[level-membership-for-neurosurgery-category]
CHAPTER 228 Dystonia in Children
Definition and Pathophysiology
Dystonia is an involuntary movement disorder characterized by sustained or intermittent muscle contractions that cause twisting and repetitive movements, abnormal postures, or both.1 Muscle agonists and antagonists are affected simultaneously, as documented by electromyography. Dystonia may occur with voluntary movement or at rest. Voluntary movement of one body part may result in dystonia not only in that part but also in overflow to other regions. Children with dystonia typically have widely varying muscle tone; at times they are described as being “loose as noodles” and at other times as “stiff as a board.” Dystonia is often misdiagnosed as spasticity or athetosis; the distinction is important because treatment and outcomes of the two movement disorders are so different.
Dystonia is attributed to abnormalities in the cortical–basal ganglia–cortical circuits and, particularly in children, is often secondary to structural lesions, especially lesions in the putamen. Such lesions result in decreased firing and more erratic firing of neurons in the internal globus pallidus (GPi) such that synchronization of reentrant circuits is altered and the supplementary motor cortex and premotor cortex are excessively stimulated.2 Output of the GPi is often minimal at rest but excessive with movement. Sensory fields are enlarged and scaling of movement is altered. Although dystonia in adults occasionally results from lesions in sites other than the basal ganglia, including the brainstem, spinal cord, and peripheral nerves, involvement of sites other than the basal ganglia is rare in children.
Classification, Grading, and Causes
Dystonia was initially described in 1911 by Oppenheim, who labeled the disorder dystonia musculorum deformans.3 The term was subsequently altered to idiopathic torsion dystonia. Pediatric dystonia may be classified by etiology or by site or sites of involvement.1,4 Primary dystonia develops without any evident cause. In children with primary dystonia, magnetic resonance imaging of the brain typically reveals no structural abnormalities, although recent 3-T scans have demonstrated subtle changes in the basal ganglia.5 Genetic testing may reveal abnormalities in the DYT gene; the DYT-1 abnormality is most common and involves a three-base GAG deletion in the gene encoding the torsin A protein.6 Primary dystonia is rare, with the incidence of early-onset dystonia estimated to range between 2 and 50 per million. Primary dystonia often begins in children 5 to 10 years old; it starts in one lower extremity as an involuntary foot deviation and progresses slowly to affect the entire leg, then the other leg, and ultimately to become generalized in more than half of patients while remaining segmental in the remainder. The earlier the onset of dystonia, the more likely that it will become generalized and severe. The age at onset of primary dystonia is younger than 15 years in 60% of patients.
The primary-plus dystonia category is sometimes used for children with dopa-responsive dystonia (DRD, Segawa’s disease) and myoclonus-dystonia syndrome.4 Their biopterin and homovanillic acid levels are often low and their dopa decarboxylation is probably disordered. Some have features of parkinsonism. DRD has a diurnal variation in 75% of patients, with increased severity in late afternoon. DRD may mimic cerebral palsy (CP), with patients exhibiting movement resembling a spastic gait (but without prematurity); the distinction is critical because DRD can virtually be cured by oral levodopa.7 Every child with dystonia who does not have a history consistent with CP (e.g., prematurity, intraventricular hemorrhage, perinatal hypoxia) should be given a trial of oral levodopa/carbidopa for 2 to 3 months to exclude the possibility of DRD.
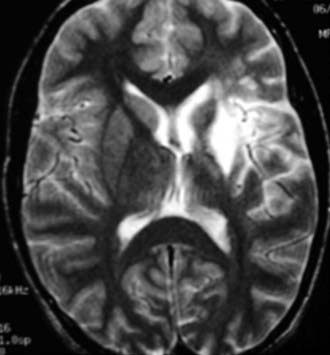
Secondary dystonia results from structural abnormalities in the brain and is the most common form of pediatric dystonia by far, in whom it accounts for 80% to 90% of cases.8–10 Most cases of secondary dystonia are associated with CP; traumatic brain injury is the second most common cause of secondary dystonia in children and stroke the third. However, any structural lesion affecting the basal ganglia, particularly cavernous malformations or tumors, may induce dystonia. The putamen is the most common site of structural abnormality (Fig. 228-1). Dystonic CP may begin in infancy or at any time during childhood, even in adolescence. It may affect the entire body or involve the upper and lower extremities while being associated with hypotonia of the neck and trunk. Dystonic CP often worsens slowly over time, although worsening is unusual after the teenage years. Posttraumatic dystonia is more likely to be focal or hemidystonic than generalized and may develop months to years after pediatric brain injury. Pediatric dystonia may also be caused by medications, including dopamine receptor blocking agents (neuroleptics), dopa mimetics (cocaine), and phenothiazines.
Primary dystonia can be graded with the Burke-Fahn-Marsden (BFM) scale, a validated scale developed to grade adults with primary dystonia.11 Secondary dystonia can be graded with the Barry-Albright Dystonia (BAD) scale, a validated scale developed to grade children with secondary dystonia.12 Neither scale is sensitive to the small changes in dystonia that may be of clinical significance in children.
Neurosurgical Treatment and Outcomes
Peripheral Rhizotomies
Dystonia in the extremities has been treated by both dorsal and ventral rhizotomies. In 1970, Kottke reported 6 children with dystonia and athetosis who improved after bilateral dorsal rhizotomies of C1-3.13 In 1973, Heimburger and coauthors reported improved dystonia, athetosis, and spasticity in 15 children treated by C1-3 dorsal rhizotomies.14 Fraoli and coworkers performed dorsal rhizotomies of C1-5 in 16 children with dystonia and athetosis and reported moderate improvement in 8, slight improvement in 5, worsening in 2, and death in 1.15 The reason why dorsal rhizotomies would improve dystonia is unclear.
Ventral rhizotomies have been reported in adults with torticollis and recently reported in children. Six children with severe generalized dystonia and spasticity in their extremities who were not candidates for intrathecal baclofen (ITB) were treated with combined dorsal and ventral rhizotomies; improvement was achieved in both dystonia and athetosis.16 In that report, approximately 85% of the ventral roots in either the cervical or lumbar regions were divided, with sustained improvement in dystonia and without adverse side effects. Ventral rhizotomies seem to be appropriate therapeutic options in children with either focal dystonia or dystonia that primarily affects the extremities but leaves the axial musculature relatively unaffected, as well as being appropriate in developing countries without access to ITB and deep brain stimulation (DBS).
Intrathecal Baclofen
The first report of ITB for dystonia was in 1991 by Narayan and colleagues, who used it to treat an 18-year-old with generalized dystonia.17 In 2001, we reported the use of ITB in 86 patients with severe, generalized dystonia; their mean age was 13 years and the dystonia was associated with CP in 71%.18 Responses to ITB were evaluated before pump implantation either by continuous ITB infusions via an external catheter (72%) or by bolus injections (17%); 91% to 93% of the patients responded to the screening doses with a 25% or greater decrease in their preoperative dystonia scores. Pumps were subsequently implanted into 77 patients, and their dystonia scores decreased significantly in comparison to their preoperative scores at a mean follow-up of 29 months. Subjectively, parents and patients reported improved ease of care and quality of life in 86%, improved speech in 33%, and improved extremity function in approximately a third. Side effects of ITB occurred in 26% and complications in 38%.
For children with severe, generalized secondary dystonia, ITB is probably the treatment of choice. Motta and coauthors reported the effects of ITB in 19 children (mean age, 8.9 years) who were at Gross Motor Function Classification System (GMFCS) level V and evaluated by the BFM and BAD scales 3, 6, and 12 months after ITB.19 Dystonia scores were decreased significantly on both scales. Woon and associates recently reported improvement after ITB in 8 dystonic children with generalized dystonia of various causes.20
Subsequent experience has confirmed the efficacy of ITB in more than 90% of children with secondary dystonia, and we no longer perform screening trials before implanting pumps. However, for children with severe, generalized heredodegenerative dystonia, the responses to ITB are more variable and we screen them with continuous infusion trials before pump implantation. Few children with primary dystonia have been treated with ITB. Although significant improvement has been reported in a few cases, their dystonia probably improves much more after DBS than after ITB.21
In our 2001 report, we noted that catheter tips positioned intrathecally at T6 or above were associated with less dystonia than were catheter tips located at T8 or below.18 Since then we have positioned catheter tips in the cervical region, often at C1-4, and—at therapeutic doses—have not observed any adverse effects involving alertness or respiration. In summary, ITB is probably the treatment of choice for children with generalized secondary dystonia.
Intraventricular infusion of baclofen (IVB) to treat dystonia in children was first reported in 2006, when it was used to treat two children with severe, generalized secondary dystonia.22 Since then, we have used it to treat an additional nine patients, eight of them children.23 The intraventricular catheters are inserted endoscopically into the third ventricle; the distal end is tunneled distally as a shunt would be and connected to infusion pumps in the usual locations. IVB doses may be somewhat lower than ITB doses, but insufficient numbers of patients have been treated to know that with any certainty. Patients treated with IVB have had no adverse side effects related to the infusion, only the same dose-related side effects seen with ITB. Two children who had not responded adequately to ITB were treated with IVB, and their responses were no better than those seen with ITB.
Children in status dystonicus (dystonic storms) have extremely severe dystonia that can be life-threatening, and it is somewhat analogous to status epilepticus.24 It can occur in children with either primary or secondary dystonia. They are treated initially with high doses of oral medications, such as dantrolene, 2 mg/kg per dose intravenously every 6 hours, combined with intravenous midazolam (up to 10 µg/kg per minute), haloperidol (1 to 2 mg every 6 hours), or both, frequently with assisted ventilation. If the storming persists despite intravenous medications, IVB is a reasonable therapeutic option, either via an external microinfusion pump or via an implanted pump. Children in whom storming continues after IVB can be treated with DBS.
Stereotactic Procedures
Pallidotomies
Pallidotomies have been reported to improve primary dystonia, but destructive procedures—which cannot be adjusted (other than enlarging the lesion) and which preclude future innovative therapies—are not ideal in children and are rarely used if DBS is available.25,26 In addition, bilateral pallidotomies have appreciable risks, including dysarthria and dysphagia, and are infrequently indicated for children.
Deep Brain Stimulation
In 1999, Coubes and coauthors published the initial report of GPi-DBS to treat dystonia in a child, an 8-year-old girl with severe generalized dystonia requiring sedation and mechanical ventilation.27 At present, DBS is the surgical treatment of choice for children with primary dystonia. It has been used in children as young as 4 years (although rarely in children younger than 11 years) and has been approved by the Food and Drug Administration for use in children as young as 7 years if they have failed medical therapy. The DBS electrode is implanted into the GPi, either under anesthesia without microelectrode recordings or, if microelectrode recordings are used, under sedation with dexmedetomidine. The technical details of GPi-DBS for dystonia have recently been reported.28 The electrode tip is positioned in the posterior-lateral GPi, about 2 mm above the optic radiation. Postoperatively, monopolar DBS is begun at parameters of 1 V, 60 to 120 Hz, and a pulse width of 120 to 450, with the parameters adjusted thereafter according to clinical response. In our experience, stimulation of two cathodes may give a better response than stimulation of one. Some children with primary dystonia respond within a few days, whereas others require several weeks.
The GPi is the anatomic target of choice for DBS in children with primary dystonia. Vercueil and colleagues observed better outcomes with stimulation of GPi than stimulation of the ventrolateral thalamic nucleus in patients with both primary and secondary dystonia.29 Coubes and coworkers reported 80% or greater reductions in dystonia scores in children with primary dystonia treated by DBS, with no significant difference in outcomes for children who are DYT-1 positive or negative.30,31 Zorzi and associates reported using GPi-DBS to treat 12 patients with childhood-onset dystonia, 9 with primary and 3 with secondary dystonia.32 Eleven of the patients improved, with a mean improvement in the BFM score of 45% ± 28%. In Alterman and Tagliati’s series of 14 patients with primary dystonia that began in childhood, the mean age at the onset of symptoms was 8 years, and the median age at surgery was 14 years.28 All patients responded; their BFM scores at 3, 6, and 12 months postoperatively improved by 31%, 70%, and 78%, respectively.
Alterman and Tagliati also treated 5 children with secondary dystonia with GPi-DBS and found less improvement—BFM scores 33% lower at 1 year postoperatively—than seen in patients with primary dystonia. We and others have observed similar findings in children with secondary dystonia. Zhang and colleagues used DBS, primarily of the subthalamic nucleus, to treat 9 patients with secondary dystonia; 3 patients with tardive or posttraumatic dystonia improved more than those with perinatal anoxia and diffuse impairment of the globus pallidus.33 In the series of 15 patients reported by Eltahawy and associates, 9 with primary and 6 with secondary dystonia, patients with primary dystonia improved in both movement and function.34 Patients with secondary dystonia had either mild or no improvement in their dystonia and no improvement in function. The authors concluded that the presence of structural changes in the bilateral basal ganglia was associated with poorer DBS outcomes.
Complications of DBS in children may occur less frequently than in adults, particularly the risk for intracerebral hemorrhage. Alterman and Tagliati reported infections requiring hardware removal in 4 of 19 patients and extension lead fractures in an additional 2 patients.28
Epidural Motor Cortex Stimulation
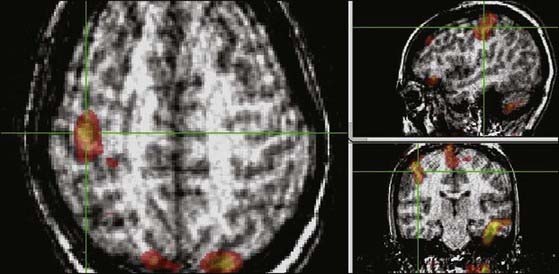
Focal dystonia in adults has been treated by epidural motor cortex stimulation (EMCS), with improvement in focal dystonia after focal ischemic events.35 EMCS may act by interrupting the abnormal cortical–basal ganglia–cortical circuits at a cortical level. In our experience, EMCS can improve focal dystonia in children, but the number of patients treated is insufficient to make any reliable prediction of outcome. The location for EMCS electrode placement can be guided by preoperative functional magnetic resonance imaging to localize the site of excessive cortical activity (Fig. 228-2).
Albright AL. Intraventricular baclofen infusion for dystonia. Report of two cases. J Neurosurg. 2006;105:101-104.
Albright AL, Barry MJ, Shafron DH, et al. Intrathecal baclofen for generalized dystonia. Dev Med Child Neurol. 2001;43:652-657.
Albright AL, Ferson SS. Intraventricular baclofen for dystonia; techniques and outcomes. J Neurosurg Pediatr. 2009;3:11-14.
Albright AL, Tyler-Kabara E. Combined dorsal and ventral rhizotomies for extremity dystonia. J Neurosurg. 2007;107:324-327.
Alterman RL, Tagliati M. Deep brain stimulation for torsion dystonia in children. Childs Nerv Syst. 2007;23:1033-1040.
Barry M, VanSwearingen J, Albright AL. Reliability and responsiveness of the Barry-Albright Dystonia Scale. Dev Med Child Neurol. 1999;41:404-411.
Bressman S. Dystonia. Curr Opin Neurol. 1998;11:363-372.
Burke RE, Fahn S, Gold A. Delayed-onset dystonia in patients with “static” encephalopathy. J Neurol Neurosurg Psychiatry. 1980;43:789-797.
Burke RE, Fahn S, Marsden D, et al. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1995;35:73-77.
Carbon M, Kingsley PB, Tang C, et al. Microstructural changes in primary torsion dystonia. Mov Disord. 2007;23:234-239.
Chutorian AM. Childhood dystonias. Acta Neuropediatr. 1996;2:33-51.
Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long-term results. J Neurosurg. 2004;101:189-194.
Coubes P, Echenne B, Roubertie A, et al. Treatment of early-onset dystonia by chronic bilateral stimulation of the internal globus pallidus. Apropos of a case. Neurochirurgie. 1999;45:139-144.
Coubes P, Roubertie A, Vayssiere N, et al. Treatment of DYT-generalized dystonia by stimulation of the internal globus pallidus. Lancet. 2000;355:2220-2221.
Eltahawy HA, Saint-Cyr J, Giladi N, et al. Primary dystonia is more responsive than secondary dystonia to pallidal interventions: outcome after pallidotomy or pallidal deep brain stimulation. Neurosurgery. 2004;54:613-621.
Fahn S. Concept and classification of dystonia. Adv Neurol. 1988;50:1-8.
Fraoli B, Nucci F, Baldassarre L. Bilateral cervical posterior rhizotomy: effects on dystonia and athetosis, or respiration and other autonomic functions. Appl Neurophysiol. 1977/78;40:26-40.
Hartmann A, Pogarell O, Oertel WH. Secondary dystonias. J Neurol. 1998;245:511-518.
Heimburger RF, Slominski A, Griswold P. Cervical posterior rhizotomies for reducing spasticity in cerebral palsy. J Neurosurg. 1973;39:30-34.
Kottke FJ. Modification of athetosis by denervation of the tonic neck reflexes. Dev Med Child Neurol. 1970;12:236-237.
Lozano AM, Kumar R, Gross RE, et al. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord. 1997;12:865-870.
Mariotti P, Fasano A, Contarino MF, et al. Management of status dystonicus: our experience and review of the literature. Mov Disord. 2007;22:963-968.
Motta F, Stignani C, Antonello CE. Effect of intrathecal baclofen on dystonia in children with cerebral palsy and the use of functional scales. J Pediatr Orthop. 2008;28:213-217.
Narayan RK, Loubser PG, Jankovic J, et al. Intrathecal baclofen for intractable axial dystonia. Neurology. 1991;41:1141-1142.
Nygaard TG, Marsden CD, Fahn S. Dopa-responsive dystonia: long-term treatment response and prognosis. Neurology. 1991;41:174-181.
Oppenheim H. Uber eine eigenartige Krampfkrankheit des Kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia musculorum deformans). Neurol Centralbl. 1911;30:1090-1107.
Priori A, Lefaucheur JP. Chronic epidural motor cortex stimulation for movement disorders. Lancet Neurol. 2007;6:279-286.
Saint Hilaire M-H, Burke RE, Bressman SB, et al. Delayed-onset dystonia due to perinatal or early childhood asphyxia. Neurology. 1991;41:216-222.
Vercueil L, Pollak P, Fraix V, et al. Deep brain stimulation in the treatment of severe dystonia. J Neurol. 2001;248:695-700.
Vitek JL. Pathophysiology of dystonia: a neuronal model. Mov Disord. 2002;17:S49-S62.
Walker RH, Danishi FO, Swope DM, et al. Intrathecal baclofen for dystonia: benefits and complications during six years of experience. Mov Disord. 2001;15:1242-1247.
Woon K, Tsegaye M, Vloeberghs MH. The role of intrathecal baclofen in the management of primary and secondary dystonia in children. Br J Neurosurg. 2007;21:355-358.
Yoshor D, Hamilton WJ, Ondo W, et al. Comparison of thalamotomy and pallidotomy for the treatment of dystonia. Neurosurgery. 2001;48:818-826.
Zhang J-G, Zhang K, Want Z-C, et al. Deep brain stimulation in the treatment of secondary dystonia. Chin Med J. 2006;119:1069-1074.
Zorzi G, Marras C, Nardocci N, et al. Stimulation of the globus pallidus internus for childhood-onset dystonia. Mov Disord. 2005;20:1194-1200.
1 Fahn S. Concept and classification of dystonia. Adv Neurol. 1988;50:1-8.
2 Vitek JL. Pathophysiology of dystonia: a neuronal model. Mov Disord. 2002;17:S49-S62.
3 Oppenheim H. Uber eine eigenartige Krampfkrankheit des Kindlichen und jugendlichen Alters (Dysbasia lordotica progressiva, Dystonia musculorum deformans). Neurol Centralbl. 1911;30:1090-1107.
4 Chutorian AM. Childhood dystonias. Acta Neuropediatr. 1996;2:33-51.
5 Carbon M, Kingsley PB, Tang C, et al. Microstructural changes in primary torsion dystonia. Mov Disord. 2007;23:234-239.
6 Bressman S. Dystonia. Curr Opin Neurol. 1998;11:363-372.
7 Nygaard TG, Marsden CD, Fahn S. Dopa-responsive dystonia: long-term treatment response and prognosis. Neurology. 1991;41:174-181.
8 Burke RE, Fahn S, Gold A. Delayed-onset dystonia in patients with “static” encephalopathy. J Neurol Neurosurg Psychiatry. 1980;43:789-797.
9 Hartmann A, Pogarell O, Oertel WH. Secondary dystonias. J Neurol. 1998;245:511-518.
10 Saint Hilaire M-H, Burke RE, Bressman SB, et al. Delayed-onset dystonia due to perinatal or early childhood asphyxia. Neurology. 1991;41:216-222.
11 Burke RE, Fahn S, Marsden D, et al. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1995;35:73-77.
12 Barry M, VanSwearingen J, Albright AL. Reliability and responsiveness of the Barry-Albright Dystonia Scale. Dev Med Child Neurol. 1999;41:404-411.
13 Kottke FJ. Modification of athetosis by denervation of the tonic neck reflexes. Dev Med Child Neurol. 1970;12:236-237.
14 Heimburger RF, Slominski A, Griswold P. Cervical posterior rhizotomies for reducing spasticity in cerebral palsy. J Neurosurg. 1973;39:30-34.
15 Fraoli B, Nucci F, Baldassarre L. Bilateral cervical posterior rhizotomy: effects on dystonia and athetosis, or respiration and other autonomic functions. Appl Neurophysiol. 1977/78;40:26-40.
16 Albright AL, Tyler-Kabara E. Combined dorsal and ventral rhizotomies for extremity dystonia. J Neurosurg. 2007;107:324-327.
17 Narayan RK, Loubser PG, Jankovic J, et al. Intrathecal baclofen for intractable axial dystonia. Neurology. 1991;41:1141-1142.
18 Albright AL, Barry MJ, Shafron DH, et al. Intrathecal baclofen for generalized dystonia. Dev Med Child Neurol. 2001;43:652-657.
19 Motta F, Stignani C, Antonello CE. Effect of intrathecal baclofen on dystonia in children with cerebral palsy and the use of functional scales. J Pediatr Orthop. 2008;28:213-217.
20 Woon K, Tsegaye M, Vloeberghs MH. The role of intrathecal baclofen in the management of primary and secondary dystonia in children. Br J Neurosurg. 2007;21:355-358.
21 Walker RH, Danishi FO, Swope DM, et al. Intrathecal baclofen for dystonia: benefits and complications during six years of experience. Mov Disord. 2001;15:1242-1247.
22 Albright AL. Intraventricular baclofen infusion for dystonia. Report of two cases. J Neurosurg. 2006;105:101-104.
23 Albright AL, Ferson SS. Intraventricular baclofen for dystonia; techniques and outcomes. J Neurosurg Pediatr. 2009;2:11-14.
24 Mariotti P, Fasano A, Contarino MF, et al. Management of status dystonicus: our experience and review of the literature. Mov Disord. 2007;22:963-968.
25 Lozano AM, Kumar R, Gross RE, et al. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord. 1997;12:865-870.
26 Yoshor D, Hamilton WJ, Ondo W, et al. Comparison of thalamotomy and pallidotomy for the treatment of dystonia. Neurosurgery. 2001;48:818-826.
27 Coubes P, Echenne B, Roubertie A, et al. Treatment of early-onset dystonia by chronic bilateral stimulation of the internal globus pallidus. Apropos of a case. Neurochirurgie. 1999;45:139-144.
28 Alterman RL, Tagliati M. Deep brain stimulation for torsion dystonia in children. Childs Nerv Syst. 2007;23:1033-1040.
29 Vercueil L, Pollak P, Fraix V, et al. Deep brain stimulation in the treatment of severe dystonia. J Neurol. 2001;248:695-700.
30 Coubes P, Roubertie A, Vayssiere N, et al. Treatment of DYT-generalized dystonia by stimulation of the internal globus pallidus. Lancet. 2000;355:2220-2221.
31 Coubes P, Cif L, El Fertit H, et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: long-term results. J Neurosurg. 2004;101:189-194.
32 Zorzi G, Marras C, Nardocci N, et al. Stimulation of the globus pallidus internus for childhood-onset dystonia. Mov Disord. 2005;20:1194-1200.
33 Zhang J-G, Zhang K, Want Z-C, et al. Deep brain stimulation in the treatment of secondary dystonia. Chin Med J. 2006;119:1069-1074.
34 Eltahawy HA, Saint-Cyr J, Giladi N, et al. Primary dystonia is more responsive than secondary dystonia to pallidal interventions: outcome after pallidotomy or pallidal deep brain stimulation. Neurosurgery. 2004;54:613-621.
35 Priori A, Lefaucheur JP. Chronic epidural motor cortex stimulation for movement disorders. Lancet Neurol. 2007;6:279-286.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 228 Dystonia in Children
Definition and Pathophysiology
Dystonia is an involuntary movement disorder characterized by sustained or intermittent muscle contractions that cause twisting and repetitive movements, abnormal postures, or both.1 Muscle agonists and antagonists are affected simultaneously, as documented by electromyography. Dystonia may occur with voluntary movement or at rest. Voluntary movement of one body part may result in dystonia not only in that part but also in overflow to other regions. Children with dystonia typically have widely varying muscle tone; at times they are described as being “loose as noodles” and at other times as “stiff as a board.” Dystonia is often misdiagnosed as spasticity or athetosis; the distinction is important because treatment and outcomes of the two movement disorders are so different.
Dystonia is attributed to abnormalities in the cortical–basal ganglia–cortical circuits and, particularly in children, is often secondary to structural lesions, especially lesions in the putamen. Such lesions result in decreased firing and more erratic firing of neurons in the internal globus pallidus (GPi) such that synchronization of reentrant circuits is altered and the supplementary motor cortex and premotor cortex are excessively stimulated.2 Output of the GPi is often minimal at rest but excessive with movement. Sensory fields are enlarged and scaling of movement is altered. Although dystonia in adults occasionally results from lesions in sites other than the basal ganglia, including the brainstem, spinal cord, and peripheral nerves, involvement of sites other than the basal ganglia is rare in children.
Classification, Grading, and Causes
Dystonia was initially described in 1911 by Oppenheim, who labeled the disorder dystonia musculorum deformans.3 The term was subsequently altered to idiopathic torsion dystonia. Pediatric dystonia may be classified by etiology or by site or sites of involvement.1,4 Primary dystonia develops without any evident cause. In children with primary dystonia, magnetic resonance imaging of the brain typically reveals no structural abnormalities, although recent 3-T scans have demonstrated subtle changes in the basal ganglia.5 Genetic testing may reveal abnormalities in the DYT gene; the DYT-1 abnormality is most common and involves a three-base GAG deletion in the gene encoding the torsin A protein.6 Primary dystonia is rare, with the incidence of early-onset dystonia estimated to range between 2 and 50 per million. Primary dystonia often begins in children 5 to 10 years old; it starts in one lower extremity as an involuntary foot deviation and progresses slowly to affect the entire leg, then the other leg, and ultimately to become generalized in more than half of patients while remaining segmental in the remainder. The earlier the onset of dystonia, the more likely that it will become generalized and severe. The age at onset of primary dystonia is younger than 15 years in 60% of patients.
The primary-plus dystonia category is sometimes used for children with dopa-responsive dystonia (DRD, Segawa’s disease) and myoclonus-dystonia syndrome.4 Their biopterin and homovanillic acid levels are often low and their dopa decarboxylation is probably disordered. Some have features of parkinsonism. DRD has a diurnal variation in 75% of patients, with increased severity in late afternoon. DRD may mimic cerebral palsy (CP), with patients exhibiting movement resembling a spastic gait (but without prematurity); the distinction is critical because DRD can virtually be cured by oral levodopa.7 Every child with dystonia who does not have a history consistent with CP (e.g., prematurity, intraventricular hemorrhage, perinatal hypoxia) should be given a trial of oral levodopa/carbidopa for 2 to 3 months to exclude the possibility of DRD.
Secondary dystonia results from structural abnormalities in the brain and is the most common form of pediatric dystonia by far, in whom it accounts for 80% to 90% of cases.8–10 Most cases of secondary dystonia are associated with CP; traumatic brain injury is the second most common cause of secondary dystonia in children and stroke the third. However, any structural lesion affecting the basal ganglia, particularly cavernous malformations or tumors, may induce dystonia. The putamen is the most common site of structural abnormality (Fig. 228-1). Dystonic CP may begin in infancy or at any time during childhood, even in adolescence. It may affect the entire body or involve the upper and lower extremities while being associated with hypotonia of the neck and trunk. Dystonic CP often worsens slowly over time, although worsening is unusual after the teenage years. Posttraumatic dystonia is more likely to be focal or hemidystonic than generalized and may develop months to years after pediatric brain injury. Pediatric dystonia may also be caused by medications, including dopamine receptor blocking agents (neuroleptics), dopa mimetics (cocaine), and phenothiazines.
Primary dystonia can be graded with the Burke-Fahn-Marsden (BFM) scale, a validated scale developed to grade adults with primary dystonia.11 Secondary dystonia can be graded with the Barry-Albright Dystonia (BAD) scale, a validated scale developed to grade children with secondary dystonia.12 Neither scale is sensitive to the small changes in dystonia that may be of clinical significance in children.
Neurosurgical Treatment and Outcomes
Peripheral Rhizotomies
Dystonia in the extremities has been treated by both dorsal and ventral rhizotomies. In 1970, Kottke reported 6 children with dystonia and athetosis who improved after bilateral dorsal rhizotomies of C1-3.13 In 1973, Heimburger and coauthors reported improved dystonia, athetosis, and spasticity in 15 children treated by C1-3 dorsal rhizotomies.14 Fraoli and coworkers performed dorsal rhizotomies of C1-5 in 16 children with dystonia and athetosis and reported moderate improvement in 8, slight improvement in 5, worsening in 2, and death in 1.15 The reason why dorsal rhizotomies would improve dystonia is unclear.
Ventral rhizotomies have been reported in adults with torticollis and recently reported in children. Six children with severe generalized dystonia and spasticity in their extremities who were not candidates for intrathecal baclofen (ITB) were treated with combined dorsal and ventral rhizotomies; improvement was achieved in both dystonia and athetosis.16 In that report, approximately 85% of the ventral roots in either the cervical or lumbar regions were divided, with sustained improvement in dystonia and without adverse side effects. Ventral rhizotomies seem to be appropriate therapeutic options in children with either focal dystonia or dystonia that primarily affects the extremities but leaves the axial musculature relatively unaffected, as well as being appropriate in developing countries without access to ITB and deep brain stimulation (DBS).
Intrathecal Baclofen
The first report of ITB for dystonia was in 1991 by Narayan and colleagues, who used it to treat an 18-year-old with generalized dystonia.17 In 2001, we reported the use of ITB in 86 patients with severe, generalized dystonia; their mean age was 13 years and the dystonia was associated with CP in 71%.18 Responses to ITB were evaluated before pump implantation either by continuous ITB infusions via an external catheter (72%) or by bolus injections (17%); 91% to 93% of the patients responded to the screening doses with a 25% or greater decrease in their preoperative dystonia scores. Pumps were subsequently implanted into 77 patients, and their dystonia scores decreased significantly in comparison to their preoperative scores at a mean follow-up of 29 months. Subjectively, parents and patients reported improved ease of care and quality of life in 86%, improved speech in 33%, and improved extremity function in approximately a third. Side effects of ITB occurred in 26% and complications in 38%.
For children with severe, generalized secondary
[/not-level-membership-for-neurosurgery-category]
