus (1590) and J.F.C. Corvinius (1749).2 Since then, numerous case reports and case series have been collected, the epidemiology and regional variations have been delineated, and the long-term outcomes of children treated in the latter portion of the 20th century have been documented. The past half century has witnessed advancements in the basic science of neural development, improvements in prenatal imaging and care, more global recognition of these developmental anomalies, and better access to quality care for children in developing countries, where the incidence of this condition is higher. This chapter presents our best understanding of the classification schemes, embryopathogenesis, epidemiology, management, outcomes, and prognosis for children with encephalocele.
Classification
Classification of encephaloceles is based on the anatomic location of the skull defect, as detailed in Table 176-1. The two main groups are anterior and posterior: anterior encephaloceles are divided into sincipital (from sinciput, or forehead) and basal, whereas posterior encephaloceles are divided into occipital, occipitocervical, and parietal. Detailed computed tomography (CT) and magnetic resonance imaging (MRI) in the modern era can demonstrate the precise location and anatomic contents of the herniation,4 thereby allowing further subclassification (see Table 176-1) than previously established through clinical experience.5
| POSTERIOR ENCEPHALOCELES |
Embryology
The pathogenesis of encephalocele is poorly understood at a molecular level. Although encephalocele may represent defective closure of the neural tube in gross pathology, it does not appear to occur through defective neurulation. Primary neurulation, the process by which the future brain and the majority of the spinal cord form, occurs between the third and fourth gestational weeks through a process of embryonic folding and fusion of the midline (reviewed by Copp6) (Fig. 176-1). The initial closure point of the neural tube occurs at the region of the future occiput, whereas a second closure point takes place at the junction of the future forebrain and midbrain, with creation of a rostral (cranial) and caudal (spinal) neuropore. The rostral neuropore closes in a bidirectional fashion from the initial rhombencephalic, occipital closure (caudorostral), as well as in a rostrocaudal direction from a third closure site at the chiasmatic plate, with the extreme anterior end of the body axis corresponding to the future foramen caecum (see Fig. 176-1).7 As epithelial fusion occurs to create a neural tube, the dorsal midline tissues separate into an outer surface ectoderm (future epidermis) and inner neuroepithelium (future brain and spinal cord).8 Because encephaloceles are typically covered with normal skin, the initial closure event has already taken place, and thus the defect must arise after primary neurulation. Theories regarding encephalocele formation are focused on this stage of neural development, after the first month of gestation.
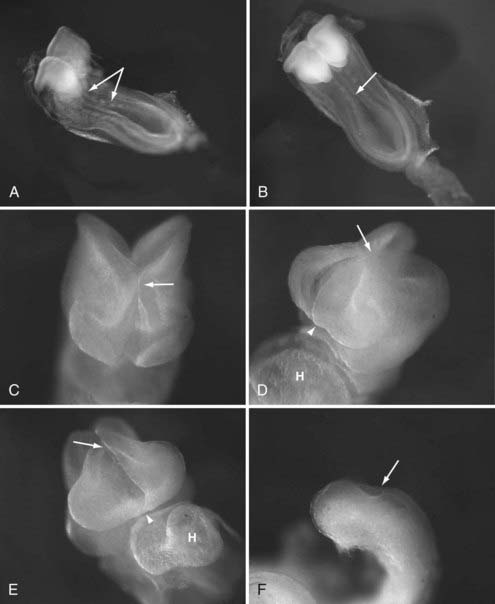
The most widely accepted theory is derived from that of Etienne Geoffroy Sain Hilaire (1827), the French naturalist whose work influenced Charles Darwin’s, which states that encephalocele is caused by an error in mesodermal differentiation.9 The paraxial mesoderm, from which the meninges and skull will form, migrates in between the ectodermal layers. In cases of encephalocele formation, a neuroschisis (fissure) develops after primary neurulation, which leads to scarring and subsequent adhesion between the cutaneous and the neuroectoderm and prevents interposition of the mesoderm. Pathologic specimens appear to follow these patterns in encephalocele formation, and a skull defect is present through which variably abnormal meninges and neural elements herniate.
Epidemiology and Further Theory Regarding Embryopathogenesis
The overall worldwide incidence of encephalocele is impossible to quantify because the majority of congenital encephaloceles lead to spontaneous abortion10 and collection of epidemiologic data in developing nations, where many of these lesions occur, is challenging.11 For unknown reasons, the prevalence of encephalocele in live births varies according to geographic location and race. In North America, Europe, and northern Asia, occipital encephaloceles predominate at a frequency varying between 0.8 and 4.0 per 10,000 live births, and sincipital encephaloceles are considerably more rare.10 By contrast, in Southeast Asia, sincipital encephaloceles are the predominant type and occur in 1 in 5000 live births, and occipital encephaloceles are much less common.12,13 Congenital basal encephaloceles represent just 10% of all encephaloceles in most series.14
Both remote and more recent epidemiologic data on the effect of seasonal variations and environmental exposure on the development of sincipital encephalocele have added credence to a toxicologic theory of this birth defect.13–15 A higher incidence of frontoethmoidal encephaloceles occurs in newborns conceived during the rainy season in Burma and Cambodia, when fungal molds are found in rice. A well-documented and ubiquitous fungal teratogen such as aflatoxin could combine with genetic factors to cause this condition.13,14 Most recently, a higher incidence of sincipital encephalocele was identified in a population of Assamese tea workers. An environmental factor such as a pesticide has been postulated to play a role.15 However, almost all congenitally acquired encephaloceles—and sincipital encephaloceles in particular—are sporadic, and familial association, though reported, is unusual.16 The genetic factors that would predispose an embryo to sincipital encephalocele are unknown.
Unlike sincipital encephaloceles, occipital encephaloceles are included in several rare but severe autosomal recessive and teratogenic syndromes. In syndromic cases, occipital encephaloceles are seen most commonly in Meckel-Gruber (or Meckel) syndrome (MGS), one of the cerebello-oculo-renal syndromes. Although MGS is a genetically heterogeneous disorder, proteins related to the syndrome mediate ciliary formation and epithelial morphogenesis at an early and critical developmental stage17 as a result of impairments in Sonic Hedgehog signaling.18 Impairments in neural tube patterning, governed by these processes, could in theory lead to the neuroschisis and subsequent failure of mesodermal interposition that occur after neural tube closure in St. Hilaire’s model of encephalocele formation. Beyond these scant details, the molecular pathogenesis of encephalocele formation remains unknown, but the details encoded in these developmental signaling pathways are certain to provide answers in the coming years.
Clinical Findings, Diagnosis, Management, and Outcome
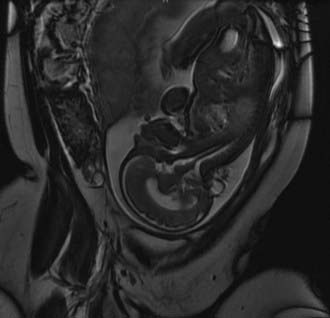
Despite their varied demographics and outcome, the principles of management of sincipital, cranial vault, and basal encephaloceles are similar and focus on removal of the sac, preservation of functional neural tissue, protection from leakage of CSF by closure of the meninges and nondysplastic skin, and correction of the cosmetic deformity. Prenatal diagnosis with amniocentesis (α-fetoprotein and acetylcholinesterase levels may vary from normal to abnormal, depending on epithelialization of the lesion), fetal ultrasound, and fetal MRI (Fig. 176-2) is commonplace today and allows accurate characterization of the location and type of encephalocele expected.
Postnatal MRI is the diagnostic study of choice to classify the encephalocele and predict the degree of functional tissue inside the sac and the extent of hydrocephalus, if any. Furthermore, concomitant magnetic resonance angiography/venography (MRA/MRV) may be useful in defining the lesion with respect to its surrounding vasculature. Volumetric (three-dimensional) CT can help in planning craniofacial reconstruction in infants with sincipital encephaloceles, although the need for such an extensive work-up has been called into question in recent series in locations where access to such studies is limited and surgical familiarity with the pathologies expected and encountered make them superfluous.11,19 Given the comparative rarity of sincipital encephaloceles in the Western hemisphere and the complex management issues that they present, a comprehensive work-up can certainly be warranted.
Posterior Encephaloceles
In the Western hemisphere, occipital encephaloceles are encountered most frequently, and with the advent of better prenatal imaging, they are becoming more frequently diagnosed during pregnancy.20 In the prenatal period, family counseling and a discussion of management and outcome must take place based on adequate imaging, knowledge of the historical and more recent literature concerning the long-term results after encephalocele repair10,20–22 (discussed later), and knowledge of the parents’ ethical and religious considerations.
Occipital encephaloceles occur between the lambda and foramen magnum, typically in the midline, and are divided into supratorcular and infratorcular, depending on their proximity to the confluence of the sinuses. Occipitocervical encephaloceles incorporate a cranial and cervical spinal bony defect. Parietal encephaloceles exist at any point between and including the lambda and bregma. As mentioned earlier, well-detailed MRI and MRA sequences will show the involvement of neurovascular structures within or near the sac, associated hydrocephalus, and other related abnormalities in the craniocervical axis. Occipital encephaloceles vary in their appearance, size, and contents, from small, atretic lesions (Fig. 176-3) with little or no brain elements to large lesions (Figs. 176-4 to 176-6) that may have portions of functional cerebellum, cortex, and brainstem contained within them. Associated central nervous system findings in patients with occipital encephaloceles include hydrocephalus, kinking of the brainstem, and an absent, rudimentary, or inverted cerebellum with the brainstem herniated posteriorly and the cerebellar vermis ventrally. A small posterior fossa or posterior fossa cysts reminiscent of a Dandy-Walker variant, along with abnormal displacement of the torcular, transverse sinuses, and tentorium, may be seen. Cortical dysplasia and callosal agenesis are frequently present.
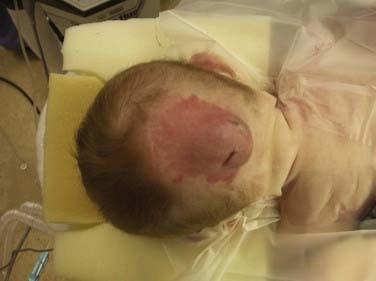
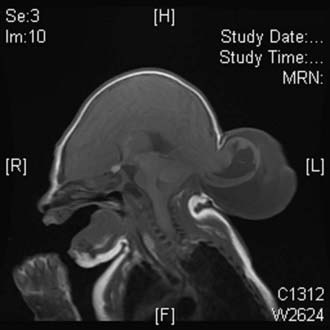
FIGURE 176-4 This male neonate was born with a large supratorcular encephalocele containing dysplastic occipital lobe and cerebellar tissue. There was no associated hydrocephalus (also see Fig. 176-6).
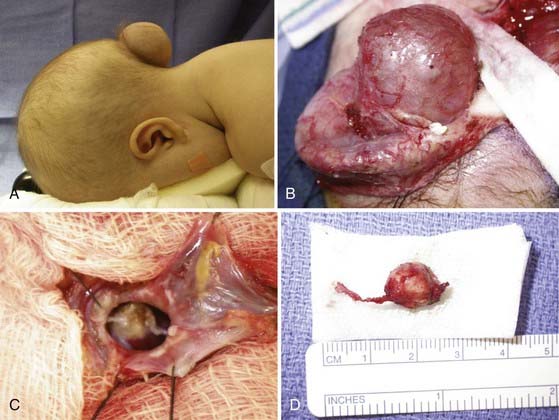
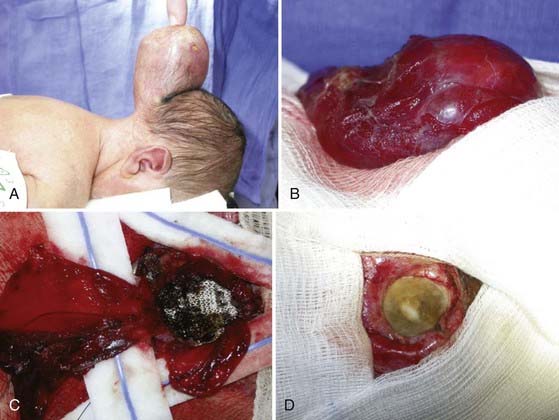
FIGURE 176-6 A, A male neonate in whom magnetic resonance imaging demonstrated a large supratorcular encephalocele underwent resection of the lesion through stepwise dissection of the epithelium from the cyst (B). The cyst walls were opened and the contents amputated (C). The dura was repaired primarily and augmented with a collagenous matrix (D) (also see Fig. 176-4).
Cranial abnormalities may include microphthalmia, facial clefts, and a sloping forehead because of caudal displacement of the frontal, temporal, and occipital lobes. In contrast to sincipital encephaloceles, occipital encephaloceles may occur in association with other extracranial anomalies such as myelomeningocele and cardiac, renal, limb, and genital anomalies (see discussion of MGS earlier). The association between occipital encephalocele and other systemic developmental anomalies suggests a global developmental migration process gone awry, as recent genetic work in syndromic encephalocele appears to indicate.17,18
Surgical Management
Typically, the infant is positioned prone on a horseshoe headrest (see Figs. 176-3, 176-5A, and 176-6A), with the face and body pressure points carefully padded. I use Reston Foam (3M, St Paul, MN) applied to the forehead, maxillary eminences, chest, iliac crests, and knees. The skin incision can be either horizontal or vertical, but it will ultimately extend elliptically around the lesion. Using Colorado tip monopolar cautery (Bovie Medical Corporation, Clearwater, FL) at a low setting, a dissection plane is created between the skin and abnormal epithelium (see Figs. 176-5B and 176-6B), and the dissection is carried to the pericranium to approach the skull defect and neck of the sac. Care is taken to delineate the dural margin at the bone defect, decompress the contents of the sac by removing CSF, and assess the normalcy of the tissue that it contains. Typically, gliotic and malformed neural elements can be amputated, with care taken to avoid major normal neurovascular structures (see Figs. 176-5C and 176-6C). The dura can be repaired primarily with fine Prolene suture and augmented with collagen-based dural substitute (see Fig. 176-6D), and the skin can be approximated by removal of redundant skin to form a cosmetically acceptable closure. Usually, the bone defect is small enough to not warrant intervention in the neonatal period; if necessary, closure can be done with a split-thickness graft when calvarial development is sufficient. If early repair is necessary, as with aplasia cutis congenita, an autologous graft can be harvested from a more normal part of the calvaria to cover and protect the midline defect and dural closure.
Management of hydrocephalus in the postoperative period is critical to preserve a watertight dural closure. Daily wound assessment to look for evidence of leakage or the development of pseudomeningocele and monitoring of ventricular and head circumference enlargement with serial ultrasound and measurements, respectively, are usually sufficient. A ventriculoperitoneal shunt is inserted if hydrocephalus becomes clinically significant and is preferably placed before leakage of CSF and wound compromise. The incidence of hydrocephalus appears to be higher with posterior encephaloceles.22
Outcome
To adequately counsel the parents of children with occipital encephalocele, an understanding of its comorbidity and outcome is essential. Historically, a worse outcome has been predicted for children with posterior encephaloceles (occipital, occipitocervical, parietal) than for those with anterior lesions (sincipital, basal), and the presence of hydrocephalus, other brain anomalies, a seizure disorder, and functional brain tissue within the sac has also been associated with a poor neurological outcome. Hydrocephalus and seizure disorder occur more commonly with occipital encephalocele, and a large cohort from a North American institution showed that patients with occipital encephaloceles fared worse than did their sincipital counterparts in an outcome survey that assessed physical, emotional, cognitive, and overall health outcomes.22 Hydrocephalus and seizure disorder were found to have significant additive adverse prognostic effects. The authors asserted that only half of all patients with occipital encephalocele will be able to live independently and functionally in society.22 A recent multivariate analysis from a separate, large North American series of children treated during a similar period revealed that location was not a significant predictor of outcome; instead, hydrocephalus and associated intracranial abnormalities had cumulative predictive effects for developmental delay.20
Anterior Encephaloceles
Sincipital encephaloceles, which involve the region of the foramen caecum, are present at birth but may not be manifested until later in childhood or adulthood, depending on their subclassification. Sincipital encephaloceles (Figs. 176-7 to 176-9) are further classified according to their bone defect and the course of the encephalocele sac and are designated nasofrontal, nasoethmoidal, and naso-orbital. Interfrontal encephaloceles (Figs. 176-10 and 176-11), which are located between the nasion and bregma, more closely resemble other cranial vault encephaloceles and are less common. All but the nasoethmoidal encephaloceles are visible at birth as a facial swelling that enlarges with crying or a Valsalva maneuver. The differential diagnosis for masses in the naso-orbital area includes dermoid cyst or teratoma with or without a dermal sinus tract, but these growths are typically in the midline, in contrast to the eccentric naso-orbital or nasofrontal lesions, are not pulsatile, and do not enlarge with crying. Nasoethmoidal encephaloceles, which may be manifested as a nasal mass later in childhood and even in adulthood, can be confused with nasal polyps. The two conditions, however, are distinguishable by epidemiology, location, and diagnostic signs. Polyps are extremely rare in the pediatric population,23 evolve from the turbinates and therefore do not emanate from the midline, do not swell with jugular compression (positive Furstenberg sign), and do not cause widening of the nasal bridge23,24 as sincipital encephaloceles do.
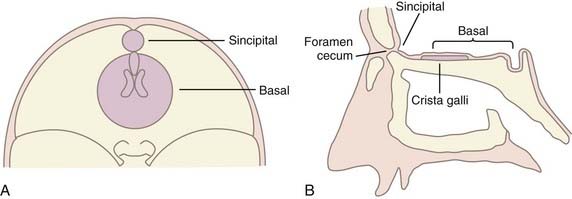
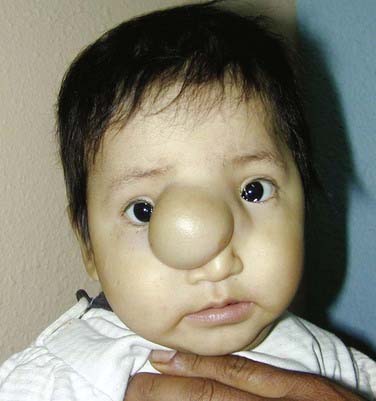
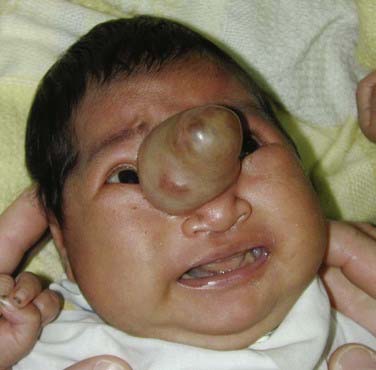
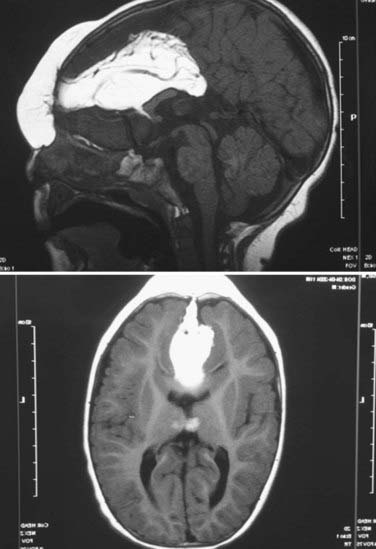
FIGURE 176-10 Magnetic resonance images of an interfrontal encephalocele with associated lipoma of the soft tissues and corpus callosum (see also Fig. 176-11).
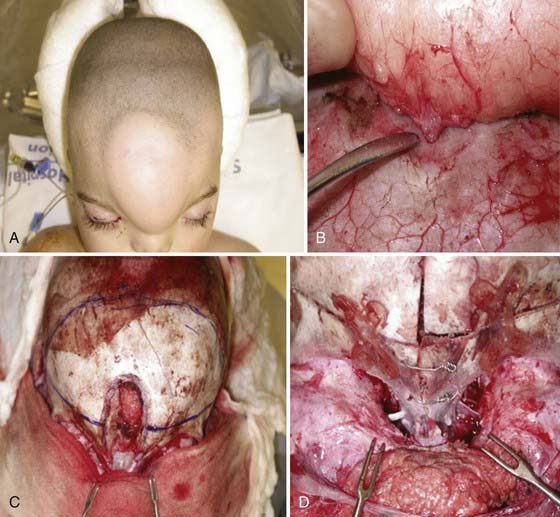
FIGURE 176-11 Operative correction of interfrontal encephalocele with an associated lipoma (see also Fig. 176-10). The patient is a 1-year-old boy with facial deformity and a large forehead lipoma (A). Widening of the nasion is evident (A and C). The soft tissue lipoma was attached by a slender stalk through a small opening in the bone (B), which was divided. Craniofacial reconstruction was then performed with remodeling of the frontal, nasal, and orbital bones. Marked telecanthus is visible in D, which was then treated by bilateral canthopexy.
Basal encephaloceles are situated further posteriorly in the anterior cranial fossa, closer to the suprasellar cistern, and are therefore more likely to contain viable neural structures such as the hypothalamus, pituitary gland and stalk, optic chiasm, and vessels of the anterior circle of Willis. These encephaloceles are also classified according to the location of the herniated stalk: transethmoidal and transsphenoidal (Fig. 176-12). Teratoma should be considered in the differential diagnosis of lesions in this region as well. The initial symptoms may be associated with nasal obstruction, but CSF rhinorrhea and repeated bouts of meningitis can also herald the presence of this skull base lesion.25
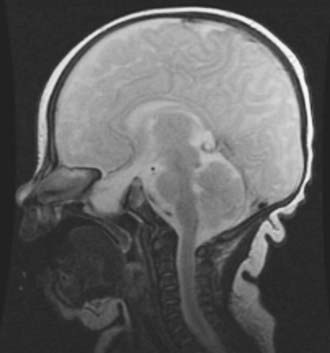
FIGURE 176-12 Magnetic resonance image of a sphenoidal encephalocele in a 3-year-old with symptoms of nasal obstruction.
(Courtesy of Neil Feldstein, M.D.)
Associated deformities with anterior encephaloceles are usually limited to the cranial region, in contrast to those associated with occipital encephaloceles. Interfrontal encephaloceles can be associated with large lipomas (see Figs. 176-10 and 176-11) in the subcutaneous tissues of the forehead and corpus callosum. Sincipital encephaloceles are accompanied by telecanthus (see Fig. 176-11D) (widening of the nasion because of displacement of the medial orbital walls) more frequently than hypertelorism (where the interpupillary distance is increased), cleft lip and palate, craniosynostosis, and ocular deformities.26
Surgical Management
A variety of protocols for timing, staging, and determining the anatomic approach to anterior encephaloceles have been proposed, depending on the surgeon’s experience and resources and the patient population being treated. As with posterior encephaloceles, the three main goals of surgery are (1) correction of the deformity, (2) prevention of CSF leaks, and (3) preservation of functional neurovascular elements. Aesthetic and naso-orbital considerations, however, require that the surgeon be trained in the management of complex craniofacial abnormalities with attention to correction of telecanthus and maintenance of the horizontal ocular axis, correction of hypertelorism when present, preservation of patent nasolacrimal ducts, and reconstruction of nasal abnormalities as necessary. In the absence of CSF leaks, these lesions, which are usually covered with skin, can be treated electively in early infancy to avoid the consequences of a craniofacial deformity that will progress in the setting of a pulsatile encephalocele. Staging is a matter of choice after weighing the risks, benefits, and resources. Analogous to the treatment of single-suture synostosis in early infancy,27 single, early operations for repair of anterior encephalocele correct the deformity caused by the encephalocele and rely on neurological and craniofacial development in the first few years of childhood to model the skeleton for optimal aesthetic results. Arguments against this approach and in favor of a staged reconstruction later in childhood include the small blood volume and danger of blood loss in an infant, interference with anterior facial growth, and re-formation of the telecanthus after early repair.28 Intracranial repair is thought to be safer than an extracranial approach because of the ability to mobilize the dural defects and their contents and achieve a watertight closure free of contamination within the nasal cavity.14,15,19,29–33 With the advent of improved optics, endoscopic instruments, techniques of dural closure, and surgical experience, a minimally invasive endoscopic approach is gaining popularity.34–37
In the open, transcranial approach, the patient is positioned supine with the head on a horseshoe headrest. I prefer the use of a lumbar drain when a complex dural repair is anticipated (large or posteriorly placed skull base defect) because this aids in brain relaxation, minimizes the effects of retraction on the immature brain, and facilitates dural healing. A zigzag bicoronal incision is carried out with meticulous attention to ensure hemostasis. A large, vascularized, pericranial flap is preserved and reflected anteriorly, followed by bifrontal craniotomy just above the floor of the anterior cranial fossa. Depending on the need for preservation of the contents of the encephalocele, the herniated structure is ligated or mobilized, the dura repaired primarily when possible, and the closure augmented with a vascularized pericranial flap. If there is a need for reconstruction of the skull base because of a large bone defect, this can be done with an autologous bone graft from the infant’s calvaria or a split-thickness graft in an older child. The nasal, facial, and orbital deformities are then treated (see Fig. 176-11C and D). In most sincipital encephaloceles, medial canthopexy is performed; in cases of hypertelorism, orbital translocation is carried out with the techniques outlined by Tessier.28 The nasolacrimal ducts must be preserved, and when blocked, they should be cannulated or dacryocystorhinostomy considered to avoid the development of dacryocystitis during the postoperative period.38 Aesthetic reconstruction of the facial soft tissues and normalization of the naso-orbital appearance are then performed by narrowing the intercanthal distance (as earlier) and contouring the frontal bone flap and nasal bones to form a more normal nasofrontal angle (see Fig. 176-11D).11
In the endoscopic technique, meticulous preoperative imaging35 and frameless stereotaxis are important adjuncts to adequate endoscopic equipment (optics, irrigation, instruments, and cautery), and they facilitate repair of the dura and skull base.35–37,39–41 The approaches are typically transnasal to obviate the need for a large coronal incision and bifrontal craniotomy and can be used to successfully treat sincipital and basal encephaloceles that are not associated with a significant craniofacial deformity.
Outcome
Although anterior encephaloceles have been associated with better outcomes than posterior encephaloceles, historically, the recent data presented earlier show that location of the encephalocele has less to do with outcome than do associated hydrocephalus and seizure disorder.20 Fortunately, these complications are less common with sincipital and basal encephaloceles than with occipital and parietal lesions. Nevertheless, superior outcomes are obtained in cases in which a better cosmetic outcome is achieved.42 Visual abnormalities and anosmia are other common long-term morbidities associated with anterior encephaloceles.32
Bui Bui CJ, Tubbs RS, Shannon CN, et al. Institutional experience with cranial vault encephaloceles. J Neurosurg. 2007;107(suppl 1):22-25.
Castelnuovo Castelnuovo P, Bignami M, Pistochini A, et al. Endoscopic endonasal management of encephaloceles in children: an eight-year experience. Int J Pediatr Otorhinolarngol. 2009;73:1132-1136.
Copp Copp AJ. Neurulation in the cranial region—normal and abnormal. J Anat. 2005;207:623-635.
Copp Copp AJ, Brook FA, Estibeiro JP, et al. The embryonic development of mammalian neural tube defects. Prog Neurobiol. 1990;35:363-403.
Lo Lo BWY, Kulkarni AV, Rutka JT, et al. Clinical predictors of developmental outcome in patients with cephaloceles. J Neurosurg Pediatr. 2008;2:254-257.
Roux Roux FE, Lauwers F, Oucheng N, et al. Treatment of frontoethmoidal meningoencephalocele in Cambodia: a low-cost procedure for developing countries. J Neurosurg. 2007;107(suppl 1):11-21.
1 Heinecke W. Die chirurgischen Krankheiten des Kopfes. Stuttgart, Billroth and Luecke’s Deutsche Chirurgie. Lief. 1882;331:224.
2 Ballantyne JW. Manual of Antenatal Pathology and Hygiene. The Embryo. Edinburgh: W. Green & Sons; 1904.
3 Emery JL, Kalhan SC. The pathology of exencephalus. Dev Med Child Neurol. 1970;12(supp 22):51-64.
4 Hedlund G. Congenital frontonasal masses: developmental anatomy, malformations, and MR imaging. Pediatr Radiol. 2006;36:647-662.
5 Suwanwela C, Suwanwela N. A morphological classification of sincipital encephalomeningoceles. J Neurosurg. 1972;36:201-211.
6 Copp AJ. Neurulation in the cranial region—normal and abnormal. J Anat. 2005;207:623-635.
7 O’Rahilly R, Muller F. Bidirectional closure of the rostral neuropore in the human embryo. Am J Anat. 1989;184:249-268.
8 Copp AJ, Brook FA, Estibeiro JP, et al. The embryonic development of mammalian neural tube defects. Prog Neurobiol. 1990;35:363-403.
9 Saint-Hilaire EG. Des adhérences de l’exterieur du foetus, considérées comme le principal fait occasionnel de la monstruosité, et observations nouvelles à l’appui de cette théorie. Archives Générales de Médecine. Journal Publié par une Société de Médecins. 1827;14:392-406.
10 Mealey J, Dzenitis AJ, Hockey AA. The prognosis of encephaloceles. J Neurosurg. 1970;32:209-218.
11 Roux FE, Lauwers F, Oucheng N, et al. Treatment of frontoethmoidal meningoencephalocele in Cambodia: a low-cost procedure for developing countries. J Neurosurg. 2007;107(suppl 1):11-21.
12 Suwanwela C. Geographical distribution of frontoethmoidal encephalomeningocele. Br J Prev Soc Med. 1972;26:193-198.
13 Thu H, Kyu H. Epidemiology of frontoethmoidal encephalocele in Burma. J Epidemiol Commun Health. 38-39, 1984.
14 Roux FE, Oucheng N, Lauwers-Cances V, et al. Seasonal variations in frontoethmoidal meningoencephalocele births in Cambodia. J Neurosurg Pediatr. 2009;4:553-556.
15 Dutta HK, Deori P. Anterior encephaloceles in children of Assamese tea workers. J Neurosurg Pediatr. 2010;5:80-84.
16 Martinez-Lage JF, Martinez Robledo A, Poza M, et al. Familial occurrence of atretic cephaloceles. Pediatr Neurosurg. 1996;25:260-264.
17 Dawe HR, Smith UM, Cullinane AR, et al. The Meckel-Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16:173-186.
18 Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565-4575.
19 Pinzer T, Lauer G, Gollogly J, et al. A complex therapy for treatment of frontoethmoidal meningoencephalocele in a developing third world country: neurosurgical aspects. J Neurosurg. 2006;104(suppl 5):326-331.
20 Lo BWY, Kulkarni AV, Rutka JT, et al. Clinical predictors of developmental outcome in patients with cephaloceles. J Neurosurg Pediatr. 2008;2:254-257.
21 Ingraham FD, Swan H. Spina bifida and cranium bifida. I. A survey of five hundred and forty-six cases. N Engl J Med. 1943;228:559-563.
22 Bui CJ, Tubbs RS, Shannon CN, et al. Institutional experience with cranial vault encephaloceles. J Neurosurg. 2007;107(suppl 1):22-25.
23 Robinson RG. Anterior encephalocele. Br J Surg. 1958;45:36-40.
24 Hoving EW. Nasal encephaloceles. Childs Nerv Syst. 2000;16:702-706.
25 Blumenfeld R, Skolnik EM. Intranasal encephaloceles. Acta Otolaryngol (Stockh). 1965;82:527-531.
26 Rapport RL, Dunn AC, Alhady F. Anterior encephalocele. J Neurosurg. 1981;54:213-219.
27 Jimenez DF, Barone CM, Cartwright CC, et al. Early management of craniosynostosis using endoscopic-assisted strip craniectomies and cranial orthotic molding therapy. Pediatrics. 2002;110:97-104.
28 Tessier P. Experience in the treatment of orbital hypertelorism. Plast Reconstr Surg. 1974;53:1-18.
29 Mahapatra AK, Suri A. Anterior encephaloceles: a study of 92 cases. Pediatr Neurosurg. 2002;36:113-118.
30 Mahapatra AK, Agrawal D. Anterior encephaloceles: a series of 103 cases over 32 years. J Clin Neurosci. 2006;13:536-539.
31 Rutka JT, Carlotti C, Iantosca M. Encephaloceles. In: Winn HR, editor. Youmans Neurological Surgery. 5th ed. Philadelphia: Saunders; 2004:3198-3213.
32 Macfarlane R, Rutka JT, Armstrong D, et al. Encephaloceles of the anterior cranial fossa: management and outcome. Pediatr Neurosurg. 1995;23:148-158.
33 Matson DD. Neurosurgery of Infancy and Childhood. Springfield, IL: Charles C Thomas; 1969.
34 Marton E, Billeci D, Schiesari E, et al. Transnasal endoscopic repair of cerebrospinal fluid fistulas and encephaloceles: surgical indications and complications. Minim Invasive Neurosurg. 2005;48:175-181.
35 Castelnuovo P, Bignami M, Pistochini A, et al. Endoscopic endonasal management of encephaloceles in children: an eight-year experience. Int J Pediatr Otorhinolarngol. 2009;73:1132-1136.
36 Nyquist GG, Anand VK, Mehra S, et al. Endoscopic endonasal repair of anterior skull base non-traumatic cerebrospinal fluid leaks, meningoceles, and encephaloceles. J Neurosurg. 2010;113:961-966.
37 Tabaee A, Anand VK, Cappabianca P, et al. Endoscopic management of spontaneous meningoencephalocele of the lateral sphenoid sinus. J Neurosurg. 2010;112:1070-1077.
38 Jimenez DF, Barone CM. Encephaloceles, meningoceles, and dermal sinuses. In: Albright AL, Pollack I, Adelson PD, et al, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:189-208.
39 Ismail AS, Costantino PD, Sen C. Transnasal transsphenoidal endoscopic repair of CSF leakage using multilayer acellular dermis. Skull Base. 2007;17:125-132.
40 Leng LZ, Brown S, Anand VK, et al. “Gasket-seal” watertight closure in minimal-access endoscopic cranial base surgery. Neurosurgery. 2008;62(5 suppl 2):ONSE342-ONSE343.
41 Shah RN, Surowitz JB, Patel MR, et al. Endoscopic pedicled nasoseptal flap reconstruction for pediatric skull base defects. Laryngoscope. 2009;119:1067-1075.
42 Pertschuk MK, Whitaker LA. Psychosocial outcome of craniofacial surgery in children. In: Marchac D, editor. Craniofacial Surgery. Berlin: Springer; 1987:486-487.






