Drugs Used in Renal Disease
DRUG CONSIDERATIONS IN PATIENTS WITH RENAL DYSFUNCTION
Influence of Drugs on Renal Function
All anaesthetic agents may cause a generalized depression of renal function which is transient and clinically insignificant. However, nephrotoxic drugs can impair renal function permanently. For example, they may lead to severe sodium and water depletion, reduction in renal blood supply, direct renal damage or renal obstruction (Table 10.1). Some drugs impair renal function by more than one mechanism.
TABLE 10.1
Mechanisms of Drug-Induced Renal Damage
Sodium and water depletion
Reduced renal perfusion
Direct renal toxicity
Urinary obstruction
VASOACTIVE DRUGS USED IN RENAL DYSFUNCTION





Adenosine
Adenosine also slows the heart rate and impairs atrioventricular conduction and is used in the treatment of supraventricular tachycardias (Ch 47).
Calcium Channel Blockers
Calcium channel blockers (CCBs) (Ch 8) are predominantly used in the treatment of hypertension. They act selectively on calcium channels in the cellular membrane of cardiac and vascular smooth muscle (VSM) cells. Free calcium within the VSM enhances vascular tone and contributes to vasoconstriction. CCBs reduce the transmembrane calcium influx in VSM cells, causing vasodilatation. In addition, CCBs have a direct diuretic effect that contributes to their long-term antihypertensive action. Nifedipine increases the urine volume and sodium excretion, and may inhibit aldosterone release. This diuretic action is independent of any change in renal blood flow or GFR.
Noradrenaline/Adrenaline/Phenylephrine
Although noradrenaline and adrenaline also have β-receptor effects, all three of these drugs are very potent vasoconstrictors acting on the vascular α-receptors (Ch 8). Their use is often accompanied by fear of inducing decreases in renal blood flow, GFR and renal function.
DIURETICS
Diuretics are classified according to their mechanism and site of action on the nephron (see Fig. 10.1):
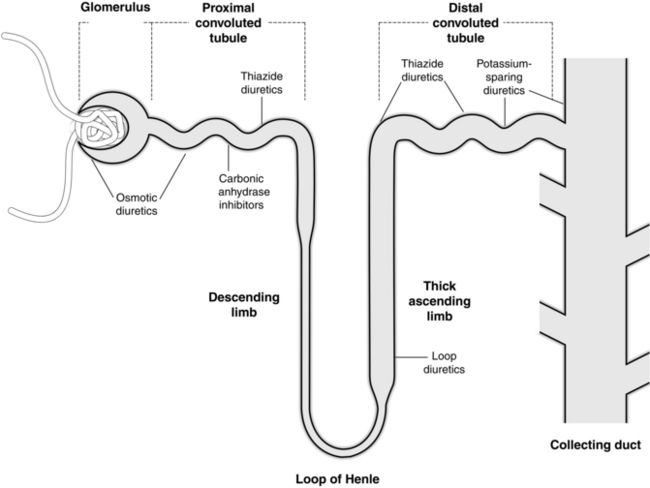
 glomerulus and proximal renal tubule – e.g. osmotic diuretics, carbonic anhydrase inhibitors
glomerulus and proximal renal tubule – e.g. osmotic diuretics, carbonic anhydrase inhibitors
 ascending limb of the loop of Henle – e.g. loop diuretics
ascending limb of the loop of Henle – e.g. loop diuretics


Osmotic Diuretics
DRUGS AND RENAL TRANSPLANTATION
Immunosuppression
Mycophenolate Mofetil
There are a variety of substances with potent immunosuppressant properties which are usually used in combination with each other. Despite their powerful immunosuppressant effects, it is unlikely that any of the new or existing drugs may be used as monotherapy. The calcineurin inhibitors (CNIs), ciclosporin and tacrolimus, have revolutionized the overall success of renal transplantation through reduction in early immunological injury and acute rejection rates. However, the CNIs have a significant adverse effect on renal function and cardiovascular disease, and extended long-term graft survival has not been achieved. CNI minimization using mycophenolate mofetil or sirolimus may be associated with a modest increase in creatinine clearance and a decrease in serum creatinine in the short term. A typical perioperative regimen for renal transplantation is shown in Table 10.2.
TABLE 10.2
A Typical Perioperative Regimen for Renal Transplant
| Preoperative | Induction immunosuppression (ciclosporin-Neoral, FK506/tacrolimus, rapamycin, or sirolimus and mycophenolate mofetil (MMF) Heparin 5000 units subcutaneously Ranitidine 150 mg orally (stress ulcer prophylaxis) Nifedipine 20 mg orally (vasodilator, free radical scavenger) |
| At induction | Antibiotic prophylaxis – co-amoxiclav (augmentin) 1.2 g intravenously methylprednisolone 0.5 g i.v. |
| During vascular anastomosis | Mannitol 0.5 g kg–1 intravenously ± dopamine 3–5 μg kg–1 min–1 |
| Postoperative | Antibiotic prophylaxis Heparin 5000 units subcutaneously, twice daily Aspirin 150 mg orally Ranitidine 150 mg orally, twice daily Co-trimoxazole 480 mg orally, daily (prophylaxis against Pneumocystis carinii) Immunosuppression (‘triple therapy’) |
Fischereder, M., Kretzler, M. New immunosuppressive strategies in renal transplant recipients. J. Nephrol. 2004;17:9–18.
Flechner, S.M., Kobashigawa, J., Klintmalm, G. Calcineurin inhibitor-sparing regimens in solid organ transplantation: focus on improving renal function and nephrotoxicity. Clin. Transplant. 2008;22(1):1–15.
Garwood, S. Cardiac surgery-associated acute renal injury: new paradigms and innovative therapies. J. Cardiothorac. Vasc. Anesth. 2010;24(6):990–1001.
Gordon, A.C., Russell, J.A., Whalley, K.R., et al. The effects of vasopressin on acute kidney injury in septic shock. Intensive Care Med. 2010;36(1):83–91.
Gottlieb, S.S. Adenosine A1 antagonists and the cardiorenal syndrome. Curr. Heart Fail. Rep. 2008;5(2):105–109.
Guyton, A.C., Hall, J.E. Diuretics, kidney diseases (Ch 31). In: Guyton A.C., Hall J.E., eds. Textbook of Medical Physiology. eleventh ed. Philadelphia: WB Saunders; 2011:397–409.
Huang, D.T., Clermont, G., Dremsizov, T.T., et al. Implementation of early goal-directed therapy for severe sepsis and septic shock: a decision analysis. Crit. Care Med. 2007;35(9):2090–2100.
Lindvall, G., Sartipy, U., Ivert, T., et al. Aprotinin is not associated with postoperative renal impairment after primary coronary surgery. Ann. Thorac. Surg. 2008;86:13–19.
Schrier, R.W., Wang, W. Acute renal failure and sepsis. N. Engl. J. Med. 2004;351:59–69.