1 Embryology of the Spine
KEY POINTS
Gastrulation
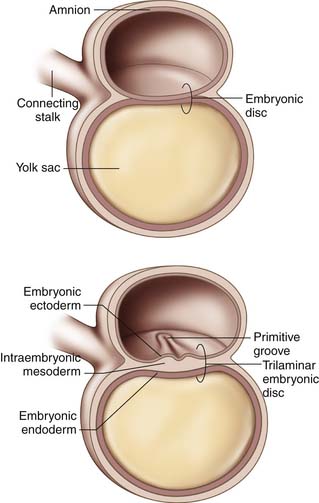
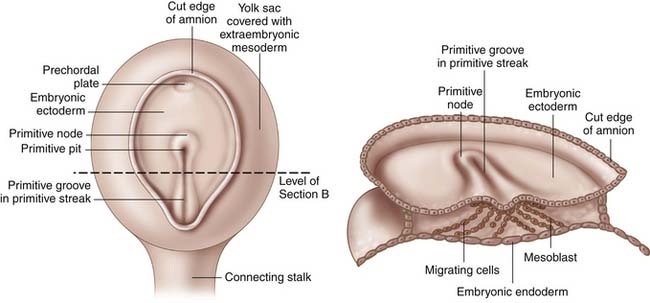
Gastrulation begins in the third week of gestation and gives rise to three distinct germ layers, the ectoderm, the mesoderm, and the endoderm. The initial phase of gastrulation begins with formation of the primitive streak, which is sometimes named the primitive groove (Figure 1-1). This midline thickening of the germinal disc terminates in the primitive node. Under control of embryonic growth factors, cells of the epiblast layer migrate inward to form the mesoderm and the endoderm through the process of invagination. Cells migrating farthest from the epiderm and closest to the yolk sac become the endoderm. The remaining epiblast cells will eventually differentiate into the ectoderm (Figure 1-2). The migrating cells that are sandwiched between the endoderm and ectoderm layers will become the mesoderm. Control of these migrations is maintained through various cell-signaling pathways that also contribute to establishment of the body axes in all planes. The signaling pathways, or organizer genes, are secreted by the primitive streak and mesoderm. The cranial direction of the embryonic disc is established by a specialized area of cells, referred to as the anterior visceral endoderm, that expresses genes required for formation of the head and cerebrum. The dorsal-ventral axis is regulated by growth factors in the TGF-β family including bone morphogenic protein-4, fibroblast growth factor, and the sonic hedgehog gene. Control of sidedness is regulated by fibroblast growth factor-8, Nodal, and Lefty-2, all of which are secreted on the left side of the germinal disc. An additional protein, Lefty-1, is secreted to prevent migration of the left sided growth factors across the midline.1
Somite Period
The presence of the notochord induces proliferation of the mesoderm. At approximately 17 days of gestation the mesoderm thickens into two masses, each located directly adjacent to the notochord. This initial layer, termed the paraxial mesoderm, continues to spread laterally to eventually differentiate into three distinct areas, paraxial mesoderm, intermediate mesoderm, and lateral mesoderm. During the somite period, lasting from approximately 19 to 30 days post fertilization, the paraxial mesoderm will develop into segmental bulbs of tissue on either side of the notochord (Figure 1-3). The first pair of somites will appear adjacent to the notochord, and they will continue to develop in a cranial to caudal direction until a total of 42 to 44 pairs of somites appear by the end of the fifth week of gestation. The first 24 somite segments are responsible for the cervical, thoracic, and lumbar spine. Somites 25 through 29 contribute to formation of the sacrum, while pairs 30 through 35 are responsible for coccyx formation. The rest of the 42 to 44 somite pairs disappear through a process of regression, which occurs at approximately 6 weeks of gestation.
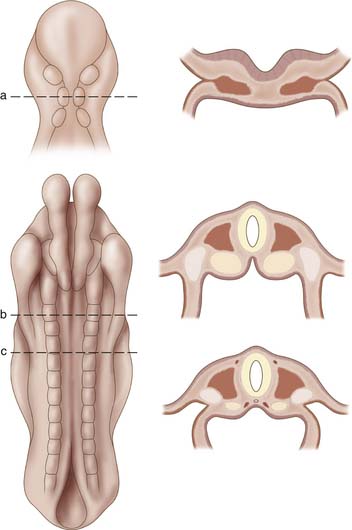
The somites continue to differentiate into two distinct tissues. Ventromedial cells develop into the sclerotome, while dorsolateral cells develop into the dermatomyotome. The latter cells will eventually give rise to the integument system and dorsal musculature of the body, while the sclerotome will migrate to surround the notochord and give rise to the vertebral column. Regulation of sclerotome formation is controlled by proteins coded by the sonic hedgehog gene, which is expressed by cells of the notochord. This process of sclerotome migration will begin by the fourth week of gestation. Each sclerotome will be divided by an intersegmental vessel and a loose area of intersegmental mesenchyme. In addition, a pair of myotomes and accompanying segmental nerves will be associated with each sclerotome.
As the process of differentiation continues, each sclerotome will divide into a cranial region of relatively loosely packed cells and a caudal portion of rapidly proliferating and densely packed cells. At this point in spinal development, classic embryology texts describe a phenomenon by which the pace of proliferation is so great that the caudal part of the one sclerotome begins to overgrow into the cranial portion of the adjacent sclerotome and thereby fusing to create a single mass of tissue destined to become the precartilaginous vertebral body. Parke (The Spine, 1999) suggests that this theory of “resegmentation” may not be accurate, and provides compelling evidence toward an alternate route of vertebral body formation. In his summary of the recent evidence, Parke outlines a pathway of spinal development which begins with a uniform layer of axial mesenchyme surrounding the notochord. The sclerotomal organization is still maintained with an intersegmental vessel, a nerve, and a peripheral layer of dermatomyotome associated with each segment. However, the uniform mesenchyme undergoes a period of differentiation by which densities develop within the loose tissues. These dense regions will develop into the intervertebral discs and eventually pinch off the notochord which will be trapped within the dense tissue to become the nucleus pulposus.2 The loose tissues between the discs form the cartilaginous centrum, which is the precursor of the vertebral bodies. The caudal portion of the centrum undergoes rapid proliferation, and cells migrate peripherally to surround the neural tube, forming the membranous neural arches which will serve to protect the neural elements. In total, each bony vertebral segment will consist of five ossification centers, one centrum, two neural arches, and two costal elements.
Ossification of the vertebral bodies occurs around the ninth week of gestation and begins at the thoracolumbar junction. Ossification then proceeds in both cranial and caudal directions, with the caudal segments demonstrating a quicker rate of ossification compared with the cranial segments. Ossification of the posterior arches begins at approximately the same time but begins in the cervical vertebrae and proceeds in a caudal direction. As the two neural arch centers approach midline, they begin to fuse, forming the lamina and spinous process. Fusion of the neural arches first occurs in the lumbar segments during the first year of life and proceeds cranially. Fusion is not completed until ages of 5 to 8 years. The costal ossification centers have a variable role in vertebral body formation. In the cervical spine, these centers have a minimal contribution and may contribute to part of the foramen transversarium. In the thoracic spine, these ossification centers are the precursors of the ribs. In the lumbosacral spine, the costal ossification centers are responsible for formation of the transverse processes and the anterolateral portion of the sacrum.2
Upper Cervical Spine
A detailed anatomic study of cervical spine anatomy was presented by O’Rahilly and Meyer in a serial time reconstruction of human embryos ranging from 8 to approximately 16 weeks of gestation.3 It is generally believed that the most cranial 4 or 5 pairs of somites are responsible for the occipital-atlas complex. Development of this junction is regulated by growth factors derived from the notochord as it crosses into the cranium. The notochord travels through the middle or slightly anterior portion of each centrum, and up through the future dens at the level of the axis. It then makes an anterior directed turn to enter the skull just above the level of the dens. At this time, each centrum is divided by a thickening of the notochord that will develop into the nucleus pulposus. The true boundary between the spine and cranium is not fully understood, with some authors suggesting that the atlas is a standalone accessory cranial bone with the true head-neck boundary being between the C1 and C2 articulation.
O’Rahilly and Meyer describe the centrum of the axis as being composed of three axial columns which they termed X, Y, and Z. The first and most cranial column, X, will develop an articulation with the anterior tubercle of C1, forming the atlanto-dens joint space.4 By approximately 9 weeks of gestation, the X column, or future dens, is already bounded posteriorly by the transverse ligament and anchored into the occipital condyles by the alar ligamentous complex. Columns Y and Z are separated by the remnants of an intervertebral disc that may persist well into birth. Although it is generally accepted that column Z will form the centrum of the axis, the fate of column Y remains uncertain. Some believe that it is incorporated into the axis centrum, while other embryologists believe, based on reptilian studies, that it is incorporated into the centrum of the atlas. Calcifications in the three columns are readily visible by the time the embryo reached a length of 120 mm; however, fusion will not take place until 6 to 8 years of age. In some instances, the tip of the dens may calcify independently without fusion to the remaining axis; this is termed os odontoideum.
Neural Development
The neural elements likewise form during gastrulation. Under control of growth factors secreted by the prechordal plate, the ectoderm begins to thicken at the cranial end to form the neural plate and the lateral edges of the germinal disc fold to form the neural crests. This process is primary neurulation. As previously discussed, the neural crests meet in the midline to form the neural tube. The tube has two open ends, the cranial and caudal neuropores, both of which communicate with the amniotic cavity. This communication allows for prenatal detection of central nervous system markers in amniotic fluid if neural tube closure has failed to complete. There are likely multiple foci at which neural tube closure initiates and progresses both cranially and caudally in a zipperlike fashion. The cranial neuropore is generally first to close, and final closure is not complete until about 25 days post gestation.1 The caudal neuropore closes approximately 2 days later. Failure of neuropore closure at the cranial end results in anencephaly, a deficiency of skull, scalp, and forebrain. Failure of caudal neuropore closure results in spina bifida. Once closure is completed, the neural tube must separate from the ectoderm. This process is termed dysjunction; premature separation during this step of neurulation may pull primitive mesenchyme tissues inside the developing neural tube, resulting in a lipomeningocele or lipomyelomeningocele.5 Incomplete separation may lead to cutaneous sinuses that communicate with the spinal canal.
The spinal nerves begin to form in the fourth week after gestation. Each nerve is composed of a ventral motor root and a dorsal sensory root. Axons of the ventral motor horn cells project through the marginal zone and coalesce outside of the neural tube into the ventral motor root. These axons will continue to the motor endplates of the muscles formed from its respective sclerotome.
Sacrum and Conus Medullaris Development
Development of the neural structures in the caudal terminus of the spine deserves some special attention. At their respective most distal points, the neural tube and notochord coalesce into an undifferentiated cellular mass that will develop into the coccyx, sacrum, and fifth lumbar vertebrae. This process is the beginning of secondary neurulation. A single canal will form within this mass through a process called canalization. Debate exists in the literature as to whether this newly formed neural tube is initially continuous with the primary neural tube or whether the two coalesce at a later point in development. It is known that the chick embryo develops two distinct neural tubes that anastomose in the sacral region, while in a mouse embryo, the secondary neural tube forms as an extension of the primary neural tube. The pathway of secondary neurulation in humans is not yet elucidated; however, it is known that the distal portion of the tube and central canal will regress in a cephalic direction via a process called retrogressive differentiation. This will give rise to the conus medullaris and will leave behind a thin film of pia mater tissue called the filum terminale.5 Nerve root compression may result when an abnormally thick filum terminale is present (usually greater than 2 mm in diameter).
Associated Anomalies
While discussing spinal embryology, it is important to remember that spinal development is not an isolated event. Multiple organ systems are developing in parallel with the spine and often share the same germinal tissue source. Any internal or external insult to the developing embryo may affect other organ systems. The mesoderm is particularly involved in the genesis of several organs. Paraxial mesoderm, the precursor of the centrum and vertebral column, is also responsible for formation of the dermis, skeletal muscle, and the connective tissue of the head.6 The intermediate and lateral mesoderm is responsible for formation of the urogenital, cardiac, and renal systems. In children with known congenital spinal defects, the incidence of associated anomalies has been reported as high as 30% to 60%.7 The most common organ system to be affected is the genitourinary system. Mesoderm tissues that make up the spinal column also contribute to formation of the mesonephros. While it is the medial region of the mesoderm that forms the vertebrae, the ventrolateral region forms the genitourinary organs.8 The cardiopulmonary system is also commonly involved in conjunction with a congenital spinal abnormality. These anomalies may be fatal and should be diagnosed and treated before their associated problems progress. Diagnosis of both congenital spinal defects and associated anomalies may be made on prenatal ultrasound examination.
The timing of insult during fetal development also affects the rate of associated anomalies. Tsou (1980) divided a group of 144 patients with congenital spinal anomalies into two groups: embryonic anomalies, defined as those that occurred in the first 56 days post fertilization, and fetal anomalies, defined as those that occurred from day 57 of gestation to birth.9 They found that the rate of associated defects was 7% in the fetal group as compared with 35% in the embryonic group. Associated orthopedic anomalies included Klippel-Feil syndrome, acetabular dysplasia, clubfoot, congenital short leg, Sprengel deformity, coxa vara, radial clubhand, and thumb aplasia. Nonorthopedic associated anomalies included dextrocardia, hypospadia, microtia, lung aplasia, pulmonary arterial stenosis, imperforate anus, mandibular anomalies, cleft palate, and hemidiaphragm.9
Congenital Spinal Anomalies
Defects of Formation
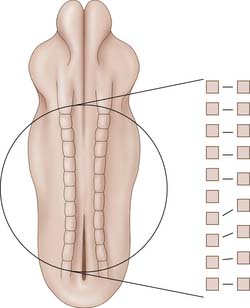
Although it is generally agreed that hemivertebrae are the result of the failure of formation, the exact pathophysiology has not yet been elucidated. It is helpful to separate failures of formation as those occurring during the embryonic period and those that occur in the fetal period. During the embryonic stage, most authors propose a theory of “segmental shift,” which occurs during the sclerotomic pairing phase of embryogenesis.9 As the somites join in the midline, it is assumed that each somite is in the same developmental phase as its counterpart across the midline. This development usually proceeds in a predictable pattern from a cranial to caudal direction. Asynchronous development of one somite in a hemimetameric pair may prevent normal midline fusion and result in a caudal shift of the column such that the two contralateral pairs are in a synchronous phase of development. This would leave an isolated out-of-phase hemivertebra without a cross-midline counterpart (Figure 1-4). This segmental shift theory is further supported by the presence of double-balanced hemivertebra where each of the asynchronous hemi-vertebra is found on one side of the midline. The most caudal hemivertebra is commonly found at the lumbosacral junction where there is no further room for compensation from the somite below. Another mechanism for hemivertebra formation may result from a physiologic insult to the somite precursor during the embryonic period. Although midline fusion occurs between corresponding somites, the injured hemimetameric pair may undergo growth retardation of variable severity. Mild growth retardation may result in a hypoplastic hemivertebra in which the growth plates are formed, but the rate of growth is not equal to the opposite side. More severe forms of sclerotome growth retardation may result in a failure of segmentation and will be discussed later.
Insults to the growing spine during the embryonic stage tend to globally affect the vertebral segment, including both posterior and anterior elements. Insults occurring during the fetal period tend to be more specific and only affect a portion of the vertebra, the centrum being the area most commonly afflicted. Centrum hypoplasia and aplasia is described by Tsou as a spectrum of growth retardation that occurs from 2 to 7 months post fertilization during a period of normally rapid vertebral growth.9 A vascular etiology for centrum aplasia and hypoplasia was proposed by Schmorl and Junghanns; however, this has not yet been proven. Identification of these failures of formation is clinically important as they may often result in structural deformity of the spine. Centrum aplasia and posterior hemicentrum have been shown to cause an isolated kyphotic deformity, while wedge vertebra, posterior corner hemivertebra, and a lateral hemicentrum more commonly cause a mixed kyphoscoliotic deformity.6
Defects of Segmentation
Segmentation defects may also occur during formation of the intervertebral disc or the adjacent articulations. By the late embryonic period, mesodermal cells have migrated around the notochord and formed dense collections of tissues which will form the annulus fibrosus. In a more common form of segmentation defect, the anterior portion of the annulus undergoes first what Tsou describes as a cartilaginous transformation, followed by osseous metaplasia.9 A bony bar forms between two or more adjacent vertebral bodies as ossification continues into childhood. This anterior tether may result in a severe kyphotic deformity that worsens with continued growth.
Spina Bifida
Derived from the Latin term bifidus, spina bifida literally means a spine split in two. Although the severity of the disease may range from a benign incidental finding on x-ray to severe neurologic damage, the etiology remains the same, a failure of the embryonic vertebral arches to fuse. Causes for this lack of fusion are multifactorial. Mitchell (1997) suggested a weak genetic component by demonstrating an increased risk in siblings of affected children and even further increased risk with multiple affected siblings.10 Environmental factors also play a role in the etiology of spina bifida. Mitchell correlated incidence with time of season, geographic location, ethnicity, race, socioeconomic status, maternal age and parity, and maternal nutritional status, specifically the dietary intake of folic acid and alcohol. Although the mechanism by which folic acid aids in neural tube closure is unknown, the role of folic acid as a substrate in DNA synthesis has been well described. An enzyme called methyl tetra hydroxy folate reductase (MTHFR) is involved in folate metabolism during DNA synthesis. Genetic alterations in this enzyme may lead to decreased enzymatic activity and increase the dietary folate requirements for proper DNA synthesis. As neural tube closure has been shown to begin early in the embryonic period, it is vital to begin folate supplementation as early as possible in the prenatal period and encourage dietary supplementation during family planning.
Spina bifida cystica refers to a more severe form of spina bifida; it can be broken down into several subgroups based on the degree of the involved tissue layers.6 The first group, spina bifida with meningocele, involves the meninges as well as the posterior arches. A cystic pouch is present within the meninges without involvement of the spinal cord or nerve roots. Patients are typically spared neurologically. Physical exam findings may be similar to those of spina bifida occulta, but also include subcutaneous lipomas and hemangiomas adjacent to the lesion. Spina bifida with myelomeningocele is the next most severe form of spina bifida. This disease results from failure of fusion in the posterior arches with involvement of the spinal cord and meninges. By definition, in spina bifida with myelomeningocele, the neural elements are not exposed to the external environment and are covered by a membranous cerebrospinal fluid–filled sac. This disease typically presents with neurological disorder based on the neurological level of the lesion. Associated anomalies include Arnold-Chiari malformation, hydrocephalus, scoliosis, and kyphosis. The most severe manifestation of spina bifida cystica is myeloschisis. In this severe presentation the neural elements are completely exposed. Neurologic injury is certain and infections are common.
Conclusion
Clinicians treating spinal disorders may benefit from an understanding of the processes that drive spinal embryogenesis and the origin of common disorders affecting the spine. The process of embryogenesis is extremely complicated, but incredibly well synchronized. Multiple events happen in series and in parallel, all under the control of signaling pathways that are just now becoming understood. Spine development has been widely elucidated using human and animal data; however there remain many unknowns, especially on a molecular level. It is vital to remember that disorders of spinal development may not be isolated events; other organ systems are often affected. Awareness and early intervention may be required for optimal patient care.
1. Sadler T.W. Medical embryology, ninth ed. Baltimore: Lippincott Williams & Wilkins; 2004.
2. Herkowitz H.N., Garfin S.R., Eismont F.J., Bell G.R. Rothman-Simeone the spine. Philadelphia: WB Saunders; 1999.
3. O’Rahilly R., Meyer D.B. The timing and sequence of events in the development of the human vertebral column during the embryonic period proper. Anat. Embryol. (Berl). 1979;157(2):167-176.
4. O’Rahilly R., Muller F., Meyer D.B. The human vertebral column at the end of the embryonic period proper. 2. The occipitocervical region. J. Anat.. 1983;136(1):181-195.
5. Grimme J.D., Castillo M. Congenital anomalies of the spine. Neuroimaging Clin. N. Am.. 2007;17(1):1-16.
6. Kaplan K.M., Spivak J.M., Bendo J.A. Embryology of the spine and associated congenital abnormalities. Spine J.. 2005;5(5):564-576.
7. Jaskwhich D., et al. Congenital scoliosis. Curr. Opin. Pediatr.. 2000;12(1):61-66.
8. MacEwen G.D., Winter R.B., Hardy J.H. Evaluation of kidney anomalies in congenital scoliosis. J. Bone Joint Surg. Am.. 1972;54(7):1451-1454.
9. Tsou P.M: Embryology of congenital kyphosis, Clin. Orthop. Relat. Res., 128:1977, 18-25.
10. Mitchell L.E. Genetic epidemiology of birth defects: nonsyndromic cleft lip and neural tube defects. Epidemiol. Rev.. 1997;19(1):61-68.