[level-membership-for-pediatrics-category]
Chapter 52 Electrolyte and Acid-Base Disorders
52.1 Composition of Body Fluids
Total Body Water
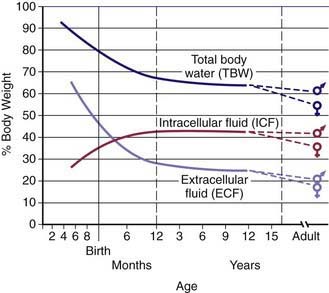
Water is the most plentiful constituent of the human body. Total body water (TBW) as a percentage of body weight varies with age (Web  Fig. 52-1). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and basically remains at this level until puberty. At puberty, the fat content of females increases more than that of males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and, thus, is a smaller percentage of body weight.
Fig. 52-1). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and basically remains at this level until puberty. At puberty, the fat content of females increases more than that of males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and, thus, is a smaller percentage of body weight.
Fluid Compartments
TBW is divided between 2 main compartments: intracellular fluid (ICF) and extracellular fluid (ECF). In the fetus and newborn, the ECF volume is larger than the ICF volume (see Web Fig. 52-1). The normal postnatal diuresis causes an immediate decrease in the ECF volume. This is followed by continued expansion of the ICF volume, which results from cellular growth. By 1 yr of age, the ratio of the ICF volume to the ECF volume approaches adult levels. The ECF volume is approximately 20-25% of body weight, and the ICF volume is approximately 30-40% of body weight, close to twice the ECF volume (Web Fig. 52-2). With puberty, the increased muscle mass of males causes them to have a higher ICF volume than females. There is no significant difference in the ECF volume between postpubertal females and males.
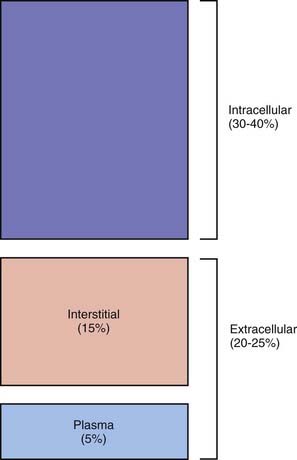
Web Figure 52-2 Compartments of total body water, expressed as percentages of body weight, in an older child or adult.
The ECF is further divided into the plasma water and the interstitial fluid (see Web Fig. 52-2). The plasma water is 5% of body weight. The blood volume, given a hematocrit of 40%, is usually 8% of body weight, although it is higher in newborns and young infants; in premature newborns, it is approximately 10% of body weight. The volume of plasma water can be altered by pathologic conditions, including dehydration, anemia, polycythemia, heart failure, abnormal plasma osmolality, and hypoalbuminemia. The interstitial fluid, normally 15% of body weight, can increase dramatically in diseases associated with edema, such as heart failure, protein-losing enteropathy, liver failure, nephrotic syndrome, and sepsis. An increase in interstitial fluid also occurs in patients with ascites or pleural effusions.
Electrolyte Composition
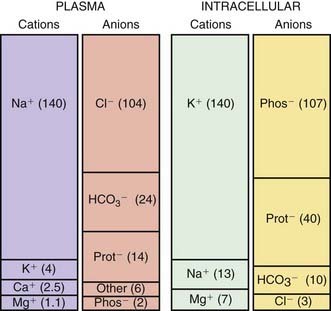
The composition of the solutes in the ICF and ECF are very different (Web Fig. 52-3). Sodium and chloride are the dominant cation and anion, respectively, in the ECF. The sodium and chloride concentrations in the ICF are much lower. Potassium is the most abundant cation in the ICF, and its concentration within the cells is approximately 30 times higher than in the ECF. Proteins, organic anions, and phosphate are the most plentiful anions in the ICF. The dissimilarity between the anions in the ICF and the ECF is largely determined by the presence of intracellular molecules that do not cross the cell membrane, the barrier separating the ECF and the ICF. In contrast, the difference in the distribution of cations—sodium and potassium—is due to the activity of the Na+,K+-ATPase pump, which uses cellular energy to actively extrude sodium from cells and move potassium into cells. The chemical gradient between the intracellular potassium concentration and the extracellular potassium concentration creates the electrical gradient across the cell membrane. The concentration-dependent movement of potassium out of the cell makes the intracellular space negative relative to the extracellular space.
Osmolality
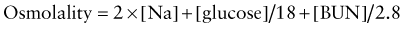
Glucose and blood urea nitrogen (BUN) are measured in mg/dL. Division of these values by 18 and 2.8, respectively, as shown, converts the units into mmol/L. Multiplication of the sodium value by 2 accounts for its accompanying anions, principally chloride and bicarbonate. The calculated osmolality is usually slightly lower than the measured osmolality.
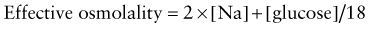
The effective osmolality (also called the tonicity) determines the osmotic force that is mediating the shift of water between the ECF and the ICF.
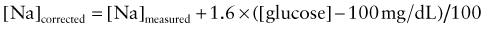
where [Na]measured = sodium concentration measured by the clinical laboratory and [Na]corrected = corrected sodium concentration (the sodium concentration if the glucose concentration were normal and its accompanying water moved back into the cells). The [Na]corrected is the more reliable indicator of the patient’s true ratio of total body sodium to TBW, the normal determinant of the sodium concentration.
Garibaldi BT, Cameron SJ, Choi M. Pseudohyponatremia in a patient with HIV and hepatitis C coinfection. J Gen Intern Med. 2008;23:202-205.
Nguyen MK, Ornekian V, Butch AW, et al. A new method for determining plasma water content: application in pseudohyponatremia. Am J Physiol Renal Physiol. 2007;292:F1652-F1656.
Pellegrino B, Parravani A, Cook L, et al. Ethylene glycol intoxication: disparate findings of immediate versus delayed presentation. W V Med J. 2006;102:32-34.
52.2 Regulation of Osmolality and Volume
Regulation of Osmolality
Osmoreceptors in the hypothalamus sense the plasma osmolality (Chapter 552). An elevated effective osmolality leads to secretion of antidiuretic hormone (ADH) by neurons in the supraoptic and paraventricular nuclei in the hypothalamus. The axons of these neurons terminate in the posterior pituitary. Circulating ADH binds to its V2 receptors in the collecting duct cells of the kidney, and, via the generation of cyclic adenosine monophosphate, causes insertion of water channels (aquaporin-2) into the renal collecting ducts. This produces increased permeability to water, permitting resorption of water into the hypertonic renal medulla. The end result is that the urine concentration increases and water excretion decreases. Urinary water losses cannot be completely eliminated because there is obligatory excretion of urinary solutes, such as urea and sodium. The regulation of ADH secretion is tightly linked to plasma osmolality, responses being detectable with a 1% change in the osmolality. ADH secretion virtually disappears when the plasma osmolality is low, allowing excretion of maximally dilute urine. The consequent loss of free water (water without sodium) corrects the plasma osmolality. ADH secretion is not an all-or-nothing response; there is a graded adjustment as the osmolality changes.
A number of conditions can limit the kidney’s ability to excrete adequate water to correct low plasma osmolality. In the syndrome of inappropriate antidiuretic hormone (SIADH), ADH continues to be produced despite a low plasma osmolality. In the presence of ADH, urinary dilution does not occur, and sufficient water is not excreted (Chapters 52.3 and 553).
The normal response to increased plasma osmolality is conservation of water by the kidney. In central diabetes insipidus, this does not occur because of an absence of ADH secretion (Chapter 552.1). Patients with nephrogenic diabetes insipidus have an inability to respond to ADH and produce dilute urine despite an increase in plasma osmolality (Chapters 52.3, 524, and 552).
Regulation of Volume
Sodium resorption occurs throughout the nephron (Chapter 522). Whereas the majority of filtered sodium is resorbed in the proximal tubule and the loop of Henle, the distal tubule and the collecting duct are the main sites for precise regulation of sodium balance. Approximately 65% of the filtered sodium is reclaimed in the proximal tubule, which is the major site for resorption of bicarbonate, glucose, phosphate, amino acids, and other substances that are filtered by the glomerulus. The transport of all these substances is linked to sodium resorption by cotransporters, or a sodium-hydrogen exchanger in the case of bicarbonate. This link is clinically important for bicarbonate and phosphate because their resorption parallels sodium resorption. In patients with metabolic alkalosis and volume depletion, correction of the metabolic alkalosis requires urinary loss of bicarbonate, but the volume depletion stimulates sodium and bicarbonate retention, preventing correction of the alkalosis. Volume expansion causes increased urinary losses of phosphate, even when there is phosphate depletion. Resorption of uric acid and urea occurs in the proximal tubule and increases when sodium retention increases. This arrangement accounts for the elevated uric acid and BUN measurements that often accompany dehydration, which is a stimulus for sodium retention in the proximal tubule. The cells of the proximal tubule are permeable to water; thus, water resorption in this segment parallels sodium resorption.
Volume depletion usually occurs when sodium losses exceed intake. The most common etiology in children is gastroenteritis. Excessive losses of sodium may also occur from the skin in children with burns, in sweat from patients with cystic fibrosis, or after vigorous exercise. Inadequate intake of sodium is uncommon except in neglect, in famine, or with an inappropriate choice of liquid diet in a child who cannot take solids. Urinary sodium wasting may occur in a range of renal diseases, from renal dysplasia to tubular disorders, such as Bartter syndrome. The neonate, especially if premature, has a mild impairment in the ability to conserve sodium. Iatrogenic renal sodium wasting takes place during diuretic therapy. Renal sodium loss occurs as a result of derangement in the normal regulatory systems. An absence of aldosterone, seen most commonly in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency, causes sodium wasting (Chapter 570).
Ball SG. Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem. 2007;44:417-431.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19:1076-1078.
Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519-531.
Nielsen S, Kwon TH, Frokiaer J, et al. Regulation and dysregulation of aquaporins in water balance disorders. J Intern Med. 2007;261:53-64.
Schrier RW. Aquaporin-related disorders of water homeostasis. Drug News Perspect. 2007;20:447-453.
Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol. 2007;18:2028-2031.
52.3 Sodium
Sodium Metabolism
Body Content and Physiologic Function
Sodium is the dominant cation of the ECF (see  Web Fig. 52-3), and it is the principal determinant of extracellular osmolality. Sodium is therefore necessary for the maintenance of intravascular volume. Less than 3% of sodium is intracellular. More than 40% of total body sodium is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular sodium concentration, approximately 10 mEq/L, is maintained by Na+,K+-ATPase, which exchanges intracellular sodium for extracellular potassium.
Web Fig. 52-3), and it is the principal determinant of extracellular osmolality. Sodium is therefore necessary for the maintenance of intravascular volume. Less than 3% of sodium is intracellular. More than 40% of total body sodium is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular sodium concentration, approximately 10 mEq/L, is maintained by Na+,K+-ATPase, which exchanges intracellular sodium for extracellular potassium.
Intake
Sodium is readily absorbed throughout the gastrointestinal tract. Mineralocorticoids increase sodium transport into the body, although this effect has limited clinical significance. The presence of glucose enhances sodium absorption owing to the presence of a cotransport system. This is the rationale for including sodium and glucose in oral rehydration solutions (Chapter 332).
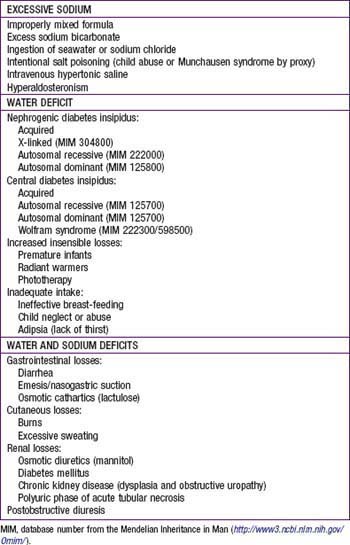
Hypernatremia
Etiology and Pathophysiology
There are 3 basic mechanisms of hypernatremia (Table 52-1). Sodium intoxication is frequently iatrogenic in a hospital setting as a result of correction of metabolic acidosis with sodium bicarbonate. Baking soda, a putative home remedy for upset stomach, is another source of sodium bicarbonate; the hypernatremia is accompanied by a profound metabolic alkalosis. In hyperaldosteronism, there is renal retention of sodium and resultant hypertension; the hypernatremia is usually mild.
Table 52-1 CAUSES OF HYPERNATREMIA
EXCESSIVE SODIUM
WATER DEFICIT
WATER AND SODIUM DEFICITS
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
The classic causes of hypernatremia from a water deficit are nephrogenic and central diabetes insipidus (Chapters 524 and 552). Hypernatremia develops in diabetes insipidus only if the patient does not have access to water or cannot drink adequately because of immaturity, neurologic impairment, emesis, or anorexia. Infants are at high risk because of their inability to control their own water intake. Central diabetes insipidus and the genetic forms of nephrogenic diabetes insipidus typically cause massive urinary water losses and very dilute urine. The water losses are less dramatic, and the urine often has the same osmolality as plasma when nephrogenic diabetes insipidus is secondary to disease (obstructive uropathy, renal dysplasia, sickle cell disease).
When hypernatremia occurs in conditions with deficits of sodium and water, the water deficit exceeds the sodium deficit. This occurs only if the patient is unable to ingest adequate water. Diarrhea results in depletion of both sodium and water. Because diarrhea is hypotonic—typical sodium concentration of 35-65 mEq/L—water losses exceed sodium losses, potentially leading to hypernatremia. Most children with gastroenteritis do not have hypernatremia because they drink enough hypotonic fluid to compensate for stool water losses (Chapter 332). Fluids such as water, juice, and formula are more hypotonic than the stool losses, allowing correction of the water deficit and potentially even causing hyponatremia. Hypernatremia is most likely to occur in the child with diarrhea who has inadequate intake because of emesis, lack of access to water, or anorexia.
Clinical Manifestations
Most children with hypernatremia are dehydrated and show the typical clinical signs and symptoms (Chapter 54). Children with hypernatremic dehydration tend to have better preservation of intravascular volume because of the shift of water from the intracellular space to the extracellular space. This shift maintains blood pressure and urine output and allows hypernatremic infants to be less symptomatic initially and potentially to become more dehydrated before medical attention is sought. Breast-fed infants with hypernatremia are often profoundly dehydrated, with failure to thrive. Probably because of intracellular water loss, the pinched abdominal skin of a dehydrated, hypernatremic infant has a “doughy” feel.
Diagnosis
When there is isolated water loss, the signs of volume depletion are usually less severe initially because much of the loss is from the intracellular space. When pure water loss causes signs of dehydration, the hypernatremia and water deficit are usually severe. In the child with renal water loss, either central or nephrogenic diabetes insipidus, the urine is inappropriately dilute and urine volume is not low. The urine is maximally concentrated and urine volume is low if the losses are extrarenal or due to inadequate intake. With extrarenal causes of loss of water, the urine osmolality should be >1,000 mOsm/kg. When diabetes insipidus is suspected, the evaluation may include measurement of ADH and a water deprivation test, including a trial of desmopressin acetate (synthetic ADH analog) to differentiate between nephrogenic diabetes insipidus and central diabetes insipidus (Chapter 552.1). A water deprivation test is unnecessary if the patient has simultaneous documentation of hypernatremia and poorly concentrated urine (osmolality lower than that of plasma). In children with central diabetes insipidus, administration of desmopressin acetate increases the urine osmolality above the plasma osmolality, although maximum osmolality does not occur immediately because of the decreased osmolality of the renal medulla due to the chronic lack of ADH. In children with nephrogenic diabetes insipidus, there is no response to desmopressin acetate.
Treatment
As hypernatremia develops, the brain generates idiogenic osmoles to increase the intracellular osmolality and prevent the loss of brain water. This mechanism is not instantaneous and is most prominent when hypernatremia has developed gradually. If the serum sodium concentration is lowered rapidly, there is movement of water from the serum into the brain cells to equalize the osmolality in the 2 compartments (Fig. 52-1). The resultant brain swelling manifests as seizures or coma.
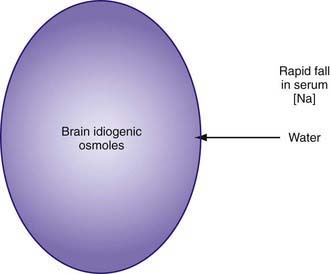
In the child with hypernatremic dehydration, as in any child with dehydration, the first priority is restoration of intravascular volume with isotonic fluid (Chapter 54). Normal saline is preferable to lactated Ringer solution because the lower sodium concentration of the latter can cause the serum sodium to decrease too rapidly, especially if multiple fluid boluses are given. Repeated boluses of normal saline (10-20 mL/kg) may be required to treat hypotension, tachycardia, and signs of poor perfusion (peripheral pulses, capillary refill time) (Chapters 54 and 64).
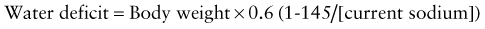
This calculation is equivalent to 3-4 mL of water per kg for each 1 mEq that the current sodium level exceeds 145 mEq. The utility of such formulas has never been proven in clinical practice. Most patients with hypernatremic dehydration do well with a fluid sodium concentration of approximately half-normal saline, but with a fluid rate that is only 20-30% greater than maintenance fluid. Use of this concentration prevents excessive delivery of free water and too rapid a decrease in the serum sodium level. Patients with pure water loss may require a more hypotonic fluid (0.2 normal saline). Excessive water and sodium losses may also need to be replaced. If signs or symptoms of volume depletion develop, the patient receives additional boluses of isotonic saline. Monitoring of the rate of decrease of the serum sodium concentration permits adjustment in the rate and sodium concentration of the fluid that the patient is receiving, avoiding overly rapid correction of the hypernatremia. Many patients with mild to moderate hypernatremic dehydration due to gastroenteritis can be managed with oral rehydration (Chapter 332).
It is important to address the underlying cause of the hypernatremia, if possible. The child with central diabetes insipidus should receive desmopressin acetate. Because this treatment reduces renal excretion of water, excessive intake of water must consequently be avoided to prevent both overly rapid correction of the hypernatremia and the development of hyponatremia. Over the long term, reduced sodium intake and the use of medications can somewhat ameliorate the water losses in nephrogenic diabetes insipidus (Chapter 524). The daily water intake of a child who is receiving tube feeding may need to be increased to compensate for high losses. The patient with significant ongoing losses, such as through diarrhea, may need supplemental water and electrolytes (Chapter 53). Sodium intake is reduced if it contributed to the hypernatremia.
Hyponatremia
Etiology and Pathophysiology
The causes of hyponatremia are listed in Table 52-2. Pseudohyponatremia is a laboratory artifact that is present when the plasma contains very high concentrations of protein (multiple myeloma, intravenous immunoglobulin infusion) or lipid (hypertriglyceridemia, hypercholesterolemia). It does not occur when a direct ion-selective electrode determines the sodium concentration in undiluted plasma, a technique that is used by the instruments used for measuring arterial blood gases. In true hyponatremia, the measured osmolality is low, whereas it is normal in pseudohyponatremia. Hyperosmolality, as may occur with hyperglycemia, causes a low serum sodium concentration because water moves down its osmotic gradient from the intracellular space into the extracellular space, diluting the sodium concentration. However, because the manifestations of hyponatremia are due to the low plasma osmolality, patients with hyponatremia resulting from hyperosmolality do not have symptoms of hyponatremia. When the etiology of the hyperosmolality resolves, such as hyperglycemia in diabetes mellitus, water moves back into the cells and the sodium concentration rises to its “true” value. Mannitol or sucrose, a component of intravenous immunoglobulin preparations, may cause hyponatremia due to hyperosmolality.
Table 52-2 CAUSES OF HYPONATREMIA
PSEUDOHYPONATREMIA
EXTRARENAL LOSSES
RENAL LOSSES
EUVOLEMIC HYPONATREMIA
HYPERVOLEMIC HYPONATREMIA
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* Most cases of proximal renal tubular acidosis are not due to this primary genetic disorder. Proximal renal tubular acidosis is usually part of Fanconi syndrome, which has multiple etiologies.
Renal salt wasting occurs in hereditary kidney diseases, such as juvenile nephronophthisis and autosomal recessive polycystic kidney disease. Obstructive uropathy, most commonly a consequence of posterior urethral valves, produces salt wasting, but patients with the disease may also have hypernatremia as a result of impaired ability to concentrate urine and high water loss. Acquired tubulointerstitial nephritis, usually secondary to either medications or infections, may cause salt wasting, along with other evidence of tubular dysfunction. CNS injury may produce cerebral salt wasting, which is theoretically due to the production of a natriuretic peptide that causes renal salt wasting. In type II renal tubular acidosis (RTA), usually associated with Fanconi syndrome (Chapter 523), there is increased excretion of sodium and bicarbonate in the urine. Patients with Fanconi syndrome also have glycosuria, aminoaciduria, and hypophosphatemia due to renal phosphate wasting.
In SIADH, the secretion of ADH is not inhibited by either low serum osmolality or expanded intravascular volume (Chapter 553). The result is that the child with SIADH is unable to excrete water. This results in dilution of the serum sodium and hyponatremia. The expansion of the extracellular volume due to the retained water causes a mild increase in intravascular volume. The kidney increases sodium excretion in an effort to decrease intravascular volume to normal; thus, the patients has a mild decrease in body sodium. SIADH most commonly occurs with disorders of the CNS (infection, hemorrhage, trauma, tumor, thrombosis), but lung disease (infection, asthma, positive pressure ventilation) and malignant tumors (producing ADH) are other potential causes. A variety of medications may cause SIADH, including recreational use of 3,4-methylenedioxymethylamphetamine (MDMA, or “Ecstasy”), opiates, antiepileptic drugs (carbamazepine, oxcarbamazepine, valproate), tricyclic antidepressants, vincristine, cytoxan, and selective serotonin reuptake inhibitors. The diagnosis of SIADH is one of exclusion, because other causes of hyponatremia must be eliminated (Table 52-3). Because SIADH is a state of intravascular volume expansion, low serum uric acid and BUN levels are supportive of the diagnosis.
Diagnosis
The history usually points to a likely etiology of the hyponatremia. Most patients with hyponatremia have a history of volume depletion. Diarrhea and diuretic use are very common causes of hyponatremia in children. A history of polyuria, perhaps with enuresis, and/or salt craving is present in children with primary kidney diseases or absence of aldosterone effect. Children may have signs or symptoms suggesting a diagnosis of hypothyroidism or adrenal insufficiency (Chapters 559 and 569). Brain injury raises the possibility of SIADH or cerebral salt wasting, with the caveat that SIADH is much more likely. Liver disease, nephrotic syndrome, renal failure, or congestive heart failure may be acute or chronic. The history should include a review of the patient’s intake, both intravenous and enteral, with careful attention to the amounts of water, sodium, and protein.
In patients with true hyponatremia, the next step in the diagnostic process is to clinically evaluate the volume status. Patients with hyponatremia can be hypovolemic, hypervolemic, or euvolemic. The diagnosis of volume depletion relies on the usual findings with dehydration (Chapter 54), although subtle volume depletion may not be clinically apparent. In a patient with subtle volume depletion, a fluid bolus results in a decrease in the urine osmolality and an increase in the serum sodium concentration. Children with hypervolemia are edematous on physical examination. They may have ascites, pulmonary edema, pleural effusion, or hypertension.
Hypovolemic hyponatremia can have renal or nonrenal causes. The urine sodium concentration is very useful in differentiating between renal and nonrenal causes. When the losses are nonrenal and the kidney is working properly, there is renal retention of sodium, a normal homeostatic response to volume depletion. Thus, the urinary sodium concentration is low, typically <10 mEq/L, although sodium conservation in neonates is less avid. When the kidney is the cause of the sodium loss, the urine sodium concentration is >20 mEq/L, reflecting the defect in renal sodium retention. The interpretation of the urine sodium level is challenging with diuretic therapy because it is high when diuretics are being used but low after the diuretic effect is gone. This becomes an issue only when diuretic use is surreptitious. The urine sodium concentration is not useful if a metabolic alkalosis is present; the urine chloride concentration must be used instead (Chapter 52.7).
Treatment
The child with hypovolemic hyponatremia has a deficiency in sodium and may have a deficiency in water. The cornerstone of therapy is to replace the sodium deficit and any water deficit that is present. The 1st step in treating any dehydrated patient is to restore the intravascular volume with isotonic saline. Ultimately, complete restoration of intravascular volume suppresses ADH production, thereby permitting excretion of the excess water. Chapter 54 discusses the management of hyponatremic dehydration.
Children with iatrogenic hyponatremia due to the administration of hypotonic intravenous fluids should receive 3% saline if they are symptomatic. Subsequent management is dictated by the patient’s volume status. The hypovolemic child should receive isotonic intravenous fluids. The child with nonphysiologic stimuli for ADH production should undergo fluid restriction. Prevention of this iatrogenic complication requires judicious use of intravenous fluids (Chapter 53).
Centers for Disease Control and Prevention (CDC). Application of lower sodium intake recommendations to adults—United States, 1999-2006. MMWR Morb Mortal Wkly Rep. 2009;58:281-283.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet. 2008;371:1624-1632.
Don M, Valerio G, Korppi M, et al. Hyponatremia in pediatric community-acquired pneumonia. Pediatr Nephrol. 2008;23:2247-2253.
Ellison DH, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064-2072.
Fenske W, Maier SKG, Blechschmidt A, et al. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123:652-657.
Karakas HM, Erdem G, Yakinci C. Osmotic demyelination syndrome in a 40-day-old infant. Diagn Interv Radiol. 2007;13:121-124.
Leung C, Chang WC, Yeh SJ. Hypernatremic dehydration due to concentrated infant formula: report of two cases. Pediatr Neonatol. 2009;50:70-73.
Linshaw MA. Back to basics: congenital nephrogenic diabetes insipidus. Pediatr Rev. 2007;28:372-380.
Modi N. Avoiding hypernatraemic dehydration in healthy term infants. Arch Child. 2007;92:474-475.
Rianthavorn P, Cain JP, Turman MA. Use of conivaptan to allow aggressive hydration to prevent tumor lysis syndrome in a pediatric patient with large-cell lymphoma and SIADH. Pediatr Nephrol. 2008;23:1367-1370.
Rivkees SA. Differentiating appropriate antidiuretic hormone secretion, inappropriate antidiuretic hormone secretion and cerebral salt wasting: the common, uncommon, and misnamed. Curr Opin Pediatr. 2008;20:448-452.
Verbalis JG, Goldsmith SR, Greenberg A, et al. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120:S1-S21.
52.4 Potassium
Potassium Metabolism
Body Content and Physiologic Function
The intracellular concentration of potassium, approximately 150 mEq/L, is much higher than the plasma concentration (see Web Fig. 52-3). The majority of body potassium is contained in muscle. As muscle mass increases, there is an increase in body potassium. There is thus an increase in body potassium during puberty, and it is more significant in males. The majority of extracellular potassium is in bone; <1% of total body potassium is in plasma.
mEq/L, is much higher than the plasma concentration (see Web Fig. 52-3). The majority of body potassium is contained in muscle. As muscle mass increases, there is an increase in body potassium. There is thus an increase in body potassium during puberty, and it is more significant in males. The majority of extracellular potassium is in bone; <1% of total body potassium is in plasma.
Hyperkalemia
Etiology and Pathophysiology
Three basic mechanisms cause hyperkalemia (Table 52-4). In the individual patient, the etiology is sometimes multifactorial.
Table 52-4 CAUSES OF HYPERKALEMIA
SPURIOUS LABORATORY VALUE
INCREASED INTAKE
TRANSCELLULAR SHIFTS
DECREASED EXCRETION
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
Normal doses of succinylcholine or β-blockers and fluoride or digitalis intoxication all cause a shift of potassium out of the intracellular compartment. Succinylcholine should not be used during anesthesia in patients at risk for hyperkalemia. β-Blockers prevent the normal cellular uptake of potassium mediated by binding of β-agonists to the β2-adrenergic receptors. Potassium release from muscle cells occurs during exercise, and levels can increase by 1-2 mEq/L with high activity. With an increased plasma osmolality, water moves from the intracellular space and potassium follows. This process occurs with hyperglycemia, although in nondiabetic patients, the resultant increase in insulin causes potassium to move intracellularly. In diabetic ketoacidosis, the absence of insulin causes potassium to leave the intracellular space, and the problem is compounded by the hyperosmolality. The effect of hyperosmolality causes a transcellular shift of potassium into the extracellular space after mannitol or hypertonic saline infusions. Malignant hyperthermia, which is triggered by some inhaled anesthetics, causes muscle release of potassium (Chapter 603.2). Hyperkalemic periodic paralysis is an autosomal dominant disorder caused by a mutated sodium channel. It results in episodic cellular release of potassium and attacks of paralysis (Chapter 603.1).
A wide range of primary adrenal disorders, both hereditary and acquired, can cause decreased production of aldosterone, with secondary hyperkalemia (Chapters 569 and 570). Patients with these disorders typically have metabolic acidosis and salt wasting with hyponatremia. Children with more subtle adrenal insufficiency may have electrolyte problems only during acute illnesses. The most common form of congenital adrenal hyperplasia, 21-hydroxylase deficiency, typically manifests in male infants as hyperkalemia, metabolic acidosis, hyponatremia, and volume depletion. Females with this disorder usually are diagnosed as newborns because of their ambiguous genitalia; treatment prevents the development of electrolyte problems.
Renin, via angiotensin II, stimulates aldosterone production. A deficiency in renin, a result of kidney damage, can lead to decreased aldosterone production. Hyporeninemia occurs in many kidney diseases, with some of the more common pediatric causes listed in Table 52-4. These patients typically have hyperkalemia and a metabolic acidosis, without hyponatremia. Some of these patients have impaired renal function, partially accounting for the hyperkalemia, but the impairment in potassium excretion is more extreme than expected for the degree of renal insufficiency.
A variety of renal tubular disorders impair renal excretion of potassium. Children with pseudohypoaldosteronism type 1 have hyperkalemia, metabolic acidosis, and salt wasting leading to hyponatremia and volume depletion; aldosterone values are elevated. In the autosomal recessive variant, there is a defect in the renal sodium channel that is normally activated by aldosterone. Patients with this variant have severe symptoms, beginning in infancy. Patients with the autosomal dominant form have a defect in the aldosterone receptor, and the disease is milder, often remitting in adulthood. Pseudohypoaldosteronism type 2, also called Gordon syndrome, is an autosomal dominant disorder characterized by hypertension due to salt retention and impaired excretion of potassium and acid, leading to hyperkalemia and metabolic acidosis. Activating mutations in either WNK1 or WNK4, both serine-threonine kinases located in the distal nephron, cause Gordon syndrome. In Bartter syndrome due to mutations in the potassium channel ROMK (type 2 Bartter syndrome), there can be transient hyperkalemia in neonates, but hypokalemia subsequently develops (Chapter 525).
Diagnosis
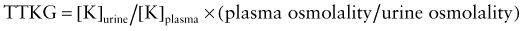
where [K]urine is urine potassium concentration and [K]plasma is plasma potassium concentration. For the result to be valid, the urine osmolality must be greater than the serum osmolality. The TTKG normally varies widely, ranging from 5-15. The TTKG should be >10 in the setting of hyperkalemia, assuming normal renal excretion of potassium. A TTKG <8 during hyperkalemia suggests a defect in renal potassium excretion, which is usually due to lack of aldosterone or an inability to respond to aldosterone. Measurement of aldosterone is useful for differentiating these possible mechanisms. Patients with a lack of aldosterone respond to fludrocortisone, an oral mineralocorticoid, by increasing urinary potassium and decreasing serum potassium. An appropriate TTKG with normal kidney function argues for a nonrenal cause of hyperkalemia.
Treatment
The 1st action in a child with a concerning elevation of plasma potassium is to stop all sources of additional potassium (oral, intravenous) (Chapter 529). Washed red blood cells can be used for patients who require blood transfusions. If the potassium level is >6.0-6.5 mEq/L, an ECG should be obtained to help assess the urgency of the situation. Peak T waves are the first sign of hyperkalemia followed by a prolonged PR interval and, when most severe, a prolonged QRS complex. Life-threatening ventricular arrhythmias may also develop. The treatment of hyperkalemia has 2 basic goals: (1) to stabilize the heart to prevent life-threatening arrhythmias and (2) to remove potassium from the body. The treatments that acutely prevent arrhythmias all have the advantage of working quickly (within minutes) but do not remove potassium from the body. Calcium stabilizes the cell membrane of heart cells, preventing arrhythmias. It is given intravenously over a few minutes, and its action is almost immediate. Calcium should be given over 30 min in a patient receiving digitalis, because otherwise the calcium may cause arrhythmias. Bicarbonate causes potassium to move intracellularly, lowering the plasma potassium level. It is most efficacious in a patient with a metabolic acidosis. Insulin causes potassium to move intracellularly but must be given with glucose to avoid hypoglycemia. The combination of insulin and glucose works within 30 min. Nebulized albuterol, by stimulation of β1-receptors, leads to rapid intracellular movement of potassium. This has the advantage of not requiring an intravenous route of administration, allowing it to be given concurrently with the other measures.
Long-term management of hyperkalemia includes reducing intake via dietary changes and eliminating or reducing medications that cause hyperkalemia (Chapter 529). Some patients require medications to increase potassium excretion, such as sodium polystyrene sulfonate and loop or thiazide diuretics. Some infants with chronic renal failure may need to start dialysis to allow adequate caloric intake without hyperkalemia. It is unusual for an older child to require dialysis principally to control chronic hyperkalemia. The disorders that are due to a deficiency in aldosterone respond to replacement therapy with fludrocortisone.
Hypokalemia
Hypokalemia is common in children, with most cases related to gastroenteritis.
Etiology and Pathophysiology
There are 4 basic mechanisms of hypokalemia (Table 52-5). Spurious hypokalemia occurs in patients with leukemia and very elevated white blood cell counts if plasma for analysis is left at room temperature, permitting the white blood cells to take up potassium from the plasma. With a transcellular shift, there is no change in total body potassium, although there may be concomitant potassium depletion resulting from other factors. Decreased intake, extrarenal losses, and renal losses are all associated with total body potassium depletion.
Table 52-5 CAUSES OF HYPOKALEMIA
SPURIOUS
High white blood cell count
TRANSCELLULAR SHIFTS
DECREASED INTAKE
Anorexia nervosa
EXTRARENAL LOSSES
RENAL LOSSES
EAST, epilepsy, ataxia, sensorineural hearing loss, and tubulopathy; MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* Most cases of proximal renal tubular acidosis are not due to this primary genetic disorder. Proximal renal tubular acidosis is usually part of Fanconi syndrome, which has multiple etiologies.
Because the intracellular potassium concentration is much higher than the plasma level, a significant amount of potassium can move into cells without markedly changing the intracellular potassium concentration. Alkalemia is one of the more common causes of a transcellular shift. The effect is much greater with a metabolic alkalosis than with a respiratory alkalosis. The impact of exogenous insulin on potassium movement into the cells is substantial in patients with diabetic ketoacidosis. Endogenous insulin may be the cause when a patient is given a bolus of glucose. Both endogenous (epinephrine in stress) and exogenous (albuterol) β-adrenergic agonists stimulate cellular uptake of potassium. Theophylline overdose, barium intoxication, administration of cesium chloride (a homeopathic cancer remedy), and toluene intoxication from paint or glue sniffing can cause a transcellular shift hypokalemia, often with severe clinical manifestations. Children with hypokalemic periodic paralysis, a rare autosomal dominant disorder, have acute cellular uptake of potassium (Chapter 603). Thyrotoxic periodic paralysis, which is more common in Asians, is an unusual initial manifestation of hyperthyroidism. Affected patients have dramatic hypokalemia as a result of a transcellular shift of potassium. Hypokalemia can occur during refeeding syndrome (Chapter 330.08).
Urinary potassium wasting may be accompanied by a metabolic acidosis (proximal or distal RTA). In diabetic ketoacidosis, although it is often associated with normal plasma potassium caused by transcellular shifts, there is significant total body potassium depletion from urinary losses due to the osmotic diuresis, and the potassium level may decrease dramatically with insulin therapy (Chapter 583). Both the polyuric phase of acute tubular necrosis and postobstructive diuresis cause transient, highly variable potassium wasting and may be associated with a metabolic acidosis. Tubular damage, which occurs either directly from medications or secondary to interstitial nephritis, is often accompanied by other tubular losses of nutrients, including magnesium, sodium, and water. Such tubular damage may cause a secondary RTA with a metabolic acidosis. Isolated magnesium deficiency causes renal potassium wasting. Penicillin is an anion that is excreted in the urine, resulting in increased potassium excretion because the penicillin anion must be accompanied by a cation. Hypokalemia from penicillin therapy occurs only with the sodium salt of penicillin, not with the potassium salt.
The combination of metabolic alkalosis, hypokalemia, a high urine chloride level, and normal blood pressure is characteristic of Bartter syndrome, Gitelman syndrome, and current diuretic use. Patients with any of these conditions have high urinary losses of potassium and chloride, despite a state of relative volume depletion with secondary hyperaldosteronism. Bartter and Gitelman syndromes are autosomal recessive disorders caused by defects in tubular transporters (Chapter 525). Bartter syndrome is usually associated with hypercalciuria, and often with nephrocalcinosis, whereas children with Gitelman syndrome have low urinary calcium losses but hypomagnesemia due to urinary magnesium losses. Some patients with Bartter syndrome have hypomagnesemia.
In the presence of high aldosterone levels, there is urinary loss of potassium, hypokalemia, metabolic alkalosis, and an elevated urinary chloride level. Also ,renal retention of sodium leads to hypertension. Primary hyperaldosteronism caused by adenoma or hyperplasia is much less common in children than in adults (Chapter 572). Glucocorticoid-remediable aldosteronism, an autosomal dominant disorder that leads to high levels of aldosterone, is often diagnosed in childhood, although hypokalemia is not always present.
A variety of disorders causes hypertension and hypokalemia without increased aldosterone levels. Some are due to increased levels of mineralocorticoids other than aldosterone. Such increases occur in 2 forms of congenital adrenal hyperplasia (Chapter 570). In 11β-hydroxylase deficiency, which is associated with virilization, the value of 11-deoxycorticosterone (DOC) is elevated, causing variable hypertension and hypokalemia. A similar mechanism, increased DOC, occurs in 17α-hydroxylase deficiency, but patients with this disorder are more uniformly hypertensive and hypokalemic, and they have a defect in sex hormone production. Cushing syndrome, frequently associated with hypertension, less commonly causes metabolic alkalosis and hypokalemia. This is secondary to the mineralocorticoid activity of cortisol. In 11β-hydroxysteroid dehydrogenase deficiency, an autosomal recessive disorder, the enzymatic defect prevents the conversion of cortisol to cortisone in the kidney. Because cortisol binds to and activates the aldosterone receptor, children with this deficiency have all the features of excessive mineralocorticoids, including hypertension, hypokalemia, and metabolic alkalosis. Patients with this disorder, which is also called apparent mineralocorticoid excess, respond to spironolactone therapy, which blocks the mineralocorticoid receptor. An acquired form of 11β-hydroxysteroid dehydrogenase deficiency occurs from the ingestion of substances that inhibit this enzyme. A classic example is glycyrrhizic acid, which is found in natural licorice. Liddle syndrome is an autosomal dominant disorder that results from an activating mutation of the distal nephron sodium channel that is normally upregulated by aldosterone. Patients have the characteristics of hyperaldosteronism—hypertension, hypokalemia, and alkalosis—but low serum aldosterone levels. These patients respond to the potassium-sparing diuretics (triamterene and amiloride) that inhibit this sodium channel (Chapter 525.3).
Clinical Manifestations
The heart and skeletal muscle are especially vulnerable to hypokalemia. ECG changes include a flattened T wave, a depressed ST segment, and the appearance of a U wave, which is located between the T wave (if still visible) and the P wave. Ventricular fibrillation and torsades de pointes may occur, although usually only in the context of underlying heart disease. Hypokalemia makes the heart especially susceptible to digitalis-induced arrhythmias, such as supraventricular tachycardia, ventricular tachycardia, and heart block (Chapter 429).
Diagnosis
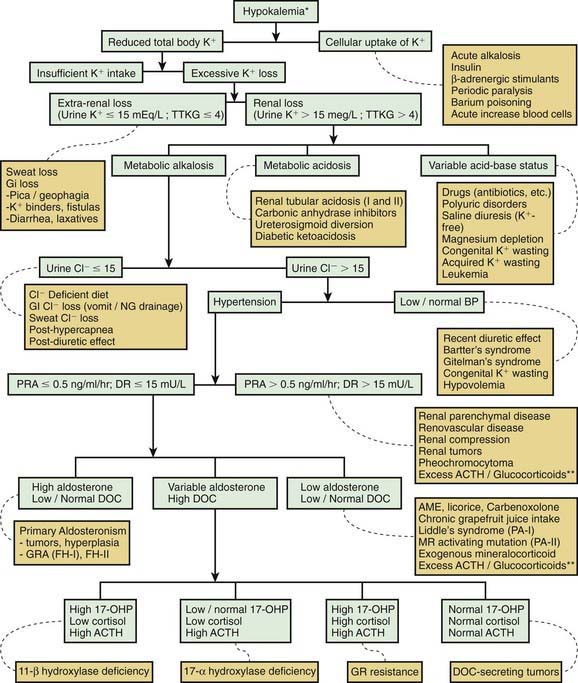
Most causes of hypokalemia are readily apparent from the history. It is important to review the child’s diet, gastrointestinal losses, and medications. Both emesis and diuretic use can be surreptitious. The presence of hypertension suggests excess mineralocorticoids. Concomitant electrolyte abnormalities are useful clues. The combination of hypokalemia and metabolic acidosis is characteristic of diarrhea and of distal and proximal RTA. A concurrent metabolic alkalosis is characteristic of emesis or nasogastric losses, aldosterone excess, use of diuretics, and Bartter and Gitelman syndromes. An approach to persistent hypokalemia is shown in Figure 52-2.
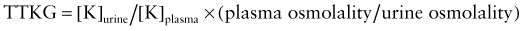
where [K]urine = urine potassium concentration and [K]plasma = plasma potassium concentration.
Antoniou T, Gomes T, Juurlink DN, et al. Trimethoprim-sulfamethoxazole–induced hyperkalemia in patients receiving inhibitors of the renin-angiotensin system. Arch Intern Med. 2010;170(12):1045-1049.
Atabek ME, Pirgon O, Sert A. Hypokalemic rhabdomyolysis in a child with 11-hydroxylase deficiency. J Pediatr Endocrinol. 2008;21:93-96.
Austin JD, Hofman P, Anderson BJ. Life-threatening hyperkalemia following partial pancreatectomy for neonatal hyperinsulinism. Pediatr Crit Care Med. 2008;9:e17-e19.
Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960-1970.
Garcia E, Nakhleh N, Simmons D, et al. Profound hypokalemia: unusual presentation and management in a 12-year-old boy. Pediatr Emerg Care. 2008;24:157-160.
Greenlee M, Wingo CS, McDonough AA, et al. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med. 2009;150:619-625.
Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18:2649-2652.
Kose M, Pekcan S, Ozcelik U, et al. An epidemic of pseudo-Bartter syndrome in cystic fibrosis patients. Eur J Pediatr. 2008;167:115-116.
Lin SH, Halperin ML. Hypokalemia: a practical approach to diagnosis and its genetic basis. Curr Med Chem. 2007;14:1551-1565.
Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330.
Nyirenda MJ, Tang JI, Padfield PL, et al. Hyperkalaemia. BMJ. 2009;339:1019-1024.
Proesmans W. Threading through the mizmaze of Bartter syndrome. Pediatr Nephrol. 2006;21:896-902.
Shoemaker LR, Eaton BV, Buchino JJ. A three-year-old with persistent hypokalemia. J Pediatr. 2007;151:696-699.
Smellie WSA. Spurious hyperkalaemia. BMJ. 2007;334:693-695.
Sood MM, Sood AR, Richardson R. Emergency management and commonly encountered outpatient scenarios in patients with hyperkalemia. Mayo Clin Proc. 2007;82:1553-1561.
52.5 Magnesium
Magnesium Metabolism
Body Content and Physiologic Function
Magnesium is the 4th most common cation in the body and the 3rd most common intracellular cation (see  Web Fig. 52-3). Between 50% and 60% of body magnesium is in bone, where it serves as a reservoir because 30% is exchangeable, allowing movement to the extracellular space. Most intracellular magnesium is bound to proteins; only approximately 25% is exchangeable. Because cells with higher metabolic rates have higher magnesium concentrations, most intracellular magnesium is present in muscle and liver.
Web Fig. 52-3). Between 50% and 60% of body magnesium is in bone, where it serves as a reservoir because 30% is exchangeable, allowing movement to the extracellular space. Most intracellular magnesium is bound to proteins; only approximately 25% is exchangeable. Because cells with higher metabolic rates have higher magnesium concentrations, most intracellular magnesium is present in muscle and liver.
The normal plasma magnesium concentration is 1.5-2.3 mg/dL (1.2-1.9 mEq/L; 0.62-0.94 mmol/L), with some variation among clinical laboratories. In the USA, serum magnesium is reported as mg/dL (Web Table 52-1). Infants have slightly higher plasma magnesium concentrations than older children and adults. Only 1% of body magnesium is extracellular (60% ionized; 15% complexed; 25% protein bound).
Hypomagnesemia
Etiology and Pathophysiology
Gastrointestinal and renal losses are the major causes of hypomagnesemia (Web Table 52-2). Diarrheal fluid contains up to 200 mg/L of magnesium; gastric contents have only approximately 15 mg/L, but high losses can cause depletion. Steatorrhea causes magnesium loss as a result of the formation of magnesium-lipid salts; restriction of dietary fat can decrease losses.
Web Table 52-2 CAUSES OF HYPOMAGNESEMIA
GASTROINTESTINAL DISORDERS
RENAL DISORDERS
MISCELLANEOUS CAUSES
EAST, epilepsy, ataxia, sensorineural hearing loss, and tubulopathy; MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* This disorder is also associated with renal magnesium wasting.
A number of rare genetic diseases cause renal magnesium loss. Gitelman and Bartter syndromes, both autosomal recessive disorders, are the most common entities (Chapter 525). Gitelman syndrome, which is due to a defect in the thiazide-sensitive Na+-Cl− cotransporter in the distal tubule, is usually associated with hypomagnesemia. Hypomagnesemia occurs in a minority of patients with Bartter syndrome, which can be caused by mutations in at least 5 different genes. In both disorders, there is hypokalemic metabolic alkalosis. Typically, hypomagnesemia is not severe and is asymptomatic, although tetany due to hypomagnesemia occasionally occurs.
EAST syndrome is due to mutations in a potassium channel, and patients with this autosomal recessive disorder have hypokalemia, metabolic alkalosis, and hypomagnesemia. Autosomal dominant hypoparathyroidism is caused by an activating mutation in the calcium-sensing receptor, which also senses magnesium levels in the kidney (Chapter 565). The mutated receptor inappropriately perceives that magnesium and calcium levels are elevated, leading to urinary wasting of both cations. Hypomagnesemia, if present, is usually mild. A mutation in a mitochondrially encoded transfer RNA is associated with hypomagnesemia, hypertension, and hypercholesterolemia. Hypomagnesemia is occasionally present in children with other mitochondrial disorders.
Transient hypomagnesemia in newborns, which is sometimes idiopathic, is more commonly seen in infants of diabetic mothers, presumably as a result of maternal depletion from osmotic losses. Other maternal diseases that cause magnesium losses predispose infants to hypomagnesemia. Hypomagnesemia is more common in infants with intrauterine growth restriction. Hypomagnesemia may develop in newborn infants who require exchange transfusions because of magnesium removal by the citrate in banked blood (Chapter 100).
Clinical Manifestations
Hypomagnesemia causes secondary hypocalcemia by impairing the release of PTH by the parathyroid gland and through blunting of the tissue response to PTH. Thus, hypomagnesemia is part of the differential diagnosis of hypocalcemia (Chapter 565). It usually occurs only at magnesium levels <0.7 mg/dL. The dominant manifestations of hypomagnesemia are due to hypocalcemia: tetany, presence of Chvostek and Trousseau signs, and seizures. However, with severe hypomagnesemia, these same signs and symptoms may be present despite normocalcemia. Persistent hypocalcemia due to hypomagnesemia is a rare cause of rickets.
Diagnosis
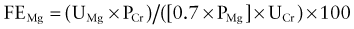
where UMg is urinary magnesium concentration, PCr is plasma creatinine concentration, PMg is plasma magnesium concentration, and UCr is urinary magnesium concentration. The plasma magnesium concentration is multiplied by 0.7 because approximately 30% is bound to albumin and not filtered at the glomerulus.
Hypermagnesemia
Clinically significant hypermagnesemia is almost always secondary to excessive intake. It is unusual, except in neonates born to mothers who are receiving intravenous magnesium for preeclampsia or eclampsia (Chapter 100).
Adalat S, Woolf AS, Johnstone KA, et al. HNF1β mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol. 2009;20:1123-1131.
Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360:1960-1970.
Fakih M. Management of anti-EGFr-targeting monoclonal antibody-induced hypomagnesemia. Oncology (Williston). 2008;22:74-76.
Huang C-L, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18:2649-2652.
Knoers NVAM. Inherited forms of renal hypomagnesemia: an update. Pediatr Nephrol. 2009;24:697-705.
Kutsal E, Aydemir C, Eldes N, et al. Severe hypermagnesemia as a result of excessive cathartic ingestion in a child without renal failure. Pediatr Emerg Care. 2007;23:570-572.
Moe SM. Disorders involving calcium, phosphorus, and magnesium. Prim Care. 2008;35:215-237.
Naderi AS, Reilly RFJr. Hereditary etiologies of hypomagnesemia. Nat Clin Pract Nephrol. 2008;4:80-89.
52.6 Phosphorus
Approximately 65% of plasma phosphorus is in phospholipids, but these compounds are insoluble in acid and are not measured by clinical laboratories. It is the phosphorus content of plasma phosphate that is determined. The result is reported as either phosphate or phosphorus, although even when the term phosphate is used, it is actually the phosphorus concentration that is measured and reported. The result is that the terms phosphate and phosphorus are often used interchangeably. The term phosphorus is preferred when one is referring to the plasma concentration. Conversion from the units used in the USA (mg/dL) to mmol/L is straightforward (see Web Table 52-1 ).
).
Phosphorus Metabolism
Body Content and Physiologic Function
More than that of any other electrolyte, the phosphorus concentration varies with age (Table 52-6). The teleologic explanation for the high concentration during childhood is the need for phosphorus to facilitate growth. There is diurnal variation in the plasma phosphorus concentration, with the peak during sleep.
| AGE | PHOSPHORUS LEVEL (mg/dL) |
|---|---|
| 0-5 day | 4.8-8.2 |
| 1-3 yr | 3.8-6.5 |
| 4-11 yr | 3.7-5.6 |
| 12-15 yr | 2.9-5.4 |
| 16-19 yr | 2.7-4.7 |
Hypophosphatemia
Because of the wide variation in normal plasma phosphorus levels, the definition of hypophosphatemia is age-dependent (see Table 52-6). The normal range reported by a laboratory may be based on adult normal values and, therefore, may be misleading in children. A serum phosphorus level of 3 mg/dL, a normal value in an adult, indicates clinically significant hypophosphatemia in an infant.
Etiology and Pathophysiology
A variety of mechanisms cause hypophosphatemia (Table 52-7). A transcellular shift of phosphorus into cells occurs with processes that stimulate cellular usage of phosphorus (glycolysis). Usually, this shift causes only a minor, transient decrease in plasma phosphorus, but if intracellular phosphorus deficiency is present, the plasma phosphorus level can decrease significantly, producing symptoms of acute hypophosphatemia. Glucose infusion stimulates insulin release, leading to entry of glucose and phosphorus into the cells. Phosphorus is then used during glycolysis and other metabolic processes. A similar phenomenon can occur during the treatment of diabetic ketoacidosis, and patients with this disorder are typically phosphorus-depleted owing to urinary phosphorus losses. Refeeding of patients with protein-calorie malnutrition causes anabolism, which leads to significant cellular demand for phosphorus. The increased phosphorus uptake for incorporation into newly synthesized compounds containing phosphorus leads to hypophosphatemia, which can be severe and symptomatic. Refeeding hypophosphatemia occurs frequently during treatment of severe anorexia nervosa. It can occur during treatment of children with malnutrition due to any cause, such as cystic fibrosis, Crohn disease, burns, neglect, chronic infection, or famine. Hypophosphatemia usually occurs within the 1st 5 days of refeeding and is prevented by a gradual increase in nutrition with appropriate phosphorus supplementation (Chapter 43). Total parenteral nutrition without adequate phosphorus can cause hypophosphatemia.
Table 52-7 CAUSES OF HYPOPHOSPHATEMIA
TRANSCELLULAR SHIFTS
DECREASED INTAKE
RENAL LOSSES
MULTIFACTORIAL
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
Excessive renal losses of phosphorus occur in a variety of inherited and acquired disorders. Because PTH inhibits the resorption of phosphorus in the proximal tubule, hyperparathyroidism causes hypophosphatemia (Chapter 567). The dominant clinical manifestation, however, is hypercalcemia, and the hypophosphatemia is usually asymptomatic. The phosphorus level in hyperparathyroidism is not extremely low, and there is no continued loss of phosphorus because a new steady state is achieved at the lower plasma phosphorus level. Renal excretion, therefore, does not exceed intake over the long term. There are occasional malignancies that produce PTH-related peptide, which has the same actions as PTH and causes hypophosphatemia and hypercalcemia.
A variety of diseases cause renal phosphate wasting, hypophosphatemia, and rickets due to excess phosphatonin (Chapter 48). These disorders include X-linked hypophosphatemic rickets, tumor-induced osteomalacia, autosomal dominant hypophosphatemic rickets, and autosomal recessive hypophosphatemic rickets. Heterozygous mutations in a phosphate transporter or a regulator of proximal tubule phosphate transport cause hypophosphatemia, osteoporosis and nephrolithiasis (hypophosphatemic nephrolithiasis/osteoporosis type 1 or 2).
Fanconi syndrome is a generalized defect in the proximal tubule leading to urinary wasting of bicarbonate, phosphorus, amino acids, uric acid, and glucose (Chapter 523). The clinical sequelae are due to the metabolic acidosis and hypophosphatemia. In children, an underlying genetic disease, most commonly cystinosis, often causes Fanconi syndrome, but it can be secondary to a variety of toxins and acquired diseases. Some patients have incomplete Fanconi syndrome, and phosphorus wasting may be one of the manifestations.
Dent disease, an X-linked disorder, can cause renal phosphorus wasting and hypophosphatemia, although the latter is not present in most cases. Other possible manifestations of Dent disease include tubular proteinuria, hypercalciuria, nephrolithiasis, rickets, and chronic renal failure. Dent disease may be secondary to mutations in a gene that encodes a chloride channel or the OCRL1 gene, which may also cause Lowe syndrome (Chapter 523.1). Hypophosphatemic rickets with hypercalciuria is a rare disorder, principally described in kindreds from the Middle East. Mutations in a sodium-phosphate cotransporter cause hypophosphatemia in this disorder, and complications may include nephrolithiasis and osteoporosis; the disorder is autosomal dominant.
Both acquired and genetic causes of vitamin D deficiency are associated with hypophosphatemia (Chapter 48). The pathogenesis is multifactorial. Vitamin D deficiency, by impairing intestinal calcium absorption, causes secondary hyperparathyroidism that leads to increased urinary phosphorus wasting. An absence of vitamin D decreases intestinal absorption of phosphorus and directly decreases renal resorption of phosphorus. The dominant clinical manifestation is rickets, although some patients have muscle weakness that may be related to phosphorus deficiency.
Clinical Manifestations
There are acute and chronic manifestations of hypophosphatemia. Rickets occurs in children with long-term phosphorus deficiency. The clinical features of rickets are described in Chapter 48.
Diagnosis
The history and basic laboratory evaluation often suggest the etiology of hypophosphatemia. The history should investigate nutrition, medications, and familial disease. Hypophosphatemia and rickets in an otherwise healthy young child suggests a genetic defect in renal phosphorus conservation, Fanconi syndrome, inappropriate use of antacids, poor nutrition, vitamin D deficiency, or a genetic defect in vitamin D metabolism. The patient with Fanconi syndrome usually has metabolic acidosis, glycosuria, aminoaciduria, and a low plasma uric acid level. Measurement of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, calcium, and PTH differentiates among the various vitamin D deficiency disorders and primary renal phosphate wasting (Chapter 48). Hyperparathyroidism is easily distinguished by the presence of elevated plasma PTH and calcium values.
Treatment
Increasing dietary phosphorus is the only intervention needed in infants with inadequate intake. Other patients may also benefit from increased dietary phosphorus, usually from dairy products. Phosphorus-binding antacids should be discontinued in patients with hypophosphatemia. Certain diseases require specific therapy (Chapter 48).
Hyperphosphatemia
Etiology and Pathophysiology
Renal insufficiency is the most common cause of hyperphosphatemia, with the severity proportional to the degree of kidney impairment (Chapter 529). This occurs because gastrointestinal absorption of the large dietary intake of phosphorus is unregulated, and the kidneys normally excrete this phosphorus. As renal function deteriorates, increased excretion of phosphorus is able to compensate. When kidney function is <30% of normal, hyperphosphatemia usually develops, although the time of its development may vary considerably according to dietary phosphorus absorption. Many of the other causes of hyperphosphatemia are more likely to develop in the setting of renal insufficiency (Table 52-8).
Table 52-8 CAUSES OF HYPERPHOSPHATEMIA
TRANSCELLULAR SHIFTS
INCREASED INTAKE
DECREASED EXCRETION
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
The absence of PTH in hypoparathyroidism or PTH responsiveness in pseudohypoparathyroidism causes hyperphosphatemia because of increased resorption of phosphorus in the proximal tubule of the kidney (Chapters 565 and 566). The associated hypocalcemia is responsible for the clinical symptoms. The hyperphosphatemia in hyperthyroidism or acromegaly is usually minor. It is secondary to increased resorption of phosphorus in the proximal tubule due to the actions of thyroxine or growth hormone. Excessive thyroxine can also cause bone resorption, which may contribute to the hyperphosphatemia and cause hypercalcemia. Patients with familial tumoral calcinosis, a rare autosomal recessive disorder, have hyperphosphatemia due to decreased renal phosphate excretion and heterotopic calcifications. The disease may be secondary to mutations in the genes for a glycosyltransferase, the phosphatonin FGF-23, or the gene for Klotho, which encodes the co-receptor for FGF-23.
Abdullah S, Diezi M, Sung L, et al. Sevelamer hydrochloride: a novel treatment of hyperphosphatemia associated with tumor lysis syndrome in children. Pediatr Blood Cancer. 2008;51:59-61.
Bastepe M, Juppner H. Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord. 2008;9:171-180.
Biebl A, Grillenberger A, Schmitt K. Enema-induced severe hyperphosphatemia in children. Eur J Pediatr. 2009;168:111-112.
Blanchard A, Vargas-Poussou R, Peyrard S, et al. Effect of hydrochlorothiazide on urinary calcium excretion in dent disease: an uncontrolled trial. Am J Kidney Dis. 2008;52:1084-1095.
Brunelli SM, Goldfarb S. Hypophosphatemia: clinical consequences and management. J Am Soc Nephrol. 2007;18:1999-2003.
Mehanna HM, Moledina J, Travis J. Refeeding syndrome: what it is, and how to prevent and treat it. BMJ. 2008;336:1495-1498.
Santana e Meneses JF, Leite HP, de Carvalho WB, et al. Hypophosphatemia in critically ill children: prevalence and associated risk factors. Pediatr Crit Care Med. 2009;10:234-238.
Wesseling K, Bakkaloglu S, Salusky I. Chronic kidney disease mineral and bone disorder in children. Pediatr Nephrol. 2008;23:195-207.
52.7 Acid-Base Balance
Acid-Base Physiology
Introduction and Terminology
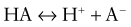
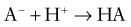
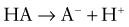
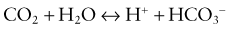
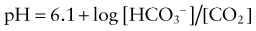
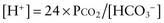
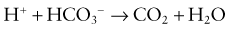
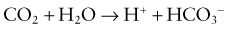
This would increase the concentration of hydrogen ions, limiting the buffering capacity of bicarbonate. However, because the lungs excrete the excess carbon dioxide, the reverse reaction does not increase; this fact enhances the buffering capacity of bicarbonate. The same principle holds with the addition of base, because the lungs decrease carbon dioxide excretion and prevent the level of carbon dioxide from falling. The lack of change in carbon dioxide concentration dramatically increases the buffering capacity of bicarbonate.
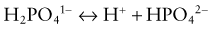
Normal Acid-Base Balance
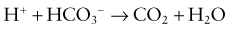
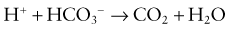
Renal Mechanisms
The kidneys regulate the serum bicarbonate concentration by modifying acid excretion in the urine. This requires a 2-step process. First, the renal tubules resorb the bicarbonate that is filtered at the glomerulus. Second, there is tubular secretion of hydrogen ions. The urinary excretion of hydrogen ions generates bicarbonate that neutralizes endogenous acid production. The tubular actions necessary for renal acid excretion occur throughout the nephron (Web Fig. 52-4).
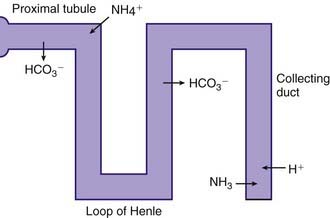
The proximal tubule reclaims approximately 85% of the filtered bicarbonate. The final 15% is reclaimed beyond the proximal tubule, mostly in the ascending limb of the loop of Henle (Web Fig. 52-5). Bicarbonate molecules are not transported from the tubular fluid into the cells of the proximal tubule. Rather, hydrogen ions are secreted into the tubular fluid, leading to conversion of filtered bicarbonate into carbon dioxide and water. The secretion of hydrogen ions by the cells of the proximal tubule is coupled to generation of intracellular bicarbonate, which is transported across the basolateral membrane of the proximal tubule cell and enters the capillaries. The bicarbonate produced in the cell replaces the bicarbonate filtered at the glomerulus.
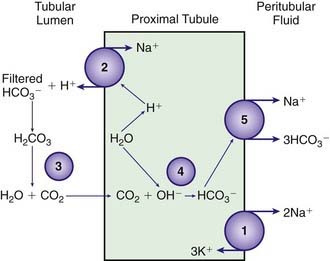
Stimuli that decrease bicarbonate resorption in the proximal tubule may cause a decrease in the serum bicarbonate concentration. A decrease in the PCO2 (respiratory alkalosis) decreases proximal tubule bicarbonate resorption, partially mediating the decrease in serum bicarbonate concentration that compensates for a respiratory alkalosis. PTH decreases proximal tubule bicarbonate resorption; hyperparathyroidism may cause a mild metabolic acidosis. A variety of medications and diseases cause a metabolic acidosis by impairing bicarbonate resorption in the proximal tubule. Examples are the medication acetazolamide, which directly inhibits carbonic anhydrase, and the many disorders that cause proximal RTA (Chapter 523).
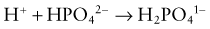
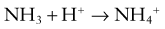
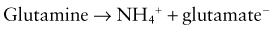
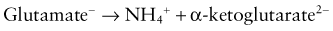
The metabolism of glutamine generates 2 ammonium ions. In addition, the metabolism of α-ketoglutarate generates 2 bicarbonate molecules. The ammonium ions are secreted into the lumen of the proximal tubule, whereas the bicarbonate molecules exit the proximal tubule cells via the basolateral Na+,3HCO3− cotransporter (see Web Fig. 52-4). This arrangement would seem to accomplish the goal of excreting hydrogen ions (as NH4+) and regenerating bicarbonate molecules. However, the ammonium ions secreted in the proximal tubule do not remain within the tubular lumen. Cells of the TAL of the loop of Henle resorb the ammonium ions. The result is that there is a high medullary interstitial concentration of ammonia, but the tubular fluid entering the collecting duct does not have significant amounts of ammonium ions. Moreover, the hydrogen ions that were secreted with ammonia, as ammonium ions, in the proximal tubule enter the bloodstream, canceling the effect of the bicarbonate generated in the proximal tubule. The excretion of ammonium ions, and hence of hydrogen ions, depends on the cells of the collecting duct.
The cells of the collecting duct secrete hydrogen ions and regenerate bicarbonate, which is returned to the bloodstream (Web Fig. 52-6). This bicarbonate neutralizes endogenous acid production. Phosphate and ammonia buffer the hydrogen ions secreted by the collecting duct. Ammonia is an effective buffer because of the high concentrations in the medullary interstitium and because the cells of the collecting duct are permeable to ammonia but not to ammonium. As ammonia diffuses into the lumen of the collecting duct, the low urine pH causes almost all of the ammonia to be converted into ammonium. This process maintains a low luminal ammonia concentration. Because the luminal pH is lower than the pH in the medullary interstitium, there is a higher concentration of ammonia within the medullary interstitium than in the tubular lumen, favoring movement of ammonia into the tubular lumen. Even though the concentration of ammonium in the tubular lumen is higher than in the interstitium, the cells of the collecting duct are impermeable to ammonium, preventing back-diffusion of ammonium out of the tubular lumen and permitting ammonia to be an effective buffer.
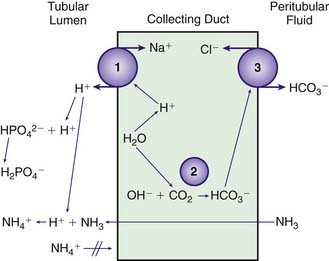
In patients with an increased pH, the kidney has 2 principal mechanisms for correcting the problem. First, less bicarbonate is resorbed in the proximal tubule, leading to an increase in urinary bicarbonate losses. Second, in a limited number of specialized cells, the process for secretion of hydrogen ions by the collecting duct (see Web Fig. 52-6) can be reversed, leading to secretion of bicarbonate into the tubular lumen and secretion of hydrogen ions into the peritubular fluid, where they enter the bloodstream.
Clinical Assessment of Acid-Base Disorders
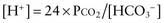
Terminology
There are formulas for calculating the appropriate metabolic or respiratory compensation for the 6 primary simple acid-base disorders (Table 52-9). The appropriate compensation is expected in a simple disorder; it is not optional. If a patient does not have the appropriate compensation, then a mixed acid-base disorder is present. A patient has a primary metabolic acidosis with a serum bicarbonate concentration of 10m Eq/L. The expected respiratory compensation is a carbon dioxide concentration of 23 mm Hg ± 2 (1.5 × 10 + 8 ± 2 = 23 ± 2; see Table 52-9). If the patient’s carbon dioxide concentration is >25 mm Hg, a concurrent respiratory acidosis is present; the carbon dioxide concentration is higher than expected. A patient may have a respiratory acidosis despite a carbon dioxide level below the “normal” value of 35-45 mm Hg. In this example, a carbon dioxide concentration <21 mm Hg indicates a concurrent respiratory alkalosis; the carbon dioxide concentration is lower than expected.
Table 52-9 APPROPRIATE COMPENSATION DURING SIMPLE ACID-BASE DISORDERS
| DISORDER | EXPECTED COMPENSATION |
|---|---|
| Metabolic acidosis | PCO2 = 1.5 × [HCO3−] + 8 ± 2 |
| Metabolic alkalosis | PCO2 increases by 7 mm Hg for each 10-mEq/L increase in serum [HCO3−] |
| Respiratory acidosis | |
| Acute | [HCO3−] increases by 1 for each 10–mm Hg increase in PCO2 |
| Chronic | [HCO3−] increases by 3.5 for each 10–mm Hg increase in PCO2 |
| Respiratory alkalosis | |
| Acute | [HCO3−] falls by 2 for each 10–mm Hg decrease in PCO2 |
| Chronic | [HCO3−] falls by 4 for each 10–mm Hg decrease in PCO2 |
Diagnosis
A systematic evaluation of an arterial blood gas sample, combined with the clinical history, can usually explain the patient’s acid-base disturbance. Assessment of an arterial blood gas sample requires knowledge of normal values (Table 52-10). In most cases, this is accomplished via a 3-step process (Fig. 52-3):
| pH | 7.35-7.45 |
| [HCO3−] | 20-28 mEq/L |
| PCO2 | 35-45 mm Hg |
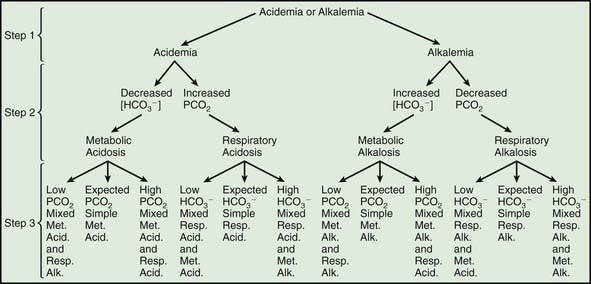
Figure 52-3 Three-step process for interpreting acid-base disturbances. In step 1, determine whether the pH is low (acidemia) or high (alkalemia). In step 2, establish an explanation for the acidemia or alkalemia. In step 3, calculate the expected compensation (see Table 52-9) and determine whether a mixed disturbance is present. Met. alk., metabolic alkalosis; Met. Acid., metabolic acidosis; Resp. Alk., respiratory alkalosis; Resp. Acid., respiratory acidosis.
The 2nd step requires inspection of the serum bicarbonate and carbon dioxide concentrations to determine a cause of the abnormal pH (see Fig. 52-3). In most cases, there is only 1 obvious explanation for the abnormal pH. In some mixed disorders, however, there may be 2 possibilities (a high PCO2 and a low [HCO3−] in a patient with acidemia). In such cases, the patient has 2 causes for abnormal pH (a metabolic acidosis and a respiratory acidosis, in this instance), and it is unnecessary to proceed to the 3rd step.
The 3rd step requires determining whether the patient’s compensation is appropriate. It is assumed that the primary disorder was diagnosed in the 2nd step, and the expected compensation is calculated (see Table 52-9). If the compensation is appropriate, then a simple acid-base disorder is present. If the compensation is not appropriate, then a mixed disorder is present. The identity of the 2nd disorder is determined by deciding whether the compensation is too little or too much compared with what was expected (see Fig. 52-3).
Metabolic Acidosis
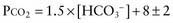
Etiology and Pathophysiology
There are many causes of a metabolic acidosis (Table 52-11), which occur via 3 basic mechanisms:
Table 52-11 CAUSES OF METABOLIC ACIDOSIS
NORMAL ANION GAP
INCREASED ANION GAP
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* Along with these genetic disorders, distal renal tubular acidosis may be secondary to renal disease or medications.
† Most cases of proximal RTA are not due to this primary genetic disorder. Proximal RTA is usually part of Fanconi syndrome, which has multiple etiologies.
‡ Hyperkalemic RTA can be secondary to a genetic disorder (some of the more common are listed) or other etiologies.
There are 3 forms of renal tubular acidosis (RTA): distal (type I), proximal (type II), and hyperkalemic (type IV) [Chapter 523]. In distal RTA, children may have accompanying hypokalemia, hypercalciuria, nephrolithiasis, and nephrocalcinosis. Failure to thrive due to chronic metabolic acidosis is the most common presenting complaint. Patients with distal RTA cannot acidify their urine and, thus, have a urine pH >5.5 despite a metabolic acidosis.
Proximal RTA is rarely present in isolation. In most patients, proximal RTA is part of Fanconi syndrome, a generalized dysfunction of the proximal tubule. The dysfunction leads to glycosuria, aminoaciduria, and excessive urinary losses of phosphate and uric acid. The presence of a low serum uric acid level, glycosuria, and aminoaciduria is helpful diagnostically. Chronic hypophosphatemia leads to rickets in children (Chapter 48). Rickets and/or failure to thrive may be the presenting complaint. The ability to acidify the urine is intact in proximal RTA; thus, untreated patients have a urine pH <5.5. However, bicarbonate therapy increases bicarbonate losses in the urine, and the urine pH increases.
Lactic acidosis most commonly occurs when inadequate oxygen delivery to the tissues leads to anaerobic metabolism and excess production of lactic acid. Lactic acidosis may be secondary to shock, severe anemia, or hypoxemia. When the underlying cause of the lactic acidosis is alleviated, the liver is able to metabolize the accumulated lactate into bicarbonate, correcting the metabolic acidosis. There is normally some tissue production of lactate that is metabolized by the liver. In children with severe liver dysfunction, impairment of lactate metabolism may produce a lactic acidosis. Rarely, a metabolically active malignancy grows so fast that its blood supply becomes inadequate, with resultant anaerobic metabolism and lactic acidosis. Patients who have short bowel syndrome due to small bowel resection can have bacterial overgrowth. In these patients, excessive bacterial metabolism of glucose into D-lactic acid can cause a lactic acidosis. Lactic acidosis occurs in a variety of inborn errors of metabolism, especially those affecting mitochondrial oxidation (Chapter 81.4). Finally, medications can cause lactic acidosis. Nucleoside reverse transcriptase inhibitors that are used to treat HIV infection inhibit mitochondrial replication; lactic acidosis is a rare complication, although elevated serum lactate concentrations without acidosis are quite common. Metformin, commonly used for treating type 2 diabetes mellitus, is most likely to cause a lactic acidosis in patients with renal insufficiency. High dosages and prolonged use of propofol can cause lactic acidosis.
In insulin-dependent diabetes mellitus, inadequate insulin leads to hyperglycemia and diabetic ketoacidosis (Chapter 583). Production of acetoacetic acid and β-hydroxybutyric acid causes the metabolic acidosis. Administration of insulin corrects the underlying metabolic problem and permits conversion of acetoacetate and β-hydroxybutyrate into bicarbonate, which helps correct the metabolic acidosis. However, in some patients, urinary losses of acetoacetate and β-hydroxybutyrate may be substantial, preventing rapid regeneration of bicarbonate. In these patients, full correction of the metabolic acidosis requires renal regeneration of bicarbonate, a slower process. The hyperglycemia causes an osmotic diuresis, usually producing volume depletion, along with substantial losses of potassium, sodium, and phosphate.
A variety of toxic ingestions (Chapter 58) can cause a metabolic acidosis. Salicylate intoxication is now much less common because aspirin is no longer recommended for fever control in children. Acute salicylate intoxication occurs after a large overdose. Chronic salicylate intoxication is possible with gradual buildup of the drug. Especially in adults, respiratory alkalosis may be the dominant acid-base disturbance. In children, the metabolic acidosis is usually the more significant finding. Other symptoms of salicylate intoxication are fever, seizures, lethargy, and coma. Hyperventilation may be particularly marked. Tinnitus, vertigo, and hearing impairment are more likely with chronic salicylate intoxication.
Many inborn errors of metabolism cause a metabolic acidosis (Chapters 78–81). The metabolic acidosis may be due to excessive production of keto acids, lactic acid, and/or other organic anions. Some patients have accompanying hypoglycemia or hyperammonemia. In most patients, the acidosis occurs episodically, only during acute decompensations, which may be precipitated by ingestion of specific dietary substrates, the stress of a mild illness, or poor compliance with dietary or medical therapy. In a few inborn errors of metabolism, patients have a chronic metabolic acidosis.
Clinical Manifestations
The underlying disorder usually produces most of the signs and symptoms in children with a mild or moderate metabolic acidosis. The clinical manifestations of the acidosis are related to the degree of acidemia; patients with appropriate respiratory compensation and less severe acidemia have fewer manifestations than those with a concomitant respiratory acidosis. At a serum pH <7.20, there may be impaired cardiac contractility and an increased risk of arrhythmias, especially if underlying heart disease or other predisposing electrolyte disorders are present. With acidemia, there may be a decrease in the cardiovascular response to catecholamines, potentially exacerbating hypotension in children with volume depletion or shock. Acidemia causes vasoconstriction of the pulmonary vasculature, which is especially problematic in newborn infants with persistent pulmonary hypertension (Chapter 95.7).
Diagnosis
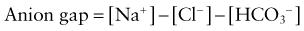
A normal anion gap is 4-11, although there is variation among laboratories. The number of serum anions must equal the number of serum cations to maintain electrical neutrality (Fig. 52-4). The anion gap is the difference between the measured cation (sodium) and the measured anions (chloride + bicarbonate). The anion gap is also the difference between the unmeasured cations (potassium, magnesium, calcium) and the unmeasured anions (albumin, phosphate, urate, sulfate). An increased anion gap occurs when there is an increase in unmeasured anions. With a lactic acidosis, there is endogenous production of lactic acid, which is composed of positively charged hydrogen ions and negatively charged lactate anions. The hydrogen ions are largely buffered by serum bicarbonate, resulting in a decrease in the bicarbonate concentration. The hydrogen ions that are not buffered by bicarbonate cause the serum pH to decrease. The lactate anions remain, causing the increase in the anion gap.
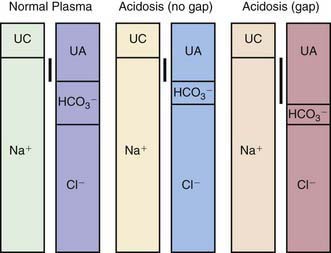
An increase in unmeasured anions, along with hydrogen ion generation, is present in all causes of an increased gap metabolic acidosis (see Table 52-11). In diabetic ketoacidosis, the keto acids β-hydroxybutyrate and acetoacetate are the unmeasured anions. In renal failure, there is retention of unmeasured anions, including phosphate, urate, and sulfate. The increase in unmeasured anions in renal failure is usually less than the decrease in the bicarbonate concentration. Renal failure is thus a mix of an increased gap and a normal gap metabolic acidosis. The normal gap metabolic acidosis is especially prominent in children with renal failure due to tubular damage, as occurs with renal dysplasia or obstructive uropathy, because these patients have a concurrent RTA. The unmeasured anions in toxic ingestions vary: formate in methanol intoxication, glycolate in ethylene glycol intoxication, and lactate and keto acids in salicylate intoxication. In inborn errors of metabolism, the unmeasured anions depend on the specific etiology and may include keto acids, lactate, and other organic anions. In a few inborn errors of metabolism, the acidosis occurs without generation of unmeasured anions; thus, the anion gap is normal.
A normal anion gap metabolic acidosis occurs when there is a decrease in the bicarbonate concentration without an increase in the unmeasured anions. With diarrhea, there is a loss of bicarbonate in the stool, causing a decrease in the serum pH and bicarbonate concentration; the serum chloride concentration increases to maintain electrical neutrality (see Fig. 52-4). Hyperchloremic metabolic acidosis is an alternative term for a normal anion gap metabolic acidosis. Calculation of the anion gap is more precise than using the chloride concentration to differentiate between a normal and an increased gap metabolic acidosis, in that the anion gap directly determines the presence of unmeasured anions. Electrical neutrality dictates that the chloride concentration increases or decreases according to the serum sodium concentration, making the chloride concentration a less reliable predictor of unmeasured anions than the more direct measure, calculation of the anion gap.
Metabolic Alkalosis
Etiology and Pathophysiology
The etiologies of a metabolic alkalosis are divided into 2 categories on the basis of urinary chloride level (Table 52-12). The alkalosis in patients with a low urinary chloride level is maintained by volume depletion; thus, volume repletion is necessary for correction of the alkalosis. The volume depletion in these patients is due to losses of sodium and potassium, but the loss of chloride is usually greater than the losses of sodium and potassium combined. Because chloride losses are the dominant cause of the volume depletion, these patients require chloride to correct the volume depletion and metabolic alkalosis; they are said to have chloride-responsive metabolic alkalosis. In contrast, the alkalosis in a patient with an elevated urinary chloride concentration does not respond to volume repletion and so is termed chloride-resistant metabolic alkalosis.
Table 52-12 CAUSES OF METABOLIC ALKALOSIS
CHLORIDE-RESPONSIVE (URINARY CHLORIDE < 15 mEq/L)
CHLORIDE-RESISTANT (URINARY CHLORIDE > 20 mEq/L)
EAST, epilepsy, ataxia, sensorineural hearing loss, and tubulopathy; MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
Most patients with diarrhea have a metabolic acidosis due to stool losses of bicarbonate. In chloride-losing diarrhea, an autosomal recessive disorder, there is a defect in the normal intestinal exchange of bicarbonate for chloride, causing excessive stool losses of chloride (Chapter 330). In addition, stool losses of hydrogen ions and potassium cause metabolic alkalosis and hypokalemia, both of which are exacerbated by increased renal hydrogen and potassium losses due to volume depletion. Treatment is with oral supplements of potassium and sodium chloride. Use of a gastric proton pump inhibitor, by decreasing gastric HCl production, reduces both the volume of diarrhea and the need for electrolyte supplementation.
An infant formula with extremely low chloride content has led to chloride deficiency and volume depletion. The infants fed this formula, which is no longer available, had a metabolic alkalosis and hypokalemia. Cystic fibrosis can rarely cause metabolic alkalosis, hypokalemia, and hyponatremia due to excessive losses of sodium chloride in sweat (Chapter 395). The volume depletion causes the metabolic alkalosis and hypokalemia through increased urinary losses, whereas the hyponatremia, a less common finding, is secondary to sodium loss combined with renal water conservation in an effort to protect the intravascular volume (“appropriate” ADH production).
In glucocorticoid-remediable aldosteronism, an autosomal dominant disorder, there is excess production of aldosterone owing to the presence of an aldosterone synthase gene that is regulated by adrenocorticotropic hormone (ACTH) (Chapter 570.8). Glucocorticoids effectively treat this disorder by inhibiting ACTH production by the pituitary, downregulating the inappropriate aldosterone production. Renovascular disease and renin-secreting tumors both cause excessive renin, leading to an increase in aldosterone, although hypokalemia and metabolic alkalosis are less common findings than hypertension. In 2 forms of congenital adrenal hyperplasia, 11β-hydroxylase deficiency and 17α-hydroxylase deficiency, there is excessive production of the mineralocorticoid 11-deoxycorticosterone (Chapters 570.2 and 570.4). Hypertension, hypokalemia, and metabolic alkalosis are more likely in 17α-hydroxylase deficiency than in 11β-hydroxylase deficiency. These disorders respond to glucocorticoids because the excess production of 11-deoxycorticosterone is under the control of ACTH.
Cortisol can bind to the mineralocorticoid receptors in the kidney and function as a mineralocorticoid. This binding normally does not occur because 11β-hydroxysteroid dehydrogenase in the kidney converts cortisol to cortisone, which does not bind to the mineralocorticoid receptor. In 11β-hydroxysteroid dehydrogenase deficiency, also called apparent mineralocorticoid excess, however, cortisol is not converted in the kidney to cortisone. Cortisol is therefore available to bind to the mineralocorticoid receptor in the kidney and act as a mineralocorticoid. Patients with this deficiency, despite low levels of aldosterone, are hypertensive and hypokalemic, and they have a metabolic alkalosis. The same phenomenon can occur with excessive intake of natural licorice, a component of which, glycyrrhizic acid, inhibits 11β-hydroxysteroid dehydrogenase. The autosomal dominant disorder Liddle syndrome is secondary to an activating mutation of the sodium channel in the distal nephron (Chapter 525.3). Upregulation of this sodium channel is 1 of the principal actions of aldosterone. Because this sodium channel is continuously open, children with Liddle syndrome have the features of hyperaldosteronism, including hypertension, hypokalemia, and metabolic alkalosis, but low serum levels of aldosterone.
Bartter syndrome and Gitelman syndrome are autosomal recessive disorders associated with normal blood pressure, elevations of urinary chloride, metabolic alkalosis, and hypokalemia (Chapter 525). In Bartter syndrome, patients have a defect in sodium and chloride resorption in the loop of Henle. This leads to excessive urinary losses of sodium and chloride, and as in patients receiving loop diuretics, volume depletion and secondary hyperaldosteronism occur, causing hypokalemia and metabolic alkalosis. Gitelman syndrome is usually milder than Bartter syndrome. Patients have renal sodium and chloride wasting with volume depletion due mutations in the gene encoding the thiazide-sensitive sodium-chloride transporter in the distal tubule. As in patients receiving a thiazide diuretic, affected patients have volume depletion and secondary hyperaldosteronism with hypokalemia and metabolic alkalosis. Children with Gitelman syndrome have hypocalciuria and hypomagnesemia. Some patients with autosomal dominant hypoparathyroidism have hypokalemia and metabolic alkalosis due to impaired sodium and chloride resorption in the loop of Henle. EAST syndrome (epilepsy, ataxia, sensorineural deafness and tubulopathy) causes hypokalemia, metabolic alkalosis and hypokalemia.
Clinical Manifestations
Alkalemia causes potassium to shift into the intracellular space, producing a decrease in the extracellular potassium concentration. Alkalemia leads to increased urinary losses of potassium. Increased potassium losses are present in many of the conditions that cause a metabolic alkalosis. Therefore, most patients with a metabolic alkalosis have hypokalemia, and their symptoms may be related to the hypokalemia (Chapter 52.4).
Diagnosis
Diuretics and gastric losses are the most common causes of metabolic alkalosis and are usually readily apparent from the patient history. Occasionally, metabolic alkalosis, usually with hypokalemia, may be a clue to the presence of bulimia or surreptitious diuretic use (Chapter 26). Patients with bulimia have a low urine chloride level, indicating that they have volume depletion as a result of an extrarenal etiology, but there is no alternative explanation for their volume depletion. Surreptitious diuretic use may be diagnosed by obtaining a urine toxicology screen for diuretics. The urine chloride level is increased while a patient is using diuretics but is low when the patient stops taking them. Rarely, children with mild Bartter syndrome or Gitelman syndrome are misdiagnosed as having bulimia or abusing diuretics. The urine chloride value is always elevated in Bartter syndrome and Gitelman syndrome, and the urine toxicology screen for diuretics has a negative result. Metabolic alkalosis with hypokalemia is occasionally the initial manifestation of cystic fibrosis. An elevated sweat chloride finding is diagnostic.
Measuring serum concentrations of renin and aldosterone differentiates children with a metabolic alkalosis, a high urinary chloride level, and elevated blood pressure. Both renin and aldosterone are elevated in children with either renovascular disease or a renin-secreting tumor. Aldosterone is high and renin is low in patients with adrenal adenomas or hyperplasia and glucocorticoid-remediable aldosteronism. Renin and aldosterone are low in children with Cushing syndrome, Liddle syndrome, licorice ingestion, 17α-hydroxylase deficiency, 11β-hydroxylase deficiency, and 11β-hydroxysteroid dehydrogenase deficiency. An elevated 24-hr urine cortisol value is diagnostic of Cushing syndrome, which is suspected from the presence of the other classic features of this disease (Chapter 571). Elevations of 11-Deoxycorticosterone values are seen in 17α-hydroxylase deficiency and 11β-hydroxylase deficiency.
Respiratory Acidosis
Etiology and Pathophysiology
The causes of a respiratory acidosis are either pulmonary or nonpulmonary (Table 52-13). CNS disorders can decrease the activity of the central respiratory center, reducing ventilatory drive. A variety of medications and illicit drugs suppress the respiratory center. The signals from the respiratory center need to be transmitted to the respiratory muscles via the nervous system. Respiratory muscle failure can be secondary to disruption of the signal from the CNS in the spinal cord, the phrenic nerve, or the neuromuscular junction. Disorders directly affecting the muscles of respiration can prevent adequate ventilation, causing a respiratory acidosis.
Table 52-13 CAUSES OF RESPIRATORY ACIDOSIS
CENTRAL NERVOUS SYSTEM DEPRESSION
DISORDERS OF THE SPINAL CORD, PERIPHERAL NERVES, OR NEUROMUSCULAR JUNCTION
RESPIRATORY MUSCLE WEAKNESS
PULMONARY DISEASE
UPPER AIRWAY DISEASE
MISCELLANEOUS
Diagnosis
For a patient with respiratory acidosis, calculation of the gradient between the alveolar oxygen concentration and the arterial oxygen concentration, the A-aO2 gradient, is useful for distinguishing between poor respiratory effort and intrinsic lung disease. The A-aO2 gradient is increased if the hypoxemia is due to intrinsic lung disease (Chapter 365).
Treatment
All patients with an acute respiratory acidosis are hypoxic and therefore need to receive supplemental oxygen. Mechanical ventilation is necessary in some children with a respiratory acidosis. Children with a significant respiratory acidosis due to a CNS disease usually require mechanical ventilation because such a disorder is unlikely to respond quickly to therapy. In addition, hypercarbia causes cerebral vasodilation, and the increase in intracranial pressure can be dangerous in a child with an underlying CNS disease. Readily reversible CNS depression, such as from a narcotic overdose, may not require mechanical ventilation. Decisions on mechanical ventilation for other patients depend on a number of factors. Patients with severe hypercarbia—PCO2 > 75 mm Hg—usually require mechanical ventilation (Chapters 65 and 366). The threshold for intubation is lower if there is concomitant metabolic acidosis, a slowly responsive underlying disease, or hypoxia that responds poorly to oxygen, or if the patient appears to be tiring and respiratory arrest seems likely.
Respiratory Alkalosis
Etiology and Pathophysiology
A variety of stimuli can increase the ventilatory drive and cause a respiratory alkalosis (Table 52-14). Arterial hypoxemia or tissue hypoxia stimulates peripheral chemoreceptors to signal the central respiratory center in the medulla to increase ventilation. The resultant greater respiratory effort increases the oxygen content of the blood but depresses the PCO2. The effect of hypoxemia on ventilation begins when the oxygen saturation decreases to approximately 90% (PO2 = 60 mm Hg), and hyperventilation increases as hypoxemia worsens. Acute hypoxia is a more potent stimulus for hyperventilation than chronic hypoxia; thus, chronic hypoxia, as occurs in cyanotic heart disease, causes a much less severe respiratory alkalosis than an equivalent degree of acute hypoxia. There are many causes of hypoxemia or tissue hypoxia, including primary lung disease, severe anemia, and carbon monoxide poisoning.
Table 52-14 CAUSES OF RESPIRATORY ALKALOSIS
HYPOXEMIA OR TISSUE HYPOXIA
LUNG RECEPTOR STIMULATION
CENTRAL STIMULATION
Fry AC, Karet FE. Inherited renal acidoses. Physiology. 2007;22:202-211.
Gabriely I, Leu JP, Barzel US. Clinical problem-solving: back to basics. N Engl J Med. 2008;358:1952-1956.
Kellum JA. Disorders of acid-base balance. Crit Care Med. 2007;35:2630-2636.
Kraut JA, Kurtz I. Toxic alcohol ingestions: clinical features, diagnosis, and management. Clin J Am Soc Nephrol. 2008;3:208-225.
Kraut JA, Madias NE. Serum anion gap: its uses and limitations in clinical medicine. Clin J Am Soc Nephrol. 2007;2:162-174.
Kurtz I, Kraut J, Ornekian V, et al. Acid-base analysis: a critique of the Stewart and bicarbonate-centered approaches. Am J Physiol Renal Physiol. 2008;294:F1009-F1031.
Manini AF, Hoffman RS, Nelson LS. Alcoholic ketoacidosis in an 11-year-old boy. Pediatr Emerg Care. 2008;24:170-171.
Marano M, Iodice F, Stoppa F, et al. Treatment of severe diabetic acidosis with tris-hydroxymethyl aminomethane in a thirteen-year-old child. Minerva Anestesiol. 2008;74:93-95.
Palmer BF. Approach to fluid and electrolyte disorders and acid-base problems. Prim Care. 2008;35:195-213.
Schimert P, Bernet-Buettiker V, Rutishauser C, et al. Transplacental metabolic alkalosis. J Paediatr Child Health. 2007;43:851-853.
Stacpoole PW, Gilbert LR, Neiberger RE, et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121:e1223-e1228.
Walsh SB, Shirley DG, Wrong OM, et al. Urinary acidification assessed by simultaneous furosemide and fludrocortisone treatment: an alternative to ammonium chloride. Kidney Int. 2007;71:1310-1316.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 52 Electrolyte and Acid-Base Disorders
52.1 Composition of Body Fluids
Total Body Water
Water is the most plentiful constituent of the human body. Total body water (TBW) as a percentage of body weight varies with age (Web Fig. 52-1). The fetus has very high TBW, which gradually decreases to approximately 75% of birthweight for a term infant. Premature infants have higher TBW than term infants. During the 1st yr of life, TBW decreases to approximately 60% of body weight and basically remains at this level until puberty. At puberty, the fat content of females increases more than that of males, who acquire more muscle mass than females. Because fat has very low water content and muscle has high water content, by the end of puberty TBW in males remains at 60%, but TBW in females decreases to approximately 50% of body weight. The high fat content in overweight children causes a decrease in TBW as a percentage of body weight. During dehydration, TBW decreases and, thus, is a smaller percentage of body weight.
Fluid Compartments
TBW is divided between 2 main compartments: intracellular fluid (ICF) and extracellular fluid (ECF). In the fetus and newborn, the ECF volume is larger than the ICF volume (see Web Fig. 52-1). The normal postnatal diuresis causes an immediate decrease in the ECF volume. This is followed by continued expansion of the ICF volume, which results from cellular growth. By 1 yr of age, the ratio of the ICF volume to the ECF volume approaches adult levels. The ECF volume is approximately 20-25% of body weight, and the ICF volume is approximately 30-40% of body weight, close to twice the ECF volume (Web Fig. 52-2). With puberty, the increased muscle mass of males causes them to have a higher ICF volume than females. There is no significant difference in the ECF volume between postpubertal females and males.
Web Figure 52-2 Compartments of total body water, expressed as percentages of body weight, in an older child or adult.
The ECF is further divided into the plasma water and the interstitial fluid (see Web Fig. 52-2). The plasma water is 5% of body weight. The blood volume, given a hematocrit of 40%, is usually 8% of body weight, although it is higher in newborns and young infants; in premature newborns, it is approximately 10% of body weight. The volume of plasma water can be altered by pathologic conditions, including dehydration, anemia, polycythemia, heart failure, abnormal plasma osmolality, and hypoalbuminemia. The interstitial fluid, normally 15% of body weight, can increase dramatically in diseases associated with edema, such as heart failure, protein-losing enteropathy, liver failure, nephrotic syndrome, and sepsis. An increase in interstitial fluid also occurs in patients with ascites or pleural effusions.
Electrolyte Composition
The composition of the solutes in the ICF and ECF are very different (Web Fig. 52-3). Sodium and chloride are the dominant cation and anion, respectively, in the ECF. The sodium and chloride concentrations in the ICF are much lower. Potassium is the most abundant cation in the ICF, and its concentration within the cells is approximately 30 times higher than in the ECF. Proteins, organic anions, and phosphate are the most plentiful anions in the ICF. The dissimilarity between the anions in the ICF and the ECF is largely determined by the presence of intracellular molecules that do not cross the cell membrane, the barrier separating the ECF and the ICF. In contrast, the difference in the distribution of cations—sodium and potassium—is due to the activity of the Na+,K+-ATPase pump, which uses cellular energy to actively extrude sodium from cells and move potassium into cells. The chemical gradient between the intracellular potassium concentration and the extracellular potassium concentration creates the electrical gradient across the cell membrane. The concentration-dependent movement of potassium out of the cell makes the intracellular space negative relative to the extracellular space.
Osmolality
Glucose and blood urea nitrogen (BUN) are measured in mg/dL. Division of these values by 18 and 2.8, respectively, as shown, converts the units into mmol/L. Multiplication of the sodium value by 2 accounts for its accompanying anions, principally chloride and bicarbonate. The calculated osmolality is usually slightly lower than the measured osmolality.
The effective osmolality (also called the tonicity) determines the osmotic force that is mediating the shift of water between the ECF and the ICF.
where [Na]measured = sodium concentration measured by the clinical laboratory and [Na]corrected = corrected sodium concentration (the sodium concentration if the glucose concentration were normal and its accompanying water moved back into the cells). The [Na]corrected is the more reliable indicator of the patient’s true ratio of total body sodium to TBW, the normal determinant of the sodium concentration.
Garibaldi BT, Cameron SJ, Choi M. Pseudohyponatremia in a patient with HIV and hepatitis C coinfection. J Gen Intern Med. 2008;23:202-205.
Nguyen MK, Ornekian V, Butch AW, et al. A new method for determining plasma water content: application in pseudohyponatremia. Am J Physiol Renal Physiol. 2007;292:F1652-F1656.
Pellegrino B, Parravani A, Cook L, et al. Ethylene glycol intoxication: disparate findings of immediate versus delayed presentation. W V Med J. 2006;102:32-34.
52.2 Regulation of Osmolality and Volume
Regulation of Osmolality
Osmoreceptors in the hypothalamus sense the plasma osmolality (Chapter 552). An elevated effective osmolality leads to secretion of antidiuretic hormone (ADH) by neurons in the supraoptic and paraventricular nuclei in the hypothalamus. The axons of these neurons terminate in the posterior pituitary. Circulating ADH binds to its V2 receptors in the collecting duct cells of the kidney, and, via the generation of cyclic adenosine monophosphate, causes insertion of water channels (aquaporin-2) into the renal collecting ducts. This produces increased permeability to water, permitting resorption of water into the hypertonic renal medulla. The end result is that the urine concentration increases and water excretion decreases. Urinary water losses cannot be completely eliminated because there is obligatory excretion of urinary solutes, such as urea and sodium. The regulation of ADH secretion is tightly linked to plasma osmolality, responses being detectable with a 1% change in the osmolality. ADH secretion virtually disappears when the plasma osmolality is low, allowing excretion of maximally dilute urine. The consequent loss of free water (water without sodium) corrects the plasma osmolality. ADH secretion is not an all-or-nothing response; there is a graded adjustment as the osmolality changes.
A number of conditions can limit the kidney’s ability to excrete adequate water to correct low plasma osmolality. In the syndrome of inappropriate antidiuretic hormone (SIADH), ADH continues to be produced despite a low plasma osmolality. In the presence of ADH, urinary dilution does not occur, and sufficient water is not excreted (Chapters 52.3 and 553).
The normal response to increased plasma osmolality is conservation of water by the kidney. In central diabetes insipidus, this does not occur because of an absence of ADH secretion (Chapter 552.1). Patients with nephrogenic diabetes insipidus have an inability to respond to ADH and produce dilute urine despite an increase in plasma osmolality (Chapters 52.3, 524, and 552).
Regulation of Volume
Sodium resorption occurs throughout the nephron (Chapter 522). Whereas the majority of filtered sodium is resorbed in the proximal tubule and the loop of Henle, the distal tubule and the collecting duct are the main sites for precise regulation of sodium balance. Approximately 65% of the filtered sodium is reclaimed in the proximal tubule, which is the major site for resorption of bicarbonate, glucose, phosphate, amino acids, and other substances that are filtered by the glomerulus. The transport of all these substances is linked to sodium resorption by cotransporters, or a sodium-hydrogen exchanger in the case of bicarbonate. This link is clinically important for bicarbonate and phosphate because their resorption parallels sodium resorption. In patients with metabolic alkalosis and volume depletion, correction of the metabolic alkalosis requires urinary loss of bicarbonate, but the volume depletion stimulates sodium and bicarbonate retention, preventing correction of the alkalosis. Volume expansion causes increased urinary losses of phosphate, even when there is phosphate depletion. Resorption of uric acid and urea occurs in the proximal tubule and increases when sodium retention increases. This arrangement accounts for the elevated uric acid and BUN measurements that often accompany dehydration, which is a stimulus for sodium retention in the proximal tubule. The cells of the proximal tubule are permeable to water; thus, water resorption in this segment parallels sodium resorption.
Volume depletion usually occurs when sodium losses exceed intake. The most common etiology in children is gastroenteritis. Excessive losses of sodium may also occur from the skin in children with burns, in sweat from patients with cystic fibrosis, or after vigorous exercise. Inadequate intake of sodium is uncommon except in neglect, in famine, or with an inappropriate choice of liquid diet in a child who cannot take solids. Urinary sodium wasting may occur in a range of renal diseases, from renal dysplasia to tubular disorders, such as Bartter syndrome. The neonate, especially if premature, has a mild impairment in the ability to conserve sodium. Iatrogenic renal sodium wasting takes place during diuretic therapy. Renal sodium loss occurs as a result of derangement in the normal regulatory systems. An absence of aldosterone, seen most commonly in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency, causes sodium wasting (Chapter 570).
Ball SG. Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem. 2007;44:417-431.
Berl T. Impact of solute intake on urine flow and water excretion. J Am Soc Nephrol. 2008;19:1076-1078.
Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519-531.
Nielsen S, Kwon TH, Frokiaer J, et al. Regulation and dysregulation of aquaporins in water balance disorders. J Intern Med. 2007;261:53-64.
Schrier RW. Aquaporin-related disorders of water homeostasis. Drug News Perspect. 2007;20:447-453.
Schrier RW. Decreased effective blood volume in edematous disorders: what does this mean? J Am Soc Nephrol. 2007;18:2028-2031.
52.3 Sodium
Sodium Metabolism
Body Content and Physiologic Function
Sodium is the dominant cation of the ECF (see Web Fig. 52-3), and it is the principal determinant of extracellular osmolality. Sodium is therefore necessary for the maintenance of intravascular volume. Less than 3% of sodium is intracellular. More than 40% of total body sodium is in bone; the remainder is in the interstitial and intravascular spaces. The low intracellular sodium concentration, approximately 10 mEq/L, is maintained by Na+,K+-ATPase, which exchanges intracellular sodium for extracellular potassium.
Intake
Sodium is readily absorbed throughout the gastrointestinal tract. Mineralocorticoids increase sodium transport into the body, although this effect has limited clinical significance. The presence of glucose enhances sodium absorption owing to the presence of a cotransport system. This is the rationale for including sodium and glucose in oral rehydration solutions (Chapter 332).
Hypernatremia
Etiology and Pathophysiology
There are 3 basic mechanisms of hypernatremia (Table 52-1). Sodium intoxication is frequently iatrogenic in a hospital setting as a result of correction of metabolic acidosis with sodium bicarbonate. Baking soda, a putative home remedy for upset stomach, is another source of sodium bicarbonate; the hypernatremia is accompanied by a profound metabolic alkalosis. In hyperaldosteronism, there is renal retention of sodium and resultant hypertension; the hypernatremia is usually mild.
Table 52-1 CAUSES OF HYPERNATREMIA
EXCESSIVE SODIUM
WATER DEFICIT
WATER AND SODIUM DEFICITS
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
The classic causes of hypernatremia from a water deficit are nephrogenic and central diabetes insipidus (Chapters 524 and 552). Hypernatremia develops in diabetes insipidus only if the patient does not have access to water or cannot drink adequately because of immaturity, neurologic impairment, emesis, or anorexia. Infants are at high risk because of their inability to control their own water intake. Central diabetes insipidus and the genetic forms of nephrogenic diabetes insipidus typically cause massive urinary water losses and very dilute urine. The water losses are less dramatic, and the urine often has the same osmolality as plasma when nephrogenic diabetes insipidus is secondary to disease (obstructive uropathy, renal dysplasia, sickle cell disease).
When hypernatremia occurs in conditions with deficits of sodium and water, the water deficit exceeds the sodium deficit. This occurs only if the patient is unable to ingest adequate water. Diarrhea results in depletion of both sodium and water. Because diarrhea is hypotonic—typical sodium concentration of 35-65 mEq/L—water losses exceed sodium losses, potentially leading to hypernatremia. Most children with gastroenteritis do not have hypernatremia because they drink enough hypotonic fluid to compensate for stool water losses (Chapter 332). Fluids such as water, juice, and formula are more hypotonic than the stool losses, allowing correction of the water deficit and potentially even causing hyponatremia. Hypernatremia is most likely to occur in the child with diarrhea who has inadequate intake because of emesis, lack of access to water, or anorexia.
Clinical Manifestations
Most children with hypernatremia are dehydrated and show the typical clinical signs and symptoms (Chapter 54). Children with hypernatremic dehydration tend to have better preservation of intravascular volume because of the shift of water from the intracellular space to the extracellular space. This shift maintains blood pressure and urine output and allows hypernatremic infants to be less symptomatic initially and potentially to become more dehydrated before medical attention is sought. Breast-fed infants with hypernatremia are often profoundly dehydrated, with failure to thrive. Probably because of intracellular water loss, the pinched abdominal skin of a dehydrated, hypernatremic infant has a “doughy” feel.
Diagnosis
When there is isolated water loss, the signs of volume depletion are usually less severe initially because much of the loss is from the intracellular space. When pure water loss causes signs of dehydration, the hypernatremia and water deficit are usually severe. In the child with renal water loss, either central or nephrogenic diabetes insipidus, the urine is inappropriately dilute and urine volume is not low. The urine is maximally concentrated and urine volume is low if the losses are extrarenal or due to inadequate intake. With extrarenal causes of loss of water, the urine osmolality should be >1,000 mOsm/kg. When diabetes insipidus is suspected, the evaluation may include measurement of ADH and a water deprivation test, including a trial of desmopressin acetate (synthetic ADH analog) to differentiate between nephrogenic diabetes insipidus and central diabetes insipidus (Chapter 552.1). A water deprivation test is unnecessary if the patient has simultaneous documentation of hypernatremia and poorly concentrated urine (osmolality lower than that of plasma). In children with central diabetes insipidus, administration of desmopressin acetate increases the urine osmolality above the plasma osmolality, although maximum osmolality does not occur immediately because of the decreased osmolality of the renal medulla due to the chronic lack of ADH. In children with nephrogenic diabetes insipidus, there is no response to desmopressin acetate.
Treatment
As hypernatremia develops, the brain generates idiogenic osmoles to increase the intracellular osmolality and prevent the loss of brain water. This mechanism is not instantaneous and is most prominent when hypernatremia has developed gradually. If the serum sodium concentration is lowered rapidly, there is movement of water from the serum into the brain cells to equalize the osmolality in the 2 compartments (Fig. 52-1). The resultant brain swelling manifests as seizures or coma.
In the child with hypernatremic dehydration, as in any child with dehydration, the first priority is restoration of intravascular volume with isotonic fluid (Chapter 54). Normal saline is preferable to lactated Ringer solution because the lower sodium concentration of the latter can cause the serum sodium to decrease too rapidly, especially if multiple fluid boluses are given. Repeated boluses of normal saline (10-20 mL/kg) may be required to treat hypotension, tachycardia, and signs of poor perfusion (peripheral pulses, capillary refill time) (Chapters 54 and 64).
This calculation is equivalent to 3-4 mL of water per kg for each 1 mEq that the current sodium level exceeds 145 mEq. The utility of such formulas has never been proven in clinical practice. Most patients with hypernatremic dehydration do well with a fluid sodium concentration of approximately half-normal saline, but with a fluid rate that is only 20-30% greater than maintenance fluid. Use of this concentration prevents excessive delivery of free water and too rapid a decrease in the serum sodium level. Patients with pure water loss may require a more hypotonic fluid (0.2 normal saline). Excessive water and sodium losses may also need to be replaced. If signs or symptoms of volume depletion develop, the patient receives additional boluses of isotonic saline. Monitoring of the rate of decrease of the serum sodium concentration permits adjustment in the rate and sodium concentration of the fluid that the patient is receiving, avoiding overly rapid correction of the hypernatremia. Many patients with mild to moderate hypernatremic dehydration due to gastroenteritis can be managed with oral rehydration (Chapter 332).
It is important to address the underlying cause of the hypernatremia, if possible. The child with central diabetes insipidus should receive desmopressin acetate. Because this treatment reduces renal excretion of water, excessive intake of water must consequently be avoided to prevent both overly rapid correction of the hypernatremia and the development of hyponatremia. Over the long term, reduced sodium intake and the use of medications can somewhat ameliorate the water losses in nephrogenic diabetes insipidus (Chapter 524). The daily water intake of a child who is receiving tube feeding may need to be increased to compensate for high losses. The patient with significant ongoing losses, such as through diarrhea, may need supplemental water and electrolytes (Chapter 53). Sodium intake is reduced if it contributed to the hypernatremia.
Hyponatremia
Etiology and Pathophysiology
The causes of hyponatremia are listed in Table 52-2. Pseudohyponatremia is a laboratory artifact that is present when the plasma contains very high concentrations of protein (multiple myeloma, intravenous immunoglobulin infusion) or lipid (hypertriglyceridemia, hypercholesterolemia). It does not occur when a direct ion-selective electrode determines the sodium concentration in undiluted plasma, a technique that is used by the instruments used for measuring arterial blood gases. In true hyponatremia, the measured osmolality is low, whereas it is normal in pseudohyponatremia. Hyperosmolality, as may occur with hyperglycemia, causes a low serum sodium concentration because water moves down its osmotic gradient from the intracellular space into the extracellular space, diluting the sodium concentration. However, because the manifestations of hyponatremia are due to the low plasma osmolality, patients with hyponatremia resulting from hyperosmolality do not have symptoms of hyponatremia. When the etiology of the hyperosmolality resolves, such as hyperglycemia in diabetes mellitus, water moves back into the cells and the sodium concentration rises to its “true” value. Mannitol or sucrose, a component of intravenous immunoglobulin preparations, may cause hyponatremia due to hyperosmolality.
Table 52-2 CAUSES OF HYPONATREMIA
PSEUDOHYPONATREMIA
EXTRARENAL LOSSES
RENAL LOSSES
EUVOLEMIC HYPONATREMIA
HYPERVOLEMIC HYPONATREMIA
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
* Most cases of proximal renal tubular acidosis are not due to this primary genetic disorder. Proximal renal tubular acidosis is usually part of Fanconi syndrome, which has multiple etiologies.
Renal salt wasting occurs in hereditary kidney diseases, such as juvenile nephronophthisis and autosomal recessive polycystic kidney disease. Obstructive uropathy, most commonly a consequence of posterior urethral valves, produces salt wasting, but patients with the disease may also have hypernatremia as a result of impaired ability to concentrate urine and high water loss. Acquired tubulointerstitial nephritis, usually secondary to either medications or infections, may cause salt wasting, along with other evidence of tubular dysfunction. CNS injury may produce cerebral salt wasting, which is theoretically due to the production of a natriuretic peptide that causes renal salt wasting. In type II renal tubular acidosis (RTA), usually associated with Fanconi syndrome (Chapter 523), there is increased excretion of sodium and bicarbonate in the urine. Patients with Fanconi syndrome also have glycosuria, aminoaciduria, and hypophosphatemia due to renal phosphate wasting.
In SIADH, the secretion of ADH is not inhibited by either low serum osmolality or expanded intravascular volume (Chapter 553). The result is that the child with SIADH is unable to excrete water. This results in dilution of the serum sodium and hyponatremia. The expansion of the extracellular volume due to the retained water causes a mild increase in intravascular volume. The kidney increases sodium excretion in an effort to decrease intravascular volume to normal; thus, the patients has a mild decrease in body sodium. SIADH most commonly occurs with disorders of the CNS (infection, hemorrhage, trauma, tumor, thrombosis), but lung disease (infection, asthma, positive pressure ventilation) and malignant tumors (producing ADH) are other potential causes. A variety of medications may cause SIADH, including recreational use of 3,4-methylenedioxymethylamphetamine (MDMA, or “Ecstasy”), opiates, antiepileptic drugs (carbamazepine, oxcarbamazepine, valproate), tricyclic antidepressants, vincristine, cytoxan, and selective serotonin reuptake inhibitors. The diagnosis of SIADH is one of exclusion, because other causes of hyponatremia must be eliminated (Table 52-3). Because SIADH is a state of intravascular volume expansion, low serum uric acid and BUN levels are supportive of the diagnosis.
Diagnosis
The history usually points to a likely etiology of the hyponatremia. Most patients with hyponatremia have a history of volume depletion. Diarrhea and diuretic use are very common causes of hyponatremia in children. A history of polyuria, perhaps with enuresis, and/or salt craving is present in children with primary kidney diseases or absence of aldosterone effect. Children may have signs or symptoms suggesting a diagnosis of hypothyroidism or adrenal insufficiency (Chapters 559 and 569). Brain injury raises the possibility of SIADH or cerebral salt wasting, with the caveat that SIADH is much more likely. Liver disease, nephrotic syndrome, renal failure, or congestive heart failure may be acute or chronic. The history should include a review of the patient’s intake, both intravenous and enteral, with careful attention to the amounts of water, sodium, and protein.
In patients with true hyponatremia, the next step in the diagnostic process is to clinically evaluate the volume status. Patients with hyponatremia can be hypovolemic, hypervolemic, or euvolemic. The diagnosis of volume depletion relies on the usual findings with dehydration (Chapter 54), although subtle volume depletion may not be clinically apparent. In a patient with subtle volume depletion, a fluid bolus results in a decrease in the urine osmolality and an increase in the serum sodium concentration. Children with hypervolemia are edematous on physical examination. They may have ascites, pulmonary edema, pleural effusion, or hypertension.
Hypovolemic hyponatremia can have renal or nonrenal causes. The urine sodium concentration is very useful in differentiating between renal and nonrenal causes. When the losses are nonrenal and the kidney is working properly, there is renal retention of sodium, a normal homeostatic response to volume depletion. Thus, the urinary sodium concentration is low, typically <10 mEq/L, although sodium conservation in neonates is less avid. When the kidney is the cause of the sodium loss, the urine sodium concentration is >20 mEq/L, reflecting the defect in renal sodium retention. The interpretation of the urine sodium level is challenging with diuretic therapy because it is high when diuretics are being used but low after the diuretic effect is gone. This becomes an issue only when diuretic use is surreptitious. The urine sodium concentration is not useful if a metabolic alkalosis is present; the urine chloride concentration must be used instead (Chapter 52.7).
Treatment
The child with hypovolemic hyponatremia has a deficiency in sodium and may have a deficiency in water. The cornerstone of therapy is to replace the sodium deficit and any water deficit that is present. The 1st step in treating any dehydrated patient is to restore the intravascular volume with isotonic saline. Ultimately, complete restoration of intravascular volume suppresses ADH production, thereby permitting excretion of the excess water. Chapter 54 discusses the management of hyponatremic dehydration.
Children with iatrogenic hyponatremia due to the administration of hypotonic intravenous fluids should receive 3% saline if they are symptomatic. Subsequent management is dictated by the patient’s volume status. The hypovolemic child should receive isotonic intravenous fluids. The child with nonphysiologic stimuli for ADH production should undergo fluid restriction. Prevention of this iatrogenic complication requires judicious use of intravenous fluids (Chapter 53).
Centers for Disease Control and Prevention (CDC). Application of lower sodium intake recommendations to adults—United States, 1999-2006. MMWR Morb Mortal Wkly Rep. 2009;58:281-283.
Decaux G, Soupart A, Vassart G. Non-peptide arginine-vasopressin antagonists: the vaptans. Lancet. 2008;371:1624-1632.
Don M, Valerio G, Korppi M, et al. Hyponatremia in pediatric community-acquired pneumonia. Pediatr Nephrol. 2008;23:2247-2253.
Ellison DH, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;356:2064-2072.
Fenske W, Maier SKG, Blechschmidt A, et al. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;123:652-657.
Karakas HM, Erdem G, Yakinci C. Osmotic demyelination syndrome in a 40-day-old infant. Diagn Interv Radiol. 2007;13:121-124.
Leung C, Chang WC, Yeh SJ. Hypernatremic dehydration due to concentrated infant formula: report of two cases. Pediatr Neonatol. 2009;50:70-73.
Linshaw MA. Back to basics: congenital nephrogenic diabetes insipidus. Pediatr Rev. 2007;28:372-380.
Modi N. Avoiding hypernatraemic dehydration in healthy term infants. Arch Child. 2007;92:474-475.
Rianthavorn P, Cain JP, Turman MA. Use of conivaptan to allow aggressive hydration to prevent tumor lysis syndrome in a pediatric patient with large-cell lymphoma and SIADH. Pediatr Nephrol. 2008;23:1367-1370.
Rivkees SA. Differentiating appropriate antidiuretic hormone secretion, inappropriate antidiuretic hormone secretion and cerebral salt wasting: the common, uncommon, and misnamed. Curr Opin Pediatr. 2008;20:448-452.
Verbalis JG, Goldsmith SR, Greenberg A, et al. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120:S1-S21.
52.4 Potassium
Potassium Metabolism
Body Content and Physiologic Function
The intracellular concentration of potassium, approximately 150 mEq/L, is much higher than the plasma concentration (see Web Fig. 52-3). The majority of body potassium is contained in muscle. As muscle mass increases, there is an increase in body potassium. There is thus an increase in body potassium during puberty, and it is more significant in males. The majority of extracellular potassium is in bone; <1% of total body potassium is in plasma.
Hyperkalemia
Etiology and Pathophysiology
Three basic mechanisms cause hyperkalemia (Table 52-4). In the individual patient, the etiology is sometimes multifactorial.
Table 52-4 CAUSES OF HYPERKALEMIA
SPURIOUS LABORATORY VALUE
INCREASED INTAKE
TRANSCELLULAR SHIFTS
DECREASED EXCRETION
MIM, database number from the Mendelian Inheritance in Man (http://www3.ncbi.nlm.nih.gov/Omim/).
Normal doses of succinylcholine or β-blockers and fluoride or digitalis intoxication all cause a shift of potassium out of the intracellular compartment. Succinylcholine should not be used during anesthesia in patients at risk for hyperkalemia. β-Blockers prevent the normal cellular uptake of potassium mediated by binding of β-agonists to the β2-adrenergic receptors. Potassium release from muscle cells occurs during exercise, and levels can increase by 1-2 mEq/L with high activity. With an increased plasma osmolality, water moves from the intracellular space and potassium follows. This process occurs with hyperglycemia, although in nondiabetic patients, the resultant increase in insulin causes potassium to move intracellularly. In diabetic ketoacidosis, the absence of insulin causes potassium to leave the intracellular space, and the problem is compounded by the hyperosmolality. The effect of hyperosmolality causes a transcellular shift of potassium into the extracellular space after mannitol or hypertonic saline infusions. Malignant hyperthermia, which is triggered by some inhaled anesthetics, causes muscle release of potassium (Chapter 603.2). Hyperkalemic periodic paralysis is an autosomal dominant disorder caused by a mutated sodium channel. It results in episodic cellular release of potassium and attacks of paralysis (Chapter 603.1).
A wide range of primary adrenal disorders, both hereditary and acquired, can cause decreased production of aldosterone, with secondary hyperkalemia (Chapters 569 and 570). Patients with these disorders typically have metabolic acidosis and salt wasting with hyponatremia. Children with more subtle adrenal insufficiency may have electrolyte problems only during acute illnesses. The most common form of congenital adrenal hyperplasia, 21-hydroxylase deficiency, typically manifests in male infants as hyperkalemia, metabolic acidosis, hyponatremia, and volume depletion. Females with this disorder usually are diagnosed as newborns because of their ambiguous genitalia; treatment prevents the development of electrolyte problems.
Renin, via angiotensin II, stimulates aldosterone production. A deficiency in renin, a result of kidney damage, can lead to decreased aldosterone production. Hyporeninemia occurs in many kidney diseases, with some of the more common pediatric causes listed in Table 52-4. These patients typically have hyperkalemia and a metabolic acidosis, without hyponatremia. Some of these patients have impaired renal function, partially accounting for the hyperkalemia, but the impairment in potassium excretion is more extreme than expected for the degree of renal insufficiency.
A variety of renal tubular disorders impair renal excretion of potassium. Children with pseudohypoaldosteronism type 1 have hyperkalemia, metabolic acidosis, and salt wasting leading to hyponatremia and volume depletion; aldosterone values are elevated. In the autosomal recessive variant, there is a defect in the renal sodium channel that is normally activated by aldosterone. Patients with this variant have severe symptoms, beginning in infancy. Patients with the autosomal dominant form have a defect in the aldosterone receptor, and the disease is milder, often remitting in adulthood. Pseudohypoaldosteronism type 2, also called Gordon syndrome, is an autosomal dominant disorder characterized by hypertension due to salt retention and impaired excretion of potassium and acid, leading to hyperkalemia and metabolic acidosis. Activating mutations in either WNK1 or WNK4, both serine-threonine kinases located in the distal nephron, cause Gordon syndrome. In Bartter syndrome due to mutations in the potassium channel ROMK (type 2 Bartter syndrome), there can be transient hyperkalemia in neonates, but hypokalemia subsequently develops (Chapter 525).
[/not-level-membership-for-pediatrics-category]
