Chapter 72 Emerging Therapies for Spinal Cord Injury
The reversal of paralysis following spinal cord injury (SCI) represents one of the greatest challenges in all of neuroscience. Despite significant improvements in the early medical and surgical management of SCI, along with a greatly improved understanding of SCI pathophysiology, there remains no effective treatment to improve neurologic outcomes following SCI. SCI destroys neuronal connectivity by severing descending motor and ascending sensory axonal pathways. Such damage often results in permanent paralysis and loss of sensation below the level of injury. Although SCI causes loss of neurons and glial cells at the lesion site, functional deficits result primarily from loss of white matter axons by direct trauma and from progressive damage to initially intact axons by complex secondary injury mechanisms.1 To date, numerous potential therapies have been investigated in prospective, randomized, controlled clinical trials, yet all have failed to demonstrate neurologic benefit despite evidence that these agents demonstrated benefit in preclinical animal studies of SCI. This chapter will review previous clinical trials as well as major recent preclinical advances, in pharmacologic and cell-based therapeutic approaches to SCI.
Spinal Cord Injury Epidemiology
SCI is associated with severe physical, psychological, social, and economic burdens on patients and their families. With an annual incidence rate of 15 to 40 persons per million, it has been estimated that at least 10,000 North Americans will suffer an SCI each year.2 With the average acute-care and rehabilitation charges of approximately $60,000 for each of these phases of care, the societal expense associated with the medical management, as well as lost earnings of this disorder over the course of one’s life, is significant.3 It has been estimated that the lifetime cost of medical care and other injury-related expenses for a 25-year-old patient with SCI who suffers high cervical quadriplegia is approximately $3 million.4 SCI occurs predominantly in young, otherwise healthy individuals, with injury occurring with the greatest frequency in those between 15 and 25 years of age; the male-to-female ratio for SCI is approximately 4 to 1.5 Common causes of SCI in the United States are motor vehicle accidents (50%), falls and work-related injuries (30%), violent crime (11%), and sports-related injuries (9%).6 Injuries most commonly occur in the cervical spine and are associated with the most devastating neurologic impairments (e.g., quadriplegia). A recent report from the U.S. National Spinal Cord Injury Database found that 56% of all SCI cases occur in the cervical spine.
Pathophysiology of Spinal Cord Injury
A detailed understanding of the pathophysiologic processes that occur following SCI is paramount to the development of effective therapies for SCI. The pathophysiology of SCI is best described as biphasic, consisting of a primary and a secondary phase of injury.7 The primary phase involves the initial, immediate mechanical injury during which failure of the spinal column occurs, and includes compression, contusion, shear, or laceration due to penetrating injury and acute stretching of the spinal cord as a result of vertebral distraction or sudden acceleration-deceleration of the spinal column. The most common underlying primary injury mechanism results from the acute compression and contusion of the spinal cord due to bone or disc displacement within the spinal column during fracture-dislocation or burst fracture of the spine.2 The primary mechanical trauma to the spinal cord, along with subsequent persistent compression, triggers a complex and delayed pathologic cascade, termed the secondary injury phase, which involves vascular dysfunction, edema, ischemia, excitotoxicity, electrolyte shifts, free radical production, inflammation, and delayed apoptotic cell death. Although neurologic deficits are present immediately following the initial injury, the secondary injury phase results in a protracted period of neuroinflammation and tissue destruction. The spatial extent of secondary injury events spreads both radially and longitudinally along the spinal cord in a rostral-to-caudal manner, resulting in neuronal and glial cell death. The end result is cavitation of central gray matter along with partial or complete loss of adjacent white matter tracts.
The pathophysiology of SCI has been extensively studied in many experimental animal models, including mice, rats, cats, and primates, in order to understand the cellular and molecular mechanisms of tissue damage and the consequent disability.2 Traumatic SCI initiates a series of destructive cellular inflammatory processes which accentuate tissue damage at and beyond the original site of trauma, and cystic cavitation inevitably occurs due to these secondary injury events. Contusion SCI in animals produces a predictable pattern of progressive injury resulting in neuronal and glial cell death, vascular injury, axonal destruction, and demyelination that is analogous to the pattern in human spinal cord contusion injury, the most common type of the human SCI (Fig. 72-1).7,8 The progressive expansion of the injury from gray to white matter causes secondary damage to initially intact axons within hours to several weeks after injury. The cellular inflammatory response, predominated by macrophages, has been implicated as the primary mediator of progressive secondary injury. The major targets for spinal cord repair include severed and/or demyelinated axons, inflammatory cells and proinflammatory cytokines, and glial scar components.
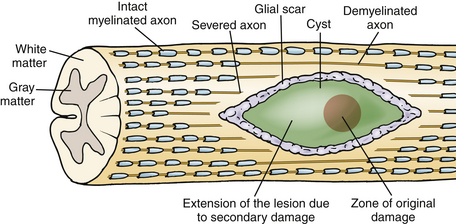
Emerging Preclinical Spinal Cord Injuries Therapies
The failure of axonal regeneration after CNS injury is the result of two distinct processes: the limited intrinsic regenerative potential of CNS neurons and the inhibitory extrinsic environment of the injured CNS. However, CNS neurons may regenerate when provided with a “permissive” substrate that promotes axonal growth. The ability of CNS neurons to regenerate over long distances after axotomy was first established through pioneering work in which injured CNS axons were found to regenerate into and through growth-permissive peripheral nerve (PN) grafts.9 The therapeutic potential of PN grafts for use in SCI was then demonstrated by Cheng et al. in 1996,10 who reported that transplantation of multiple PN grafts stabilized with fibrin glue containing acidic fibroblast growth factor into a rat spinal cord transection model promoted hindlimb functional recovery.
Experimental therapies for SCI can be grouped into several subtypes, based on their general mechanism of action. These general treatment subcategories include therapies that (1) promote neuroprotection, (2) stimulate intrinsic axonal regrowth, (3) enhance remyelination, and (4) remove or block inhibitory molecules within damaged myelin and within the astroglial scar. To date, researchers have identified and characterized a number of molecular signals associated with axonal growth following injury and have developed drug therapies that are capable of stimulating axonal regeneration. Several regeneration-associated genes have been found to be up-regulated after axonal injury, including L1, c-fos, and c-jun, and the 43-kD growth-associated protein (GAP43). Despite these findings, the degree and extent of their up-regulation is apparently insufficient to promote a strong regenerative response in the CNS. Numerous reports have implicated a decrease in the levels of intracellular cAMP after CNS injury as a major cause of intrinsic regenerative failure. The elevation of intracellular cAMP levels by cAMP analogues and/or the phosphodiesterase inhibitor, rolipram, has been shown to increase axonal sprouting and reduce the effects of myelin-associated inhibitors.11,12
Multiple inhibitory molecules exist that make the injured CNS a nonpermissive environment for axonal growth. These molecules may be categorically divided into myelin-associated inhibitors and inhibitors associated with the astrocytic glial scar. Recently, major advances in molecular neuroscience have led to the identification of several growth-inhibitory molecules associated with myelin (Nogo, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein) and the glial scar (chondroitin sulfate proteoglycans, CSPGs), as well as their receptors and downstream signaling pathways, which make up the inhibitory extrinsic environment of the injured CNS. This chapter will review several recently developed targeted molecular strategies that enable axons to overcome inhibitory influences of myelin-associated proteins and astroglial scar components.
Considering the multifactorial nature of secondary pathologic events attributed to SCI, drug cocktails with multifaceted modes of neuroprotection and axonal growth promotion will likely be useful in preventing or limiting secondary injury progression. Because of the numerous complex pathophysiologic changes that occur after SCI, successful repair may involve strategies that combine the use of neuroprotective drug therapies with those that promote axonal regeneration, such as cell transplantation, genetic engineering to increase neurotrophic factors, neutralization of inhibitory factors, limiting glial scar formation, and neurorehabilitation. Furthermore, neuroprotective drug therapies that function at later time points following SCI will likely have greater clinical relevance than those with small therapeutic windows. There is also a requirement to establish methods for the systematic evaluation of preclinical therapeutic outcomes from the various research SCI centers. In the evaluation of future potential SCI therapies, a clinical basis for the implementation of novel therapies will be required. Refined clinical outcome measures as well as neurophysiologic measures are needed for a precise qualitative and quantitative assessment of spinal cord function in SCI patients, especially at an early stage after injury. This will allow for better detection of improvements in functional recovery and will enable clinicians to precisely monitor the effects of novel therapies.3,13
Neuroprotective Strategies
Methylprednisolone
The most extensively investigated pharmacologic intervention for SCI is the glucocorticoid methylprednisolone sodium succinate (MPSS, Solu-Medrol). To date, three major clinical trials evaluating the use of MPSS in acute SCI have been performed. The National Acute Spinal Cord Injury Study (NASCIS I) compared the efficacy of administering an intravenous (IV) loading dose of MPSS (100 or 1000 mg) and a dose of either 25 or 250 mg every 6 hours thereafter for 10 days to SCI patients who had motor or sensory deficits below the level of injury.14 This study demonstrated that higher-dose MPSS was no more effective than the lower dose in reducing neurologic deficits between the two groups at 6 weeks, 6 months, and 1 year after injury. Although a placebo group was not included in the NASCIS I because of ethical concerns, the use of MPSS became much less prevalent after this trial.
Subsequent animal studies demonstrated that the effective neuroprotective dose of MPSS was much higher than that used in NASCIS I.15 Therefore, following the NASCIS I study, the NASCIS II trial was conducted to compare the efficacy of high-dose MPSS administered as a 30-mg/kg bolus over the first hour, followed by an infusion of 5.4 mg/kg/hour over the next 23 hours, to that of placebo and that of the opioid receptor antagonist naloxone.16,17 The trial was also designed to examine the effects of different timing schedules of drug administration following SCI. An analysis of all patients entered into the trial failed to demonstrate a significant difference in motor and sensory function among the treatments. However, upon stratification of the data on the basis of time to loading dose (<8 hours or >8 hours from SCI) and adjustment for the severity of SCI (complete vs. incomplete), analysis of the results from the NASCIS II study at the 6-week and 6-month follow-up revealed that patients who were treated with MPSS within the first 8 hours of injury had significantly improved motor and sensory function, compared to patients who received placebo, naloxone, or MPSS at later times. Furthermore, these differences remained significant 1 year after injury.18 None of the differences in patients taking naloxone or in patients treated with MPSS more than 8 hours after SCI were statistically significant.
Despite the beneficial therapeutic effects demonstrated with MPSS treatment, the results from the NASCIS II trial have not been universally accepted.19,20 Concerns about the small sample population size for the groups showing beneficial effects, nonstandardized performance of medical and surgical protocols by participating centers, and lack of defined functional outcome measures to assess whether the improvements seen with MPSS treatments were correlated with clinical significance have reduced enthusiasm for the indication of MPSS for acute SCI. Additional criticisms remained because of concerns about the adverse effects of steroids and the small therapeutic effects that were noted over the short (6-month) follow-up period used as the basis for conclusions about the trial, as well as the differential loss to follow-up between MPSS-treated and untreated patients. The NASCIS III trial resolved some of these issues with the inclusion of a functional independence measure.21 All patients in this study received an initial MPSS bolus of 30 mg/kg before being randomized to one of three treatment arms: (1) MPSS infusion of 5.4 mg/kg/hour over 24 hours, (2) MPSS infusion of 5.4 mg/kg/hour over 48 hours, or (3) tirilizad mesylate (TM, a lazaroid with inhibitory effects on lipid peroxidation without glucocorticoid side effects) 2.5-mg/kg bolus every 6 hours for 48 hours. The results from this third study concluded that no benefit was associated with extending MPSS treatment beyond 24 hours if MPSS had been administered within the first 3 hours of SCI, while patients who started MPSS therapy at 3 to 8 hours demonstrated improvements in motor capabilities if drug infusion was continued for 48 hours in comparison to 24 hours of infusion.
Research on the neuroprotective mechanism of MPSS in experimental SCI models has demonstrated that in addition to possessing antioxidant properties, MPSS reduces tumor necrosis factor–α (TNF-α) protein synthesis and nuclear factor kappa B (NF-κB) activity, effects that may account for the anti-inflammatory actions exerted by MPSS.22 However, MPSS treatment is not without adverse effects; high-dose MPSS has been associated with an increased prevalence of wound infections, pneumonia, sepsis, and death due to respiratory complications.16,21 Furthermore, the anti-inflammatory actions of high-dose MPSS may also have detrimental effects on neuronal regeneration and axonal sprouting while exacerbating postischemic necrosis. These adverse effects may explain the ineffectiveness of MPSS treatment started beyond 8 hours post-SCI observed in the NASCIS II study. The guidelines committee of the American Association of Neurological Surgeons and Congress of Neurological Surgeons Joint Section on Disorders of the Spine and Peripheral Nerves, upon reviewing the evidence regarding the use of MPSS in the treatment of acute SCI in adults, concluded that its use could be supported only at the level of a treatment option (Joint Section on Disorders of the Spine and Peripheral Nerves of the AANS/CNS, 2002). However, criticisms directed toward the NASCIS II and III trials must be balanced by the current lack of alternative neuroprotective strategies for acute SCI. Therefore, MPSS administration may remain justified for acute SCI (within 8 hours) in nondiabetic and nonimmunocompromised patients, given the severity of SCI deficits and current lack of alternatives. Although many neurosurgeons continue to administer steroids, the rationale for this administration appears to have changed over recent years, according to a survey published in 2006, which revealed that the majority of respondents continue to administer methylprednisolone but that this is motivated predominantly by fear of litigation.23
21-Aminosteroids
The 21-aminosteroids (lazaroids) are synthetic glucocorticoid analogues that are capable of inhibiting lipid peroxidation without activating glucocorticoid receptors. The 21-aminosteroid TM utilizes three neuroprotective mechanisms: antioxidation, preservation of endogenous vitamin E, and membrane stabilization through the inhibition of lipid peroxidation.15 TM was administered in comparison to MPSS in the NASCIS III clinical trial and was shown to be as effective as the 24-hour MPSS regimen.21 However, because TM was not shown to have superior efficacy in comparison to MPSS, and given the absence of proven benefit over placebo controls, 21-aminosteroids have not been adopted for neuroprotection in clinical SCI.
GM-1 Ganglioside
The gangliosides are a group of sialic acid–containing glycosphingolipids located in high concentrations in the outer membranes of nervous tissue. A small randomized, placebo-controlled, double-blind trial, the Maryland monosialotetrahexosylganglioside (GM-1) Ganglioside Study, was conducted to investigate the efficacy of GM-1 in patients with cervical and thoracic SCIs.24 The test drug protocol consisted of either 100 mg of GM-1 or placebo control administered once daily via IV infusion for a total of 18 to 32 doses, the first dose beginning within 72 hours of the onset of SCI. Results from this study demonstrated that GM-1 improved the motor and sensory recovery of lower extremities only, suggesting that GM-1 had an ability to enhance the function of axons traversing the site of injury but had no effect on the gray matter at the level of trauma. Furthermore, unlike the dosing regimen of MPSS, which appears to be effective when initiated within the first 8 hours of SCI, GM-1 demonstrated neuroprotective effects following initiation of treatment 48 hours post-SCI. The results from this study led to the initiation of a larger clinical trial comparing the effects of low- and high-dose GM-1 to placebo.
The randomized double-blind Sygen Multicenter Acute Spinal Cord Injury Study was completed in the late 1990s and reported in 2001.25 In this landmark clinical trial, all 797 patients received a 30-mg/kg bolus of MPSS, followed by a 5.4-mg/kg/hour infusion of MPSS for 23 hours initiated within the first 8 hours following SCI, as the standard of care based on the initial report from the NASCIS II. Placebo, low-dose GM-1 (300 mg loading dose followed by 100 mg/day for 56 days), or high-dose GM-1 (600 mg loading dose followed by 200 mg/day for 56 days) was then randomly assigned following the completion of corticosteroid administration to avoid unwanted drug interactions. Recovery patterns were measured by using the American Spinal Injury Association (ASIA) and modified Benzel classification neurologic examination scales.26 The primary efficacy assessment assigned to this trial was termed marked recovery, defined as the proportion of patients who demonstrated a two-grade improvement from baseline examination. Although the primary outcome for this trial was negative, patients who received either low- or high-dose Sygen showed an accelerated motor recovery over the first 3 months postinjury, regardless of their initial severity. There was also a trend toward improved bowel/bladder function, sacral sensation, and anal contraction.26 Interestingly, the placebo group of this trial failed to confirm the neurologic improvements noted with MPSS for the NASCIS II trial. To our knowledge, no future clinical trials with Sygen are currently planned.
Opioid Antagonists
Following SCI, the release of the endogenous opioid peptide dynorphin A is increased at the injury site along with kappa opioid receptor binding capacity. Although dynorphin A and other related opiates have well-defined roles as inhibitory neurotransmitters in pain reduction, evidence also exists to support the hypothesis that exposure to high concentrations of dynorphins can induce hyperalgesia and allodynia and may contribute to neurodegeneration. Moreover, sustained exposure to dynorphin-derived peptides is neurotoxic, and activation of the kappa-opioid receptor subtype has been associated with reduced spinal cord blood flow, which may contribute to secondary injury after SCI.27 The use of opioid antagonists for treatment of SCI is controversial. While the nonselective opioid receptor antagonist naloxone has demonstrated an ability to improve spinal cord conduction, reduce edema, and decrease allodynia in animal models of SCI, results from the NASCIS II trial showed that naloxone given as an IV bolus of 5.4 mg/kg followed by an infusion of 4 mg/kg for 23 hours failed to provide benefit.16 However, further analysis of the effects of timing for initiating naloxone treatment in patients with incomplete SCI showed that naloxone dosing within the first 8 hours of SCI resulted in significant motor recovery below the level of injury.28 Therefore, further refinement of naloxone-dosing regimens in experimental SCI models may be beneficial.
Thyrotropin-Releasing Hormone
TRH is a tripeptide (Glu-His-Pro) that possesses numerous physiologic actions, in addition to its well-characterized actions on the pituitary gland. TRH and its analogues are capable of antagonizing the effects of endogenous opioids, platelet-activating factor, peptidoleukotrienes, and excitatory amino acids, all of which have been implicated in the biochemical events of secondary SCI.29 In response to the overwhelming therapeutic benefit demonstrated by TRH in animal models of SCI, a small clinical trial consisting of 20 patients was designed to assess the safety and potential efficacy of TRH in patients with complete and incomplete SCI.30 The patients were randomized on the basis of the severity of their SCI (complete or incomplete) in a double-blinded fashion to receive either a 0.2-mg/kg bolus of TRH followed by a 0.2-mg/kg/hour infusion of TRH over 6 hours or an equal volume of saline vehicle placebo. The results of the 4-month follow-up revealed statistically significant improvements in neurologic and sensory function. However, this study was hampered by the small patient population that was included in the study and the high variability of neurologic scores obtained within the placebo group. Results for complete SCI patients treated with TRH were negative. Although 1-year data were available, a relatively high number of patients were lost to follow-up, and the data were deemed inconclusive. Considering the amount of evidence in support of neuroprotective effects generated by TRH and TRH analogues from previous preclinical studies and the results obtained from this small clinical trial, larger and more extensive trials with TRH may be beneficial.
Glutamate Receptor Antagonists
The class of glutamate receptor antagonists readily crosses the blood–spinal cord barrier following systemic administration. However, given the fact that glutamate is a key neurotransmitter possessing a variety of physiologic functions, blockade of glutamatergic synaptic transmission results in unwanted psychomimetic effects. Gacyclidine, a potent and specific phencyclidine analogue noncompetitive N-methyl D-aspartate receptor antagonist, was previously shown to provide neuroprotection through improvement of the functional, histologic, and electrophysiologic status of the injured rat spinal cord following experimental contusion with an absence of dose-related adverse effects.31 These findings prompted initiation of a randomized double-blind phase II clinical trial with more than 200 patients recruited to test the efficacy of 0.005, 0.01, and 0.2 mg/kg gacyclidine administered via IV infusion within 2 hours of SCI and with a second dose given within the next 4 hours.32 The subjects of this trial were divided into four strata based on the level of SCI (cervical or thoracic) and the severity of injury (complete or incomplete). The study results, reported for the 1-month follow-up, demonstrated improved overall ASIA scores. However, results for the follow-up at 12 months posttreatment failed to support long-term benefit on neurologic scores. Despite evidence of a trend for improved motor function in the incomplete cervical strata receiving higher gacyclidine dosing, further development of this drug for SCI has been halted.
Calcium Channel Blockade
Calcium influx is believed to be an important mediator of excitotoxic intracellular damage and vasospasm-induced ischemia. Consequently, Ca2+-channel blockers are prescribed in the treatment of systemic hypertension, coronary artery disease, stroke, cardiac arrhythmias, and vasospasm. Vasospasm and the release of endogenous vasoactive amines have been suggested as important contributing factors to the pathobiology of SCI.2 The dihydropyridine-sensitive (L-type) Ca2+-channel blocke nimodipine represents the most widely investigated therapy for vasospasm. In a rat model of severe spinal cord compression, nimodipine, alone or in combination with dextran, was examined for effects on spinal cord blood flow and spinal cord axonal function.33 The results of this study demonstrated that the administration of nimodipine alone was associated with systemic hypotension due to vasodilation. However, the combination of nimodipine with dextran resulted in increased cord blood flow within the lesion epicenter and improved axonal function in the motor and somatosensory tracts of the cord.
In 1996, a randomized clinical trial of 100 acute SCI patients was initiated, comparing the safety, efficacy, and neurologic outcome following administration of nimodipine (0.015 mg/kg/hour over 2 hours followed by 0.03 mg/kg/hour for 7 days), MPSS (NASCIS II dosing regimen), both treatments, or placebo control.34 Results from the 1-year follow-up demonstrated no neurologic benefit beyond the natural course of recovery within the placebo group. The authors also examined the results based on the timing of surgical spinal decompression and stabilization and found no correlation with recovery of neurologic function and the timing of surgery, although the patients were not randomized on the basis of timing of surgery. Although the data from this SCI clinical trial were not suggestive of nimodipine-mediated therapeutic effects, the conclusions made by the authors were susceptible to type II errors owing to the small patient population.
Sodium Channel Blockade
After SCI, there is a deleterious accumulation of intracellular sodium.35 Injury causes failure of the Na+/K+-ATPase and accumulation of axoplasmic sodium through noninactivating Na+ channels, which, together with membrane depolarization, promotes reverse Na-Ca exchange and axonal calcium overload. Thus, administration of sodium channel blockers may prevent the ensuing calcium-induced cell injury or death. Pharmacologic antagonism of voltage-gated Na+ channels has been demonstrated to prevent axonal degeneration, preserve the function of injured spinal cord white matter tracts, and reduce damage to myelin.36 For example, the focal administration of tetrodotoxin directly into the contused rodent spinal cord significantly reduced axonal loss and axoplasmic pathology, as compared to vehicle-treated animals.36 The benzothiazole anticonvulsant Na+ channel blocker riluzole is neuroprotective and promotes functional neurologic recovery following SCI in rodents.37 Riluzole exerts neuroprotective properties in the injured spinal cord following systemic administration by sparing gray and white matter rostral-to-caudal to the injury epicenter,37 a property that appears to be due to its ability to decrease the levels of intracellular sodium and calcium. The use of riluzole as a therapy for SCI is potentially feasible, as it has already received approval from the Food and Drug Administration (FDA) for treatment of amyotrophic lateral sclerosis. Furthermore, riluzole has been used as an adjunctive therapy in combination with MPSS.38 However, despite the current in vivo evidence in support of riluzole as a therapeutic drug for SCI, efforts to proceed with clinical testing of this drug are lacking.
Potassium Channel Blockade
Demyelination of intact and injured axons is a prominent feature of SCI. Axonal conduction deficits that arise from demyelination appear to contribute to the neurologic outcome following SCI, although the underlying mechanisms remain unclear.39,40 It has been suggested that focal demyelination of intact axons, along with altered activity of ion channels, plays an important role in the loss of axonal conduction.41 The exposure of K+ channels as a result of demyelination results in a reduced safety factor of action potential propagation (i.e., the ratio of action current generated by an impulse to the minimum amount of action current needed to maintain conduction) across the demyelinated region of the axon. Under normal physiologic conditions, myelinated axons have a high density of voltage-gated Na+ channels at the nodes of Ranvier, while rapidly inactivating voltage-gated K+ channels are clustered at the paranodal and internodal regions beneath the myelin sheath. As such, these K+ channels normally do not play a prominent role in repolarization of the action potential, as their outward current is constrained by myelin. However, when demyelination occurs, there is no myelin to shield the capacitance of the internode, and shunting of the action current occurs due to these fast voltage-gated K+ channels. Subsequently, the action potential progressively declines, resulting in conduction failure, marked slowing of the conduction velocity, and/or the inability to sustain repetitive discharges.42
Experimental studies using K+ channel blockers on animal nerve preparations have provided the rationale for undertaking clinical trials to assess the safety and efficacy of 4-AP, a blocker of rapidly inactivating voltage-gated K+ channels, as a therapy in SCI patients. Electrophysiologic studies have shown that 4-AP is capable of restoring conduction in focally demyelinated axons, enhancing synaptic transmission in many types of neurons, and potentiating muscle contraction.42 Several preclinical trials of intravenously administered 4-AP have demonstrated transient improvements in neurologic function in patients with chronic SCI.43–45 To date, three multicenter phase II randomized clinical trials of fampridine-SR, a sustained-release oral form of the K+ channel-blocking compound 4-AP, have been conducted with SCI patients. The first of these studies was a randomized crossover study, which demonstrated that fampridine-SR improved spasticity (modified Ashworth scores), ASIA motor and sensory scores, erectile dysfunction, and bowel function compared to placebo.46 In another crossover trial involving 60 patients, there was a nonsignificant trend toward reduced erectile dysfunction and decreased spasticity.47 A double-blind, randomized, placebo-controlled, parallel-group phase II clinical trial was conducted at 11 academic rehabilitation research centers in the United States to assess the safety and efficacy of fampridine-SR in subjects with chronic SCI.48 The study enrolled 91 patients with motor-incomplete SCI, randomized to three arms: fampridine-SR 25 mg twice daily (group I), fampridine-SR 40 mg twice daily (group II), and placebo (group III) for 8 weeks. Outcome measures included ASIA and Ashworth scores, bladder and bowel management questionnaires, and Subject Global Impression (SGI). A higher discontinuation rate was seen in group II patients, compared to group I and group III patients, due to more frequent adverse side effects such as hypertonia, generalized spasm, insomnia, dizziness, pain, constipation, headache, and seizures. Overall, group I patients showed significant improvement in SGI, and subgroup analysis showed improvement in spasticity in the lower-dose fampridine-SR group compared to the placebo group.48
Erythropoietin
Erythropoietin (EPO) is a 34-kD hematopoietic glycoprotein that binds to its receptor (EPOR) to induce signals promoting survival, differentiation, and proliferation of erythroid progenitor cells.49 The expression of EPO and EPOR is widely distributed within the developing and adult human brain and spinal cord, and is up-regulated in the adult brain after injury. Peripherally administered EPO crosses the blood-brain barrier, stimulates neurogenesis and neuronal differentiation, and activates neurotrophic, antiapoptotic, antioxidant, and anti-inflammatory signaling pathways.50 EPO is the only hematopoietic growth factor whose production is regulated by hypoxia, in which low oxygen tension activates hypoxia-inducing factor-1 to up-regulate EPO gene transcription.
Endogenous and exogenously administered EPO, and EPOR, have been reported to play important roles in SCI.51 EPO expression is up-regulated after SCI as part of the physiologic response to hypoxia. EPO has been shown to be neuroprotective in vitro and is capable of protecting neuronal cells from hypoxia-induced apoptosis and from excitotoxic cell death.52 The administration of exogenous recombinant human EPO (rhEPO) has been reported to produce substantial neuroprotection in animal models of SCI, spinal nerve root crush injury, transient spinal cord ischemia, and spinal cord inflammation in experimental autoimmune encephalitis. Although the mechanisms by which EPO exhibits its neuroprotective effects are not fully understood, EPO is capable of preventing apoptosis, reducing inflammation, and restoring vascular integrity. The preventive effects of EPO on neuronal apoptosis have also been demonstrated in a spinal cord compression model in rats, in which EPO administration results in inhibition of caspase-1 and caspase-3 and induction of survival proteins such as Bcl-xL. EPO administration also results in a reduction in neutrophil infiltration after SCI and has been shown to delay the postinjury increase in TNF-α, decrease interleukin-6 (IL-6) levels, and reduce apoptotic cell death.53–55 EPO has also been shown to prevent endothelial cell apoptosis, stimulate mitogenesis, and promote angiogenesis by restricting vascular endothelial growth factor–induced permeability and strengthening endothelial tight junctions. In addition to the anti-inflammatory properties of rhEPO, the inhibition of lipid peroxidation may contribute to its neuroprotective effects.56 Results of studies in animal models suggest that treatment with rhEPO may be beneficial after SCI, even when rhEPO was administered up to 24 hours after the initial injury. The delivery of rhEPO appears to protect ventral spinal cord motor neurons in an ischemic injury model in rabbits.57 Although a study of rat thoracic spinal cord contusion and clip compression injury reported substantial tissue sparing and recovery in locomotor function,55 an independent SCI research group failed to reproduce similar results, as delivery of rhEPO in the same injury paradigm failed to decrease secondary injury and cystic cavitation or to improve locomotor function.58
In a recent phase II clinical trial, intravenously administered rhEPO was shown to be safe and demonstrated a strong trend to reduce infarct size and improve clinical outcome in stroke patients.59 Although there has been great interest in conducting an SCI clinical trial with rhEPO, given the significance of initial preclinical results following experimental SCI, the use of rhEPO in clinical trials of human SCI deserves caution, and requires further investigation before implementing clinical trials. A major concern of EPO is the inadvertent and unwanted stimulation of hematopoietic activity, increasing the risk for thrombosis. Several EPO analogues have been developed in attempts to address this concern. The administration of the short-lived asialo-erythropoietin, in which sialic acid residues have been removed, has been shown to be neuroprotective in animal models of stroke, SCI, and peripheral neuropathy without causing erythrocytosis.60 In addition, asialo-erythropoietin was as effective as rhEPO in normalizing motor function after experimental SCI using a clip compression model. Other EPO analogues such as carbamylated EPO do not bind to the EPO receptor, a property that confers a loss of hematopoietic activity.61 Carbamylated EPO is capable of maintaining its associated neuroprotective properties, resulting in reduced neurologic deficit in comparison to saline or EPO in a chronic rodent model of SCI, and remained effective even when treatment was delayed for 24 hours.61
Cyclosporin-A
Agents that suppress the systemic immune response have also demonstrated beneficial effects within the injured spinal cord. Cyclosporin-A (CsA) is a potent immunosuppressive drug that is capable of inhibiting mitochondrial permeability transition, limiting constitutive and inducible nitric oxide synthase activity and expression (thereby diminishing free radical production), and reducing lipid peroxidation.62 For example, a systemic low dose (2.5 mg/kg) of CsA was shown to reduce lipid peroxidation when administered within the first 6 hours of SCI in rats.63 The delayed (2, 6, and 12 hours post-SCI) intraperitoneal injection of CsA was shown to inhibit inducible nitric oxide synthase activity, presumably via inhibition of the calcium-dependent calcineurin.64 Comparison of the efficacy of low-dose CsA alone and in combination with high-dose (30 mg/kg) MPSS demonstrated that CsA alone was superior to MPSS following SCI, as CsA was able to reduce lipid peroxidation to the same extent as that mediated by high-dose MPSS while yielding greater survival.65 Furthermore, CsA was demonstrated to inhibit autoimmune-mediated demyelination and neuronal cell death and resulted in improved motor outcome following SCI.66 However, conflicting experimental results have been obtained in various laboratories examining the potential beneficial effects of CsA after SCI. Using a stereologic method to assess lesion volume, Rabchevsky et al.67 reported that CsA treatment 15 minutes after a moderate experimental contusion SCI failed to alter the amount of spared white matter and did not improve locomotor recovery. However, when CsA was administered 4 days after moderate contusion SCI injury, motor functional recovery was observed in CsA-treated animals 3 weeks postinjury, although no significant difference in lesion volume was observed between CsA-treated and control groups.68
A nonimmunosuppressive CsA derivative, termed NIM811, also inhibits the mitochondrial permeability transition pore and is significantly less cytotoxic than CsA is. The effects of NIM811 on apoptosis, lesion size, and tissue sparing have been examined following contusion SCI and oral administration of either 20 mg/kg NIM811 or vehicle 15 minutes postinjury. NIM811 reduced apoptosis during the first 24 hours following SCI and reduced the lesion volume and enhanced the degree of spared gray and white matter at 7 days postinjury.69 Together, these findings support the need for continued experimental investigation of CsA and its derivatives as potential neuroprotective therapies in animal models of SCI.
Minocycline
Minocycline, a highly lipophilic semisynthetic derivative of tetracycline, is capable of crossing the blood-brain barrier. Minocycline possesses anti-inflammatory properties, which are distinct from its ability to inhibit bacterial protein synthesis. Minocycline can inhibit excitotoxicity, oxidative stress, caspase-dependent and caspase-independent pathways of neuronal death, and proinflammatory mediators released by activated microglia.70 The anti-inflammatory properties of minocycline include reduction in the expression or activity of inflammatory cytokines, free radicals, and matrix metalloproteinases.71
Neuroprotection by minocycline has been demonstrated in animal models of SCI. In these studies, systemically administered minocycline demonstrated convincing neuroprotective ability by decreasing apoptosis of oligodendrocytes, diminishing microglial cell activation, reducing lesion size, and improving neurologic deficit.70,71 In addition, minocycline was shown to be a superior therapeutic agent to MPSS and maintained neuroprotective efficacy when administered 1 hour following experimental SCI.71 Minocycline has been shown to improve functional recovery after clip compression SCI in mice and after contusion SCI in rats, in which it inhibits release of cytochrome c from mitochondria.70–73 After intraperitoneal administration of minocycline in a rat contusion model of SCI, minocycline exerts neuroprotective and anti-inflammatory effects, leading to a reduction in caspase-3 activation, reduced neuronal apoptosis, and improved recovery early after SCI.74 The proposed mechanisms of minocycline-mediated neuroprotection within the injured spinal cord also include increased mRNA levels of the anti-inflammatory cytokine IL-10 and decreased TNF-α production.70 Although positive effects with minocycline have been reported in several animal models of injury with different drug administration schemes, an independent replication of the study by Lee et al.72 using minocycline after experimental contusion SCI did not lead to significant functional or histopathologic improvements.75 Although minocycline has been demonstrated to have a good safety profile with prolonged use in humans, the use of minocycline following contusive SCI may require further experimental investigation before clinical trials are implemented.
Despite mixed results of minocycline in SCI animal models, a recent double-blind randomized controlled pilot study was initiated in SCI patients presenting within 12 hours after nonpenetrating injury.76 Patients were randomized to receive IV minocycline 200 mg twice daily, IV minocycline 400 mg twice daily after an 800-mg loading dose, or placebo for 7 days. There were no adverse treatment-related effects. Minocycline treatment improved ASIA motor scores in complete and incomplete cervical SCI patients through 1 year and improved the Functional Independence Measure and SF-36 scores.76 However, no differences in ASIA motor scores were observed for patients with thoracic complete SCI. On the basis of these results, large-scale clinical trials with minocycline may be anticipated.
Early Surgical Decompression
There is substantial experimental evidence that persistent compression of the spinal cord is a potentially reversible form of secondary injury.7 The severity of SCI in experimental models appears to vary depending on several factors, including the force of compression, duration of compression, spinal cord displacement, impulse, and kinetic energy. The severity of the pathologic changes and the degree of recovery appear to be directly related to the duration of acute compression, as demonstrated by experimental studies in which longer compression times produced less demonstrable clinical recovery. Furthermore, experimental studies of spinal cord decompression performed after SCI, using both kinetic and static compression models, have demonstrated that neurologic recovery is enhanced by early decompressive surgery.77 However, it is difficult to determine a time window for the effective application of surgical decompressive intervention in the clinical setting from these animal models. Results of studies on secondary injury mechanisms suggest that early intervention within hours after SCI is critical to attain a neuroprotective effect, but whether the same time window applies to surgical treatment remains unclear.
Clinical studies assessing surgical decompression for SCI have not provided convincing evidence that surgical decompression influences patients’ neurologic outcome after SCI or a clear consensus as to the appropriate timing of surgical intervention. Although suggestive evidence is presented in these studies that early decompressive surgery in selected patients may enhance neurologic recovery, most studies were uncontrolled, and any beneficial effects must be considered in the context of spontaneous recovery, which can occur in nonoperatively managed patients with SCI. Several retrospective studies have shown improvement in neurologic function after delayed decompressive surgery in patients with cervical or thoracolumbar SCI whose recovery has plateaued, and there have been reports documenting recovery of neurologic function after performance of delayed decompressive surgery months to years postinjury.78 Taken together, these studies have suggested that ongoing spinal cord compression is an important contributing factor to neurologic dysfunction.
To better define the role of decompressive surgery in the management of acute SCI, a prospective randomized controlled trial (the Surgical Treatment of Acute Spinal Cord Injury Study, STASCIS) was planned in 2003. This trial was designed to be randomized; however, resistance to randomizing patients to an intentionally delayed decompression led to restructuring as a prospective observational study. This study recruited 170 consecutive patients with subaxial cervical SCI and imaging evidence of spinal cord compression from 10 centers in the United States and Canada. To date, results of this ongoing prospective study suggest that early decompressive surgery significantly improves outcomes and reduces complication rates in patients with SCI. One-year results from the STASCIS trial showed 24% of patients who received decompressive surgery within 24 hours of their injury experienced a two-grade or greater improvement on the ASIA scale, compared with 4% of those in the delayed-treatment group.3 However, major barriers still exist in achieving early decompression in the SCI population, likely because of delays in hospital transfer and challenges with obtaining appropriate neuroimaging and accessing operating room facilities. Thus, implementation of the STASCIS protocol will require major efforts to influence public policy.
Hypothermia
Hypothermia has long been explored for its putative neuroprotective effects, despite associated risks that include coagulopathy, sepsis, and cardiac dysrhythmia. In addition to reducing the metabolic rate, hypothermia appears to reduce extracellular glutamate, vasogenic edema, apoptosis, neutrophil and macrophage invasion and activation, and oxidative stress. In animal models of traumatic SCI, both regional (epidural) and systemic hypothermia have been studied and have demonstrated inconsistent results. Recent studies have demonstrated the benefits of mild systemic hypothermia (33°C) in promoting tissue sparing and functional recovery in animal models of thoracic and cervical contusive SCI.79–81
The effects of modest and severe hypothermia have also been tested in SCI patients.80 Modest systemic hypothermia was reported to be safe in severely injured SCI patients. In a recently published study, the clinical application of modest hypothermia was evaluated in 14 patients with acute cervical SCI.82 This safety study included a retrospective analysis on a subset of patients with acute cervical SCI over a 2-year period. An FDA-approved intravascular catheter was utilized according to established guidelines to deliver a rapid and stable hypothermic state in patients. Patients were intubated and sedated by using muscle relaxants and were cooled to the target temperature (33°C) for 48 hours, after which they were rewarmed to 37°C at a controlled rate. Compared with a control group of age- and injury-matched patients, modest systemic hypothermia appears to be relatively safe, with an incidence of potential risk factors, including respiratory complications, pulmonary embolism, and myocardial infarction, comparable to that in the control group. However, because of a lack of sufficient randomized clinical trial data with the use of modest hypothermia, the AANS/CNS Joint Section on Disorders of the Spine and Peripheral Nerves recently decided that not enough evidence is available to recommend for or against the practice of therapeutic hypothermia as a treatment for SCI. Therefore, continued clinical investigations into this experimental therapy will be required.
Cell Transplantation Strategies
Activated Autologous Macrophages
Based on the premise that the relative inability of the CNS to regenerate can be largely attributed to the insufficient recruitment and activation of macrophages within the immune-privileged injured CNS, preclinical studies using transplantation of activated autologous macrophages after experimental SCI were performed.83,84 Studies from several laboratories have demonstrated the ability for peripheral macrophages to synthesize nerve growth factor following sciatic nerve lesion as well as the capability of these cells to phagocytose myelin,85,86 providing additional rationale for using hematogenous macrophages to repair the injured spinal cord. In preclinical experiments, local injection of autologous macrophages, activated by incubation with autologous PN or skin, induced partial motor recovery after spinal cord transection in adult rats.87 In these studies, sciatic nerve- or skin-coincubated macrophages were reported to demonstrate a distinctive profile of cytokine secretion and cell-surface markers indicative of antigen-presenting activity, such as enhanced synthesis of the IL-1α and brain-derived neurotrophic factor, expression of major histocompatibility complex (MHC) class II molecules, and decreased synthesis of the proinflammatory cytokine TNF-α.88,89 Furthermore, this treatment resulted in a significant recovery of motor function and reduced cystic cavity formation in animal models of contusion SCI.88
Based on positive results in preclinical studies, a phase I clinical SCI trial of activated macrophage transplantation was performed in Israel. The treatment, termed ProCord (Proneuron Biotechnologies, Inc., New York, NY), consisted of a single injection of autologous blood-derived macrophages (activated by coincubation with skin) directly into the epicenter of the injured spinal cord. The initial phase I study was a nonrandomized, open-label study that enrolled 16 patients with complete SCI.89 No adverse treatment-related events were reported. On the basis of these findings, ProNeuron initiated the recruitment of patients into a phase II international multicenter randomized clinical trial to evaluate the safety and efficacy of ProCord for complete SCI. The study enrolled acute complete SCI patients with an injury between C5 and T11 within 14 days of injury. All control and treatment patients underwent SCI rehabilitation and underwent follow-up testing for 1 year. However, the phase II ProCord trial was stopped prior to completion because of several major drawbacks in the design of the clinical trial, including the following: (1) The majority of patients were treated relatively late after injury (day 14 or later), (2) the injection of the macrophages often necessitated a second surgery after an initial surgery for decompression, (3) it was difficult to accurately identify the location of the lesion border at the time of macrophage injection, and (4) the expense was too great.
Human Embryonic Stem Cells and Oligodendrocyte Progenitor Cells
Numerous preclinical studies suggest that embryonic and adult stem cells, along with their lineage-specific progenitors, may improve the outcome after experimental SCI. Transplanted stem cells may potentially act through several proposed mechanisms, which include (1) providing trophic support to promote the survival and regrowth of host tissue, (2) acting as a cellular scaffold to permit axonal elongation through the site of injury, and/or (3) the replacement of lost or damaged cells (e.g., oligodendrocytes). Demyelination of intact axons is a prominent feature of SCI and contributes to loss of function after injury. Therefore, potential therapeutic strategies may involve the replacement of myelin-producing cells through the transplantation of embryonic stem cells, various organ-specific adult stem cells, or lineage-restricted progenitor cells. ES cells provide novel prospects for cellular replacement strategies because of their ability to provide seemingly unlimited numbers of stem cell in vitro, their ability to undergo genetic modification, and their broad developmental capacity.90 McDonald et al.91 reported that transplantation of neural differentiated mouse embryonic stem cells into a contusion SCI in rats improved functional recovery and suggested remyelination as a likely mechanism underlying the effect. Despite these findings, the progress of human embryonic stem cells (hESCs) research has been hampered by numerous scientific issues and ethical concerns. One of the many scientific challenges facing hESC research is the production of high-purity cell lineages from pluripotent hESCs, an issue that has become paramount because of the potential of these cells to differentiate into teratomas. As a result, many researchers have turned to pursuing the use of ESC-derived lineage-restricted progenitor cells. Recently, researchers have successfully differentiated hESCs along the oligodendrocyte lineage, obtaining highly purified oligodendrocyte progenitor cells (OPCs). 92 The transplantation of human hESC-derived OPCs into adult rat spinal cord injuries has been shown to enhance remyelination and promote improvement of motor function.93 In this study, transplantation of OPCs 1 week after SCI resulted in widespread oligodendrocyte remyelination throughout the white matter. The total number of remyelinated axons in the acute transplant group increased by 136% compared to endogenous remyelination in controls, and remyelination in the acute OPC-transplanted group was approximately double the amount of endogenous remyelination observed 1 year after injury in nontransplanted controls. In addition, histologic studies confirmed that substantial remyelination was performed by transplanted OPCs. Transplantation of hESC-derived OPCs 10 months after SCI did not result in increased remyelination compared with control animals that did not receive OPCs, a finding that is paradoxical, considering that a significantly greater density of demyelinated axons is present as compared to the acute injury group. Histopathologic analysis of the chronic transplant group revealed widespread astrogliosis and engulfment of axons by astrocyte processes, suggesting the presence of an inhibitor of remyelination after chronic injury. Several studies have suggested that an established, but not necessarily ongoing, glial scar is largely responsible for the failure of remyelination. Overall, these landmark studies demonstrated the feasibility of predifferentiating hESCs into functional OPCs and demonstrated their therapeutic potential at early time points after experimental SCI. However, it remains unclear whether the improved functional recovery that has been seen in preclinical studies is due to enhanced remyelination, to the secretion of trophic factors by OPCs, or to other neuroprotective effects of OPC transplantation that have yet to be characterized.
Schwann Cells and Peripheral Nerve Grafting
The Schwann cell is one of the most widely studied cell types for repair of the spinal cord.94–97 These cells play a crucial role in endogenous repair of PNs because of their ability to dedifferentiate, migrate, proliferate, express growth-promoting factors and extracellular matrix molecules, and myelinate regenerating axons.98 Following SCI, Schwann cells migrate from the periphery into the injury site, where they participate in endogenous repair processes.8,39 For transplantation into the spinal cord, large numbers of Schwann cells are necessary to fill injury-induced cystic cavities. Several culture systems have been developed that provide large, highly purified populations of Schwann cells, and the development of in vitro systems to harvest human Schwann cells has created the opportunity for autologous transplantation. In experimental SCI models, grafting of Schwann cells or PN into the lesion site has been shown to promote axonal regeneration and myelination.9,94,95 However, axons do not regenerate beyond the transplant, owing to the inhibitory nature of the glial scar surrounding the injury.95 Although Schwann cells have great potential for repair of the injured spinal cord, their combination with other interventions is needed to maximize axonal regeneration and functional recovery. To overcome the glial scar inhibition, additional approaches need to be incorporated into therapeutic strategies, such as increasing the intrinsic capacity of axons to regenerate, by using trophic factors or elevating cAMP levels, and removing growth-inhibitory molecules associated with the astroglial scar and damaged myelin.98
PN grafting, first described by Richardson et al. in the 1980s,9 represents a promising treatment strategy for spinal cord repair. In 1996, Cheng et al.10 performed transplantation of autologous intercostal nerve transplants affixed with fibrin glue containing acidic fibroblast growth factor. These grafts spanned the injury site and joined the rostral white matter to the distal gray matter. Regeneration of the corticospinal tract and recovery of hindlimb function was seen over a 6-month period. However, despite multiple attempts by several independent groups to replicate these results, only one could obtain similar, albeit smaller, effects 6 years later.99 A second group observed some axonal regeneration in primates, but this did not occur beyond the lesion border and did not result in functional recovery.100 Despite incomplete preclinical evidence, this strategy has been used to treat patients with SCI. A case report of a single patient with chronic incomplete SCI has shown that autologous sural nerve grafts could improve both motor and sensory function.101 A similar study was conducted in Brazil over a 5-year period in eight patients in which no motor or sensory recovery was seen.102 Hence, there is insufficient data to recommend this strategy as a treatment.
• The regenerating axons can be directed toward a specific target area.
• The number and source of regenerating axons are easily determined by tracing techniques.
• The graft can be used for electrophysiologic experiments to measure functional recovery associated with axons in the graft.
• Functional recovery due to axonal regeneration within the PN bridge may be confirmed by lesioning experiments.
• Autologous nerve grafts may be used, reducing the possibility of graft rejection.
In this paradigm, regenerated axons that reach the distal end of the PN graft fail to extend back into the spinal cord when PN grafts are used alone. However, regenerated axons have been shown to reenter the distal spinal cord after additional treatment with chondroitinase ABC(ChABC), a bacterial enzyme that degrades inhibitory CSPGs present within the glial scar at the distal graft-host interface. Previous studies have shown that delivery of ChABC, either via an osmotic minipump or by microinjection into the distal lesion site, resulted in extensive CSPG degradation, enhanced axonal regeneration, and functional recovery in the PN grafting-bridging model.103 Furthermore, the delivery of exogenous growth and neurotrophic factors, through genetic modification of transplanted or host cells or by direct protein delivery, encourages longer-distance axonal regrowth into the spinal cord. The PN grafting approach appears to be effective in promoting axonal regeneration of both acute and chronically injured neurons.
Axonal Regeneration
Phosphodiesterase Inhibitors
The elevation of intracellular cAMP levels represents a promising therapeutic strategy for inducing neurons to overcome myelin inhibitory signals.11,12 Although there is no spontaneous regeneration of mammalian CNS axons after injury, dorsal root ganglion (DRG) axons have been shown to regenerate if the peripheral branch of these neurons is lesioned prior to CNS injury (termed a preconditioning lesion). The axonal regeneration that is seen in preconditioned DRGs is associated with marked elevations in the level of intracellular cAMP. Similarly, the injection of dibutryl cAMP (db-cAMP), a nonhydrolyzable cAMP analogue, into neuronal cell bodies can mimic a preconditioning lesion.104 Prophylactic administration of cAMP is an impractical clinical therapy for SCI, however, and several strategies have been employed to increase intracellular cAMP levels postinjury. These strategies involve direct injection of db-cAMP or the use of indirect methods such as stimulating adenylate cyclase with forskolin. The transcription factor cAMP response element binding protein (CREB) is activated by elevated cAMP levels in a protein kinase A–dependent fashion and serves as the primary mediator of cAMP-induced transcription.105 CREB activity has been shown to be essential for overcoming neurite outgrowth inhibition of cerebellar neurons by myelin, as the expression of dominant-negative CREB blocked these effects of cAMP.106 Several cAMP-regulated gene products have been identified that play a role in overcoming myelin inhibition, including arginase I (Arg I) and IL-6. Arg I expression is increased in cerebellar neurons in response to cAMP, and overexpression of Arg I is sufficient to overcome inhibition by myelin-associated glycoprotein (MAG).107 Arg I hydrolyzes arginine to ornithine and urea, stimulating the synthesis of polyamines, such as putrescine, spermidine, and spermine. The priming of DRG neurons with polyamines has been shown to enhance neurite outgrowth on MAG, and this effect is lost when pharmacologic inhibitors of polyamine synthesis are administered with cAMP.107
As an alternative approach to using cAMP analogues, inhibition of the cAMP-degrading enzyme phosphodiesterase has been extensively studied in animal models of SCI. Rolipram, a specific inhibitor of type IV phosphodiesterase, readily crosses the blood-brain barrier and may be either delivered orally or injected subcutaneously, thereby allowing for simple and clinically relevant means of drug delivery. The therapeutic potential of rolipram has been assessed in several SCI studies.11,108–110 Rolipram, delivered through a preconditioned subcutaneous priming method, was shown to overcome myelin inhibition in vitro, as DRG neurons from preconditioned animals demonstrated increased neurite outgrowth on inhibitory myelin substrates.109 The efficacy of rolipram in vivo was assessed after subcutaneous delivery for 10 days via mini-osmotic pumps implanted 2 weeks after spinal cord hemisection lesion, along with acute transplantation of embryonic spinal cord tissue at the injury site. In animals that received rolipram, there was significantly more axonal regrowth into the transplants, particularly that of serotonergic fibers. Rolipram-treated animals also had significantly greater functional recovery, measured by using forelimb paw placement testing. In addition, rolipram treatment resulted in a decrease in astrocytic GFAP expression adjacent to the lesion, indicative of reduced glial scarring.109 In a contusion SCI model, delivery of rolipram has been shown to prevent a drop in cAMP levels seen in the rostral spinal cord, sensorimotor cortex, and brainstem after injury.11 Furthermore, the combined treatment of rolipram and Schwann cell transplantation promoted significant supraspinal and propriospinal axonal sparing and myelination.11 Injection of db-cAMP adjacent to the graft further elevated cAMP levels beyond those in uninjured controls. The combination of rolipram, Schwann cell grafts, and db-cAMP delivered after contusion SCI resulted in enhanced axonal sparing and myelination, promoted axonal regrowth of serotonergic fibers into and beyond the graft site, and improved functional recovery.11 Rolipram has also been shown to increase phrenic nerve output ipsilateral to an experimental C2 hemisection lesion. Intravenous rolipram restored respiratory-related activity to the phrenic nerve ipsilateral to the injury and significantly enhanced phrenic nerve inspiratory burst activity in both normal and C2 hemisected animals. These results provided evidence that elevating cAMP levels by using a phosphodiesterase inhibitor may enhance phrenic nerve output and restore respiratory-related phrenic nerve function after high cervical SCI.111
Although rolipram has been shown to promote axonal regeneration following experimental SCI, recent studies also suggest that the drug promotes robust neuroprotection in experimental models of SCI.110,112,113 The delivery of rolipram after contusive SCI resulted in significant white matter sparing at the injury epicenter and increased the number of oligodendrocyte-myelinated axons in ventral white matter months after injury.11,110 Rolipram has been shown to decrease the production of the potent proinflammatory mediators TNF-α and IL-1α, promote myelinated tissue sparing, and improve locomotor function after experimental contusion SCI.11,110,113 It appears likely that rolipram will enter clinical trials in the future, pending FDA approval based on preclinical studies.
Removal and Blockade of Inhibitory Substrates
Chondroitinase ABC
It is well accepted that significant neuronal regeneration fails to occur following injury to the CNS. The cause is multifactorial, due in part to CSPGs within the forming glial scar and throughout the perineuronal net. The formation of the glial scar after CNS injury presents both a chemical barrier and a physical barrier to axonal regeneration. The up-regulation of axonal growth inhibitors, such as CSPGs, ephrins, and semaphorins, within the glial scar represents a major impediment for axonal regeneration.114,115 CSPGs function as potent inhibitory extracellular matrix molecules, consisting of a protein core to which many large, sulfated glycosaminoglycan (GAG) chains are covalently attached. These glycosaminoglycans confer most of the inhibitory properties of CSPGs. The CSPGs form a large family of molecules, including aggrecan, brevican, neurocan, NG2, phosphacan, and versican.
The degrading enzyme chondroitinase ABC (ChABC) cleaves the inhibitory glycosaminoglycans from the protein core of CSPGs, thereby removing the axonal growth-inhibitory properties of intact CSPGs.116 Several studies have demonstrated that axons are able to extend over long distances in vitro following ChABC treatment. Culture of adult sensory DRG neurons on a gradient of inhibitory CSPGs resulted in the formation of dystrophic end-bulbs, which mimics regeneration failure in vivo. Combining inflammation-induced preconditioning of DRG in vivo before harvest with ChABC digestion of proteoglycans in vitro resulted in significant axonal regeneration across a once potently inhibitory substrate.117 The degradation of CSPGs using ChABC renders the environment of the damaged CNS more permissive to axon regeneration, and overcoming proteoglycan inhibition using ChABC has been shown to promote axonal growth past the lesion and enhance functional recovery, making it a promising strategy for repair of the injured rat spinal cord.118–121 The presence of CSPGs within the lesion penumbra appears to be a major factor preventing the regeneration of axons.122,123 The role of CSPGs in limiting axonal regeneration has been studied in a transgenic mouse model in which the gfap promoter was used to express ChABC in astrocytes.121 In this study, corticospinal axons entered the lesion site but did not extend caudally across a dorsal hemisection lesion in transgenic mice. Accordingly, no significant improvement in motor functional recovery was observed in this model. In contrast, functionally significant sensory axon regeneration was observed, suggesting that ChABC acts on spatially distinct axonal pathways. Intrathecal delivery of ChABC has been shown to degrade CSPGs at the site of injury, up-regulate a regeneration-associated protein (GAP-43), promote regeneration of ascending sensory projections and descending corticospinal axons, and improve functional recovery.124,125 In addition, microinjection of ChABC induces collateral sprouting in the cuneate nucleus after cervical SCI through digestion of the perineuronal net.126
The administration of ChABC has also been used as an adjunctive strategy to promote axonal regeneration in combination with neurotrophic factors and various cell types, such as Schwann cells, PN grafts, olfactory-ensheathing cells, and fetal spinal cord tissue.103,120,127,128 Recent studies have shown that delivery of ChABC into the rostral and caudal ends of the lesion site enhanced the ability of regenerating axons to enter, as well as exit, PN grafts transplanted into CNS lesions.103 Despite these findings, recent evidence suggests that axonal sprouting might not be responsible for the functional recovery that is seen after ChABC delivery and that neuroprotective effects of ChABC may exist.129
Currently, there are no ongoing clinical trials of ChABC for the treatment of SCI. However, on the basis of an increasing number of preclinical studies supporting a role for ChABC in spinal cord repair, it is likely that ChABC will enter pilot studies and clinical trials for SCI in the near future (Fig. 72-2).
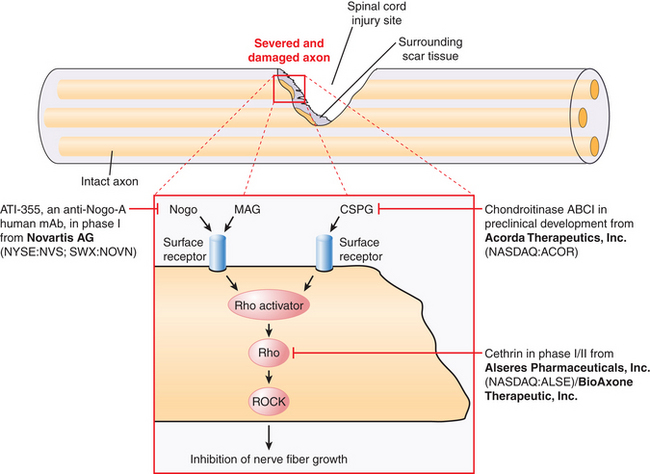
Anti-Nogo-A Antibody (ATI-355)
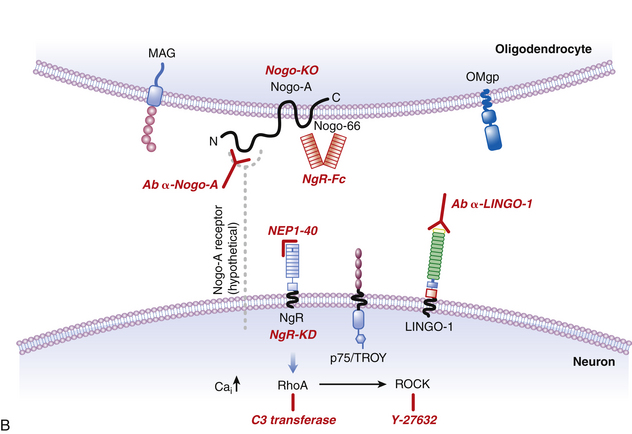
In the late 1980s, pioneering work by Caroni and Schwab130,131 demonstrated that oligodendrocyte myelin was a major inhibitor of axonal growth within the CNS. The myelin was then biochemically separated into 35- and 250-kD inhibitory fractions (termed NI-35 and NI-250), and a monoclonal antibody (termed IN-1) was developed that could block their inhibitory properties in vitro.130 Subsequent in vivo application of IN-1 in rodents resulted in substantial axonal sprouting and long-distance corticospinal axonal regeneration within the adult mammalian CNS and was associated with improved functional recovery. The IN-1 antibody was also used in the characterization and protein sequencing of its target antigen, which has led to the identification of several myelin-associated inhibitors, known as Nogo-A; myelin-associated glycoprotein (MAG); and oligodendrocyte myelin glycoprotein (OMgp). Recent advances in molecular neuroscience have also led to the identification of their receptor complex (consisting of the Nogo receptor (NgR), LINGO-1, and p75NTR/TROY), and a common downstream signaling pathway involving two key proteins, Rho-A GTPase and Rho kinase (ROCK). These landmark findings have prompted the development of targeted strategies aimed at halting the signaling cascade, thereby enabling axons to overcome myelin inhibition and regeneration failure. To date, several experimental and preclinical strategies have been employed, including the development of the following:
• NgR-Fc, a soluble Nogo receptor fusion protein that blocks Nogo-A
• A humanized anti-Nogo-A neutralizing antibody
• NEP1–40, a Nogo-66 receptor antagonist
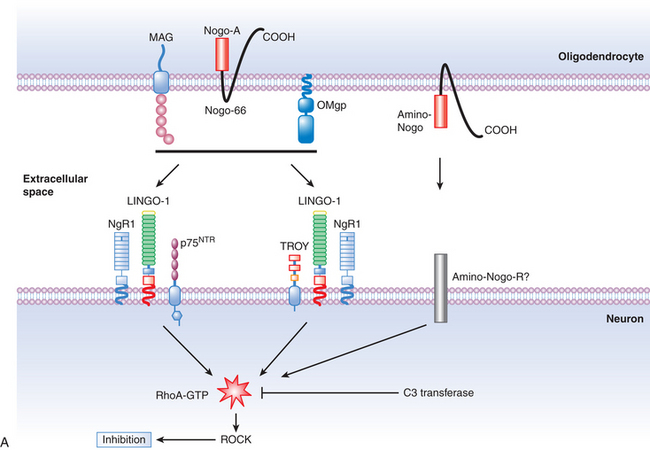
The myelin-associated inhibitors (Nogo-A, MAG, and OMgp) utilize a common receptor, the Nogo-66 receptor (NgR), which transduces signals, resulting in the inhibition of axonal regeneration. Recently, a novel vaccine approach has been used to stimulate the production of an anti-NgR antibody to overcome NgR-mediated growth inhibition after SCI.79 Adult rats immunized with recombinant NgR produced high titers of the anti-NgR antibody, and antisera that was obtained from the immunized rats promoted neurite outgrowth of rat cerebellar neurons on the inhibitory myelin substrate MAG in vitro. In a spinal cord dorsal hemisection model, NgR immunization promoted regeneration of lesioned corticospinal tract axons beyond the lesion site. In a contusive SCI model, NgR immunization markedly reduced the total lesion volume and improved hindlimb locomotor recovery.79 Thus, the NgR vaccine approach may represent a promising repair strategy to promote recovery following SCI.
The blockade of Nogo-A through the use of a humanized monoclonal neutralizing antibody, termed ATI-355, has been shown to promote axonal sprouting and functional recovery following SCI in numerous animal models, including primates, and represents a clinically relevant and promising strategy to overcome myelin inhibition (see Fig. 72-2). The delivery of anti-Nogo A antibody has been shown to stimulate axonal sprouting caudal to the site of experimental SCI. The axonal sprouting that is seen after treatment with the Nogo-A antibody is accompanied by enhanced functional recovery of manual dexterity in primates, as compared to control antibody.132 Furthermore, delivery of anti-Nogo-A antibody also reduced retrograde axonal degeneration (axonal dieback) in anti-Nogo-A antibody–treated monkeys. In the cervical cord, anti-Nogo-A treatment enhanced axonal sprouting of corticospinal fibers rostral to the site of injury, and some of these fibers grew around the lesion and into the caudal spinal segments.
Following preclinical studies that demonstrated the safety and effectiveness of humanized anti-Nogo-A antibody, a large-scale phase I clinical trial was initiated by Novartis Pharma in close collaboration with the European and North American Clinical Trial Networks for SCI to assess the safety, feasibility, and pharmacokinetics of this antibody in patients with complete SCI between C5 and T12 at 4 to 14 days postinjury.13 The agent is being administered via continuous intrathecal infusion, and patients are being enrolled in four increasing dose regimens, the highest dose being delivered over 28 days. However, the FDA expressed concerns about the external nature of the infusion pump, and the clinical evaluation has subsequently been limited to Europe and Canada. In addition, neutropenia was recently reported as a severe adverse event associated with this therapy. The results of this trial are currently pending.
Rho Antagonists
The microenvironment of the lesioned spinal cord is not conducive for axonal regeneration, owing in large part to the presence of myelin-associated inhibitory proteins, which include Nogo-A, MAG, and OMgp, and the presence of CSPGs, semaphorins, and ephrins within the glial scar. Many of these inhibitors have been shown to activate a common signaling pathway within neurons, consisting of the RhoA GTPase and its downstream effector, the serine/threonine kinase, Rho-associated coiled kinase (ROCK) (Fig. 72-3A).133 The Rho/ROCK pathway is an important determinant in the response of axons to growth inhibitory proteins, which exert growth-inhibitory effects through the regulation of the actin-myosin network, leading to stimulation of actin-myosin contractility via myosin light chain phosphorylation, inhibition of myosin phosphatase, and inactivation of the actin-depolymerizing factor cofilin.133 These molecular events eventually lead to the induction of neurite retraction and subsequently to growth cone collapse.134
Rho activity has been shown to increase extensively following transection of the rat spinal cord, and numerous studies have demonstrated that inhibition of the Rho/ROCK pathway results in enhanced axonal regeneration and functional recovery in animal models of SCI.135,136 Several pharmacologic methods have been used to inhibit the Rho/ROCK pathway, including the delivery of the Clostridium botulinum–derived Rho antagonist (C3 ribosyltransferase, dominant negative Rho, or use of the pyridine-derivative Y-27632, a specific inhibitor of ROCK.)137 In vitro experiments with Rho and ROCK inhibitors have shown that these drugs are capable of preventing the inhibition of neurite outgrowth that is observed on typical inhibitory substrates present within the glial scar and in white matter.133,138,139 Intravenous and intrathecal delivery of Y-27632 has been shown to enhance sprouting of corticospinal and dorsal column axons and to accelerate locomotor recovery after corticospinal lesions in adult rats.140,141 The regenerative effects of C3 transferase appear to be greater than those of Y-27632, suggesting the presence of other effectors of the Rho signaling pathway that are inactivated by C3 transferase but not by Y-27632.138 The administration of Rho pathway antagonists for up to 24 hours following spinal cord transection in mice resulted in a rapid improvement in locomotion, with progressive improvement in forelimb-hindlimb coordination, suggesting that C3 transferase may be a promising option for SCI with a clinically relevant therapeutic window.138 Neuroprotective actions have also been attributed to the Rho pathway antagonists. For example, C3-mediated neuroprotection appears to involve an ability to enhance the half-life of vascular endothelial nitric oxide synthase mRNA, thereby improving blood flow to ischemic regions.142
Following a multitude of preclinical studies in support of axonal regenerative and neuroprotective properties of Rho pathway antagonists, BioAxone Therapeutic, Inc., initiated a phase I/IIa multicenter open-label, dose-escalating clinical trial to assess the pharmacokinetics and evaluate the safety, tolerability, and neurologic status of patients following administration of a single extradural application of the rho antagonist Cethrin (see Fig. 72-2). Cethrin is a recombinant fusion protein composed of C3 ribosyltransferase in combination with a membrane transport sequence, which allows the protein to cross cellular membranes, where it inhibits RhoA activity by ADP-ribosylation. The patient population in the phase I/IIa Cethrin trial consisted of males and females aged 16 to 70 years of age with an acute thoracic or cervical SCI corresponding to an ASIA grade A who were scheduled to undergo spinal decompression/stabilization within 7 days of injury. Injuries from T2 to T12 and from C4 to T1 were subject to separate analysis, and 6-month and 1-year follow-ups were planned. The study recruited 37 patients and did not include control subjects. The drug was administered an average of 53 hours postinjury, delivered in a single dose with fibrin sealant. Dose levels ranged from 0.3 mg to 9.0 mg. At 6 months follow-up, approximately 28% of the patients had improved by one or more ASIA grades (five subjects improved by two grades, and two subjects improved by three grades). The results of the study show that 19.4% of patients improved by two or more ASIA grades at 6 months, a rate that is 1.5- to 3-fold higher than rates in historical controls. Overall, cervical SCIs appeared to show greater benefit. There were no adverse events related to Cethrin. To date, it appears that local application of the Rho inhibitor Cethrin is both feasible and safe in patients with acute SCI. These results must be interpreted with caution, given the early phase of this trial and its nonrandomized nature. However, this rate of improved neurologic function appears promising, and a subsequent prospective, randomized phase II study is being conducted under the sponsorship of Alseres Pharmaceuticals.
Bracken M.B., Shepard M.J., Collins W.F., et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury: results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
Houle J.D., Tom V.J., Mayes D., et al. Combining an autologous peripheral nervous system “bridge” and matrix modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. J Neurosci. 2006;26(28):7405-7415.
Lehmann M., Fournier A., Selles-Navarro I., Dergham P., et al. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19(17):7537-7547.
Pearse D.D., Pereira F.C., Marcillo A.E., et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10(6):610-616.
Richardson P.M., McGuinness U.M., Aguayo A.J. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284(5753):264-265.
1. Schwab M.E., Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76(2):319-370.
2. Sekhon L.H., Fehlings M.G. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine (Phila Pa 1976). 2001;26(Suppl 24):S2-S12.
3. Baptiste D.C., Fehlings M.G. Update on the treatment of spinal cord injury. Prog Brain Res. 2007;161:217-233.
4. Priebe M.M., Chiodo A.E., Scelza W.M., et al. Spinal cord injury medicine: 6. Economic and societal issues in spinal cord injury. Arch Phys Med Rehabil. 2007;88:S84-S88.
5. McDonald J.W., Sadowsky C. Spinal-cord injury. Lancet. 2002;359:417-425.
6. Ho C.H., Wuermser L.A., Priebe M.M., et al. Spinal cord injury medicine: 1. Epidemiology and classification. Arch Phys Med Rehabil. 2007;88:S49-S54.
7. Tator C.H. Biology of neurological recovery and functional restoration after spinal cord injury. Neurosurgery. 1998;42(4):696-707.
8. Norenberg M.D., Smith J., Marcillo A. The pathology of human spinal cord injury: defining the problems. J Neurotrauma. 2004;21(4):429-440.
9. Richardson P.M., McGuinness U.M., Aguayo A.J. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284(5753):264-265.
10. Cheng H., Cao Y., Olson L. Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science. 1996;273(5274):510-513.
11. Pearse D.D., Pereira F.C., Marcillo A.E., et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10(6):610-616.
12. Hannila S.S., Filbin M.T. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp Neurol. 2008;209(2):321-332.
13. Curt A., Schwab M.E., Dietz V. Providing the clinical basis for new interventional therapies: refined diagnosis and assessment of recovery after spinal cord injury. Spinal Cord. 2004;42(1):1-6.
14. Bracken M.B., Collins W.F., Freeman D.F., et al. Efficacy of methylprednisolone in acute spinal cord injury. JAMA. 1984;251(1):45-52.
15. Hall E.D. Lipid antioxidants in acute central nervous system injury. Ann Emerg Med. 1993;22(6):1022-1027.
16. Bracken M.B., Shepard M.J., Collins W.F., et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury: results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322(20):1405-1411.
17. Bracken M.B. Treatment of acute spinal cord injury with methylprednisolone: results of a multicenter, randomized clinical trial. J Neurotrauma. 1991;8(Suppl 1):S47-S50. discussion S51-S52
18. Bracken M.B., Shepard M.J., Collins W.F.Jr., et al. Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up data. Results of the second National Acute Spinal Cord Injury Study. J Neurosurg. 1992;76(1):23-31.
19. Fehlings M.G. Spine Focus Panel. Summary statement: the use of methylprednisolone in acute spinal cord injury. Spine (Phila Pa 1976). 2001;26(Suppl 24):S55.
20. Hurlbert R.J. The role of steroids in acute spinal cord injury: an evidence-based analysis. Spine (Phila Pa 1976). 2001;26(Suppl 24):S39-S46.
21. Bracken M.B., Shepard M.J., Holford T.R., et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. JAMA. 1997;277(20):1597-1604.
22. Xu J., Fan G., Chen S., et al. Methylprednisolone inhibition of TNF-alpha expression and NF-kB activation after spinal cord injury in rats. Brain Res Mol Brain Res. 1998;59(2):135-142.
23. Baptiste D.C., Fehlings M.G. Pharmacological approaches to repair the injured spinal cord. J Neurotrauma. 2006;23(3-4):318-334.
24. Geisler F.H., Dorsey F.C., Coleman W.P. Recovery of motor function after spinal-cord injury: a randomized, placebo-controlled trial with GM-1 ganglioside. N Engl J Med. 1991;324(26):1829-1838.
25. Geisler F.H., Coleman W.P., Grieco G., et al. The Sygen multicenter acute spinal cord injury study. Spine (Phila Pa 1976). 2001;26(Suppl 24):S87-S98.
26. Geisler F.H., Coleman W.P., Grieco G., et al. Measurements and recovery patterns in a multicenter study of acute spinal cord injury. Spine (Phila Pa 1976). 2001;26(Suppl 24):S68-S86.
27. Long J.B., Martinez-Arizala A., Petras J.M., Holaday J.W. Endogenous opioids in spinal cord injury: a critical evaluation. Cent Nerv Syst Trauma. 1986;3(4):295-315.
28. Bracken M.B., Holford T.R. Effects of timing of methylprednisolone or naloxone administration on recovery of segmental and long-tract neurological function in NASCIS 2. J Neurosurg. 1993;79(4):500-507.
29. Dumont R.J., Okonkwo D.O., Verma S., et al. Acute spinal cord injury: part I. pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24(5):254-264.
30. Pitts L.H., Ross A., Chase G.A., Faden A.I. Treatment with thyrotropin-releasing hormone (TRH) in patients with traumatic spinal cord injuries. J Neurotrauma. 1995;12(3):235-243.
31. Gaviria M., Privat A., d’Arbigny P., et al. Neuroprotective effects of a novel NMDA antagonist, Gacyclidine, after experimental contusive spinal cord injury in adult rats. Brain Res. 2000;874(2):200-209.
32. Tadie M., D’Arbigny P., Mathé J., et al. Acute spinal cord injury: early care and treatment in a multicenter study with gacyclidine. Soc Neurosci. 1999;25:1090.
33. Fehlings M.G., Tator C.H., Linden R.D. The effect of nimodipine and dextran on axonal function and blood flow following experimental spinal cord injury. J Neurosurg. 1989;71(3):403-416.
34. Petitjean M.E., Pointillart V., Dixmerias F., et al. Medical treatment of spinal cord injury in the acute stage. Ann Fr Anesth Reanim. 1998;17(2):114-122.
35. Stys P.K. White matter injury mechanisms. Curr Mol Med. 2004;4(2):113-130.
36. Rosenberg L.J., Teng Y.D., Wrathall J.R. Effects of the sodium channel blocker tetrodotoxin on acute white matter pathology after experimental contusive spinal cord injury. J Neurosci. 1999;19(14):6122-6133.
37. Schwartz G., Fehlings M.G. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94(Suppl 2):245-256.
38. Mu X., Azbill R.D., Springer J.E. Riluzole and methylprednisolone combined treatment improves functional recovery in traumatic spinal cord injury. J Neurotrauma. 2000;17(9):773-780.
39. Guest J.D., Hiester E.D., Bunge R.P. Demyelination and Schwann cell responses adjacent to injury epicenter cavities following chronic human spinal cord injury. Exp Neurol. 2005;192(2):384-393.
40. Totoiu M.O., Keirstead H.S. Spinal cord injury is accompanied by chronic progressive demyelination. J Comp Neurol. 2005;486(4):373-383.
41. Nashmi R., Fehlings M.G. Mechanisms of axonal dysfunction after spinal cord injury: with an emphasis on the role of voltage-gated potassium channels. Brain Res Brain Res Rev. 2001;38(1-2):165-191.
42. Hayes K.C. Fampridine-SR for multiple sclerosis and spinal cord injury. Expert Rev Neurother. 2007;7(5):453-461.
43. Hansebout R.R., Blight A.R., Fawcett S., Reddy K. 4-Aminopyridine in chronic spinal cord injury: a controlled, double-blind, crossover study in eight patients. J Neurotrauma. 1993;10(1):1-18.
44. Hayes K.C., Blight A.R., Potter P.J., et al. Preclinical trial of 4-aminopyridine in patients with chronic spinal cord injury. Paraplegia. 1993;31(4):216-224.
45. Potter P.J., Hayes K.C., Hsieh J.T., et al. Sustained improvements in neurological function in spinal cord injured patients treated with oral 4-aminopyridine: three cases. Spinal Cord. 1998;36(3):147-155.
46. Potter P.J., Hayes K.C., Segal J.L., et al. Randomized double-blind crossover trial of Fampridine-SR (sustained release 4-aminopyridine) in patients with incomplete spinal cord injury. J Neurotrauma. 1998;15(10):837-849.
47. Ditunno J.F.Jr., Graziani V., Katz M.A., et al. Double-blind, placebo-controlled, dose-escalating study evaluating the safety and efficacy of oral doses of Fampridine-SR in patients with chronic spinal cord injury. J Spinal Cord Med. 2002;25(Suppl 1):S35-S36.
48. Cardenas D.D., Ditunno J., Graziani V., et al. Phase 2 trial of sustained-release fampridine in chronic spinal cord injury. Spinal Cord. 2007;45(2):158-168.
49. Jelkmann W., Metzen E. Erythropoietin in the control of red cell production. Ann Anat. 1996;178(5):391-403.
50. Erbayraktar S., Yilmaz O., Gökmen N., Brines M. Erythropoietin is a multifunctional tissue-protective cytokine. Curr Hematol Rep. 2003;2(6):465-470.
51. Grasso G., Sfacteria A., Passalacqua M., et al. Erythropoietin and erythropoietin receptor expression after experimental spinal cord injury encourages therapy by exogenous erythropoietin. Neurosurgery. 2005;56(4):821-827.
52. Brines M., Cerami A. Erythropoietin in spinal cord injury. In: Hoke A., editor. Erythropoietin and the nervous system: novel therapeutic options for neuroprotection. New York: Springer; 2006:147-164.
53. Agnello D., Bigini P., Villa P., et al. Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain Res. 2002;952(1):128-134.
54. Cerami A. Beyond erythropoiesis: novel applications for recombinant human erythropoietin. Semin Hematol. 2001;38(3 Suppl 7):33-39.
55. Gorio A., Gokmen N., Erbayraktar S., et al. Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci U S A. 2002;99(14):9450-9455.
56. Kaptanoglu E., Solaroglu I., Okutan O., et al. Erythropoietin exerts neuroprotection after acute spinal cord injury in rats: effect on lipid peroxidation and early ultrastructural findings. Neurosurg Rev. 2004;27(2):113-120.
57. Celik M., Gökmen N., Erbayraktar S., et al. Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci U S A. 2002;99(4):2258-2263.
58. Pinzon A., Marcillo A., Pabon D., et al. A re-assessment of erythropoietin as a neuroprotective agent following rat spinal cord compression or contusion injury. Exp Neurol. 2008;213(1):129-136.
59. Ehrenreich H., Hasselblatt M., Dembowski C., et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med. 2002;8(8):495-505.
60. Erbayraktar S., Grasso G., Sfacteria A., et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci U S A. 2003;100(11):6741-6746.
61. Leist M., Ghezzi P., Grasso G., et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305(5681):239-242.
62. Ibarra A., Diaz-Ruiz A. Protective effect of cyclosporin-A in spinal cord injury: an overview. Curr Med Chem. 2006;13(22):2703-2710.
63. Diaz-Ruiz A., Rios C., Duarte I., et al. Cyclosporin-A inhibits lipid peroxidation after spinal cord injury in rats. Neurosci Lett. 1999;266(1):61-64.
64. Diaz-Ruiz A., Vergara P., Perez-Severiano F., et al. Cyclosporin-A inhibits inducible nitric oxide synthase activity and expression after spinal cord injury in rats. Neurosci Lett. 2004;357(1):49-52.
65. Diaz-Ruiz A., Rios C., Duarte I., et al. Lipid peroxidation inhibition in spinal cord injury: cyclosporin-A vs methylprednisolone. Neuroreport. 2000;11(8):1765-1767.
66. Ibarra A., Correa D., Willms K., et al. Effects of cyclosporin-A on immune response, tissue protection and motor function of rats subjected to spinal cord injury. Brain Res. 2003;979(1-2):165-178.
67. Rabchevsky A.G., Fugaccia I., Sullivan P.G., Scheff S.W. Cyclosporin A treatment following spinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing. J Neurotrauma. 2001;18(5):513-522.
68. McMahon S.S., Albermann S., Rooney G.E., et al. Effect of cyclosporin A on functional recovery in the spinal cord following contusion injury. J Anat. 2009;215(3):267-279.
69. Ravikumar R., McEwen M.L., Springer J.E. Post-treatment with the cyclosporin derivative, NIM811, reduced indices of cell death and increased the volume of spared tissue in the acute period following spinal cord contusion. J Neurotrauma. 2007;24(10):1618-1630.
70. Stirling D.P., Khodarahmi K., Liu J., et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24(9):2182-2190.
71. Wells J.E., Hurlbert R.J., Fehlings M.G., et al. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126(Pt 7):1628-1637.
72. Lee S.M., Yune T.Y., Kim S.J., et al. Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma. 2003;20(10):1017-1027.
73. Teng Y.D., Choi H., Onario R.C., et al. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci U S A. 2004;101(9):3071-3076.
74. Festoff B.W., Ameenuddin S., Arnold P.M., et al. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem. 2006;97(5):1314-1326.
75. Pinzon A., Marcillo A., Quintana A., et al. A re-assessment of minocycline as a neuroprotective agent in a rat spinal cord contusion model. Brain Res. 2008;1243:146-151.
76. Casha S., Zygun D., McGowan D., et al. Neuroprotection with minocycline after spinal cord injury: results of a double-blind randomized controlled pilot study. Abstract, Congress of Neurological Surgeons. 2009. Annual Meeting
77. Shields C.B., Zhang Y.P., Shields L.B., et al. The therapeutic window for spinal cord decompression in a rat spinal cord injury model. J Neurosurg Spine. 2005;3(4):302-307.
78. Benzel E.C., Larson S.J. Functional recovery after decompressive spine operation for cervical spine fractures. Neurosurgery. 1987;20:742-746.
79. Yu C.G., Jimenez O., Marcillo A.E., et al. Beneficial effects of modest systemic hypothermia on locomotor function and histopathological damage following contusion-induced spinal cord injury in rats. J Neurosurg. 2000;93(Suppl 1):85-93.
80. Dietrich W.D.3rd. Therapeutic hypothermia for spinal cord injury. Crit Care Med. 2009;37(Suppl 7):S238-S242.
81. Lo T.P.Jr., Cho K.S., Garg M.S., et al. Systemic hypothermia improves histological and functional outcome after cervical spinal cord contusion in rats. J Comp Neurol. 2009;514(5):433-448.
82. Levi A.D., Green B.A., Wang M.Y., et al. Clinical application of modest hypothermia after spinal cord injury. J Neurotrauma. 2009;26(3):407-415.
83. Lazarov-Spiegler O., Rapalino O., Agranov G., Schwartz M. Restricted inflammatory reaction in the CNS: a key impediment to axonal regeneration? Mol Med Today. 1998;4(8):337-342.
84. Schwartz M., Lazarov-Spiegler O., Rapalino O., et al. Potential repair of rat spinal cord injuries using stimulated homologous macrophages. Neurosurgery. 1999;44(5):1041-1045.
85. Heumann R., Lindholm D., Bandtlow C., et al. Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc Natl Acad Sci U S A. 1987;84(23):8735-8739.
86. Perry V.H., Brown M.C., Gordon S. The macrophage response to central and peripheral nerve injury. A possible role for macrophages in regeneration. J Exp Med. 1987;165(4):1218-1223.
87. Rapalino O., Lazarov-Spiegler O., Agranov E., et al. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814-821.
88. Bomstein Y., Marder J.B., Vitner K., et al. Features of skin-coincubated macrophages that promote recovery from spinal cord injury. J Neuroimmunol. 2003;142(1-2):10-16.
89. Knoller N., Auerbach G., Fulga V., et al. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine. 2005;3:173-181.
90. Zhang S.C., Wernig M., Duncan I.D., et al. in vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1129-1133.
91. McDonald J.W., Liu X.Z., Qu Y., et al. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med. 1999;5(12):1410-1412.
92. Nistor G.I., Totoiu M.O., Haque N., et al. Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia. 2005;49(3):385-396.
93. Keirstead H.S., Nistor G., Bernal G., et al. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25(19):4694-4705.
94. Xu X.M., Guénard V., Kleitman N., Bunge M.B. Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. J Comp Neurol. 1995;351(1):145-160.
95. Xu X.M., Chen A., Guénard V., et al. Bridging Schwann cell transplants promote axonal regeneration from both the rostral and caudal stumps of transected adult rat spinal cord. J Neurocytol. 1997;26(1):1-16.
96. Iannotti C., Li H., Yan P., et al. Glial cell line-derived neurotrophic factor-enriched bridging transplants promote propriospinal axonal regeneration and enhance myelination after spinal cord injury. Exp Neurol. 2003;183(2):379-393.
97. Golden K.L., Pearse D.D., Blits B., et al. Transduced Schwann cells promote axon growth and myelination after spinal cord injury. Exp Neurol. 2007;207(2):203-217.
98. Oudega M., Xu X.M. Schwann cell transplantation for repair of the adult spinal cord. J Neurotrauma. 2006;23(3-4):453-467.
99. Lee Y.S., Hsiao I., Lin V.W. Peripheral nerve grafts and aFGF restore partial hindlimb function in adult paraplegic rats. J Neurotrauma. 2002;19:1203-1216.
100. Levi A.D., Dancausse H., Li X., et al. Peripheral nerve grafts promoting central nervous system regeneration after spinal cord injury in the primate. J Neurosurg. 2002;96(Suppl 2):197-205.
101. Cheng H., Liao K.K., Liao S.F., et al. Spinal cord repair with acidic fibroblast growth factor as a treatment for a patient with chronic paraplegia. Spine. 2004;29(14):E284-E288.
102. Steeves J., Fawcett J., Tuszynski M. Report of international clinical trials workshop on spinal cord injury, February 20–21, 2004, Vancouver, Canada. Spinal Cord. 2004;42(10):591-597.
103. Houle J.D., Tom V.J., Mayes D., et al. Combining an autologous peripheral nervous system “bridge” and matrix modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. J Neurosci. 2006;26(28):7405-7415.
104. Qiu J., Cai D., Dai H., et al. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34(6):895-903.
105. Lonze B.E., Ginty D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605-623.
106. Gao Y., Deng K., Hou J., et al. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44(4):609-621.
107. Cai D., Deng K., Mellado W., et al. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35(4):711-719.
108. Lu P., Yang H., Jones L.L., et al. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J Neurosci. 2004;24(28):6402-6409.
109. Nikulina E., Tidwell J.L., Dai H.N., et al. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proc Natl Acad Sci U S A. 2004;101(23):8786-8790.
110. Iannotti C., Clark M., Horn K.P., et al. A combination immunomodulatory treatment promotes neuroprotection and improves locomotor recovery following contusion SCI. Exp Neurol. 2011;230(1):3-15.
111. Kajana S., Goshgarian H.G. Systemic administration of rolipram increases medullary and spinal cAMP and activates a latent respiratory motor pathway after high cervical spinal cord injury. J Spinal Cord Med. 2009;32(2):175-182.
112. Whitaker C.M., Beaumont E., Wells M.J., et al. Rolipram attenuates acute oligodendrocyte death in the adult rat ventrolateral funiculus following contusive cervical spinal cord injury. Neurosci Lett. 2008;438(2):200-204.
113. Koopmans G.C., Deumens R., Buss A., et al. Acute rolipram/thalidomide treatment improves tissue sparing and locomotion after experimental spinal cord injury. Exp Neurol. 2009;216(2):490-498.
114. Silver J., Miller J.H. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146-156.
115. Fawcett J.W. Overcoming inhibition in the damaged spinal cord. J Neurotrauma. 2006;23(3-4):371-383.
116. Grimpe B., Silver J. A novel DNA enzyme reduces glycosaminoglycan chains in the glial scar and allows microtransplanted dorsal root ganglia axons to regenerate beyond lesions in the spinal cord. J Neurosci. 2004;24(6):1393-1397.
117. Steinmetz M.P., Horn K.P., Tom V.J., et al. Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J Neurosci. 2005;25(35):8066-8076.
118. Bradbury E.J., Moon L.D., Popat R.J., et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416(6881):636-640.
119. Caggiano A.O., Zimber M.P., Ganguly A., et al. Chondroitinase ABCI improves locomotion and bladder function following contusion injury of the rat spinal cord. J Neurotrauma. 2005;22(2):226-239.
120. Fouad K., Schnell L., Bunge M.B., et al. Combining Schwann cell bridges and olfactory-ensheathing glia grafts with chondroitinase promotes locomotor recovery after complete transection of the spinal cord. J Neurosci. 2005;25(5):1169-1178.
121. Cafferty W.B., Yang S.H., Duffy P.J., et al. Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. J Neurosci. 2007;27(9):2176-2185.
122. Davies S.J., Goucher D.R., Doller C., Silver J. Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci. 1999;19(14):5810-5822.
123. Tom V.J., Steinmetz M.P., Miller J.H., et al. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24(29):6531-6539.
124. Barritt A.W., Davies M., Marchand F., et al. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. J Neurosci. 2006;26(42):10856-10867.
125. Shields L.B., Zhang Y.P., Burke D.A., et al. Benefit of chondroitinase ABC on sensory axon regeneration in a laceration model of spinal cord injury in the rat. Surg Neurol. 2008;69(6):568-577.
126. Massey J.M., Hubscher C.H., Wagoner M.R., et al. Chondroitinase ABC digestion of the perineuronal net promotes functional collateral sprouting in the cuneate nucleus after cervical spinal cord injury. J Neurosci. 2006;26(16):4406-4414.
127. Chau C.H., Shum D.K., Li H., et al. Chondroitinase ABC enhances axonal regrowth through Schwann cell-seeded guidance channels after spinal cord injury. FASEB J. 2004;18(1):194-196.
128. Kim B.G., Dai H.N., Lynskey J.V., et al. Degradation of chondroitin sulfate proteoglycans potentiates transplant-mediated axonal remodeling and functional recovery after spinal cord injury in adult rats. J Comp Neurol. 2006;497(2):182-198.
129. Tom V.J., Kadakia R., Santi L., et al. Administration of chondroitinase ABC rostral or caudal to a spinal cord injury site promotes anatomical but not functional plasticity. J Neurotrauma. 2009;26(12):2323-2333.
130. Caroni P., Schwab M.E. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85-96.
131. Caroni P., Schwab M.E. Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol. 1988;106(4):1281-1288.
132. Freund P., Schmidlin E., Wannier T., et al. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12(7):790-792.
133. Monnier P.P., Sierra A., Schwab J.M., et al. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol Cell Neurosci. 2003;22(3):319-330.
134. Jalink K., van Corven E.J., Hengeveld T., et al. Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J Cell Biol. 1994;126(3):801-810.
135. Madura T., Yamashita T., Kubo T., et al. Activation of Rho in the injured axons following spinal cord injury. EMBO Rep. 2004;5(4):412-417.
136. Mueller B.K., Mack H., Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4(5):387-398.
137. Lehmann M., Fournier A., Selles-Navarro I., et al. Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci. 1999;19(17):7537-7547.
138. Dergham P., Ellezam B., Essagian C., et al. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22(15):6570-6577.
139. Borisoff J.F., Chan C.C., Hiebert G.W., et al. Suppression of rho-kinase activity promotes axonal growth on inhibitory CNS substrates. Mol Cell Neurosci. 2003;22(3):405-416.
140. Fournier A.E., Takizawa B.T., Strittmatter S.M. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23(4):1416-1423.
141. Chan C.C., Khodarahmi K., Liu J., et al. Dose-dependent beneficial and detrimental effects of ROCK inhibitor Y27632 on axonal sprouting and functional recovery after rat spinal cord injury. Exp Neurol. 2005;196(2):352-364.
142. Laufs U, Endres M, Stagliano N, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest 106(1):15-24, 2000.