47 Drugs in pregnancy and lactation
Drugs in pregnancy
Drugs in pregnancy
There is now a greater appreciation of the risks of drug use in pregnancy, and it is generally accepted that maternal pharmacotherapy should be avoided or minimised where possible. Nevertheless, it has been estimated that over 80% of expectant mothers take three or four drugs at some stage of pregnancy (Headley et al., 2004) with a significant number of women taking medication at the time their pregnancy is detected. Indications for drug use range from chronic illnesses such as epilepsy and depression to those commonly associated with pregnancy such as hypertension, urinary tract infections and gastro-intestinal complaints.
Drugs as teratogens
A teratogen is defined as any agent that results in structural or functional abnormalities in the fetus, or in the child after birth, as a consequence of maternal exposure during pregnancy. Examples of drugs that are known to be human teratogens are shown in Box 47.1. The teratogenic mechanism for most drugs remains unclear, but may be due to the direct effects of the drug on the fetus and/or as a consequence of indirect physiological changes in the mother or fetus. Perhaps the best known, and most widely studied teratogen is thalodimide, a mild sedative that was widely marketed as a remedy for pregnancy-related nausea and vomiting. In 1961, thalidomide was withdrawn from the UK market following numerous reports of severe anatomical birth defects in infants of mothers who took the drug in early pregnancy. Whereas external congenital anomalies such as limb abnormalities, spina bifida and hydrocephalus may be obvious at birth, some defects may take many years to manifest clinically or be identified. Examples of delayed effects of teratogens are the behavioural and intellectual disorders associated with in utero alcohol exposure and the development of clear-cell vaginal cancer in young women following maternal intake of diethylstilboestrol, used first in the 1930s for the prevention of miscarriage and preterm delivery (Herbst et al., 1971).
Critical periods in human fetal development
Pre-embryonic stage (weeks 0–2 post-conception)
The first two weeks post-conception are regarded as the pre-embryonic stage and describe the period up to implantation of the fertilised ovum. Teratogenic exposure during the pre-embryonic stage is thought to elicit an ‘all-or-nothing’ response, leading either to death of the embryo or complete recovery and normal development of the fetus. Fetal malformations following drug exposure during this period are therefore thought to be unlikely, except where the half-life of the drug is sufficient to extend exposure into the embryonic stage. A good example of the latter is isotretinoin and related vitamin A derivatives which have half-lives up to a week, and which when used systemically, for example, for the treatment of acne and psoriasis, are recognised teratogens (Nulman et al., 1998).
Fetal stage (weeks 9–38 post-conception)
During the fetal stage, the fetus continues to develop, grow and mature and, importantly, remains susceptible to some drug effects. This is especially true for the central nervous system, which can be damaged by exposure to certain drugs, for example, ethanol, at any stage of pregnancy. The external genitalia also continue to form from the seventh week until term, and consequently, danazol, which has weak androgenic properties, can cause virilisation of a female fetus if given in any trimester after the eighth week of pregnancy when the androgen receptors begin to form (Rosa, 1984).
Further examples include the angiotensin-converting enzyme (ACE) inhibitors, which if given in the second and third trimesters can result in fetal renal dysfunction and subsequent oligohydramnios, that is, reduced amniotic fluid volume (Sedman et al., 1995). The non-steroidal anti-inflammatory drugs (NSAIDs) are another important group of drugs that may cause problems specifically in the third trimester. These drugs inhibit prostaglandin synthesis in a dose-related fashion and, when given late in pregnancy, may result in premature closure of the fetal ductus arteriosus and fetal renal impairment (Koren et al., 2006). NSAIDs should therefore be avoided during the third trimester.
Principles of teratogenesis
Timing of exposure
The stage of pregnancy at which a drug exposure occurs is key to determining the likelihood, severity or nature of any adverse effect on the fetus. Risk both between and within trimesters may be variable. For example, folic acid antagonists, for example, trimethoprim, are associated with an increased risk of neural tube defects if exposure occurs before neural tube closure (third to fourth week post-conception), but not after this period (Hernandez-Diaz et al., 2001). It has also been suggested that trimethoprim should be avoided after 32 weeks’ gestation in view of the theoretical risk of severe jaundice in the neonate as a result of bilirubin displacement from protein binding, although clinical evidence to support this is lacking (Dunn, 1964). Unfortunately, the precise period of teratogenic risk is known for very few substances. One drug for which this period has been established is thalidomide, where exposure between days 20 and 36 post-conception is associated with a high risk of congenital malformation (Lenz, 1988; Newman, 1986).
Species
Teratogenicity of a drug may be species dependent. Interestingly, preclinical thalidomide studies in mice and rats did not result in congenital malformation in the offspring (Breitkreutz and Anderson, 2008; Miller et al., 2009; Vorhees et al., 2001). Birth defects or other adverse reproductive outcomes observed in animal studies cannot therefore be simply extrapolated to the human situation. Further, the drug dose and route of administration used in early animal studies may not be comparable to clinical use in humans.
Genotype and environmental interaction
Not all fetuses exposed to known teratogenic drugs show evidence of having been affected in utero. It remains undetermined as to whether this variable susceptibility to teratogenic drugs is a result of genetic differences in the exposed mothers, the fetal genotype, modifying environmental factors or a combination of all three. Malformations are reported to occur in only 20–50% of infants born to mothers exposed to thalidomide during the period of greatest risk for embryopathy, that is, days 20–36 post-fertilisation (Lenz, 1966; Newman, 1985). Similarly, maternal treatment with systemic isotretinoin during the first trimester results in fetal malformation in only 18–35% of the live born infants, with a further 30% of children exhibiting developmental delay in the absence of physical deformity (Benke, 1984; Braun et al., 1984; Hill, 1984).
Pharmacological effect
The neonate can also be adversely affected by maternal drug therapy (see Table 47.1). It is generally only at birth that signs of fetal distress are observed due to in utero drug exposure or the effects of abrupt discontinuation of the maternal drug supply. The capacity of the neonate to eliminate drugs is reduced, and this can result in significant accumulation of some drugs, leading to toxicity. Neonatal withdrawal effects may require treatment.
Table 47.1 Examples of drugs with pharmacological effects on the fetus or neonate
| Drug | Possible adverse pharmacological effect | Notes |
|---|---|---|
| ACE inhibitors | Fetal and neonatal hypoxia, hypotension, renal dysfunction, oligohydramnios and intra-uterine growth retardation | Monitor fetus if long-term therapy in the second or third trimester |
| β-Blockers, for example, atenolol | Neonatal bradycardia, hypotension and hyperglycaemia | Neonatal symptoms are usually mild and improve within 48 h. No long-term effects |
| Benzodiazepines | ‘Floppy infant syndrome’ | Risk if regular use in third trimester |
| Withdrawal reactions | Neonatal observation recommended | |
| Corticosteroid (high dose) | Fetal adrenal suppression | Dependent on dose and treatment interval |
| NSAID | Premature closure of the ductus arteriosus (affecting fetal circulation) and fetal renal impairment (decreased urine output) | Avoid repeated use after week 28. If unavoidable, fetal circulation monitored regularly |
| Opioids | Neonatal withdrawal symptoms | Risk if used long-term |
| Respiratory depression | Risk if used near term | |
| Phenothiazines | Neonatal withdrawal and transient extrapyramidal symptoms | Observation for at least 48 h. Symptoms may last for several weeks |
| Tricyclic and SSRI antidepressants | Neonatal withdrawal symptoms | Risk if used long-term and/or near term. Observation for at least 48 h |
(adapted from Schaefer et al., 2007)
Maternal pharmacokinetic changes
There are a number of maternal changes which occur during pregnancy and are summarised in Table 47.2.
Table 47.2 Summary of pharmacokinetic changes during pregnancy (adapted from Schaefer et al., 2007)
| Absorption | Change during pregnancy |
|---|---|
| Gastro-intestinal motility | ↓ |
| Lung function | ↑ |
| Skin blood circulation | ↑ |
| Distribution | |
| Plasma volume | ↑ |
| Body water | ↑ |
| Plasma protein | ↓ |
| Fat deposition | ↑ |
| Metabolism | |
| Liver activity | ↑ ↓ |
| Excretion | |
| Glomerular filtration | ↑ |
Absorption
Gastric and intestinal emptying time increases by 30–40% in the second and third trimesters (Pavek et al., 2009) and could be important in delaying absorption and time to onset of action for some drugs (Loebstein et al., 1997). There is also a reduction in gastric acid secretion in the first and second trimesters and an increase in mucus secretion. As a consequence of the increase in gastric pH, the ionisation, and hence absorption, of weak acids and bases can be affected.
Distribution
The volume of distribution of drugs may be altered because of an increase of up to 50% in blood (plasma) volume and a 30% increase in cardiac output. Renal blood flow increases by up to 50% at the end of the first trimester and uterine blood flow increases and peaks at term (36–42 L/h). There is also a mean increase of 8 L in body water (60% to placenta, fetus and amniotic fluid and 40% to maternal tissues). As a consequence, there may be increased dosage requirements for some drugs to achieve the same therapeutic effect, provided these effects are not offset by other pharmacokinetic changes. Both the total plasma and the free-drug concentrations of phenytoin, carbamazepine and valproic acid decrease during pregnancy, but the free-drug fraction (ratio of free to total plasma concentration) may increase (Pavek et al., 2009).
Metabolism
The metabolic activity of cytochrome P450 isoenzymes CYP3A4, CYP2D6, CYP 2A6 and CYP 2C9 and uridine 5′-diphosphate glucuronosyltransferase (UGT) isoenzymes (UGT1a1, UGT1A4 and UGT2B7) is increased during pregnancy. Drugs metabolised by these isoenzymes may therefore require dose adjustment. This may decrease the amount of the drug available for transfer across the placenta and thereby influence fetal exposure. In contrast, the metabolic activity of CYP1A2 and CYP2C19 is decreased during pregnancy and drugs metabolised by these isoenzymes may need dose reduction to minimise toxicity (see Table 47.3).
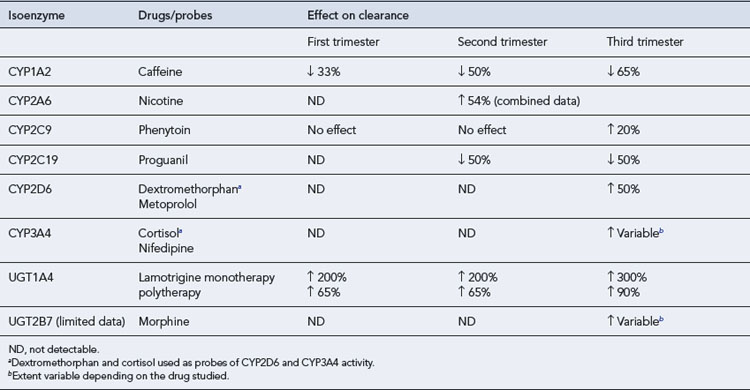
Excretion
Within the first few weeks of pregnancy, the glomerular filtration rate (GFR) increases by approximately 50%. Consequently, those drugs which are excreted primarily unchanged by the kidneys, for example, lithium, digoxin and penicillin, show enhanced elimination and lower steady-state concentrations. The following drugs have shown pregnancy-induced increases of 20–65% on their renal elimination (Anderson, 2005):
Drug selection in pregnancy
Although there are few, if any, drugs for which safe use in pregnancy can be absolutely assured, only a handful of drugs in current clinical use have been conclusively shown to be teratogenic. In general, drugs that have been used extensively in pregnant women without apparent problems are recommended in preference to new drugs for which there is less experience of use. For example, methyldopa is used rarely to treat hypertension in the non-pregnant state but has historically been preferred in pregnancy because of a long history of safe use (Schaefer et al., 2007). However, older drugs may be less effective in terms of controlling maternal illness and are often associated with an increased side-effect risk profile.
In most cases, the decision as to whether to commence or continue with a medication in pregnancy will depend on the risk–benefit analysis for that specific mother–infant pair. A frequent error made by health professionals is to apply the U.S. Food and Drug Administration (FDA) pregnancy risk categories (A (no demonstrable risk), B, C, D and X (teratogenic agents that are considered to be completely contraindicated in pregnancy) when considering whether or not to prescribe a drug in pregnancy. It is now widely accepted that these categories are oversimplified and are of little practical help in a clinical setting. The FDA has proposed that the existing categories be replaced with more detailed information sheets containing a summary of the fetal risk and the additional maternal factors that need to be taken into consideration. Importantly, the need for a detailed section discussing the available data including observed human versus animal data, the study design, dose exposure, and reported congenital malformations and/or adverse events has been emphasised (see http://www.fda.gov for up-to-date information).
Drugs in lactation
Breast milk is the best form of nutrition for young infants. It provides all the energy and nutrients required for the first 6 months of life. The World Health Organization (WHO, 2001) and the United Nations Children’s Fund (UNICEF) recommend exclusive breastfeeding for this period. Benefits of breastfeeding include protection of the infant against gastric, respiratory and urinary tract infections (Kramer and Kakuma, 2002), and reduction in rates of obesity (Horta et al., 2007), juvenile-onset diabetes (Horta et al., 2007) and atopic disease (Fewtrell, 2004). Adults who were breastfed as infants often have lower blood pressure and lower cholesterol levels (Horta et al., 2007). Maternal benefits include reduced risk of developing pre-menopausal breast cancer and delayed resumption of menstrual cycle. Breastfeeding also strengthens the mother–infant bond.
There are few contraindications to breastfeeding, although maternal HIV infection in developed countries is a notable exception. The percentage of women exclusively breastfeeding their infants after 6 months is often less than 20% (Scott et al., 2006). Reasons for early discontinuation of breastfeeding include return to work, concerns about inadequate lactation or safety of drug use.
There are two main goals to consider when formulating advice for nursing mothers. These are to protect the infant from maternal drug-related adverse effects and to allow, whenever possible, necessary maternal medication (Berlin et al., 2009).
Transfer of drugs into breast milk
Most drugs pass into breast milk to some degree although transfer is usually low. The drug ‘dose’ ingested by the infant via breast milk only rarely causes adverse effects. Examples of adverse effects observed in breastfed infants exposed to medication via breast milk are given in Table 47.4, although not all of these are proven to be directly due to the drug ingested via breast milk.
| Atenolol | Bradycardia, cyanosis, hypotension |
| Ciprofloxacin | Pseudomembranous colitis |
| Codeine | Death |
| Dapsone | Haemolytic anaemia |
| Diazepam | Lethargy, sedation, poor suckling |
| Doxepin | Sedation and respiratory arrest |
| Erythromycin | Pyloric stenosis |
| Fluoxetine | Colic, irritability, sedation |
| Indometacin | Seizures |
| Lithium | T-wave abnormalities |
| Naproxen | Prolonged bleeding, haemorrhage, anaemia |
| Phenytoin | Methaemoglobinaemia |
In the first few days of life, large gaps exist between the alveolar cells. These permit enhanced passage of drugs into milk. By the end of the first week, these gaps close under the influence of prolactin (Lawrence and Lawrence, 2011). There is greater passage of drugs into colostrum (early milk) than in mature milk as the former contains more protein and less fat. There is also some variation in fat and protein content of milk between the beginning and end of a feed, but these changes have less influence on drug passage than the physicochemical properties of the drug.
Another method by which a drug may enter milk is by a pumping system whereby energy is used to effect transfer into milk. The most important example is iodides which pass into milk in high concentrations (Hale, 2010).
Milk to plasma concentration ratio
Several methods have been proposed to determine the amount of drug transferred to breast milk. The milk to plasma (M/P) ratio is often used as a measure of the extent of drug transfer into breast milk. It is usually obtained from case reports or small clinical studies and may be based on paired concentrations or full area under the concentration–time curve (AUC) analysis. M/P ratios that are based on a pair of milk and plasma samples collected simultaneously may be inaccurate as they assume that the concentrations of drug in milk and plasma are in parallel, which may not be the case. It is better to collect multiple samples of plasma and milk across a dosing interval or until the drug is cleared from both phases after a single dose, for determination of an M/P ratio based on the respective AUCs (M/PAUC). Figure 47.1 demonstrates the markedly different estimates of M/P ratio that can be obtained via both sampling methods. The true M/P ratio may vary significantly during the same episode of breastfeeding.
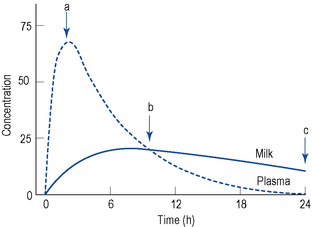
If human-derived M/P ratios are lacking for a particular drug, it may be possible to predict the extent of transfer using known physicochemical properties, for example, pKa, and a published predictive model (Atkinson and Begg, 1990; Begg et al., 1992). M/P ratios obtained from animal studies should not be used for clinical decision making, as they may not correlate well with human M/P ratios.
Calculating the infant ‘dose’ ingested via milk
When using quantitative data from milk analyses, the most accurate estimation of the infant ‘dose’ is from studies in which the milk is collected over a complete dose interval at steady state and the total dose is calculated (Fig. 47.2). Unfortunately, these studies are seldom performed. Therefore, information must be obtained from less than ideal conditions.
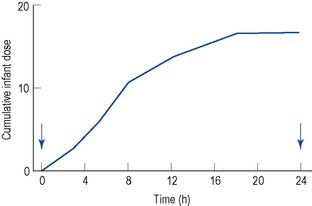
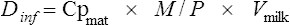
where Cpmat is the average maternal plasma concentration. M/PAUC is used in preference to a ratio based on paired concentrations when available, but this is seldom the case. The volume of milk ingested (Vmilk) is not known but is generally assumed to be around 150 mL of milk per kilogram of body weight per day. The above equation simplifies if the actual milk concentration data are available:
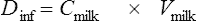
The likely infant plasma drug concentration (Cpinf) can be calculated by:
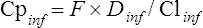
where F is oral availability and Clinf is the infant clearance. Unfortunately, neither F nor Clinf is known accurately for infants, so estimation of the likely steady-state average plasma drug concentration will be very approximate. Weight-adjusted Clinf values, that is, L/h/kg, are often significantly less than adult values in the early stages of life (Table 47.5).
Table 47.5 Approximate drug clearance by age as percentage of maternal value (Begg, 2000)
| 24–28 weeks’ post-conceptual age | 5% |
| 28–34 weeks’ post-conceptual age | 10% |
| 34–40 weeks’ post-conceptual age | 33% |
| 40–44 weeks’ post-conceptual age | 50% |
| 44–68 weeks’ post-conceptual age | 66% |
| Over 68 weeks’ post-conceptual age | 100% |
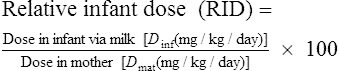
For the great majority of drugs, this calculation yields infant doses in the order of 0.1–5.0% of the weight-adjusted maternal dose expressed as a percentage (% dose). It is generally thought that relative infant dose values of less than 10% of the maternal dose are probably safe. However, the inherent toxicity of the drug should be taken into account when using this figure.
Estimated daily infant intake = Drug concentration in milk (μcg/L) × 0.15/infant weight (kg)
Some authors use the peak concentration in milk to indicate the maximum infant intake.
Variability
Recently, attention has been focused on the possible role of pharmacogenetic factors in affecting the safety of breastfed infants exposed to drugs via milk (Madadi et al., 2009). Sedation (and one death) occurred in infants of mothers with rare genotypes of cytochrome P450 2D6 leading to ultra-rapid metabolism of codeine to morphine. The incidence of these genotypes varies amongst different populations. The overall percentage of Western Europeans with the CYP2D6 ultra-rapid metaboliser phenotype is 5.4% (Ingelman-Sundberg, 2005). Higher percentages have been reported in populations from northeast Africa and the Middle East.
Assessing the risk to the infant
Many factors must be considered when assessing the risk of maternal drug therapy to the breastfeeding infant (Box 47.2).
The presence of active metabolites, for example, desmethyldiazepam, may prolong infant drug exposure and lead to drug accumulation, especially where drug clearance is low such as in the neonatal period. Similarly, drugs with long half-lives, for example, fluoxetine, may be problematic at this time. Drug clearance by the infant does not reach adult values until 6–7 months (Table 47.5). A premature infant of 30 weeks’ gestational age has a drug clearance value of about 10% of the maternal value. It is important to distinguish between gestational age and time after delivery. A 2-week-old infant born at 28 weeks will have a gestational age of 30 weeks.
Reducing risk to the breastfed infant
The maternal regimen should be simplified wherever possible. A review of therapy before delivery will help to reduce risks to the neonate. New drugs are best avoided if a therapeutic equivalent is available for which data on safe use in lactation exists. All infants exposed to drugs via breast milk should be monitored for any untoward effects. Measures to ensure the safety of the breastfed infant are summarised in Box 47.3. Some commonly used drugs thought to be safe to use in mothers of full-term healthy infants are listed in Table 47.6.
Box 47.3 Measures to ensure the safety of the breastfed infant
Table 47.6 Examples of commonly used drugs thought to be safe for use in breastfeeding mothers of full-term healthy infantsa
| Drug groups | Individual drugs |
|---|---|
| Antacids | Cetirizine |
| Bulk laxatives | Clotrimazole |
| Cephalosporins | Cromoglycate |
| Inhaled medications, for example, salbutamol | Diclofenac Heparin |
| Penicillins | Ibuprofen |
| Progestogens | Insulin |
| Vaccines (except smallpox) | Iron supplements |
| Vitamins (except high-dose A and D) | Lactulose |
| Levothyroxine | |
| Loratadine | |
| Nystatin | |
| Paracetamol | |
| Warfarin |
a This table is to be used as a guide only. Expert advice is required when the maternal dose is high, if the infant is premature, has renal or hepatic disease or G6PD deficiency.
Special situations
Drug effects on lactation
Answers
Answers
Answers
Answers
The estimated daily infant intake via milk will be 2.3 × 0.15 × 4 = 1.38 mg or 0.345 mg/kg/day.
Answers
Anderson G. Pregnancy-induced changes in pharmacokinetics: a mechanistic-based approach. Clin. Pharmacokinet.. 2005;44:989-1008.
Atkinson H.C., Begg J. Prediction of drug distribution into human milk from physicochemical characteristics. Clin. Pharmacokinet.. 1990;18:151-167.
Begg E.J. Clinical Pharmacology Essentials. The Principles Behind the Prescribing Process. Auckland: Adis International, 2000.
Begg E.J., Atkinson E.J., Duffull S.B. Prospective evaluation of a model for the prediction of milk:plasma drug concentrations from physicochemical characteristics. Br. J. Clin. Pharmacol.. 1992;33:501-505.
Benke P.J. The isotretinoin teratogen syndrome. J. Am. Med. Assoc.. 1984;251:3267-3269.
Berlin C.M., Paul I.M., Vesell E.S. Safety issues of maternal drug therapy during breastfeeding. Clin. Pharmacol. Ther.. 2009;85:20-22.
Braun J.T., Franciosi R.A., Mastri A.R., et al. Isotretinoin dysmorphic syndrome. Lancet. 1984;1:506-507.
Breitkreutz I., Anderson K.C. Thalidomide in multiple myeloma: clinical trials and aspects of drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol.. 2008;4:973-985.
Dunn P.M. The possible relationship between the maternal administration of sulphamethoxypyridazine and hyperbilirubinaemia in the newborn. J. Obstet. Gynaecol. Br. Commonw.. 1964;71:128-131.
Fewtrell M.S. The long term benefits of having been breastfed. Curr. Paediatr.. 2004;14:97-103.
Hale T.W. Medications and Mothers’ Milk, fourteenth ed. Amarillo: Hale Publishing, 2010.
Headley J., Northstone K., Simmons H., et al. Medication use during pregnancy: data from the Avon longitudinal study of parents and children. Eur. J. Clin. Pharmacol.. 2004;60:355-361.
Herbst A.L., Ulfelder H., Poskanzer D.C. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N. Engl. J. Med.. 1971;15:878-881.
Hernandez-Diaz S., Werler M.M., Walker A.M., et al. Neural tube defects in relation to use of folic acid antagonists during pregnancy. Am. J. Epidemiol.. 2001;153:961-968.
Hill R.M. Isotretinoin teratogenicity. Lancet. 1984;1:1465.
Horta B.L., Bahl R., Martines J.C., et al. Evidence on the Long-Term Effects of Breastfeeding. Geneva: WHO, 2007.
Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J.. 2005;5:6-13.
Koren G., Florescu A., Costei A.M., et al. Non-steroidal anti-inflammatory drugs during third trimester and the risk of premature closure of the ductus arteriosus: a meta-analysis. Ann. Pharmacother.. 2006;40:824-829.
Kramer M.S., Kakuma R. Optimal duration of exclusive breastfeeding. Cochrane Database of Systematic Reviews. 2002. Issue 1 Art No. CD003517 doi:10.1002/14651858.CD003517. Available at http://www2.cochrane.org/reviews/en/ab003517.html
Lawrence R.A., Lawrence R.M. Breastfeeding. A Guide for the Medical Profession, seventh ed. Philadelphia: Elsevier, 2011.
Lenz W. Malformations caused by drugs in pregnancy. Am. J. Dis. Child. 1966;112:99-106.
Lenz W. A short history of thalidomide embryopathy. Teratology. 1988;38:203-215.
Loebstein R., Lalkin A., Koren G. Pharmacokinetic changes during pregnancy and their clinical relevance. Clin. Pharmacokinet.. 1997;33:328-343.
Madadi P., Ross C.J., Hayden M.R., et al. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine during breastfeeding: a case control study. Clin. Pharmacol. Ther.. 2009;85:31-35.
Miller M.T., Ventura L., Stromland K. Thalidomide and misoprostol: ophthalmologic manifestations and associations both expected and unexpected. Birth Defects Res. Part A Clin. Mol. Teratol.. 2009;85:667-676.
Newman C.G. Teratogen update: clinical aspects of thalidomide embryopathy: a continuing preoccupation. Teratology. 1985;32:133-144.
Newman C.G. The thalidomide syndrome: risks of exposure and spectrum of malformations. Clin. Perinatol.. 1986;13:555-573.
Nulman I., Berkovitch M., Klein J., et al. Steady-state pharmacokinetics of isotretinoin and its 4-oxo metabolite: implications for fetal safety. J. Clin. Pharmacol.. 1998;38:926-930.
Pavek P., Ceckova M., Staud F. Variation of drug kinetics in pregnancy. Curr. Drug Metab.. 2009;10:520-529.
Rosa F.W. Virilization of the female fetus with maternal danazol exposure. Am. J. Obstet. Gynecol.. 1984;149:99-100.
Schaefer C., Peters P., Miller R.K., editors. Drugs During Pregnancy and Lactation: Treatment Options and Risk Assessment, second ed, Oxford: Academic Press, 2007.
Scott J.A., Binns C.W., Oddy W.H., et al. Predictors of breastfeeding duration: evidence from a cohort study. Pediatrics. 2006;117:e646-e655.
Sedman A.B., Kershaw D.B., Bunchman T.E. Recognition and management of angiotensin converting enzyme inhibitor fetopathy. Pediatr. Nephrol.. 1995;9:382-385.
Vorhees C.V., Weisenburger W.P., Minck D.R. Neurobehavioral teratogenic effects of thalidomide in rats. Neurotoxicol. Teratol.. 2001;23:255-264.
World Health Organization 2001 54th World Health Assembly. Global Strategy for Infant and Young Child Feeding. The Optimal Duration of Exclusive Breastfeeding. Geneva: WHO, 2001. Available at http://apps.who.int/gb/archive/pdf_files/WHA54/ea54id4.pdf
Department of Health 2002 Infant feeding. A Summary Report. London: DH, 2000. Available at http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_4008114
Department of Health. Infant Feeding Recommendation. London: DH, 2003. Available at http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_4096999.pdf
Hale T.W., Ilett K. Drug Therapy and Breastfeeding: From Theory to Clinical Practice. New York: Parthenon Publishing, 2002.
Lee A. Adverse Drug Reactions, second ed. London: Pharmaceutical Press, 2006.
Schaefer C., Peters P.W.J., Miller R.K., editors. Drugs During Pregnancy and Lactation, second ed, Amsterdam: Elsevier, 2007.
Tomson G., Sundwall A., Lunell N.O., et al. Transplacental passage and kinetics in the mother and newborn of oxazepam given during labour. Clin. Pharmacol. Ther.. 1979;25:74-81.
UK Teratology Information Service (UKTIS): www.uktis.org
TOXBASE: TOXBASE: www.toxbase.org
European Network of Teratology Information Services: http://www.entis-org.com
OTIS (American Organisation of Teratology Information Services): www.otispregnancy.org
Motherisk (Canadian Teratology information service): www.motherisk.org