[level-membership-for-neurosurgery-category]
CHAPTER 84 Emerging and Experimental Neurosurgical Treatments for Parkinson’s Disease
Idiopathic Parkinson’s disease (PD) affects more than 1 million North Americans, including approximately 1% of individuals older than 65 years. PD is the second most common neurodegenerative disease after Alzheimer’s, and neurodegenerative diseases as a whole are projected to surpass cancer as the second leading cause of death in the elderly by 2040.1 With a protracted course of progressive disability and dependence on others, increased mortality, and a total estimated cost of $23 billion a year in the United States, PD represents a significant ongoing burden to society.2–4
PD is diagnosed on clinical grounds, with resting tremor, rigidity, bradykinesia or akinesia, and postural instability representing the cardinal features of the disease.5 Other important manifestations can include dementia, gait disturbance, autonomic dysfunction, and depression. Neuropathologically, PD is characterized by widespread degeneration of select sites within the central and peripheral nervous systems.6,7 Although this multisystem disorder affects many brain regions and neurotransmitter systems, the loss of dopaminergic neurons within the substantia nigra pars compacta (SNc) accounts for the reduction in striatal dopaminergic innervation that underlies the symptoms of akinesia and rigidity.8 Widespread accumulation of proteinaceous Lewy bodies and Lewy neurites is also observed in PD. In the central nervous system, these deposits accumulate in predictable stages with progressive involvement of brainstem, subcortical, and neocortical regions.9 However, the relationship of these deposits to the pathogenesis of PD remains unclear.5,10 Although numerous mechanisms underlying the neurodegeneration seen in PD have been proposed, none has provided a definitive explanation of its pathogenesis.
Since their introduction more than 40 years ago, dopaminergic agents have been the mainstay of treatment for the motor symptoms of PD. More than 90% of patients initially respond well to levodopa therapy, with the majority continuing to derive some benefit throughout the course of the disease.5 Unfortunately, progressive treatment resistance, coupled with the gradual development of motor fluctuations, dyskinesias, and psychiatric disturbances, limits levodopa’s long-term use.11,12 Other agents such as dopamine agonists, anticholinergics, amantadine, and monoamine oxidase inhibitors generally yield minimal benefit and may induce unpalatable side effects.12 Clinical features of advanced PD such as postural and gait dysfunction are particularly resistant to dopamine-based therapies, suggesting a greater involvement of nondopaminergic pathways in their pathophysiology.13,14 Importantly, no pharmacotherapy has been able to alter disease progression.
The shortcomings of pharmacotherapy and the growing understanding of the pathophysiologic correlates of parkinsonism have led to a resurgence in stereotactic and functional neurosurgery for PD over the past two decades. Targeted lesioning and deep brain stimulation (DBS) at the thalamus, globus pallidus, and subthalamic nucleus have proved to be valuable additions to our therapeutic armamentarium.15 At present, surgery is reserved for medically intractable cases when tremor is a major source of disability or when levodopa-responsive patients are experiencing significant “on-off” cycling between rigidity or bradykinesia and levodopa-induced dyskinesias.16 Although highly efficacious in properly selected patients, the benefits of stereotactic surgery are limited mainly to improving rigidity, tremor, “off-state” akinesia, and levodopa-induced dyskinesias. There is little or no effect on other manifestations of the disease.15 In addition, the benefit of surgery remains symptomatic in nature, without altering the natural history of the disease.
Therefore, our present therapeutic armamentarium could be improved in a number of ways. First, making DBS more effective and safer would allow more patients to be treated and to undergo surgery earlier in the disease course. Second, new treatments could address the many symptomatic features of PD that remain poorly treated or are not affected at all by current therapies. Third, disease-modifying treatments could deter disease progression. The following sections outline experimental and emerging neurosurgical treatments whose development has been geared specifically toward closing these gaps in our current treatment options and ultimately achieving the elusive goal of modifying the natural history of PD through neuroprotection or neuroregeneration. DBS-related strategies, motor cortex stimulation, growth factor delivery, gene therapy, and cellular replenishment are discussed, with reference to currently available clinical data (Fig. 84-1).
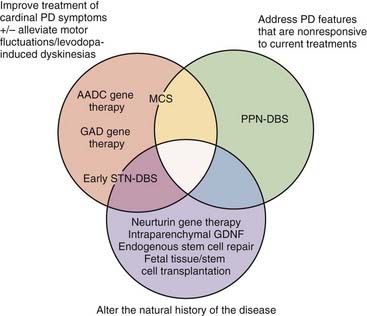
New Strategies for Deep Brain Stimulation
In properly selected patients, and when appropriate resources are available, high-frequency DBS of the subthalamic nucleus (STN) or internal globus pallidus (GPi) is the surgical treatment of choice for PD.17 Clinical outcomes and surgical techniques are now well established, with more than 30,000 patients treated worldwide.15 Armed with this baseline knowledge and experience, attempts to improve the utility of DBS in PD through early surgery and novel targets are ongoing.
Early Surgery
The observation that STN inactivation prevents nigral degeneration in rodent models of PD18,19 led investigators to consider the possibility that, in addition to its symptomatic benefits, subthalamic DBS might be neuroprotective in humans. The neuroprotective mechanism was thought to be reduced excitotoxin-induced nigral degeneration, mediated by the glutamatergic projections from the STN to the nigra.20,21 To examine this, Hilker and coworkers22 used 18F-fluorodopa (F-dopa) positron emission tomography (PET), which quantifies presynaptic dopaminergic function,23–25 to study the progression of PD after high-frequency STN-DBS. Although the study did not include controls, the annual striatal reduction in F-dopa uptake in 30 STN-DBS–treated patients with advanced PD was roughly 10% to 12%, which is consistent with previously reported rates in PD.26 This evidence, along with clinical experience, suggests that the neuroprotective effects seen in animal models cannot be readily confirmed in humans.
It remains uncertain whether a neuroprotective effect of high-frequency STN-DBS might be observed if patients were treated early in the course of their disease. Patients with advanced PD may no longer harbor the capacity to alter the course of degeneration, whereas the striatum of patients in the early stages of the disease might be more apt to gain a survival benefit from a reduction in STN-mediated glutamatergic input. In March 2006, a phase I trial of early surgery in PD began under the supervision of Dr. Charles at Vanderbilt University.27 The hypothesis of this study is that STN-DBS slows disease progression in subjects with early PD. In this single-center study, which is expected to be completed in 2010, eligible patients who have received dopaminergic therapy for less than 4 years are randomized to surgery plus medical therapy versus medical therapy alone. Motor outcomes are assessed in a single-blind fashion. A multicenter phase III trial is planned if safety and tolerability criteria are met.
Another line of investigation suggests that early surgery in patients suffering from motor fluctuations or dyskinesias may promote the maintenance of quality of life and social functioning.28 The mean time from PD diagnosis to surgery is more than 10 years; however, dopamine-related motor fluctuations and dyskinesias may arise much sooner and typically contribute to a progressive decline in social and occupational function.29 Early reports suggest that patients with a disease duration of less than 10 years who have undergone operation can maintain a baseline involvement in their work and personal lives postoperatively.28 Schüpbach and associates30 performed a matched-pairs study of 20 PD patients with a mean symptom duration of 7 years, comparing functional outcomes in patients receiving DBS versus best medical therapy. Eighteen months postoperatively, the surgical patients experienced a 24% improvement in disease-specific quality of life versus 0% in the nonsurgical patients. The principal investigator of this study, Dr. Agid, is now leading a multicenter, randomized, open-label, phase III trial (EARLYSTIM Study) examining quality of life outcomes in patients with STN-DBS versus medical therapy, with 2-year follow-up data expected in 2010.27 This promises to offer further insight into the potential benefits of early surgery.
A New Target: The Pedunculopontine Nucleus
Postural and gait dysfunction leading to falls is the leading cause of emergency room visits and is a significant cost generator in the care of PD patients.31–33 In addition, the fear of falling results in depression and a loss of independence.34 Postural and gait dysfunction is particularly resistant to current dopaminergic and surgical therapies, suggesting a greater involvement of nondopaminergic pathways and brain loci other than the pallidal and subthalamic nuclei in their pathophysiology.13,14,35–41
The pedunculopontine nucleus (PPN) is part of a brainstem locomotive center that also processes sensory and behavioral information. Running roughly between the SNc and locus caeruleus, the PPN primarily connects with basal ganglia structures and the spinal cord.42,43 Midfrequency electrical or glutamatergic stimulation of the PPN leads to increased activity of its neuronal population and produces controlled locomotion on a treadmill in decerebrate animals.44,45 Other neurotransmitters, including γ-aminobutyric acid (GABA) and acetylcholine, decrease PPN activity and inhibit locomotion.42 It has been theorized that the PPN regulates tone and locomotion by modulating lower brainstem nuclei.46,47 These nuclei, in turn, activate central pattern generators in the spinal cord, which generate the motor patterns governing locomotion and limb tone.47–50 Given the evidence for the PPN’s role in the initiation and maintenance of gait and the stability of postural tone, as well as its pharmacologic and electrophysiologic properties that appear to be reliant on cholinergic and other nondopaminergic neurotransmitters, pathologic function related to this structure could theoretically play a significant role in the gait and postural manifestations of PD.42
Although direct evidence linking PPN dysfunction to PD-related postural and gait disturbances is lacking, there are several hints at a pathophysiologic connection. The PPN in anesthetized rats with hydroxydopamine (6-OHDA) nigral lesions is characterized by abnormally enhanced activity compared with controls.51 Furthermore, some of the electrophysiologic changes recorded in the basal ganglia structures of these PD-model animals could be reversed by PPN lesions.52,53 Matsumura and Kojima54 observed a smaller degree of nigral degeneration and fewer parkinsonian symptoms if monkeys with 1-methyl, 4-phenyl, 1,2,3,6-tetrahydropyridine (MPTP) lesions were given PPN lesions. Similarly, Nandi and colleagues55 showed that microinjections of the GABAergic antagonist bicuculline into the PPN improve parkinsonian symptoms in MPTP primates. Electrophysiologic changes have been matched to metabolic alterations in the PPN of parkinsonian rodents and nonhuman primates.56,57 More important, studies of human pathology demonstrate that a significant proportion of the large cholinergic PPN neurons degenerate in PD.58–60 These individual lines of evidence collectively invoke the possibility that PD-related degeneration of nondopaminergic PPN neurons leads to electrophysiologic and metabolic disturbances that culminate in the dysregulation of PPN-mediated gait and postural functions. Therefore, medical or surgical interventions that modulate PPN activity in PD patients might normalize gait initiation and maintenance and postural stability.
Following this hypothesis, DBS of the PPN is being evaluated as a therapeutic modality in PD patients with significant gait disturbance (Fig. 84-2).61–63 Initial subjects have included PD patients with refractory and functionally significant impairment of gait initiation (debilitating freezing) and postural stability (associated with an unacceptable risk of falling). Those suffering from comorbidities that might adversely affect gait and posture, including dementia, primary affective disorders, and marked sensory impairments, are considered poor candidates for PPN stimulation.64–67
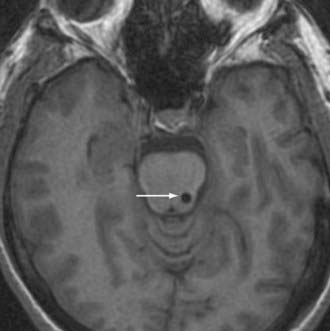
Initial reports of PPN-DBS in PD patients documented the technical aspects and electrophysiologic findings of the procedure, as well as short-term indicators of clinical outcome.61,62 A more recent study provided greater insight into the clinical outcome of six patients who underwent simultaneous bilateral PPN- and STN-DBS.63 Six months after surgery, Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores in the “off-medication–on-stimulation” condition were improved by an average of 33% with PPN-DBS alone. This was less than the improvement obtained from STN-DBS alone (54%) or simultaneous STN- and PPN-DBS (56%) in these same patients. Improvements specifically in axial symptoms (e.g., rising from a chair, posture, gait, postural stability) were similar with PPN-only, STN-only, and simultaneous PPN- and STN-DBS (60% to 70%). In patients taking medication, Stefani and colleagues63 found a greater improvement in UPDRS motor scores with combined STN and PPN stimulation compared with stimulation at either target alone. Moreover, STN-only stimulation did not improve axial symptoms in patients who were taking medication. If only axial symptoms were taken into account in the on-medication condition, both PPN-only DBS and simultaneous PPN- and STN-DBS led to a 50% to 60% improvement versus no stimulation. Importantly, no major complications were described. The only minor side effect reported was paresthesias associated with high-frequency PPN stimulation, likely resulting from the target’s proximity to the medial lemniscus.63 In summary, reports suggest the possibility of a therapeutic response to low-frequency PPN stimulation, but larger controlled studies are needed to verify these results and define its potential role in the treatment of PD.
Motor Cortex Stimulation
The concept of therapeutic modulation of cortical activity in movement disorders arose following James Parkinson’s description of the temporary abolishment of a tremor following a cortical stroke in 1824.68 Reports in the mid-1900s described improvements in posttraumatic and parkinsonian tremors following motor cortex or corticospinal tract lesioning.69,70 More recently (1979), Woolsey and colleagues71 observed immediate and temporary improvements in rigidity and tremor in PD patients undergoing intraoperative motor cortex stimulation (MCS) for the purpose of brain mapping, and several reports have described a simultaneous improvement of tremor in patients undergoing MCS for the treatment of pain.72–74
Further support for the attempt to modulate cortical activity in PD patients comes from evidence linking the disease to regional cortical dysfunction. Functional neuroimaging and electroencephalography in PD patients demonstrate hypoactivity in the supplementary motor area and dorsolateral prefrontal cortex.75–84 Interestingly, MPTP monkeys receiving chronic MCS (cMCS) exhibited a reversal of hypoactivity in the supplementary motor area and an improvement in akinesia.85 Changes in motor cortical activity are also observed in PD patients, with reduced activity early in the disease and increased activity in advanced disease.78,79,86,87 Although this variation remains poorly understood, hyperactivity may reflect compensatory cortical reorganization resulting from an increasingly deficient subcortical motor system, and it has been theorized to promote the development of levodopa-induced dyskinesias.88
Based on the observation of abnormal motor cortex activity in PD and clinical reports suggesting an acute benefit from MCS, a rationale emerged for investigating the use of cMCS in PD. Both theoretical predictions and experimental results suggested that cMCS might exert orthodromic or antidromic effects on subcortical elements of corticobasal ganglia loops (Fig. 84-3).85,89–91 STN-DBS in PD patients evokes short-latency responses, improves movement-related asynchrony, restores intracortical inhibition, and decreases resting activity in the human motor cortex.92–98 Similarly, the motor cortices can affect basal ganglia activities. Microelectrode recordings in MPTP monkeys receiving cMCS show substantial normalization of the mean firing rate and less abnormal synchronized oscillatory activity of GPi and STN neurons.85 In humans with PD, MCS using a single pulse of transcranial magnetic stimulation (TMS) evokes short-latency excitation, followed by long-lasting inhibition, of neurons located in the dorsolateral sensorimotor area of the STN.91 PET studies also demonstrate that MCS using 10-Hz TMS induces focal dopamine release within the ipsilateral dorsal striatum.89,90
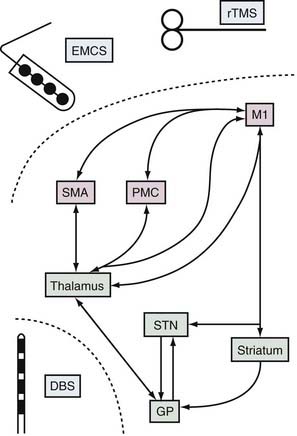
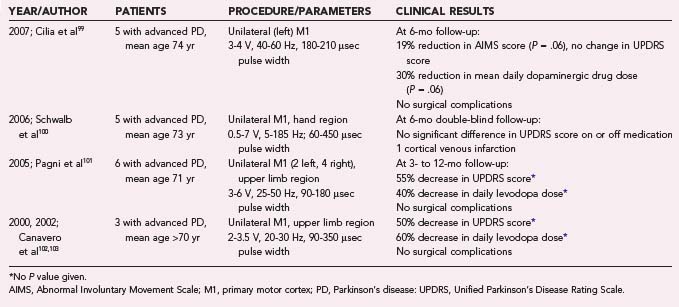
Epidural and Subdural Electrodes
Internalized epidural and subdural electrodes provide titratable systems with relatively precise target specificity, allowing focal stimulation while avoiding the greater risk of intraparenchymal placement. Since 2000, five reports have described 19 patients with advanced PD who were treated with epidural or subdural cMCS (Table 84-1).99–103 Although these publications collectively suggest a trend toward varying degrees of improvement across all clinically relevant motor domains, the modest benefits reported remain unconvincing. One poorly understood finding is the relatively consistent occurrence of bilateral improvement in symptoms despite unilateral cMCS, a phenomenon that is generally not observed with DBS. Further evidence indicates that cMCS in PD patients produces no alteration in movement-related regional cerebral blood flow as measured by PET scans, casting further doubt on its potential utility in PD.104 An additional report investigated the use of subdural cMCS for multiple system atrophy with predominant parkinsonism, a rapidly progressive neurodegenerative disease that shares some motor features with PD but for which treatments are generally ineffective.105 In the five patients studied, neither low- nor high-frequency stimulation had any benefit. Larger studies in appropriate patients may better define the efficacy and potential indications for epidural and subdural cMCS in PD. At the time of this writing, additional small phase I-II randomized, controlled, and crossover clinical trials investigating cMCS in PD are ongoing in France and Italy, with expected completion in the next couple of years.27
Transcranial Magnetic Stimulation
TMS is a noninvasive technique that induces changes in cortical activity through electromagnetic induction (see Fig. 84-3). Repetitive TMS (rTMS) delivered to the motor cortex at 5 to 25 Hz improves motor performance in PD patients.106–111 A recent systematic review of noninvasive neurostimulation in PD pooled the results from eight double-blind, sham-controlled studies of rTMS involving patients with a mean age and disease duration of 63 and 7 years, respectively. The standardized mean difference in pre- and posttreatment UPDRS scores was a modest 0.60 (95% confidence interval, 0.24 to 0.96), with no significant heterogeneity. Moreover, the longevity of this modest effect is uncertain, given the less than 2 months of follow-up.112 Frequencies of approximately 1 Hz applied to the supplementary motor area worsened motor performance but improved levodopa-induced dyskinesias.113,114 One interesting study demonstrated a concomitant increase in motor performance and relief of depression with rTMS applied to the dorsolateral prefrontal cortex, raising the possibility that modulating cortical activity might address the often debilitating comorbid depression that is often observed in patients with advancing PD.115 Other studies have failed to demonstrate any motor benefits with stimulation at the dorsolateral prefrontal cortex.116–118 Despite the appeal of its minimal invasiveness, the modest demonstrated efficacy of TMS and the practical issues involved in using it over a long period may ultimately limit its clinical utility.
Intraparenchymal Growth Factor Delivery
Glial cell line–derived neurotrophic factor (GDNF) is a neurotrophin protein that belongs to the transforming growth factor-β superfamily.119 Along with other cell types, dopaminergic neurons respond to GDNF through cell signaling pathways and react with enhanced growth, survival, differentiation, and regeneration.119 The cerebral administration of GDNF through viral vector delivery, intracerebroventricular infusion, or putaminal injection in rodent and nonhuman primate models of PD imparts neuroprotective and neuroregenerative effects.120–124 In addition, 6-OHDA rodents and MPTP primates demonstrate motor performance improvements following GDNF therapy.122,124–127
Based on these findings and the need to develop disease-modifying therapies for PD, studies have been undertaken to determine whether GDNF benefits PD patients. Cloning of the human GDNF gene led to the production of a human recombinant peptide (r-metHuGDNF, liatermin; Amgen, Thousand Oaks, CA) available for human administration. To circumvent the blood-brain barrier, targeted surgical delivery has been employed. The first randomized, controlled trial in PD patients to be published involved monthly bolus injections into the frontal horn of the lateral ventricle through an implanted cannula connected to an access port.128 Blinded outcomes in 50 patients at 8 months failed to demonstrate an improvement in UPDRS scores over a range of GDNF doses, and fairly frequent adverse effects were noted. Although these results were discouraging, an independent postmortem study of a PD patient who had received intraventricular GDNF suggested that this route of administration did not deliver GDNF to the dopamine-deficient putamen.129 Further, it appears that correct location is more important than dose when examining the efficacy of GDNF delivery in primate PD models.130
A smaller single-center, open-label trial of five PD patients receiving continuous intraputaminal infusion reported a 32% mean improvement in off-medication UPDRS motor scores at 6 months follow-up.131 There was also a significant increase in putaminal F-dopa uptake on PET imaging near the catheter tip. Most important, this route of administration was well tolerated. Slevin and colleagues132 reported similar results, documenting a mean 30% improvement in the off-medication UPDRS motor scores 6 months after the start of therapy in an open-label trial of 10 PD patients. However, in a randomized, controlled, blinded trial of intraputaminal GDNF infusion in 34 patients with idiopathic PD, no significant difference was noted in the UPDRS scores of patients receiving active GDNF and those receiving placebo.133 The fact that greater F-dopa uptake was noted on the PET scans of treated patients suggests a biologic effect that did not translate into a clinical response.
Despite the failure of intraparenchymal GDNF therapy in clinical trials, hope remains that the exciting responses of animal models of PD to growth factor delivery will eventually be translated into therapies. More basic research is required to better understand the mechanisms behind this animal-human discrepancy. Hints are beginning to emerge, including evidence that the neuroprotective action of GDNF may rely on other factors, such as transforming growth factor-β, and that drug delivery may need to be optimized to ensure ideal exposure of the targeted tissue.134,135 Indeed, the targeted surgical delivery of compounds that promote neuronal health, survival, and regeneration may one day be a reality as new data come to light.
Gene Therapy
Gene therapies for PD have long been sought as a means of providing targeted symptomatic, neuroprotective, and neuroregenerative therapy. Potential benefits include the ability to produce biologically active molecules within the brain and thus circumvent the blood-brain barrier, focus therapy in specific brain regions and thereby avoid side effects related to the exposure of other areas, and address potential underlying causes of the disorder directly. Although gene therapy has yielded encouraging preclinical results for a number of disorders, safety and technical concerns have hampered its successful translation into clinical use.136,137 Nonetheless, three gene therapy strategies have made considerable progress toward becoming viable therapies, all of which employ stereotactic delivery to specific brain targets and adeno-associated virus (AAV) vectors to mediate gene transfer. These strategies are increasing striatal dopamine production, reducing STN output, and expressing an ectopic growth factor in the striatum.
Augmenting Striatal Dopamine Production
This strategy aims to increase dopamine production in neurons and glia within the striatum by ectopically expressing the enzyme aromatic L-amino-acid decarboxylase (AADC), which catalyzes the conversion of levodopa to dopamine (Fig. 84-4). In essence, this is an extension of current pharmacologic interventions with dopaminergic drugs. Bankiewicz and associates138 theorized that declining levels of AADC not only contribute to the disease by reducing dopamine production but also lead to a backlog of exogenous levodopa, which may promote the development of levodopa-induced dyskinesias.
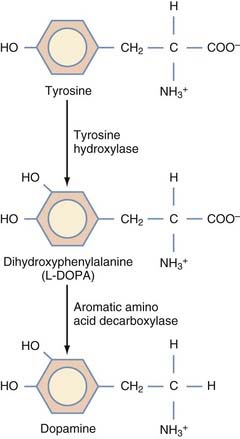
Convection-enhanced delivery of AAV-AADC to the striatum increases that nucleus’s capacity to decarboxylate exogenous levodopa in animal models of PD. Efficient and long-term expression of the transgene in the striatum restores local dopamine production and promotes behavioral recovery in 6-OHDA rats and MPTP monkeys treated with levodopa.138–142 PET data suggest that these same monkeys exhibit persistent AADC activity for at least 6 years after transfection.138,140 Similar studies using the simultaneous transduction of tyrosine hydroxylase and AADC showed a significant increase in intracellular dopamine in the striatum, producing better behavioral recovery than with the transduction of either enzyme alone; however, this multipronged approach adds complexity to the delivery.143–145
A human phase I study of AAV-AADC gene therapy for idiopathic PD is now under way.146 A noteworthy inclusion criterion is the presence of intractable motor fluctuations. Patients receive three escalating doses through bilateral stereotactic infusions into the postcommissural putamen, delivered in two 50-µL volumes spaced approximately 6 mm apart. The key outcome measures are an improvement in 18F-fluorometatyrosine PET signal at 1 and 6 months, indicating the presence of the AADC transgene, and an improvement in standard clinical rating scales. Preliminary data in five patients who received the lowest dose demonstrated increases in PET signal, suggesting successful AADC transgene expression.146 Future results, particularly from the higher dose groups, should shed light on the potential safety and efficacy of this approach.
Altering Pathologic Subthalamic Nucleus Output
The second approach to gene therapy for PD under active investigation aims to modify STN output through ectopic expression of glutamic acid decarboxylase (GAD), which catalyzes the production of GABA. The theory behind the therapy is that the local production of GAD transforms the STN projection neurons, which are naturally glutamatergic and excitatory, into inhibitory GABAergic neurons (Fig. 84-5), thereby reducing the excessive stimulatory effects of the STN on the GPi, which are thought to contribute to many parkinsonian symptoms. This follows the theory that STN-DBS generates its positive effects in PD by similarly reducing STN excitatory output.147
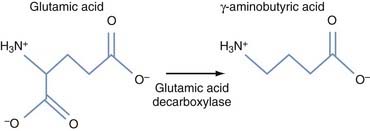
Before considering a clinical application of this concept, AAV-GAD was injected into the STN of 6-OHDA rats.148 These animals demonstrated activity-dependent GABA release from STN nerve terminals, increased survival of dopaminergic neurons, and marked behavioral recovery. Moreover, GAD transduction of the STN appeared to protect SNc dopaminergic neurons by preventing the lesion induced by 6-OHDA. A recently published study in MPTP monkeys given AAV-GAD injections found more modest improvements (about 20%) in motor behavior, although statistically significant gross motor skill and tremor improvements were noted at 1 year.149
A human phase I clinical trial of AAV-GAD gene therapy is well under way, with interim data already published.150 Twelve PD patients with reduced efficacy or tolerance of levodopa underwent unilateral stereotactic STN injections of one of three vector doses based on magnetic resonance imaging targeting. Outcome measures include changes in brain metabolism on PET scans and standard clinical rating scales. No study-related significant adverse events or withdrawals occurred, and no new antivector or anti-GAD peripheral antibodies were detected. At 1 year of follow-up, modest but significant improvements were noted in UPDRS scores, along with a trend toward superior activities of daily living scores. Further, fluorodeoxyglucose (FDG) PET demonstrated reduced thalamic metabolism in the operated hemisphere and increased local metabolism in the ipsilateral supplementary motor area, the latter being highly correlated with UPDRS score improvement.150,151 These promising clinical findings and supportive imaging studies, along with a favorable safety profile, suggest that AAV-GAD gene therapy may be beneficial in PD. Although future data from this trial will help confirm this early impression, a subsequent phase II trial will be required to judge whether the magnitude of effect is clinically meaningful.
Growth Factor Gene Therapy
Unlike the other two strategies, which are directed at controlling PD symptoms, the goal of this therapy is to prevent ongoing degeneration of dopamine neurons by ectopically expressing the growth factor neurturin throughout the striatum. Growth factors (particularly GDNF) attenuate the loss of nigrostriatal dopaminergic neurons and restore function of the nigrostriatal pathway in animal models of PD.119,152 Accordingly, targeted delivery of a gene that promotes growth factor production in the human SNc might achieve clinically meaningful neuroprotection or neurorestoration in PD patients. Owing to intellectual property issues, clinical gene therapy with GDNF is impossible at this time; however, work is proceeding with a related neurotrophin, neurturin. Neurturin is a naturally occurring structural and functional GDNF analogue and has already been used in patients.153–155
Lentivirus- and AAV-mediated GDNF gene therapy and striatal neurturin infusion have shown protective effects against the loss of dopaminergic nigral neurons and the motor consequences of MPTP intoxication in primates and 6-OHDA lesioning in rats.122,156,157 However, it was uncertain whether benefits would also be realized when nigral degeneration was quite advanced at the time of disease diagnosis. Kordower and colleagues158 performed AAV-neurturin gene therapy in MPTP monkeys and showed that neurturin expression is associated with substantial histologic and clinical neuroprotection. Importantly, these monkeys received the vector infusion following MPTP administration at a time when nigral destruction was under way. AAV-neurturin also improved motor function in aged monkeys, restoring failing nigrostrial function.159 As further confirmation of its biologic effect, AAV-neurturin provided efficient, persistent, and behaviorally relevant neuroprotection in the 6-OHDA rat model.160 These data suggest that neurturin gene therapy might benefit patients with preexisting PD.
Following the success of the phase I trial, a phase II sham-surgery controlled efficacy study enrolling 58 patients began in 2007.161 Disappointingly, despite being safe and well tolerated, there was no appreciable difference between experimental and control groups.161 Both showed an approximate 7-point improvement in UPDRS motor scores at 12 months, relative to a baseline mean of approximately 39 points.161 Recently released 18-month follow-up data from 30 patients found a clinically modest but statistically significant treatment effect.161 Further details have not been released, other than postmortem results from 2 patients indicating neurturin expression in the putamen that was not transported to the substantia nigra. These findings leave the status of further efforts in doubt.
Cellular Replenishment Strategies
Fetal Tissue Transplantation
Neural transplantation strategies seek to reverse some of the symptoms of PD by replacing the degenerating dopaminergic cells. Although it is increasingly recognized that PD involves neurodegeneration in both dopaminergic and nondopaminergic systems,162,163 attention has focused on replacing the dopaminergic neurons of the SNc, whose degeneration is PD’s hallmark. Thus far, fetal dopaminergic cells have been transplanted not into their ontogenic site in the SNc but ectopically into the putamen, because cells transplanted into the SNc failed to extend their processes to their target nuclei—the putamen and caudate nuclei.164 When assessing the potential impact of transplantation, it is important to note that an exclusive focus on repleting lost dopaminergic cells may limit the ability of these technologies to fully address the disabilities caused by PD.
Experimental neural transplantation therapies for PD are based on almost 40 years of preclinical work using animal models in which nigrostriatal dopaminergic neurons were selectively lesioned with neurotoxins. Initial studies performed in the mid-1970s were concerned mainly with histologic aspects of graft survival.165,166 Later, the functional effects of transplanted cells were assessed.164,167,168 Transplanted dopaminergic neurons have been shown to survive after transplantation,169 produce and secrete dopamine,170 form synaptic connections with host neurons,171 and attenuate both simple motor behaviors, such as asymmetric rotational behaviors induced by the administration of dopamine agonists, and more complex sensorimotor deficits, such as akinesia and motor forelimb function.172
These encouraging results provided experimental evidence that cell restoration strategies may be beneficial in the treatment of PD. Results of open-label clinical studies in which ventral mesencephalic tissue procured from 6- to 8-week fetuses was transplanted into the putamen of PD patients resemble those obtained from animal experiments. The first clinical trials of transplanted fetal dopaminergic cell therapy began in 1987.173,174 Since then more than 350 patients have received fetal dopaminergic cell transplants, although the number of centers performing the procedure is limited. Initially, solid tissue grafts were implanted via an open microsurgical technique on the ventricular surface of the caudate nucleus.175,176 Stereotactic injection of grafted tissue gradually became the preferred approach, with tissue implanted either as solid strands177 or as a dissociated single-cell suspension.178–180 Cell suspension grafts appear to develop greater vascularization,181 integration with host structures,180 and axonal reinnervation182 than their solid tissue counterparts. Autopsy studies demonstrate that transplanted cells survive (Fig. 84-6)—in some cases, for as long as a decade180,183—although the implanted cells may undergo early senescence with Lewy body inclusions.184 Graft survival and function are further supported by corresponding increases in F-dopa signal and evidence of enhanced dopamine release on PET.185 Objective measures of improved dopaminergic function are mirrored by some patients who have undergone transplantation and who exhibit significant improvements in motor performance,186–189 including reductions in bradykinesia and rigidity, improved “on” times (defined as periods of adequate disease control while on medication), and decreased daily doses of levodopa, with some patients not requiring any medication several years after transplantation.186,189,190
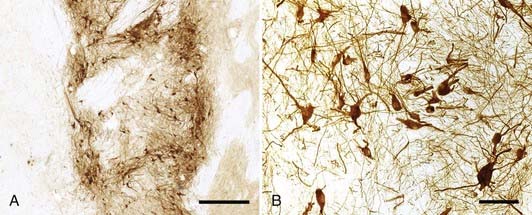
Results from two National Institutes of Health–sponsored double-blind, randomized, controlled trials undertaken to assess the effects of neural transplantation in PD were less promising than those of the open-label studies.177,191 Both were controversial, in that they included groups of patients who received placebo surgery consisting of incision and bur holes with no implantation of cells or brain penetration. In the first study, a transfrontal approach was used to stereotactically deliver strands of solid ventral mesencephalic tissue to the putamen bilaterally.177 By 1 year after transplantation, patients who received tissue grafts demonstrated an insignificant 18% improvement in the total UPDRS score in the “off” phase (defined as the clinical state while off levodopa for at least 12 hours), although a subset of patients younger than 60 years exhibited a significant 34% improvement in UPDRS motor subscores. Patients demonstrated relatively consistent graft viability as assessed by F-dopa PET imaging. In the second trial, patients were randomized to either placebo surgery or transplantation with solid ventral mesencephalic grafts derived from either one or four fetuses (i.e., a three-armed study).191 As in the first clinical trial, no significant improvement in UPDRS motor scores was observed among the groups of patients. Patients who received grafts derived from four fetuses demonstrated clinical improvement up to 9 months after transplantation, but their UPDRS scores returned to preoperative levels by the end of the study period (24 months). Aside from the lack of demonstrated benefit of fetal transplantation, the trials were also disappointing in that 15%177 and 56%191 of patients developed severe off-phase dyskinesias as a result of the transplants. Critics of these trials proposed that the suboptimal results were due to flaws in the study design and methodology, including the absence177 or only short-term use191 of immunosuppression, the culturing of fetal tissue for extended periods (up to 1 month),177 poor transplanted cell survival,177 and the inclusion of patients with more severe disease than were included in open-label trials.177,191
These disappointing results reinforce the need to optimize cell restoration strategies in experimental settings before attempting their clinical use. Optimization should include standardization of transplantation procedures, including patient selection; procurement, storage, and preparation of fetal cells; and transplantation technique.192 Absent optimization, it may be premature to dismiss transplantation entirely without due consideration of the variables that may be critical to improve its efficacy. These include transplantation targets, graft-induced dyskinesias, and cell sources.
Targets for Transplantation
Although the standard transplantation paradigm has been to implant fetal dopaminergic cells in the putamen, it is clear that such grafts do not sufficiently reconstruct the dopaminergic basal ganglia circuitry that is affected in PD. For example, metabolic activity is not normalized in other basal ganglia structures, such as the substantia nigra (SN) and STN,193 whose physiologic regulation is dependent, at least in part, on dopamine.172 To address this, fetal dopaminergic neurons have been transplanted into both the putamen and the SN in some patients, with evidence of graft survival and modest clinical benefits observed in short pilot studies.179,180 Additionally, to more fully reconstruct basal ganglia circuitry, the use of GABAergic grafts may be necessary because they have been beneficial in animal models.194
Graft-Induced Dyskinesias
The cause of the severe dyskinesias seen in patients who received transplants in the randomized clinical trials is poorly understood. Although initially hypothesized to be due to excessive dopamine production by the grafts, graft-induced dyskinesias are more likely related to graft size,195 with more pronounced abnormal involuntary movement observed in parkinsonian rats with large-volume dopaminergic grafts, and to patchy reinnervation of the caudate and putamen.196 The cellular composition of the grafts also influences the development of dyskinesias.197,198 Because only 5% to 10% of the fetal mesencephalic tissue is composed of dopaminergic neurons, the transplantation of serotoninergic cells also contained within the ventral mesencephalon may contribute to the development of dyskinesias.198 Furthermore, refinement of transplantation to include only a subset of dopaminergic neurons from the SN and not the ventral tegmental area may be necessary to prevent graft-induced dyskinesias.197
Cell Sources for Transplantation
Currently, neural transplantation relies on the availability of human fetal tissue, which limits its widespread application. Clinical data reveal that functional improvement—as measured by the UPDRS motor score, activities of daily living scores, and FDG-PET—is enhanced when patients undergo transplantation with tissue derived from at least three fetuses,177,178,180,187,188,199,200 suggesting that a mimimal number of transplanted dopaminergic cells must survive for clinical improvement to occur. Open-label autopsy studies suggest that the number is at least 100,000 cells.183 Based on these data, procurement of at least 15 fetal ventral mesencephalons is required per patient201,202 because as few as 1% of transplanted dopaminergic cells survive.186,203 Because this is logistically and politically difficult, current lines of research are investigating the use of adjunctive agents during transplantation to improve transplanted cell survival, including antiapoptotic agents and neurotrophic factors.178,189 Ultimately, dopaminergic neurons need to be produced in unlimited quantities in a standardized fashion if neural transplantation therapies for PD are to be clinically viable.204
Neural Stem Cell Transplantation
Neural stem cells are attractive because of their ability to self-renew; this allows their growth in culture, which can generate the large numbers required for widespread clinical transplantation.205,206 Moreover, their multipotential ability to differentiate into enriched populations of neurons and glia may one day allow them to be used in numerous transplantation strategies (Fig. 84-7).207
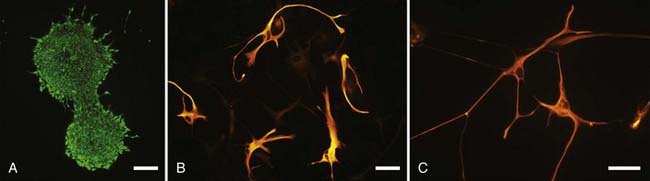
Recent studies have demonstrated the feasibility of generating dopaminergic cells from neural stem cells. Embryonic stem cells transplanted into the striatum of a parkinsonian rodent model spontaneously differentiated into dopaminergic cells, albeit in low numbers.208 The efficiency of dopaminergic differentiation has been improved using a variety of techniques, including modification of the culture environment with mitogens (e.g., sonic hedgehog),209 neurotrophic factors (e.g., fibroblast growth factor 8, GDNF, brain-derived neurotrophic factor),210 low oxygen,211 and cytokines.212 Additionally, genetic manipulation to overexpress regulatory genes of dopaminergic differentiation, such as Nurr1,209 Lmx1b,213 and Wnt5a,214 has been used to enhance dopamine cell production. Neural stem cells modified under these conditions can improve cortical activation208 and functional outcomes in parkinsonian animals, such as dopamine agonist–induced rotational asymmetry in rodents.208,209,212
Although neural stem cell–derived dopaminergic cells are a promising source of cells for neural transplantation, clinical applicability is limited by a number of factors. First, differentiation of stem cells exclusively into non-neoplastic neurons has not been consistent,208 and long-term studies of their teratogenic potential are lacking.215 Second, dopaminergic stem cells exhibit poor survival following transplantation, with some reports estimating that as few as 1% of the transplanted cells survive beyond 6 weeks.211,212 Last, although a few studies have shown that transplanted dopaminergic neural stem cells have some of the electrophysiologic properties of midbrain dopaminergic neurons,209,214,216 stem cell–derived dopaminergic cells need to demonstrate functional reinnervation of the basal ganglia, in addition to the molecular and morphologic properties of SN dopaminergic neurons, if they are ultimately to be used to replace them.217
In addition to being a source of replacement cells, stem cells may be useful for their ability to secrete factors that affect the fate of neighboring cells. These factors can support the survival of endogenous or grafted dopaminergic neurons (e.g., GDNF, sonic hedgehog).218,219 The production of an array of trophic factors, especially at physiologically relevant levels, may be important for transplantation strategies in which transplanted fetal dopaminergic neurons require exposure to a variety of neurotrophic factors than cannot be provided by cells genetically programmed to produce only one or two of them.220 Neural stem cell–produced factors are neuroprotective in parkinsonian rodent models, protecting endogenous dopaminergic neurons in the SN from MPTP-induced218 or 6-OHDA–induced221 neurotoxicity, restoring the size of caudate nucleus type 1 dopaminergic cells to control levels, and preventing the upregulation of dopaminergic cell numbers in the caudate and putamen in primates with MPTP lesions.222 Cotransplantation with murine subventricular zone neural stem cells approximately doubled the number of surviving fetal dopaminergic cells.219 All these measures of neuroprotection correlated with significant attenuation of parkinsonian motor behaviors. Therefore, the utility of neural stem cells may be as adjuncts to fetal tissue transplantation or to deliver neuroprotective agents to preserve endogenous dopaminergic nigrostriatal function.
Repair by Endogenous Stem Cells
Rather than implanting exogenous, genetically altered embryonic stem cells in the brains of PD patients, one can harness endogenous stem cells, located mainly in the subventricular zone and subgranular zone of the dentate gyrus of the adult mammalian brain,223 to repair the dopaminergic nigrostriatal system. Normally, few dopaminergic cells are present within the putamen and caudate nuclei224,225; however their numbers increase significantly in these locations in PD, suggesting a compensatory dopaminergic differentiation of endogenous neural stem cells that migrate out of the subventricular zone and into the striatum.226,227 The number of subventricular zone stem cells that proliferate, migrate into the striatum, and then differentiate into neurons can be enhanced by the intraventricular injection of trophic factors such as brain-derived neurotrophic factor, platelet-derived growth factor, and transforming growth factor-α228,229; in other studies, however, neuronal differentiation was not observed.230 Administration of agonists to the dopamine D3 receptor can also increase stem cell proliferation in the subventricular zone, striatum, SN, and ependymal layer of the third ventricle and promote their differentiation into dopaminergic cells.231,232 Whether endogenous dopamine neurogenesis occurs in the SN is still a matter of debate.233,234
The prospect of using pharmacotherapies to induce endogenous neural stem cells to proliferate, migrate to areas of dopaminergic neurodegeneration, and differentiate into dopaminergic neurons is attractive because it would avoid many of the technical and logistical difficulties associated with fetal- or stem cell–derived transplantation strategies.235 Further studies exploring endogenous brain repair are needed, although the identification of neural stem cells throughout the neuraxis also means that such therapies need to be approached with caution235 because they could contribute to the formation of tumors.236
Braak H, Ghebremedhin E, Rüb U, et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121-134.
Fiandaca M, Forsayeth J, Bankiewicz K. Current status of gene therapy trials for Parkinson’s disease. Exp Neurol. 2008;209(1):51-57.
Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710-719.
Fregni F, Simon DK, Wu A, Pascual-Leone A. Non-invasive brain stimulation for Parkinson’s disease: a systematic review and meta-analysis of the literature. J Neurol Neurosurg Psychiatry. 2005;76(12):1614-1623.
Goldman S. Disease targets and strategies for the therapeutic modulation of endogenous neural stem and progenitor cells. Clin Pharmacol Therap. 2007;82:453-460.
Isacson O. The production and use of cells as therapeutic agents in neurodegenerative diseases. Lancet Neurol. 2003;2:417-424.
Kaplitt MG, Feigin A, Tang C, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369(9579):2097-2105.
Kim J, Auerbach J, Rodríguez-Gómez J, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418:50-56.
Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459-466. Erratum in: Ann Neurol. 2006;60(6):747
Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339(15):1044-1053.
Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. N Engl J Med. 1998;339(16):1130-1143.
Marks WJJr, Ostrem JL, Verhagen L, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 2008;7(5):400-408.
Mendez I, Sanchez-Pernaute R, Cooper O, et al. Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson’s disease. Brain. 2005;128:1498-1510.
Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223-250.
Olanow CW, Goetz CG, Kordower JH, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson’s disease. Brain. 2000;123(Pt 9):1767-1783.
Perlow MJ, Freed WJ, Hoffer BJ, et al. Brain grafts reduce motor abnormalities produced by destruction of nigrostriatal dopamine system. Science. 1979;204:643-647.
Priori A, Lefaucheur JP. Chronic epidural motor cortical stimulation for movement disorders. Lancet Neurol. 2007;6(3):279-286.
Schüpbach WM, Maltête D, Houeto JL, et al. Neurosurgery at an earlier stage of Parkinson disease: a randomized, controlled trial. Neurology. 2007;68(4):267-271.
Stefani A, Lozano AM, Peppe A, et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain. 2007;130:1596-1607.
1 Lilienfeld DE, Perl DP. Projected neurodegenerative disease mortality in the United States, 1990-2040. Neuroepidemiology. 1993;12:219-228.
2 Fall PA, Saleh A, Fredrickson M, et al. Survival time, mortality, and cause of death in elderly patients with Parkinson’s disease: a 9-year follow-up. Mov Disord. 2003;18:1312-1316.
3 Morens DAM, Davis JW, Grandinetti A, et al. Epidemiologic observations on Parkinson’s disease: incidence and mortality in a prospective study of middle-aged men. Neurology. 1996;46:1044-1050.
4 Huse DM, Schulman K, Orsini L, et al. Burden of illness in Parkinson’s disease. Mov Disord. 2005;20(11):1449-1454.
5 Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339(15):1044-1053.
6 Jellinger K. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153-197.
7 Wakabayashi K, Takahashi H. Neuropathology of autonomic nervous system in Parkinson’s disease. Eur Neurol. 1997;38(Suppl 2):2-7.
8 Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283-2301.
9 Braak H, Ghebremedhin E, Rüb U, et al. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318(1):121-134.
10 Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J Neuropathol Exp Neurol. 1993;52:183-191.
11 Miyawaki E, Lyons K, Pahwa R, et al. Motor complications of chronic levodopa therapy in Parkinson’s disease. Clin Neuropharmacol. 1997;20:523-530.
12 Lang AE, Lozano AM. Parkinson’s disease. Second of two parts. N Engl J Med. 1998;339(16):1130-1143.
13 Klawans HL. Individual manifestations of Parkinson’s disease after ten or more years of levodopa. Mov Disord. 1986;1:187-192.
14 Rascol O, Payoux P, Ory F, et al. Limitations of current Parkinson’s disease therapy. Ann Neurol. 2003;53(Suppl 3):S3-S12.
15 Hamani C, Neimat J, Lozano AM. Deep brain stimulation for the treatment of Parkinson’s disease. J Neural Transm Suppl. 2006;70:393-399.
16 Moro E, Lang AE. Criteria for deep-brain stimulation in Parkinson’s disease: review and analysis. Expert Rev Neurother. 2006;6(11):1695-1705.
17 Deuschl G, Schade-Brittinger C, Krack P, et al. German Parkinson Study Group, Neurostimulation Section. A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med. 2006;355(9):896-908. Erratum in: N Engl J Med. 2006;355(12):1289
18 Piallat B, Benazzouz A, Benabid AL. Subthalamic nucleus lesion in rats prevents dopaminergic nigral neuron degeneration after striatal 6-OHDA injection: behavioural and immunohistochemical studies. Eur J Neurosci. 1996;8:1408-1414.
19 Nakao N, Nakai E, Nakai K, et al. Ablation of the subthalamic nucleus supports the survival of nigral dopaminergic neurons after nigrostriatal lesions induced by the mitochondrial toxin 3-nitropropionic acid. Ann Neurol. 1999;45:640-651.
20 Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson’s disease: a target for neuroprotection. Ann Neurol. 1998;44:S175-S188.
21 Benazzous A, Paillat B, Ni ZG, et al. Implication of the subthalamic nucleus in the pathophysiology and pathogenesis of Parkinson’s disease. Cell Transplant. 2000;9:215-221.
22 Hilker R, Portman AT, Voges J, et al. Disease progression continues in patients with advanced Parkinson’s disease and effective subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry. 2005;76(9):1217-1221.
23 Nurmi E, Ruottinen HM, Kaasinen V, et al. Progression in Parkinson’s disease: a positron emission tomography study with a dopamine transporter ligand 18F-CFT. Ann Neurol. 2000;47:804-808.
24 Morrish PK, Sawle GV, Brooks DJ. The rate of progression of Parkinson’s disease. A longitudinal 18F-dopa PET study. Adv Neurol. 1996;69:427-431.
25 Leenders KL, Palmer AJ, Quinn N, et al. Brain dopamine metabolism in patients with Parkinson’s disease measured with positron emission tomography. J Neurol Neurosurg Psychiatry. 1986;49:853-860.
26 Morrish PK, Sawle GV, Brooks DJ. An 18F-dopa-PET and clinical study of the rate of progression in Parkinson’s disease. Brain. 1996;119:585-591.
27 http://www.clinicaltrials.gov.
28 Mesnage V, Houeto JL, Welter ML, et al. Parkinson’s disease: neurosurgery at an earlier stage? J Neurol Neurosurg Psychiatry. 2002;73:778-779.
29 Volkmann J. Deep brain stimulation for the treatment of Parkinson’s disease. J Clin Neurophysiol. 2004;21:6-17.
30 Schüpbach WM, Maltête D, Houeto JL, et al. Neurosurgery at an earlier stage of Parkinson disease: a randomized, controlled trial. Neurology. 2007;68(4):267-271.
31 Bloem B, Boers I, Cramer M, Westendorp R, Gerschlager W. Falls in the elderly. I. Identification of risk factors. Wien Klin Wochenschr. 2001;113:352-362.
32 Bloem BR, Grimbergen YA, Cramer M, et al. Prospective assessment of falls in Parkinson’s disease. J Neurol. 2001;248:950-958.
33 Schrag A, Ben-Shlomo Y, Quinn N. How common are complications of Parkinson’s disease? J Neurol. 2002;249:419-423.
34 Adkin AL, Frank JS, Jog MS. Fear of falling and postural control in Parkinson’s disease. Mov Disord. 2003;18:496-502.
35 Bonnet AM, Pichon J, Vidailhet M, et al. Urinary disturbances in striatonigral degeneration and Parkinson’s disease: clinical and urodynamic aspects. Mov Disord. 1997;12:509-513.
36 Horak FB, Carlson-Kihtz P, Stephens M, et al. Effects of deep brain stimulation and levodopa on a variety of postural tasks in Parkinson’s patients. Mov Disord. 2002;17:s194.
37 Kemoun G, Defebvre L. [Gait disorders in Parkinson disease. Clinical description, analysis of posture, initiation of stabilized gait]. Presse Med. 2001;30:452-459.
38 Kemoun G, Defebvre L. [Gait disorders in Parkinson disease. Gait freezing and falls: therapeutic management]. Presse Med. 2001;30:460-468.
39 Morris ME, Iansek R, Matyas TA, Summers JJ. Stride length regulation in Parkinson’s disease. Normalization strategies and underlying mechanisms. Brain. 1996;119:551-568.
40 Robertson LT, Horak FB, Anderson VC, et al. Assessments of axial motor control during deep brain stimulation in parkinsonian patients. Neurosurgery. 2001;48:544-551.
41 Rocchi L, Chiari L, Horak FB. Effects of deep brain stimulation and levodopa on postural sway in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2002;73:267-274.
42 Pahapill PA, Lozano AM. The pedunculopontine nucleus and Parkinson’s disease. Brain. 2000;123(Pt 9):1767-1783.
43 Muthusamy KA, Aravamuthan BR, Kringelbach ML, et al. Connectivity of the human pedunculopontine nucleus region and diffusion tensor imaging in surgical targeting. J Neurosurg. 2007;107(4):814-820.
44 Garcia-Rill E. The pedunculopontine nucleus. Prog Neurobiol. 1991;36:363-389.
45 Garcia-Rill E, Skinner RD. The mesencephalic locomotor region. I. Activation of a medullary projection site. Brain Res. 1987;411:1-12.
46 Garcia-Rill E, Skinner RD. The mesencephalic locomotor region. II. Projections to reticulospinal neurons. Brain Res. 1987;411:13-20.
47 Inglis WL, Winn P. The pedunculopontine tegmental nucleus: where the striatum meets the reticular formation. Prog Neurobiol. 1995;47:1-29.
48 Mileykovskiy B, Kiyashchenko L, Kodama T, et al. Activation of pontine and medullary motor inhibitory regions reduces discharge in neurons located in the locus coeruleus and the anatomical equivalent of the midbrain locomotor region. J Neurosci. 2000;20:8551-8558.
49 Garcia-Rill E, Skinner RD, Miyazato H, Homma Y. Pedunculopontine stimulation induces prolonged activation of pontine reticular neurons. Neuroscience. 2001;104:455-465.
50 Homma Y, Skinner R, Garcia-Rill E. Effects of pedunculopontine nucleus (PPN) stimulation on caudal pontine reticular formation (PnC) neurons in vitro. J Neurophysiol. 2002;87:3033-3047.
51 Breit S, Bouali-Benazzouz R, Benabid A, Benazzouz A. Unilateral lesion of the nigrostriatal pathway induces an increase of neuronal activity of the pedunculopontine nucleus, which is reversed by the lesion of the subthalamic nucleus in the rat. Eur J Neurosci. 2001;14:1833-1842.
52 Breit S, Lessmann L, Benazzouz A, Schulz JB. Unilateral lesion of the pedunculopontine nucleus induces hyperactivity in the subthalamic nucleus and substantia nigra in the rat. Eur J Neurosci. 2005;22:2283-2294.
53 Breit S, Lessmann L, Unterbrink D, et al. Lesion of the pedunculopontine nucleus reverses hyperactivity of the subthalamic nucleus and substantia nigra pars reticulata in a 6-hydroxydopamine rat model. Eur J Neurosci. 2006;24:2275-2282.
54 Matsumura M, Kojima J. The role of the pedunculopontine tegmental nucleus in experimental parkinsonism in primates. Stereotact Funct Neurosurg. 2001;77:108-115.
55 Nandi D, Aziz TZ, Giladi N, et al. Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain. 2002;125:2418-2430.
56 Chang JW, Yang JS, Jeon MF, et al. Effect of subthalamic lesion with kainic acid on the neuronal activities of the basal ganglia of rat parkinsonian models with 6-hydroxydopamine. Acta Neurochir Suppl. 2003;87:163-168.
57 Mitchell IJ, Clarke CE, Boyce S, et al. Neural mechanisms underlying parkinsonian symptoms based upon regional uptake of 2-deoxyglucose in monkeys exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 1989;32:213-226.
58 Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A. 1987;84:5976-5980.
59 Jellinger K. The pedunculopontine nucleus in Parkinson’s disease, progressive supranuclear palsy and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1988;51:540-543.
60 Zweig RM, Jankel WR, Hedreen JC, et al. The pedunculopontine nucleus in Parkinson’s disease. Ann Neurol. 1989;26:41-46.
61 Mazzone P, Lozano A, Stanzione P, et al. Implantation of human pedunculopontine nucleus: a safe and clinically relevant target in Parkinson’s disease. Neuroreport. 2005;16:1877-1881.
62 Plaha P, Gill SS. Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson’s disease. Neuroreport. 2005;16:1883-1887.
63 Stefani A, Lozano AM, Peppe A, et al. Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson’s disease. Brain. 2007;130:1596-1607.
64 Camicioli RM, Howieson DB, Lehman S, Kaye JA. Talking while walking: the effect of a dual task in aging and Alzheimer’s disease. Neurology. 1997;48:955-958.
65 Hausdor JM, Peng CK, Goldberger AL, Stoll AL. Gait unsteadiness and fall risk in two affective disorders: a preliminary study. BMC Psychiatry. 2004;4:39.
66 Springer S, Giladi N, Peretz C, et al. Dual-tasking effects on gait variability: the role of aging, falls, and executive function. Mov Disord. 2006;21:950-957.
67 Woollacott M, Shumway-Cook A. Attention and the control of posture and gait: a review of an emerging area of research. Gait Posture. 2002;16:1-14.
68 Speelman JD, Bosch DA. Resurgence of functional neurosurgery for Parkinson’s disease: a historical perspective. Mov Disord. 1998;13:582-588.
69 Bucy PC, Case TJ. Tremor, psychologic mechanism and abolition by surgical means. Arch Neurol. 1939;41:542-567.
70 Walker AE. Cerebral peduncolotomy for the relief of involuntary movements: parkinsonian tremor. J Nerv Ment Dis. 1952;116:767-775.
71 Woolsey CN, Erickson TC, Gilson WE. Localization in somatic sensory and motor areas of human cerebral cortex as determined by direct recording of evoked potentials and electrical stimulation. J Neurosurg. 1979;51:476-506.
72 Franzini A, Ferroli P, Dones I, et al. Chronic motor cortex stimulation for movement disorders: a promising perspective. Neurol Res. 2003;25:123-126.
73 Katayama Y, Oshima H, Fukaya C, et al. Control of post-stroke movement disorders using chronic motor cortex stimulation. Acta Neurochir Suppl (Wien). 2002;79:89-92.
74 Nguyen JP, Pollin B, Feve A, et al. Improvement of action tremor by chronic cortical stimulation. Mov Disord. 1998;13:84-88.
75 Jahanshahi M, Jenkins IH, Brown RG, et al. Self-initiated versus externally triggered movements I: an investigation using measurement of regional cerebral blood flow with PET and movement-related potentials in normal and Parkinson’s disease subjects. Brain. 1995;118:913-933.
76 Playford ED, Jenkins IH, Passingham RE, et al. Impaired mesial frontal and putamen activation in Parkinson’s disease: a positron emission tomography study. Ann Neurol. 1992;32:151-161.
77 Rascol O, Sabatini U, Chollet F, et al. Supplementary and primary sensory motor area activity in Parkinson’s disease: regional cerebral blood flow changes during finger movements and effects of apomorphine. Arch Neurol. 1992;49:144-148.
78 Haslinger B, Erhard P, Kampfe N, et al. Event-related functional magnetic resonance imaging in Parkinson’s disease before and after levodopa. Brain. 2001;124:558-570.
79 Sabatini U, Boulanouar K, Fabre N, et al. Cortical motor reorganization in akinetic patients with Parkinson’s disease: a functional MRI study. Brain. 2000;123:394-403.
80 Bostantjopoulou S, Katsarou Z, Zafiriou D, et al. Abnormality of N30 somatosensory evoked potentials in Parkinson’s disease: a multidisciplinary approach. Neurophysiol Clin. 2000;30:368-376.
81 Cunnington R, Iansek R, Johnson KA, Bradshaw JL. Movement related potentials in Parkinson’s disease: motor imagery and movement preparation. Brain. 1997;120:1339-1353.
82 Cunnington R, Lalouschek W, Dirnberger G, et al. A medial to lateral shift in pre-movement cortical activity in hemi-Parkinson’s disease. Clin Neurophysiol. 2001;112:608-618.
83 Pulvermuller F, Lutzenberger W, Muller V, et al. P3 and contingent negative variation in Parkinson’s disease. Electroencephalogr Clin Neurophysiol. 1996;98:456-467.
84 Rossini PM, Babiloni F, Bernardi G, et al. Abnormalities of short-latency somatosensory evoked potentials in parkinsonian patients. Electroencephalogr Clin Neurophysiol. 1989;74:277-289.
85 Drouot X, Oshino S, Jarraya B, et al. Functional recovery in a primate model of Parkinson’s disease following motor cortex stimulation. Neuron. 2004;44:769-778.
86 Buhmann C, Glauche V, Sturenburg HJ, et al. Pharmacologically modulated fMRI-cortical responsiveness to levodopa in drug-naive hemiparkinsonian patients. Brain. 2003;126:451-461.
87 Lefaucheur JP. Motor cortex dysfunction revealed by cortical excitability studies in Parkinson’s disease: influence of antiparkinsonian treatment and cortical stimulation. Clin Neurophysiol. 2005;116:244-253.
88 Rascol O, Sabatini U, Brefel C, et al. Cortical motor overactivation in parkinsonian patients with L-dopa-induced peak-dose dyskinesia. Brain. 1998;121:527-533.
89 Strafella AP, Paus T, Barrett J, Dagher A. Repetitive transcranial magnetic stimulation of the human prefrontal cortex induces dopamine release in the caudate nucleus. J Neurosci. 2001;21:RC157.
90 Strafella AP, Paus T, Fraraccio M, Dagher A. Striatal dopamine release induced by repetitive transcranial magnetic stimulation of the human motor cortex. Brain. 2003;126:2609-2615.
91 Strafella AP, Vanderwerf Y, Sadikot AF. Transcranial magnetic stimulation of the human motor cortex influences the neuronal activity of subthalamic nucleus. Eur J Neurosci. 2004;20:2245-2249.
92 MacKinnon CD, Webb RM, Silberstein P, et al. Stimulation through electrodes implanted near the subthalamic nucleus activates projections to motor areas of the cerebral cortex in patients with Parkinson’s disease. Eur J Neurosci. 2005;21:1394-1402.
93 Devos D, Labyt E, Derambure P, et al. Subthalamic nucleus stimulation modulates motor cortex oscillatory activity in Parkinson’s disease. Brain. 2004;127:408-419.
94 Meissner W, Leblois A, Hansel D, et al. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain. 2005;128:2372-2382.
95 Haslinger B, Kalteis K, Boecker H, et al. Frequency-correlated decreases of motor cortex activity associated with subthalamic nucleus stimulation in Parkinson’s disease. Neuroimage. 2005;28:598-606.
96 Ceballos-Baumann AO, Boecker H, Bartenstein P, et al. A positron emission tomographic study of subthalamic nucleus stimulation in Parkinson disease: enhanced movement-related activity of motor association cortex and decreased motor cortex resting activity. Arch Neurol. 1999;56:997-1003.
97 Hershey T, Revilla FJ, Wernle AR, et al. Cortical and subcortical blood flow effects of subthalamic nucleus stimulation in PD. Neurology. 2003;61:816-821.
98 Payoux P, Remy P, Damier P, et al. Subthalamic nucleus stimulation reduces abnormal motor cortical overactivity in Parkinson disease. Arch Neurol. 2004;61:1307-1313.
99 Cilia R, Landi A, Vergani F, et al. Extradural motor cortex stimulation in Parkinson’s disease. Mov Disord. 2007;22(1):111-114.
100 Schwalb J, Moro E, Hamani C, et al. Open label trial of subdural motor cortex stimulation (MCS) for Parkinson’s disease. Paper presented at the Biennial Meeting of the American Society for Stereotactic and Functional Neurosurgery, Boston http://www.content.karger.com/ProdukteDB/produkte.asp?Aktion=ShowPDF&ArtikelNr=97756&Ausgabe=232359&ProduktNr=224132&filename=97756.pdf, June 1-4, 2006.
101 Pagni CA, Zeme S, Zenga F, Maina R. Extradural motor cortex stimulation in advanced Parkinson’s disease: the Turin experience: technical case report. Neurosurgery. 2005;57(4 Suppl):E402.
102 Canavero S, Paolotti R. Extradural motor cortex stimulation for advanced Parkinson’s disease: case report. Mov Disord. 2000;15(1):169-171.
103 Canavero S, Paolotti R, Bonicalzi V, et al. Extradural motor cortex stimulation for advanced Parkinson disease. Report of two cases. J Neurosurg. 2002;97(5):1208-1211.
104 Strafella AP, Lozano AM, Lang AE, et al. Subdural motor cortex stimulation in Parkinson’s disease does not modify movement-related rCBF pattern. Mov Disord. 2007;22(14):2113-2116.
105 Kleiner-Fisman G, Fisman DN, Kahn FI, et al. Motor cortical stimulation for parkinsonism in multiple system atrophy. Arch Neurol. 2003;60(11):1554-1558.
106 Bornke CH, Schulte T, Przuntek H, Muller T. Clinical effects of repetitive transcranial magnetic stimulation versus acute levodopa challenge in Parkinson’s disease. J Neural Transm Suppl. 2004;68:61-67.
107 Khedr EM, Farweez HM, Islam H. Therapeutic effect of repetitive transcranial magnetic stimulation on motor function in Parkinson’s disease patients. Eur J Neurol. 2003;10:567-572.
108 Khedr EM, Rothwell JC, Shawky OA, et al. Effect of daily repetitive transcranial magnetic stimulation on motor performance in PD. Mov Disord. 2006;21:1311-1316.
109 Lefaucheur JP, Drouot X, Von Raison F, et al. Improvement of motor performance and modulation of cortical excitability by repetitive transcranial magnetic stimulation of the motor cortex in Parkinson’s disease. Clin Neurophysiol. 2004;115:2530-2541.
110 Siebner HR, Mentschel C, Auer C, Conrad B. Repetitive transcranial magnetic stimulation has a beneficial effect on bradykinesia in Parkinson’s disease. Neuroreport. 1999;10:589-594.
111 Siebner HR, Rossmeier C, Mentschel C, et al. Short-term motor improvement after sub-threshold 5-Hz repetitive transcranial magnetic stimulation of the primary motor hand area in Parkinson’s disease. J Neurol Sci. 2000;178:91-94.
112 Fregni F, Simon DK, Wu A, Pascual-Leone A. Non-invasive brain stimulation for Parkinson’s disease: a systematic review and meta-analysis of the literature. J Neurol Neurosurg Psychiatry. 2005;76(12):1614-1623.
113 Boylan LS, Pullman SL, Lisanby SH, et al. Repetitive transcranial magnetic stimulation to SMA worsens complex movements in Parkinson’s disease. Clin Neurophysiol. 2001;112:259-264.
114 Koch G, Brusa L, Caltagirone C, et al. rTMS of supplementary motor area modulates therapy-induced dyskinesias in Parkinson disease. Neurology. 2005;65:623-625.
115 Dragasevic N, Potrebic A, Damjanovic A, et al. Therapeutic efficacy of bilateral prefrontal slow repetitive transcranial magnetic stimulation in depressed patients with Parkinson’s disease: an open study. Mov Disord. 2002;17:528-532.
116 Boggio PS, Fregni F, Bermpohl F, et al. Effect of repetitive TMS and fluoxetine on cognitive function in patients with Parkinson’s disease and concurrent depression. Mov Disord. 2005;20:1178-1184.
117 Fregni F, Santos CM, Myczkowski ML, et al. Repetitive transcranial magnetic stimulation is as effective as fluoxetine in the treatment of depression in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1171-1174.
118 Koch G, Oliveri M, Brusa L, et al. High-frequency rTMS improves time perception in Parkinson disease. Neurology. 2004;63:2405-2406.
119 Lin LF, Doherty DH, Lile JD, et al. GDNF: a glial cell line derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130-1132.
120 Beck KD, Valverde J, Alexi T, et al. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature. 1995;373(6512):339-341.
121 Choi-Lundberg DL, Lin Q, Chang YN, et al. Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science. 1997;275:838-841.
122 Kordower J, Emborg M, Bloch J, et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science. 2000;290:767-773.
123 Palfi S, Leventhal L, Chu YP, et al. Lentivirally delivered glial cell line-derived neurotrophic factor increases the number of striatal dopaminergic neurons in primate models of nigrostriatal degeneration. J Neurosci. 2002;22:4942-4954.
124 Grondin R, Zhang ZM, Yi A, et al. Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain. 2002;125:2191-2201.
125 Bilang-Bleuel A, Revah F, Colin P, et al. Intrastriatal injection of an adenoviral vector expressing glial-cell-line-derived neurotrophic factor prevents dopaminergic neuron degeneration and behavioral impairment in a rat model of Parkinson disease. Proc Natl Acad Sci U S A. 1997;94(16):8818-8823.
126 Tomac A, Lindqvist E, Lin LF, et al. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373(6512):335-339.
127 Gash DM, Zhang Z, Ovadia A, et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380:252-255.
128 Nutt JG, Burchiel KJ, Comella CL, et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60(1):69-73.
129 Kordower JH, Palfi S, Chen EY, et al. Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson’s disease. Ann Neurol. 1999;46:419-424.
130 Gash DM, Zhang Z, Ai Y, et al. Trophic factor distribution predicts functional recovery in parkinsonian monkeys. Ann Neurol. 2005;58(2):224-233.
131 Gill SS, Patel NK, Hotton GR, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589-595.
132 Slevin J, Gerhardt C, Smith C, et al. Improvement of bilateral motor function in patients with Parkinson’s disease through the unilateral infusion of glial cell-line derived neurotrophic factor. J Neurosurg. 2005;102:216-222.
133 Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol. 2006;59(3):459-466. Erratum in: Ann Neurol. 2006;60(6):747
134 Salvatore MF, Ai Y, Fischer B, et al. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp Neurol. 2006;202(2):497-505.
135 Schober A, Peterziel H, von Bartheld CS, et al. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol Dis. 2007;25(2):378-391.
136 Porras G, Bezard E. Preclinical development of gene therapy for Parkinson’s disease. Exp Neurol. 2008;209(1):72-81.
137 Somia N, Verma I. Gene therapy: trials and tribulations. Nat Rev Genet. 2000;1:91-99.
138 Bankiewicz K, Daadi M, Pivirotto P, et al. Focal striatal dopamine may potentiate dyskinesias in parkinsonian monkeys. Exp Neurol. 2006;197:363-372.
139 Bankiewicz K, Eberling JL, Kohutnicka M, et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164:2-14.
140 Bankiewicz K, Forsayeth J, Eberling JL, et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther. 2006;14:564-570.
141 Leff S, Spratt S, Snyder R, Mandel R. Long-term restoration of striatal L-aromatic amino acid decarboxylase activity using recombinant adeno-associated viral vector gene transfer in a rodent model of Parkinson’s disease. Neuroscience. 1999;92:185-196.
142 Sanchez-Pernaute R, Harvey-White J, Cunningham J, Bankiewicz K. Functional effect of adeno-associated virus mediated gene transfer of aromatic L-amino acid decarboxylase into the striatum of 6-OHDA-lesioned rats. Molec Ther. 2001;4:324-330.
143 During M, Samulski RJ, Elsworth JD, et al. In vivo expression of therapeutic human genes for dopamine production in the caudates of MPTP-treated monkeys using an AAV vector. Gene Ther. 1998;5:820-827.
144 Fan D, Ogawa M, Fujimoto KI, et al. Behavioral recovery in 6-hydroxydopamine-lesioned rats by cotransduction of striatum with tyrosine hydroxylase and aromatic L-amino acid decarboxylase genes using two separate adeno-associated virus vectors. Hum Gene Ther. 1998;9:2527-2535.
145 Hadaczek P, Kohutnicka M, Krauze M, et al. Convection-enhanced delivery of adeno-associated virus type 2 (AAV2) into the striatum and transport of AAV2 within monkey brain. Hum Gene Ther. 2006;17:291-302.
146 Fiandaca M, Forsayeth J, Bankiewicz K. Current status of gene therapy trials for Parkinson’s disease. Exp Neurol. 2008;209(1):51-57.
147 Benabid A. Deep brain stimulation for Parkinson’s disease. Curr Opin Neurobiol. 2003;13:696-706.
148 Luo J, Kaplitt M, Fitzsimons H, et al. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science. 2002;298:425-429.
149 Emborg M, Carbon M, Holden JE, et al. Subthalamic glutamic acid decarboxylase gene therapy: changes in motor function and cortical metabolism. J Cereb Blood Flow Metab. 2007;27:501-509.
150 Kaplitt MG, Feigin A, Tang C, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369(9579):2097-2105.
151 Feigin A, Kaplitt MG, Tang C, et al. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104(49):19559-19564.
152 Björklund A, Kirik D, Rosenblad C, et al. Towards a neuroprotective gene therapy for Parkinson’s disease: use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 2000;886:82-98.
153 Baloh R, Enomoto H, Johnson EJr, Milbrandt J. The GDNF family ligands and receptors—implications for neural development. Curr Opin Neurobiol. 2000;10:103-110.
154 Cik M, Masure S, Lesage AS, et al. Binding of GDNF and neurturin to human GDNF family receptor alpha 1 and 2. Influence of cRET and cooperative interactions. J Biol Chem. 2000;275:27505-27512.
155 Marks WJJr, Ostrem JL, Verhagen L, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 2008;7(5):400-408.
156 Oiwa Y, Sanchez-Pernaute R, Harvey-White J, Bankiewicz K. Progressive and extensive dopaminergic degeneration induced by convection-enhanced delivery of 6-hydroxydopamine into the rat striatum: a novel rodent model of Parkinson disease. J Neurosurg. 2003;98:136-144.
157 Eslamboli A, Georgievska B, Ridley RM, et al. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson’s disease. J Neurosci. 2005;25:769-777.
158 Kordower J, Herzog C, Dass B, et al. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006;60:706-715.
159 Herzog CD, Dass B, Holden JE, et al. Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov Disord. 2007;22(8):1124-1132.
160 Gasmi M, Brandon EP, Herzog CD, et al. AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis. 2007;27(1):67-76.
161 Ceregene, San Diego, Calif., available at http://www.ceregene.com
162 Braak H, Del Tredici K, Rub U, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197-211.
163 Lang AE, Obeso JA. Time to move beyond nigrostriatal dopamine deficiency in Parkinson’s disease. Ann Neurol. 2004;55:761-765.
164 Dunnett SB, Björklund A, Schmidt RH, et al. Intracerebral grafting of neuronal cell suspensions. IV. Behavioral recovery in rats with unilateral 6-OHDA lesions following implantation of nigral cell suspensions in different forebrain sites. Acta Physiol Scand Suppl. 1983;522:29-37.
165 Stenevi U, Björklund A, Svendgaard NA. Transplantation of central and peripheral monoamine neurons to the adult rat brain: techniques and conditions for survival. Brain Res. 1976;114:1-20.
166 Björklund A, Stenevi U, Svendgaard NA. Growth of transplanted monoaminergic neurones into the adult hippocampus along the perforant path. Nature. 1976;262:787-790.
167 Perlow MJ, Freed WJ, Hoffer BJ, et al. Brain grafts reduce motor abnormalities produced by destruction of nigrostriatal dopamine system. Science. 1979;204:643-647.
168 Björklund A, Stenevi U. Reconstruction of the nigrostriatal dopamine pathway by intracerebral nigral transplants. Brain Res. 1979;177:555-560.
169 Nishino H. Intracerebral grafting of catecholamine producing cells and reconstruction of disturbed brain function. Neurosci Res. 1993;16:157-172.
170 Triarhou LC, Stotz EH, Low WC, et al. Studies on the striatal dopamine uptake system of weaver mutant mice and effects of ventral mesencephalic grafts. Neurochem Res. 1994;19:1349-1358.
171 Dunnett SB. Functional repair of striatal systems by neural transplants: evidence for circuit reconstruction. Behav Brain Res. 1995;66:133-142.
172 Mukhida K, Baker KA, Sadi D, Mendez I. Enhancement of sensorimotor behavioral recovery in hemiparkinsonian rats with intrastriatal, intranigral, and intrasubthalamic nucleus dopaminergic transplants. J Neurosci. 2001;21:3521-3530.
173 Madrazo I, León V, Torres C, et al. Transplantation of fetal substantia nigra and adrenal medulla to the caudate nucleus in two patients with Parkinson’s disease. N Engl J Med. 1988;318:51.
174 Lindvall O, Rehncrona S, Brundin P, et al. Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and 6-month follow-up. Arch Neurol. 1989;46:615-631.
175 Madrazo I, Franco-Bourland R, Ostrosky-Solis F. Neural transplantation (autoadrenal, fetal nigral and fetal adrenal) in Parkinson’s disease: the Mexican experience. Prog Brain Res. 1990;82:593-602.
176 Molina H, Quinones AL, Ortega I. Stereotactic transplantation of foetal ventral mesencephalic cells: Cuban experiences from five patients with idiopathic Parkinson’s disease. J Neural Transplant Plast. 1992;3:338-339.
177 Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med. 2001;344:710-719.
178 Mendez I, Dagher A, Hong M, et al. Enhancement of survival of stored dopaminergic cells and promotion of graft survival by exposure of human fetal nigral tissue to glial cell line-derived neurotrophic factor in patients with Parkinson’s disease. Report of two cases and technical considerations. J Neurosurg. 2000;92:863-869.
179 Mendez I, Dagher A, Hong M, et al. Simultaneous intrastriatal and intranigral fetal dopaminergic grafts in patients with Parkinson disease: a pilot study. Report of three cases. J Neurosurg. 2002;96:589-596.
180 Mendez I, Sanchez-Pernaute R, Cooper O, et al. Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson’s disease. Brain. 2005;128:1498-1510.
181 Leigh K, Elisevich K, Rogers KA. Vascularisation and microvascular permeability in solid versus cell-suspension embryonic neural grafts. J Neurosurg. 1994;81:272-283.
182 Nikkhah G, Cunningham MG, Jödicke A, et al. Improved graft survival and striatal reinnervation by microtransplantation of fetal nigral cell suspensions in the rat Parkinson model. Brain Res. 1994;633:133-143.
183 Kordower JH, Freeman TB, Snow BJ, et al. Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson’s disease. N Engl J Med. 1995;332:1118-1124.
184 Kordower JH, Chu Y, Hauser RA, et al. Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord. 2008;15:2303-2306.
185 Piccini P, Brooks DJ, Björklund A, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson’s patient. Nat Neurosci. 1999;2:1137-1140.
186 Kordower JH, Freeman TB, Chen EY, et al. Fetal nigral grafts survive and mediate clinical benefit in a patient with Parkinson’s disease. Mov Disord. 1998;13:383-393.
187 Hagell P, Schrag A, Piccini P, et al. Sequential bilateral transplantation in Parkinson’s disease: effects of the second graft. Brain. 1999;122:1121-1132.
188 Hauser RA, Freeman TB, Snow BJ, et al. Long-term evaluation of bilateral fetal nigral transplantation in Parkinson disease. Arch Neurol. 1999;56:179-187.
189 Brundin P, Pogarell O, Hagell P, et al. Bilateral caudate and putamen grafts of embryonic mesencephalic tissue treated with lazaroids in Parkinson’s disease. Brain. 2000;123:1380-1390.
190 Wenning GK, Odin P, Morrish P, et al. Short- and long-term survival and function of unilateral intrastriatal dopaminergic grafts in Parkinson’s disease. Ann Neurol. 1997;42:95-107.
191 Olanow CW, Goetz CG, Kordower JH, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403-414.
192 Winkler C, Kirik D, Björklund A. Cell transplantation in Parkinson’s disease: how can we make it work? Trends Neurosci. 2005;28:86-92.
193 Nakao N, Ogura M, Nakai K, Itakura T. Intrastriatal mesencephalic grafts affect neuronal activity in basal ganglia nuclei and their target structures in a rat model of Parkinson’s disease. J Neurosci. 1998;18:1806-1817.
194 Winkler C, Bentlage C, Nikkhah G, et al. Intranigral transplants of GABA-rich striatal tissue induce behavioural recovery in the rat Parkinson model and promote the effects obtained by intrastriatal dopaminergic transplants. Exp Neurol. 1999;155:165-186.
195 Lane EL, Winkler C, Brundin P, Cenci MA. The impact of graft size on the development of dyskinesia following intrastriatal grafting of embryonic dopamine neurons in the rat. Neurobiol Dis. 2006;22:334-345.
196 Carlsson T, Winkler C, Lundblad M, et al. Graft placement and uneven pattern of reinnervation in the striatum is important for development of graft-induced dyskinesias. Neurobiol Dis. 2006;21:657-668.
197 Kuan WL, Lin R, Tyers P, Barker RA. The importance of A9 dopaminergic neurons in mediating the functional benefits of fetal ventral mesencephalon transplants and levodopa-induced dyskinesias. Neurobiol Dis. 2007;25:594-608.
198 Carlsson T, Carta M, Winkler C, et al. Serotonin neuron transplants exacerbate L-dopa-induced dyskinesias in a rat model of Parkinson’s disease. J Neurosci. 2007;27:8011-8022.
199 Cochen V, Ribeiro MJ, Nguyen JP, et al. Transplantation in Parkinson’s disease: PET changes correlate with the amount of grafted tissue. Mov Disord. 2003;18:928-932.
200 Nguyen JP, Remy P, Palfi S, et al. Bilateral intrastriatal grafts of fetal mesencephalic neurons in Parkinson’s disease: long-term results in 9 patients. Mov Disord. 2000;15:53-54.
201 Björklund A. Better cells for brain repair. Nature. 1993;362:414-415.
202 Isacson O, Costantini L, Schumacher JM, et al. Cell implantation therapies for Parkinson’s disease using neural stem, transgenic or xenogeneic donor cells. Parkinsonism Relat Disord. 2001;7:205-212.
203 Hagell P, Brundin P. Cell survival and clinical outcome following intrastriatal transplantation in Parkinson’s disease. J Neuropathol Exp Neurol. 2001;60:741-752.
204 Rossi F, Cattaneo E. Neural stem cell therapy for neurological diseases: dreams and reality. Nat Rev Neurosci. 2002;3:401-409.
205 Barker RA. Repairing the brain in Parkinson’s disease: where next? Mov Disord. 2002;17:233-241.
206 Dass B, Olanow CW, Kordower JH. Gene transfer of trophic factors and stem cell grafting as treatments for Parkinson’s disease. Neurology. 2006;66(Suppl 4):S89-S103.
207 Hong S, Kang UJ, Isacson O, Kim KS. Neural precursors derived from human embryonic stem cells maintain long-term proliferation without losing the potential to differentiate into all three neural lineages, including dopaminergic neurons. J Neurochem. 2008;104(2):316-324.
208 Björklund LM, Sanchez-Pernaute R, Chung S, et al. Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc Natl Acad Sci U S A. 2002;99:2344-2349.
209 Kim J, Auerbach J, Rodríguez-Gómez J, et al. Dopamine neurons derived from embryonic stem cells function in an animal model of Parkinson’s disease. Nature. 2002;418:50-56.
210 Riaz SS, Bradford HF. Factors involved in the determination of the neurotransmitter phenotype of developing neurons of the CNS: applications in cell replacement treatment for Parkinson’s disease. Prog Neurobiol. 2005;76:257-278.
211 Studer L, Csete M, Lee SH, et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377-7383.
212 Carvey PM, Ling ZD, Sortwell CE, et al. A clonal line of mesencephalic progenitor cells converted to dopamine neurons by hematopoietic cytokines: a source of cells for transplantation in Parkinson’s disease. Exp Neurol. 2001;171:98-108.
213 Smidt MP, Asbreuk CH, Cox JJ, et al. A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat Neurosci. 2000;3:337-341.
214 Parish CL, Castelo-Branco G, Rawal N, et al. Wnt5a-treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J Clin Invest. 2008;118(1):149-160.
215 Goldman S. Stem and progenitor cell-based therapy of the human central nervous system. Nat Biotechnol. 2005;23:862-871.
216 Rick CE, Ebert A, Viraq T, et al. Differentiated dopaminergic MN9D cells only partially recapitulate the electrophysiological properties of midbrain dopaminergic neurons. Dev Neurosci. 2006;28:528-537.
217 Isacson O. The production and use of cells as therapeutic agents in neurodegenerative diseases. Lancet Neurol. 2003;2:417-424.
218 Ourednik J, Ourednik V, Lynch WP, et al. Neural stem cells display an inherent mechanism for rescuing dysfunctional neurons. Nat Biotechnol. 2002;20:1103-1110.
219 Rafuse VF, Soundararajan P, Leopold C, Robertson HA. Neuroprotective properties of cultured neural progenitor cells are associated with the production of sonic hedgehog. Neuroscience. 2005;131:899-916.
220 Sortwell CE, Collier TJ, Sladek JRJr. Co-grafted embryonic striatum increases the survival of grafted embryonic dopamine neurons. J Comp Neurol. 1998;399:530-540.
221 Yasuhara T, Matsukawa N, Hara K, et al. Transplantation of human neural stem cells exerts neuroprotection in a rat model of Parkinson’s disease. J Neurosci. 2006;26:12497-12511.
222 Bjugstad KB, Redmond DEJr, Teng YD, et al. Neural stem cells implanted into MPTP-treated monkeys increase the size of endogenous tyrosine hydroxylase-positive cells found in the striatum: a return to control measures. Cell Transplant. 2005;14:183-192.
223 Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci. 2005;28:223-250.
224 Baker H, Kobayashi K, Okano H, Saino-Saito S. Cortical and striatal expression of tyrosine hydroxylase mRNA in neonatal and adult mice. Cell Mol Neurobiol. 2003;23:507-518.
225 Cossette M, Lecomte F, Parent A. Morphology and distribution of dopaminergic neurons intrinsic to the human striatum. J Chem Neuroanat. 2005;29:1-11.
226 Kay JN, Blum M. Differential response of ventral midbrain and striatal progenitor cells to lesions of the nigrostriatal dopaminergic projection. Dev Neurosci. 2000;22:56-57.
227 Porritt MJ, Batchelor PE, Hughes AJ, et al. New dopaminergic neurons in Parkinson’s disease striatum. Lancet. 2000;356:44-45.
228 Fallon J, Reid S, Kinyamu R, et al. In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci U S A. 2000;97:14686-14691.
229 Mohapel P, Frielingsdorf H, Häggblad J, et al. Platelet-derived growth factor (PDGF-BB) and brain-derived neurotrophic factor (BDNF) induce striatal neurogenesis in adult rats with 6-hydroxydopamine lesions. Neuroscience. 2005;132:767-776.
230 Cooper O, Isacson O. Intrastriatal transforming growth factor alpha delivery to a model of Parkinson’s disease induces proliferation and migration of endogenous adult neural progenitor cells without differentiation into dopaminergic neurons. J Neurosci. 2004;24:8924-8931.
231 Van Kampen JM, Robertson HA. A possible role for dopamine D3 receptor stimulation in the induction of neurogenesis in the adult rat substantia nigra. Neuroscience. 2005;136:381-386.
232 Van Kampen JM, Eckman CB. Dopamine D3 receptor agonist delivery to a model of Parkinson’s disease restores the nigrostriatal pathway and improves locomotor behavior. J Neurosci. 2006;26:7272-7280.
233 Zhao M, Momma S, Delfani K, et al. Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A. 2003;100:7925-7930.
234 Frielingsdorf H, Schwarz K, Brundin P, Mohapel P. No evidence for new dopaminergic neurons in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A. 2004;101:10177-10182.
235 Goldman S. Disease targets and strategies for the therapeutic modulation of endogenous neural stem and progenitor cells. Clin Pharmacol Therap. 2007;82:453-460.
236 Singh S, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 84 Emerging and Experimental Neurosurgical Treatments for Parkinson’s Disease
Idiopathic Parkinson’s disease (PD) affects more than 1 million North Americans, including approximately 1% of individuals older than 65 years. PD is the second most common neurodegenerative disease after Alzheimer’s, and neurodegenerative diseases as a whole are projected to surpass cancer as the second leading cause of death in the elderly by 2040.1 With a protracted course of progressive disability and dependence on others, increased mortality, and a total estimated cost of $23 billion a year in the United States, PD represents a significant ongoing burden to society.2–4
PD is diagnosed on clinical grounds, with resting tremor, rigidity, bradykinesia or akinesia, and postural instability representing the cardinal features of the disease.5 Other important manifestations can include dementia, gait disturbance, autonomic dysfunction, and depression. Neuropathologically, PD is characterized by widespread degeneration of select sites within the central and peripheral nervous systems.6,7 Although this multisystem disorder affects many brain regions and neurotransmitter systems, the loss of dopaminergic neurons within the substantia nigra pars compacta (SNc) accounts for the reduction in striatal dopaminergic innervation that underlies the symptoms of akinesia and rigidity.8 Widespread accumulation of proteinaceous Lewy bodies and Lewy neurites is also observed in PD. In the central nervous system, these deposits accumulate in predictable stages with progressive involvement of brainstem, subcortical, and neocortical regions.9 However, the relationship of these deposits to the pathogenesis of PD remains unclear.5,10 Although numerous mechanisms underlying the neurodegeneration seen in PD have been proposed, none has provided a definitive explanation of its pathogenesis.
Since their introduction more than 40 years ago, dopaminergic agents have been the mainstay of treatment for the motor symptoms of PD. More than 90% of patients initially respond well to levodopa therapy, with the majority continuing to derive some benefit throughout the course of the disease.5 Unfortunately, progressive treatment resistance, coupled with the gradual development of motor fluctuations, dyskinesias, and psychiatric disturbances, limits levodopa’s long-term use.11,12 Other agents such as dopamine agonists, anticholinergics, amantadine, and monoamine oxidase inhibitors generally yield minimal benefit and may induce unpalatable side effects.12 Clinical features of advanced PD such as postural and gait dysfunction are particularly resistant to dopamine-based therapies, suggesting a greater involvement of nondopaminergic pathways in their pathophysiology.13,14 Importantly, no pharmacotherapy has been able to alter disease progression.
The shortcomings of pharmacotherapy and the growing understanding of the pathophysiologic correlates of parkinsonism have led to a resurgence in stereotactic and functional neurosurgery for PD over the past two decades. Targeted lesioning and deep brain stimulation (DBS) at the thalamus, globus pallidus, and subthalamic nucleus have proved to be valuable additions to our therapeutic armamentarium.15 At present, surgery is reserved for medically intractable cases when tremor is a major source of disability or when levodopa-responsive patients are experiencing significant “on-off” cycling between rigidity or bradykinesia and levodopa-induced dyskinesias.16 Although highly efficacious in properly selected patients, the benefits of stereotactic surgery are limited mainly to improving rigidity, tremor, “off-state” akinesia, and levodopa-induced dyskinesias. There is little or no effect on other manifestations of the disease.15 In addition, the benefit of surgery remains symptomatic in nature, without altering the natural history of the disease.
Therefore, our present therapeutic armamentarium could be improved in a number of ways. First, making DBS more effective and safer would allow more patients to be treated and to undergo surgery earlier in the disease course. Second, new treatments could address the many symptomatic features of PD that remain poorly treated or are not affected at all by current therapies. Third, disease-modifying treatments could deter disease progression. The following sections outline experimental and emerging neurosurgical treatments whose development has been geared specifically toward closing these gaps in our current treatment options and ultimately achieving the elusive goal of modifying the natural history of PD through neuroprotection or neuroregeneration. DBS-related strategies, motor cortex stimulation, growth factor delivery, gene therapy, and cellular replenishment are discussed, with reference to currently available clinical data (Fig. 84-1).
New Strategies for Deep Brain Stimulation
In properly selected patients, and when appropriate resources are available, high-frequency DBS of the subthalamic nucleus (STN) or internal globus pallidus (GPi) is the surgical treatment of choice for PD.17 Clinical outcomes and surgical techniques are now well established, with more than 30,000 patients treated worldwide.15 Armed with this baseline knowledge and experience, attempts to improve the utility of DBS in PD through early surgery and novel targets are ongoing.
Early Surgery
The observation that STN inactivation prevents nigral degeneration in rodent models of PD18,19 led investigators to consider the possibility that, in addition to its symptomatic benefits, subthalamic DBS might be neuroprotective in humans. The neuroprotective mechanism was thought to be reduced excitotoxin-induced nigral degeneration, mediated by the glutamatergic projections from the STN to the nigra.20,21 To examine this, Hilker and coworkers22 used 18F-fluorodopa (F-dopa) positron emission tomography (PET), which quantifies presynaptic dopaminergic function,23–25 to study the progression of PD after high-frequency STN-DBS. Although the study did not include controls, the annual striatal reduction in F-dopa uptake in 30 STN-DBS–treated patients with advanced PD was roughly 10% to 12%, which is consistent with previously reported rates in PD.26 This evidence, along with clinical experience, suggests that the neuroprotective effects seen in animal models cannot be readily confirmed in humans.
It remains uncertain whether a neuroprotective effect of high-frequency STN-DBS might be observed if patients were treated early in the course of their disease. Patients with advanced PD may no longer harbor the capacity to alter the course of degeneration, whereas the striatum of patients in the early stages of the disease might be more apt to gain a survival benefit from a reduction in STN-mediated glutamatergic input. In March 2006, a phase I trial of early surgery in PD began under the supervision of Dr. Charles at Vanderbilt University.27 The hypothesis of this study is that STN-DBS slows disease progression in subjects with early PD. In this single-center study, which is expected to be completed in 2010, eligible patients who have received dopaminergic therapy for less than 4 years are randomized to surgery plus medical therapy versus medical therapy alone. Motor outcomes are assessed in a single-blind fashion. A multicenter phase III trial is planned if safety and tolerability criteria are met.
Another line of investigation suggests that early surgery in patients suffering from motor fluctuations or dyskinesias may promote the maintenance of quality of life and social functioning.28 The mean time from PD diagnosis to surgery is more than 10 years; however, dopamine-related motor fluctuations and dyskinesias may arise much sooner and typically contribute to a progressive decline in social and occupational function.29 Early reports suggest that patients with a disease duration of less than 10 years who have undergone operation can maintain a baseline involvement in their work and personal lives postoperatively.28 Schüpbach and associates30 performed a matched-pairs study of 20 PD patients with a mean symptom duration of 7 years, comparing functional outcomes in patients receiving DBS versus best medical therapy. Eighteen months postoperatively, the surgical patients experienced a 24% improvement in disease-specific quality of life versus 0% in the nonsurgical patients. The principal investigator of this study, Dr. Agid, is now leading a multicenter, randomized, open-label, phase III trial (EARLYSTIM Study) examining quality of life outcomes in patients with STN-DBS versus medical therapy, with 2-year follow-up data expected in 2010.27 This promises to offer further insight into the potential benefits of early surgery.
A New Target: The Pedunculopontine Nucleus
Postural and gait dysfunction leading to falls is the leading cause of emergency room visits and is a significant cost generator in the care of PD patients.31–33 In addition, the fear of falling results in depression and a loss of independence.34 Postural and gait dysfunction is particularly resistant to current dopaminergic and surgical therapies, suggesting a greater involvement of nondopaminergic pathways and brain loci other than the pallidal and subthalamic nuclei in their pathophysiology.13,14,35–41
The pedunculopontine nucleus (PPN) is part of a brainstem locomotive center that also processes sensory and behavioral information. Running roughly between the SNc and locus caeruleus, the PPN primarily connects with basal ganglia structures and the spinal cord.42,43 Midfrequency electrical or glutamatergic stimulation of the PPN leads to increased activity of its neuronal population and produces controlled locomotion on a treadmill in decerebrate animals.44,45 Other neurotransmitters, including γ-aminobutyric acid (GABA) and acetylcholine, decrease PPN activity and inhibit locomotion.42 It has been theorized that the PPN regulates tone and locomotion by modulating lower brainstem nuclei.46,47 These nuclei, in turn, activate central pattern generators in the spinal cord, which generate the motor patterns governing locomotion and limb tone.47–50 Given the evidence for the PPN’s role in the initiation and maintenance of gait and the stability of postural tone, as well as its pharmacologic and electrophysiologic properties that appear to be reliant on cholinergic and other nondopaminergic neurotransmitters, pathologic function related to this structure could theoretically play a significant role in the gait and postural manifestations of PD.42
Although direct evidence linking PPN dysfunction to PD-related postural and gait disturbances is lacking, there are several hints at a pathophysiologic connection. The PPN in anesthetized rats with hydroxydopamine (6-OHDA) nigral lesions is characterized by abnormally enhanced activity compared with controls.51 Furthermore, some of the electrophysiologic changes recorded in the basal ganglia structures of these PD-model animals could be reversed by PPN lesions.52,53 Matsumura and Kojima54 observed a smaller degree of nigral degeneration and fewer parkinsonian symptoms if monkeys with 1-methyl, 4-phenyl, 1,2,3,6-tetrahydropyridine (MPTP) lesions were given PPN lesions. Similarly, Nandi and colleagues55 showed that microinjections of the GABAergic antagonist bicuculline into the PPN improve parkinsonian symptoms in MPTP primates. Electrophysiologic changes have been matched to metabolic alterations in the PPN of parkinsonian rodents and nonhuman primates.56,57 More important, studies of human pathology demonstrate that a significant proportion of the large cholinergic PPN neurons degenerate in PD.58–60 These individual lines of evidence collectively invoke the possibility that PD-related degeneration of nondopaminergic PPN neurons leads to electrophysiologic and metabolic disturbances that culminate in the dysregulation of PPN-mediated gait and postural functions. Therefore, medical or surgical interventions that modulate PPN activity in PD patients might normalize gait initiation and maintenance and postural stability.
Following this hypothesis, DBS of the PPN is being evaluated as a therapeutic modality in PD patients with significant gait disturbance (Fig. 84-2).61–63 Initial subjects have included PD patients with refractory and functionally significant impairment of gait initiation (debilitating freezing) and postural stability (associated with an unacceptable risk of falling). Those suffering from comorbidities that might adversely affect gait and posture, including dementia, primary affective disorders, and marked sensory impairments, are considered poor candidates for PPN stimulation.64–67
Initial reports of PPN-DBS in PD patients documented the technical aspects and electrophysiologic findings of the procedure, as well as short-term indicators of clinical outcome.61,62 A more recent study provided greater insight into the clinical outcome of six patients who underwent simultaneous bilateral PPN- and STN-DBS.63 Six months after surgery, Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores in the “off-medication–on-stimulation” condition were improved by an average of 33% with PPN-DBS alone. This was less than the improvement obtained from STN-DBS alone (54%) or simultaneous STN- and PPN-DBS (56%) in these same patients. Improvements specifically in axial symptoms (e.g., rising from a chair, posture, gait, postural stability) were similar with PPN-only, STN-only, and simultaneous PPN- and STN-DBS (60% to 70%). In patients taking medication, Stefani and colleagues63 found a greater improvement in UPDRS motor scores with combined STN and PPN stimulation compared with stimulation at either target alone. Moreover, STN-only stimulation did not improve axial symptoms in patients who were taking medication. If only axial symptoms were taken into account in the on-medication condition, both PPN-only DBS and simultaneous PPN- and STN-DBS led to a 50% to 60% improvement versus no stimulation. Importantly, no major complications were described. The only minor side effect reported was paresthesias associated with high-frequency PPN stimulation, likely resulting from the target’s proximity to the medial lemniscus.63 In summary, reports suggest the possibility of a therapeutic response to low-frequency PPN stimulation, but larger controlled studies are needed to verify these results and define its potential role in the treatment of PD.
Motor Cortex Stimulation
The concept of therapeutic modulation of cortical activity in movement disorders arose following James Parkinson’s description of the temporary abolishment of a tremor following a cortical stroke in 1824.68 Reports in the mid-1900s described improvements in posttraumatic and parkinsonian tremors following motor cortex or corticospinal tract lesioning.69,70 More recently (1979), Woolsey and colleagues71 observed immediate and temporary improvements in rigidity and tremor in PD patients undergoing intraoperative motor cortex stimulation (MCS) for the purpose of brain mapping, and several reports have described a simultaneous improvement of tremor in patients undergoing MCS for the treatment of pain.72–74
Further support for the attempt to modulate cortical activity in PD patients comes from evidence linking the disease to regional cortical dysfunction. Functional neuroimaging and electroencephalography in PD patients demonstrate hypoactivity in the supplementary motor area and dorsolateral prefrontal cortex.75–84 Interestingly, MPTP monkeys receiving chronic MCS (cMCS) exhibited a reversal of hypoactivity in the supplementary motor area and an improvement in akinesia.85 Changes in motor cortical activity are also observed in PD patients, with reduced activity early in the disease and increased activity in advanced disease.78,79,86,87 Although this variation remains poorly understood, hyperactivity may reflect compensatory cortical reorganization resulting from an increasingly deficient subcortical motor system, and it has been theorized to promote the development of levodopa-induced dyskinesias.88
Based on the observation of abnormal motor cortex activity in PD and clinical reports suggesting an acute benefit from MCS, a rationale emerged for investigating the use of cMCS in PD. Both theoretical predictions and experimental results suggested that cMCS might exert orthodromic or antidromic effects on subcortical elements of corticobasal ganglia loops (Fig. 84-3).85,89–91 STN-DBS in PD patients evokes short-latency responses, improves movement-related asynchrony, restores intracortical inhibition, and decreases resting activity in the human motor cortex.92–98 Similarly, the motor cortices can affect basal ganglia activities. Microelectrode recordings in MPTP monkeys receiving cMCS show substantial normalization of the mean firing rate and less abnormal synchronized oscillatory activity of GPi and STN neurons.85 In humans with PD, MCS using a single pulse of transcranial magnetic stimulation (TMS) evokes short-latency excitation, followed by long-lasting inhibition, of neurons located in the dorsolateral sensorimotor area of the STN.91 PET studies also demonstrate that MCS using 10-Hz TMS induces focal dopamine release within the ipsilateral dorsal striatum.89,90
Epidural and Subdural Electrodes
Internalized epidural and subdural electrodes provide titratable systems with relatively precise target specificity, allowing focal stimulation while avoiding the greater risk of intraparenchymal placement. Since 2000, five reports have described 19 patients with advanced PD who were treated with epidural or subdural cMCS (Table 84-1).99–103 Although these publications collectively suggest a trend toward varying degrees of improvement across all clinically relevant motor domains, the modest benefits reported remain unconvincing. One poorly understood finding is the relatively consistent occurrence of bilateral improvement in symptoms despite unilateral cMCS, a phenomenon that is generally not observed with DBS. Further evidence indicates that cMCS in PD patients produces no alteration in movement-related regional cerebral blood flow as measured by PET scans, casting further doubt on its potential utility in PD.104 An additional report investigated the use of subdural cMCS for multiple system atrophy with predominant parkinsonism, a rapidly progressive neurodegenerative disease that shares some motor features with PD but for which treatments are generally ineffective.105 In the five patients studied, neither low- nor high-frequency stimulation had any benefit. Larger studies in appropriate patients may better define the efficacy and potential indications for epidural and subdural cMCS in PD. At the time of this writing, additional small phase I-II randomized, controlled, and crossover clinical trials investigating cMCS in PD are ongoing in France and Italy, with expected completion in the next couple of years.27
Transcranial Magnetic Stimulation
TMS is a noninvasive technique that induces changes in cortical activity through electromagnetic induction (see Fig. 84-3). Repetitive TMS (rTMS) delivered to the motor cortex at 5 to 25 Hz improves motor performance in PD patients.106–111 A recent systematic review of noninvasive neurostimulation in PD pooled the results from eight double-blind, sham-controlled studies of rTMS involving patients with a mean age and disease duration of 63 and 7 years, respectively. The standardized mean difference in pre- and posttreatment UPDRS scores was a modest 0.60 (95% confidence interval, 0.24 to 0.96), with no significant heterogeneity. Moreover, the longevity of this modest effect is uncertain, given the less than 2 months of follow-up.112 Frequencies of approximately 1 Hz applied to the supplementary motor area worsened motor performance but improved levodopa-induced dyskinesias.113,114 One interesting study demonstrated a concomitant increase in motor performance and relief of depression with rTMS applied to the dorsolateral prefrontal cortex, raising the possibility that modulating cortical activity might address the often debilitating comorbid depression that is often observed in patients with advancing PD.115 Other studies have failed to demonstrate any motor benefits with stimulation at the dorsolateral prefrontal cortex.116–118 Despite the appeal of its minimal invasiveness, the modest demonstrated efficacy of TMS and the practical issues involved in using it over a long period may ultimately limit its clinical utility.
Intraparenchymal Growth Factor Delivery
Glial cell line–derived neurotrophic factor (GDNF) is a neurotrophin protein that belongs to the transforming growth factor-β superfamily.119 Along with other cell types, dopaminergic neurons respond to GDNF through cell signaling pathways and react with enhanced growth, survival, differentiation, and regeneration.119 The cerebral administration of GDNF through viral vector delivery, intracerebroventricular infusion, or putaminal injection in rodent and nonhuman primate models of PD imparts neuroprotective and neuroregenerative effects.120–124 In addition, 6-OHDA rodents and MPTP primates demonstrate motor performance improvements following GDNF therapy.122,124–127
Based on these findings and the need to develop disease-modifying therapies for PD, studies have been undertaken to determine whether GDNF benefits PD patients. Cloning of the human GDNF gene led to the production of a human recombinant peptide (r-metHuGDNF, liatermin; Amgen, Thousand Oaks, CA) available for human administration. To circumvent the blood-brain barrier, targeted surgical delivery has been employed. The first randomized, controlled trial in PD patients to be published involved monthly bolus injections into the frontal horn of the lateral ventricle through an implanted cannula connected to an access port.128 Blinded outcomes in 50 patients at 8 months failed to demonstrate an improvement in UPDRS scores over a range of GDNF doses, and fairly frequent adverse effects were noted. Although these results were discouraging, an independent postmortem study of a PD patient who had received intraventricular GDNF suggested that this route of administration did not deliver GDNF to the dopamine-deficient putamen.129 Further, it appears that correct location is more important than dose when examining the efficacy of GDNF delivery in primate PD models.130
A smaller single-center, open-label trial of five PD patients receiving continuous intraputaminal infusion reported a 32% mean improvement in off-medication UPDRS motor scores at 6 months follow-up.131 There was also a significant increase in putaminal F-dopa uptake on PET imaging near the catheter tip. Most important, this route of administration was well tolerated. Slevin and colleagues132 reported similar results, documenting a mean 30% improvement in the off-medication UPDRS motor scores 6 months after the start of therapy in an open-label trial of 10 PD patients. However, in a randomized, controlled, blinded trial of intraputaminal GDNF infusion in 34 patients with idiopathic PD, no significant difference was noted in the UPDRS scores of patients receiving active GDNF and those receiving placebo.133 The fact that greater F-dopa uptake was noted on the PET scans of treated patients suggests a biologic effect that did not translate into a clinical response.
Despite the failure of intraparenchymal GDNF therapy in clinical trials, hope remains that the exciting responses of animal models of PD to growth factor delivery will eventually be translated into therapies. More basic research is required to better understand the mechanisms behind this animal-human discrepancy. Hints are beginning to emerge, including evidence that the neuroprotective action of GDNF may rely on other factors, such as transforming growth factor-β, and that drug delivery may need to be optimized to ensure ideal exposure of the targeted tissue.134,135 Indeed, the targeted surgical delivery of compounds that promote neuronal health, survival, and regeneration may one day be a reality as new data come to light.
Gene Therapy
Gene therapies for PD have long been sought as a means of providing targeted symptomatic, neuroprotective, and neuroregenerative therapy. Potential benefits include the ability to produce biologically active molecules within the brain and thus circumvent the blood-brain barrier, focus therapy in specific brain regions and thereby avoid side effects related to the exposure of other areas, and address potential underlying causes of the disorder directly. Although gene therapy has yielded encouraging preclinical results for a number of disorders, safety and technical concerns have hampered its successful translation into clinical use.136,137 Nonetheless, three gene therapy strategies have made considerable progress toward becoming viable therapies, all of which employ stereotactic delivery to specific brain targets and adeno-associated virus (AAV) vectors to mediate gene transfer. These strategies are increasing striatal dopamine production, reducing STN output, and expressing an ectopic growth factor in the striatum.
Augmenting Striatal Dopamine Production
This strategy aims to increase dopamine production in neurons and glia within the striatum by ectopically expressing the enzyme aromatic L-amino-acid decarboxylase (AADC), which catalyzes the conversion of levodopa to dopamine (Fig. 84-4). In essence, this is an extension of current pharmacologic interventions with dopaminergic drugs. Bankiewicz and associates138 theorized that declining levels of AADC not only contribute to the disease by reducing dopamine production but also lead to a backlog of exogenous levodopa, which may promote the development of levodopa-induced dyskinesias.
Convection-enhanced delivery of AAV-AADC to the striatum increases that nucleus’s capacity to decarboxylate exogenous levodopa in animal models of PD. Efficient and long-term expression of the transgene in the striatum restores local dopamine production and promotes behavioral recovery in 6-OHDA rats and MPTP monkeys treated with levodopa.138–142 PET data suggest that these same monkeys exhibit persistent AADC activity for at least 6 years after transfection.138,140 Similar studies using the simultaneous transduction of tyrosine hydroxylase and AADC showed a significant increase in intracellular dopamine in the striatum, producing better behavioral recovery than with the transduction of either enzyme alone; however, this multipronged approach adds complexity to the delivery.143–145
A human phase I study of AAV-AADC gene therapy for idiopathic PD is now under way.146 A noteworthy inclusion criterion is the presence of intractable motor fluctuations. Patients receive three escalating doses through bilateral stereotactic infusions into the postcommissural putamen, delivered in two 50-µL volumes spaced approximately 6 mm apart. The key outcome measures are an improvement in 18F-fluorometatyrosine PET signal at 1 and 6 months, indicating the presence of the AADC transgene, and an improvement in standard clinical rating scales. Preliminary data in five patients who received the lowest dose demonstrated increases in PET signal, suggesting successful AADC transgene expression.146 Future results, particularly from the higher dose groups, should shed light on the potential safety and efficacy of this approach.
Altering Pathologic Subthalamic Nucleus Output
The second approach to gene therapy for PD under active investigation aims to modify STN output through ectopic expression of glutamic acid decarboxylase (GAD), which catalyzes the production of GABA. The theory behind the therapy is that the local production of GAD transforms the STN projection neurons, which are naturally glutamatergic and excitatory, into inhibitory GABAergic neurons (Fig. 84-5), thereby reducing the excessive stimulatory effects of the STN on the GPi, which are thought to contribute to many parkinsonian symptoms. This follows the theory that STN-DBS generates its positive effects in PD by similarly reducing STN excitatory output.147
Before considering a clinical application of this concept, AAV-GAD was injected into the STN of 6-OHDA rats.148 These animals demonstrated activity-dependent GABA release from STN nerve terminals, increased survival of dopaminergic neurons, and marked behavioral recovery. Moreover, GAD transduction of the STN appeared to protect SNc dopaminergic neurons by preventing the lesion induced by 6-OHDA. A recently published study in MPTP monkeys given AAV-GAD injections found more modest improvements (about 20%) in motor behavior, although statistically significant gross motor skill and tremor improvements were noted at 1 year.149
A human phase I clinical trial of AAV-GAD gene therapy is well under way, with interim data already published.150 Twelve PD patients with reduced efficacy or tolerance of levodopa underwent unilateral stereotactic STN injections of one of three vector doses based on magnetic resonance imaging targeting. Outcome measures include changes in brain metabolism on PET scans and standard clinical rating scales. No study-related significant adverse events or withdrawals occurred, and no new antivector or anti-GAD peripheral antibodies were detected. At 1 year of follow-up, modest but significant improvements were noted in UPDRS scores, along with a trend toward superior activities of daily living scores. Further, fluorodeoxyglucose (FDG) PET demonstrated reduced thalamic metabolism in the operated hemisphere and increased local metabolism in the ipsilateral supplementary motor area, the latter being highly correlated with UPDRS score improvement.150,151 These promising clinical findings and supportive imaging studies, along with a favorable safety profile, suggest that AAV-GAD gene therapy may be beneficial in PD. Although future data from this trial will help confirm this early impression, a subsequent phase II trial will be required to judge whether the magnitude of effect is clinically meaningful.
Growth Factor Gene Therapy
Unlike the other two strategies, which are directed at controlling PD symptoms, the goal of this therapy is to prevent ongoing degeneration of dopamine neurons by ectopically expressing the growth factor neurturin throughout the striatum. Growth factors (particularly GDNF) attenuate the loss of nigrostriatal dopaminergic neurons and restore function of the nigrostriatal pathway in animal models of PD.119,152 Accordingly, targeted delivery of a gene that promotes growth factor production in the human SNc might achieve clinically meaningful neuroprotection or neurorestoration in PD patients. Owing to intellectual property issues, clinical gene therapy with GDNF is impossible at this time; however, work is proceeding with a related neurotrophin, neurturin. Neurturin is a naturally occurring structural and functional GDNF analogue and has already been used in patients.153–155
Lentivirus- and AAV-mediated GDNF gene therapy and striatal neurturin infusion have shown protective effects against the loss of dopaminergic nigral neurons and the motor consequences of MPTP intoxication in primates and 6-OHDA lesioning in rats.122,156,157 However, it was uncertain whether benefits would also be realized when nigral degeneration was quite advanced at the time of disease diagnosis. Kordower and colleagues158 performed AAV-neurturin gene therapy in MPTP monkeys and showed that neurturin expression is associated with substantial histologic and clinical neuroprotection. Importantly, these monkeys received the vector infusion following MPTP administration at a time when nigral destruction was under way. AAV-neurturin also improved motor function in aged monkeys, restoring failing nigrostrial function.159
[/not-level-membership-for-neurosurgery-category]
