[level-membership-for-basic-science-category]
Chapter 16 Drugs for inflammation and joint disease
Inflammation
Several drugs in current use act on the various stages of this inflammatory process. Antagonists of TNFα, IL-1 and IL-6 are available (see Biologic agents, p. 253). Colchicine, used in the treatment of gout, interferes with neutrophil chemotaxis, thus inhibiting their recruitment to the site of inflammation.
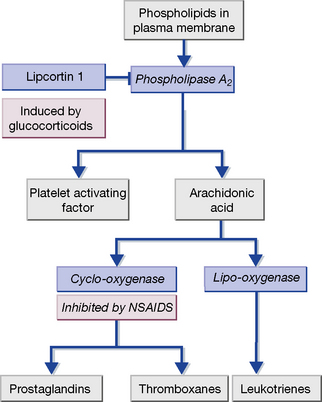
Many leucocytes, including mast cells and macrophages, as well as endothelial cells, synthesise pro-inflammatory eicosanoids and platelet-activating factor (PAF) (Fig. 16.1). These are 20-carbon unsaturated fatty acids derived from phospholipid substrates in the plasma membrane by the enzymes phospholipase A2, cyclo-oxygenase (COX) and lipo-oxygenase (which are induced by IL-1). The prostaglandins, thromboxanes and leukotrienes have diverse pro-inflammatory roles. Leukotrienes promote the activation and accumulation of leucocytes at sites of inflammation. Prostaglandins induce vasodilatation of the microcirculation and are important in pain signalling from locally inflamed tissue. Platelet-activating factor and thromboxane A2 affect the coagulation and fibrinolytic cascades. Non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, inhibit COX and hence prostaglandin and thromboxane synthesis. Glucocorticoids act by inducing the synthesis of lipocortin-1, a polypeptide that inhibits phospholipase A2, and thereby exert a broad anti-inflammatory effect. The leukotriene receptor antagonists montelukast and zafirlukast cause bronchodilatation and are used to treat asthma.
The adaptive immune response
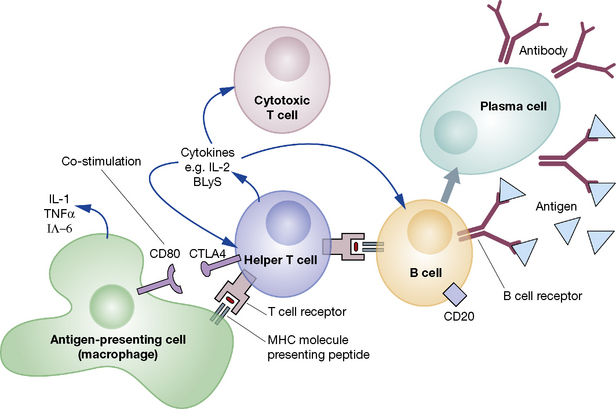
An adaptive immune response is initiated when a helper T cell recognises a peptide antigen presented on the surface of an antigen-presenting cell (APC) and is activated (Fig. 16.2). The activated helper T cell is then able to activate other types of T cell and B cells. This results in the proliferation of adaptive cellular effectors, the generation and release of antibodies by plasma cells and the production of a range of cytokines by the participating leucocytes. On occasion, amplification loops may become self-perpetuating, leading to chronic autoimmune disease.
Pharmacological manipulation of inflammatory mediators
Mode of action
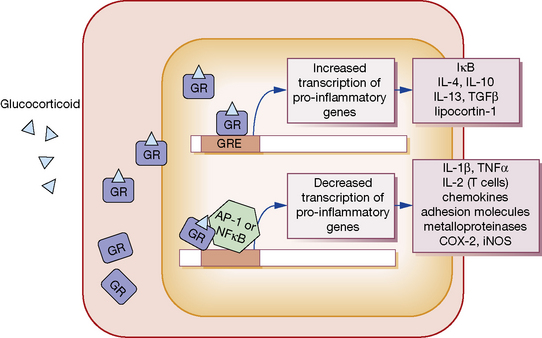
GCs, being lipophilic, diffuse across the cell membrane and bind the cytosolic glucocorticoid receptor (GR) (Fig. 16.3). Receptor polymorphisms influence the strength of the receptor interaction, and represent one source of variation in sensitivity to exogenous steroids. Once bound, the GC-GR complex translocates to the nucleus where it acts in at least two ways to alter gene transcription:
1. The GC-GR complex binds to the glucocorticoid response element within target gene promoters, increasing transcription of various anti-inflammatory genes. These include I-κB, which inhibits the activation of nuclear factor (NF)-κB, and the cytokines IL-4, IL-10, IL-13 and transforming growth factor (TGF)β, which have immunosuppressive and anti-inflammatory activity.
2. The GC-GR complex interferes with the binding of the transcription factors activating protein (AP)-1 and NF-κB to their response elements. This action decreases the transcription of a range of pro-inflammatory mediators. These include IL-1β and TNFα; IL-2, which stimulates T-cell proliferation; a range of chemokines and cellular adhesion molecules; metalloproteinases; COX-2; and inducible nitric oxide synthase.
3. GCs increase synthesis of the polypeptide lipocortin-1, which inhibits phospholipase A2 and thereby the synthesis of prostaglandins, thromboxane A2 and PAF (see Fig. 16.1).
Non-steroidal anti-inflammatory drugs (NSAIDs)
Recently concern has arisen over the effect of traditional NSAIDs and COX-2 inhibitors on the cardiovascular system, with analysis of the VIGOR1 study showing that rofecoxib in particular increases the risk of myocardial infarction (rofecoxib has since been withdrawn from use). While they retain an important role in the treatment of acute gout, inflammatory arthritis, ankylosing spondylitis and dysmenorrhea, long-term prescription should only be undertaken following a full discussion with the patient regarding the balance of risks and benefits.
Mode of action
NSAIDs inhibit cyclo-oxygenase (COX), which catalyses the synthesis of prostaglandins and thromboxane from arachidonic acid (see Fig. 16.1). There are two isoforms of COX:
• COX-1 is constitutively2 expressed in most cell types.
• COX-2 is induced when inflammatory cells (fibroblasts, endothelial cells and macrophages) are activated by cytokines such as IL-1β and TNFα. It is also often upregulated in cancer cells, and is constitutively expressed in the kidney and brain.
NSAIDs can be categorised according to their COX specificity as:
Pharmacokinetics
NSAIDs are absorbed almost completely from the gastrointestinal tract, tend not to undergo first-pass elimination (see p. 87), are highly protein bound and have small volumes of distribution. Their t½ values in plasma tend to group into short (1–5 h) or long (10–60 h). Differences in t½ are not necessarily reflected proportionately in duration of effect, as peak and trough drug concentrations at their intended site of action following steady-state dosing are much less than those in plasma. The vast majority of NSAIDs are weak organic acids and localise preferentially in the synovial tissue of inflamed joints (see pH partition hypothesis, p. 80).
Adverse effects
Gastrointestinal
NSAID-associated gastrointestinal disease appears to result from the inhibition of COX–1-mediated production of cytoprotective mucosal prostaglandins, especially PGI2 and PGE2, which inhibit acid secretion in the stomach, promote mucus production and enhance mucosal perfusion. Several large randomised controlled trials have investigated the incidence of gastrointestinal adverse effects in traditional NSAIDs compared with coxibs. The VIGOR (rofecoxib versus naproxen), CLASS3 (celecoxib versus ibuprofen and diclofenac) and TARGET4 (lumiracoxib versus ibuprofen and naproxen) studies all indicate that coxib use leads to an approximately 50% reduction of upper gastrointestinal adverse events.
Cardiovascular
The VIGOR and APPROVE5 trials reported increased thrombotic cardiovascular events in patients treated with rofecoxib, leading to concerns about a class effect of the coxibs. It was suggested that COX-2 selectivity resulted in an imbalance between prostacyclin and thromboxane production, an effect which would not be seen with traditional NSAIDs which inhibited the synthesis of both equally. Subsequent data from the prospective MEDAL6 and TARGET trials have not supported a class effect based on COX-2 selectivity. These studies suggest that treatment with either a coxib or an NSAID results in a small increase in cardiovascular risk. The risk is dose-related and rofecoxib, particularly at doses exceeding 50 mg per day, confers the highest cardiovascular risk in the majority of studies. A recent study of more than 1 million patients quantified cardiovascular risk as a composite of coronary death, non-fatal myocardial infarction and fatal and non-fatal stroke, and reported that diclofenac and rofecoxib were associated with the highest cardiovascular risk, while naproxen and perhaps celecoxib at doses ≤ 200 mg per day were the least likely to cause a cardiovascular event.7 NICE guidelines recommend that patients with pro-thrombotic risk, coronary artery or cerebrovascular disease should not be prescribed NSAIDs or a coxib. For other patients, treatment decisions should be made on an individual patient basis taking into account both cardiovascular and gastrointestinal risk factors. The medication should be prescribed for the shortest possible time and regularly reviewed.
Principal interactions
• Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers: there is a risk of renal impairment and hyperkalaemia.
• Quinolone antimicrobials: convulsions may occur if NSAIDs are co-administered.
• Anticoagulant (warfarin) and antiplatelet agents (dypiridamole, clopidogrel): increased risk of gastrointestinal bleeding with NSAIDs.
• Antihypertensives: their effect is lessened due to sodium retention by inhibition of renal prostaglandin formation.
• Ciclosporin and tacrolimus: nephrotoxic effect is exacerbated by NSAIDs.
• Cytotoxics: renal tubular excretion of methotrexate is reduced by competition with NSAIDs, with risk of methotrexate toxicity (low-dose methotrexate given weekly avoids this hazard).
• Diuretics: NSAIDs cause sodium retention and reduce diuretic efficacy and there is a risk of hyperkalaemia with potassium-sparing diuretics.
• Lithium: NSAIDs delay the excretion of lithium by the kidney and may cause lithium toxicity.
Paracetamol (acetaminophen)
Mode of action and uses
Paracetamol is an effective treatment for mild-moderate pain and for relieving fever. It does not affect platelet function or disrupt the GI mucosal barrier. Paracetamol has analgesic efficacy equivalent to aspirin, but in therapeutic doses it has only weak anti-inflammatory effects, a functional separation that reflects its differential inhibition of enzymes responsible for prostaglandin synthesis.8 For this reason, some would not class paracetamol as an NSAID.
Acute overdose
Severe hepatocellular damage and renal tubular necrosis can result from taking 150 mg/kg body-weight (about 10 or 20 tablets) in one dose.9 Patients at particular risk include:
Aspirin (acetylsalicylic acid)
Mode of action
• An antiplatelet effect due to permanent inactivation, by acetylation, of COX-1 in platelets, preventing synthesis of thromboxane A2. Platelets cannot regenerate the enzyme and the resumption of thromboxane A2 production is dependent on the entry of new platelets into the circulation (platelet lifespan is 7 days). Thus a continuous antiplatelet effect is achieved with low doses.
• Respiratory stimulation is a characteristic of aspirin intoxication and occurs both directly by stimulation of the respiratory centre and indirectly through increased carbon dioxide production.
• Although aspirin in high dose reduces renal tubular reabsorption of uric acid so increasing its elimination, other treatments for hyperuricaemia are preferred. Indeed aspirin should be avoided in gout as low doses inhibit uric acid secretion and on balance its effects on uric acid elimination are adverse.
Adverse effects
• Salicylism (the symptoms of an excessive dose): tinnitus and hearing difficulty, dizziness, headache and confusion.
• Allergy. Aspirin is a common cause of allergic or pseudoallergic symptoms and signs. Patients exhibit severe rhinitis, urticaria, angioedema, asthma or shock. Those who already suffer from recurrent urticaria, nasal polyps or asthma are more susceptible.
• Reye’s syndrome. Epidemiological evidence relates aspirin use to the development of the rare Reye’s syndrome (encephalopathy, liver injury) in children recovering from febrile viral infections. Consequently, aspirin should not be given to children aged under 15 years.
Acute overdose
• Activated charcoal 50 g by mouth prevents salicylate absorption from the GI tract. Gastric lavage or the use of an emetic is no longer recommended.
• Correction of dehydration, using dextrose 5% i.v. is often indicated.
• Acid–base disturbance. Alkalosis or mixed alkalosis/acidosis need no specific treatment. Metabolic acidosis is treated with sodium bicarbonate, which alkalinises the urine and accelerates the removal of salicylate in the urine.
• Haemodialysis may be necessary if renal failure develops or the plasma salicylate concentration exceeds 900 mg/L.
Immunomodulatory drugs
• Severity of disease: this determines the risk:benefit ratio. For example, cerebral or renal lupus is more hazardous than rash or arthritis and therefore a more potent but potentially more toxic drug regimen is justified.
• Adverse-effect profile: both the probability and severity of potential adverse effects need to be considered.
• Evidence base: this, disappointingly, is often patchy but where it exists affords greater confidence for the prescribing physician.
• Age: the risk of future malignancy is less significant in elderly patients.
• Co-morbidity: drugs causing hypertension or adverse lipid profiles may be avoided in patients with high cardiovascular risk.
• Pregnancy/breast feeding: an absolute contraindication for many immunomodulatory drugs.
• Importance of future fertility: cyclophosphamide is likely to decrease fertility; leflunomide requires a long washout period prior to pregnancy.
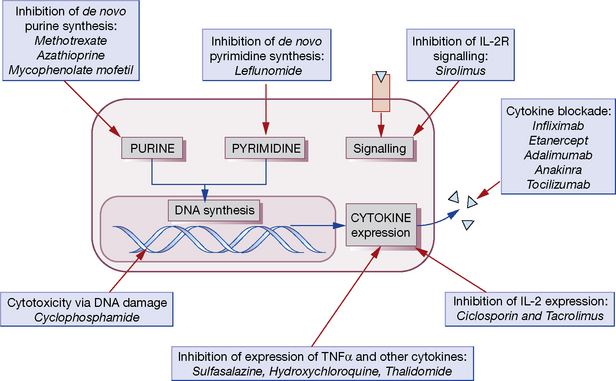
Most conventional immunomodulatory agents act by inhibiting activation or reducing proliferation of lymphocytes. Many have more than one mechanism of action and often the precise way in which they exert their effects is unknown. Moreover, their antiproliferative and cytotoxic effects are in most cases not specific to the immune system but will affect any rapidly dividing cell population. This is one of the major causes of toxicity. Figure 16.4 presents an overview of these drugs and the known mechanisms of action that are relevant to the following discussion.
Methotrexate
Mechanism of action
As a folate analogue, methotrexate inhibits folate-dependent enzymes involved in purine biosynthesis, thus reducing lymphocyte proliferation, and this was originally thought to be its principal mechanism of action (and is likely to be the source of many of its toxic effects). In recent years, however, it has become clear that methotrexate has a number of other anti-inflammatory effects, and inhibition of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase, another folate-dependent enzyme, is now thought to be the likely pathway responsible for its efficacy in chronic inflammatory conditions.10 This results in a rise in intracellular levels of AICAR, which then inhibits adenosine deaminase and prevents the degradation of adenosine. Increased plasma concentrations of adenosine are thought to mediate many anti-inflammatory effects such as decreased TNFα and IFNγ production.
Leflunomide
The active metabolite of leflunomide (A77 1726) inhibits dihydro-orotate dehydrogenase, a mitochondrial enzyme required for the synthesis of pyrimidines.11 It arrests the proliferation of activated lymphocytes and is licensed for the treatment of rheumatoid arthritis and psoriatic arthritis.
Hydroxychloroquine
Adverse effects
Hydroxychloroquine is normally very well tolerated and has a low incidence of side-effects. GI disturbances, rashes, and, rarely, blood dyscrasias may occur. Retinal toxicity may rarely occur with long-term use; measurement of visual acuity initially and at annual intervals is recommended.12
Contraindications
Hydroxychloroquine should be used with caution in hepatic or renal impairment, in neurological disorders, in glucose 6-phosphate dehydrogenase deficiency and in porphyria. The manufacturer’s instructions advise against use in pregnancy and breast feeding, but data on over 250 pregnancies in women with connective tissue disease who were taking hydroxychloroquine provide evidence that the treatment is safe in pregnancy, and probably also during breast feeding.13
Sulfasalazine
Mechanism of action
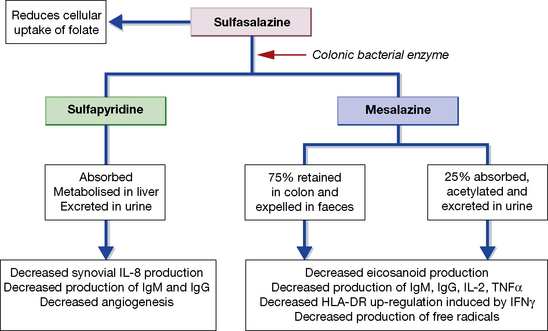
SSZ is cleaved by bacterial azoreductases in the colon to release 5-ASA and sulfapyridine (Fig. 16.5). 5-ASA is retained mostly in the colon and excreted, but 30% of intact SSZ and all sulfapyridine are absorbed. Anti-inflammatory effects of mesalazine, both in the colonic epithelial cell and in peripheral blood mononuclear cells, include inhibition of cyclo-oxygenase and lipo-oxygenase, scavenging of free radicals, and inhibition of the production of pro-inflammatory cytokines and immunoglobulins. In the treatment of inflammatory bowel disease, preparations containing 5-ASA alone have efficacy comparable to that of SSZ, but with fewer side-effects. In contrast, the sulfapyridine component appears to be the active moiety in rheumatoid arthritis. SSZ has been shown to reduce rheumatoid factor titres, inhibit IL-2-induced T-cell proliferation and inhibit macrophage IL-1 and IL-12 production, but the relative importance of these effects on its anti-inflammatory activity remains unclear.
Intravenous immunoglobulin
Intravenous immunoglobulin (IvIg) was first used in 1952 to treat primary immune deficiencies. It is composed of pooled IgG extracted from the plasma of 3000–10 000 blood donors, and contains the entire repertoire of naturally occurring antibodies. Besides its uses as a treatment for primary immune deficiencies and hypogammaglobulinaemia, it has immunomodulatory properties, making it an effective therapy for a number of autoimmune conditions. However, it has also been used in scenarios where there is no evidence of efficacy and little theoretical basis to suggest benefit. As there is a national shortage, the UK NHS has recently introduced guidelines to restrict its use only to those conditions in which there is a known benefit.14
Dapsone
Dapsone, traditionally used to treat mycobacterial (principally leprosy) and occasionally protozoal infections, also has anti-inflammatory properties. It can be used to treat discoid lupus and other inflammatory dermatological disorders characterised by neutrophil infiltration. Although it is clear that its antimicrobial activity stems from inhibition of folate synthesis, this does not appear to be the mechanism of its anti-inflammatory effects. These are less well understood, but may involve impairing neutrophil chemotaxis and stabilisation of neutrophil lysosomes, thus inhibiting the respiratory burst. Common adverse effects include rashes, haemolysis and liver dysfunction; it may also rarely cause blood dyscrasias and methaemoglobinaemia. It should be avoided in patients suffering from G-6-PD deficiency (see p. 101).
Biologic agents
Anti-TNFα therapies
Infliximab
is a chimeric monoclonal IgG1 antibody (Fig. 16.6). In rheumatoid arthritis it is administered by intravenous infusion at 3 mg/kg, repeated 2 and 6 weeks after the initial infusion and then at 8-week intervals. In ankylosing spondylitis, psoriasis and Crohn’s disease, it is used at doses of 5 mg/kg. Methotrexate is co-prescribed to limit the development of neutralising antibodies.
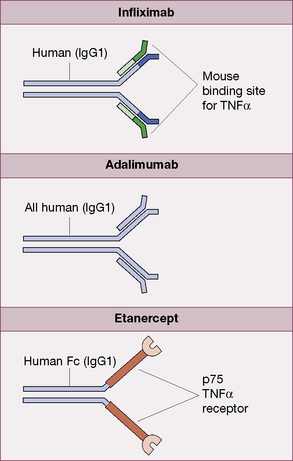
Adalimumab
is a fully human monoclonal IgG1 antibody (Fig. 16.6). The recommended dose is 40 mg by subcutaneous injection fortnightly, in combination with methotrexate. It is licensed for use in rheumatoid arthritis, ankylosing spondylitis and psoriatic arthritis.
Etanercept
is a fusion protein consisting of two human p75 TNFα receptors coupled to an Fc component of human IgG1 (Fig. 16.6). It is licensed for weekly subcutaneous injection in adult patients with rheumatoid arthritis (50 mg), for children over 4 years with polyarticular juvenile idiopathic arthritis (400 micrograms/kg; maximum dose 50 mg), psoriasis and ankylosing spondylitis.
The mechanism of action of etanercept differs from that of the monoclonal IgG antibodies in that it:
Adverse effects
The major risk with anti-TNF therapy is increased susceptibility to infection, particularly with intracellular pathogens such as Mycobacteria tuberculosis (M.Tb). In the case of M.Tb this may be reactivation of latent disease, but there is also a risk of new infection with M.Tb, other mycobacteria or intracellular pathogens such as histoplasmosis, coccidiomycosis or nocardiosis. Guidelines for assessing risk and for managing infection with Mycobacterium tuberculosis in the context of anti-TNFα agents are available.15 Chemoprophylaxis must be started prior to treatment in patients with latent infection.
Other biologic anticytokine agents
Interleukin-6 (IL-6) is another pro-inflammatory cytokine that is of critical importance to the mounting of an immune response, and is the most abundant cytokine found in the synovium of rheumatoid arthritis patients. Tocilizumab is a monocloncal antibody that binds IL-6 receptors and inhibits their intracellular signalling. It has been shown to be effective in controlling disease activity in rheumatoid arthritis,17 and is licensed for use in this condition in the UK. It is given by intravenous infusion on a monthly basis.
Management of diseases affecting the joints
Osteoarthritis
The aims of management of OA are to control pain, reduce progression of joint damage and minimise disability. The major strategies employed are non-pharmacological; there are no disease-modifying drugs in clinical use. Patient education in pain management is important to minimise the adverse affects of analgesics. NSAIDs have been shown to be marginally more effective but are associated with more adverse effects than paracetamol.18 Regular paracetamol or co-dydramol should therefore be tried before introducing a NSAID. Other options include topical NSAID gel, which may provide a modest improvement in symptoms at least in the short term. For patients with large-joint OA, occasional intra-articular injection of corticosteroid and local anaesthetic can provide some respite for up to 6 weeks.
Gout
• Over-production of uric acid occurs if there is excessive cell destruction releasing nucleic acids. This may occur when myeloproliferative and lymphoproliferative disorders are first treated.
• Under-excretion of uric acid is caused by thiazide and loop diuretics, low dose aspirin (see earlier), ethambutol, pyrazinamide, nicotinic acid, ciclosporin and alcohol (which increases uric acid synthesis and also causes a rise in serum lactic acid that inhibits tubular secretion of uric acid). Conversely, a small number of drugs have a mild uricosuric effect and increase renal clearance of urate. Losartan and fenofibrate are examples of this.
The priority in an acute attack is to relieve the intense pain by reducing the inflammatory response. NSAIDs are most commonly prescribed, but if contraindicated, colchicine (EULAR19 guidelines suggest 0.5 mg three times daily) is an alternative. A short course of oral prednisolone or intra-articular corticosteroid is also effective, although the severity of joint pain may preclude intra-articular injection during an acute attack.
Rasburicase
is a recombinant uricase which converts urate into allantoin, a more soluble metabolite which is readily excreted renally. It rapidly and profoundly lowers serum uric acid concentration and is licensed for use in tumour lysis syndrome. It has also been used in small numbers of patients with gout, but antibody development limits its effectiveness with repeated infusions, as would be needed to treat chronic tophaceous gout. A pegylated uricase (pegloticase),20 which has a longer half-life and is less immunogenic, has shown promise in recent trials.
Rheumatoid arthritis
The initial management of a patient presenting with new inflammatory polyarthritis consists of reducing systemic inflammation, joint pain and stiffness while the diagnosis is confirmed.21 This may be achieved by short-term glucocorticoids such as Depo-Medrone 120 mg i.m., combined with analgesia and an NSAID (e.g. diclofenac 50 mg three times daily).
It is particularly important to address cardiovascular risk reduction in patients with RA, given the increased risk of cardiovascular disease associated with chronic inflammation. Aggressive management of RA with DMARDs may reduce cardiovascular disease incidence at the same time as controlling inflammation. Moreover, atorvastatin has been reported to improve disease activity scores in RA.22
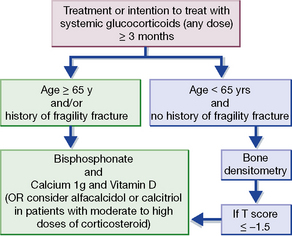
Protection against osteoporosis is particularly important for patients who receive long-term glucocortocoids; even doses less than prednisolone 7.5 mg daily increase fracture risk. The UK guideline23 for the prevention and treatment of glucocorticoid-induced osteoporosis is summarised in Figure 16.7. Bone loss is most rapid in the first few months of treatment, therefore protection must be considered at the onset of treatment, whenever the intention is to continue for more than 3 months, at any dose. Strategies include minimising the dose of corticosteroid, concomitant prescription of vitamin D 800 IU and calcium 1 g daily and advice on smoking cessation, alcohol intake, nutrition and exercise. Bisphosphonates should be prescribed for all patients aged over 65 years, or those with a history of a fragility fracture. Other patients should undergo bone densitometry and should be prescribed a bisphosphonate if the T score is − 1.5 or less24 at either lumbar spine or hip.
Psoriatic arthritis
As with RA, symptomatic management strategies include use of NSAIDs and intra-articular corticosteroids. The evidence base for the use DMARDs in PsA is generally poor, but methotrexate is most commonly used. Sulfasalazine and leflunomide can be tried if methotrexate is ineffective or contraindicated. Anti-TNFα therapy is effective in psoriatic arthritis and in severe cutaneous psoriasis,25 in contrast to the anti-T-cell biologic agents efalizumab and alefacept which, although effective for skin disease, have not been shown in trials to have dramatic benefits in PsA.
Ankylosing spondylitis
Ankylosing spondylitis (AS) is an inflammatory disorder of the spine and sacroiliac joints, sometimes associated with peripheral arthritis, anterior uveitis and aortitis, that affects 0.1% of the population. The traditional objectives in its management were to relieve pain and stiffness and maintain mobility through a combination of regular NSAIDs and physiotherapy. No DMARDs have shown efficacy in treating spinal disease, although sulfasalazine is effective for peripheral arthritis. Management of AS has been revolutionised by the discovery that anti-TNFα therapy can dramatically improve spinal disease activity.26 Evidence from serial magnetic resonance imaging indicates that bone oedema regresses following TNFα blockade.
Barnes P.J. Corticosteroids: the drugs to beat. Eur. J. Pharmacol.. 2006;533:2–14.
Baschant U., Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J. Steroid Biochem. Mol. Biol.. 2010;120:69–75.
Cohen S., Fleischmann R. Kinase inhibitors: a new approach to rheumatoid arthritis treatment. Curr. Opin. Rheumatol.. 2010;22:330–335.
D’Cruz D., Khamashta M., Hughes G. Systemic lupus erythematosus. Lancet. 2007;369:587–596.
Dasgupta B., Borg F.A., Hassan N., et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology. 2010;49:1594–1597.
Dasgupta B., Borg F.A., Hassan N., et al. BSR and BHPR guidelines for the management of polymyalgia rheumatica. Rheumatology. 2010;49:186–190.
Elwood P.C., Gallagher A.M., Duthie G.G., et al. Aspirin, salicylates, and cancer. Lancet. 2009;373:1301–1309.
Fearon D., Innate immunity: Warrell. D.A., ed. Oxford Textbook of Medicine, fourth ed, Oxford: Oxford University Press, 2003. (Chapter 5.5)
Gabay C., Lamacchia C., Palmer G. IL-1 pathways in inflammation and human diseases. Nature Reviews Rheumatology. 2010;6:232–241.
McMichael A., Principles of immunology: Warrell. D.A., Cox T.M., Firth. J.D., Benz E.J. Oxford Textbook of Medicine, fourth ed, Oxford: Oxford University Press, 2003. (Chapter. 5.1)
National Institute for Health and Clinical Excellence, Osteoarthritis: the care and management of osteoarthritis in adults. NICE Guidance CG59. Available online at: http://guidance.nice.org.uk/CG59 (accessed November 2011)
National Institute for Health and Clinical Excellence, Rheumatoid arthritis: the management of rheumatoid arthritis in adults. NICE Guidance CG79. Available online at: http://guidance.nice.org.uk/CG79 (accessed November 2011)
Østensen M., Forger F. Management of RA medications in pregnant patients. Nature Reviews Rheumatology. 2009;5:382–390.
Østensen M., Motta M. Therapy insight: the use of anti-rheumatic drugs during nursing. Nat. Clin. Pract. Rheumatol.. 2007;3:490–496.
Šenolt L., Vencovský J., Pavelka K., et al. Prospective new biological therapies for rheumatoid arthritis. Autoimmun. Rev.. 2009;9:102–107.
Terkeltaub R. Update on gout: new therapeutic strategies and options. Nature Reviews Rheumatology. 2010;6:30–38.
Zhang W., Doherty M., Bardin T., et al. EULAR evidence based recommendations for gout. Part II: management. Ann. Rheum. Dis.. 2006;65:1312–1324.
1 Bombardier C, Laine L, Reicin A et al 2000 Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR study group. New England Journal of Medicine 343:1520–1528.
2 Continuously produced, rather than depending on the presence of an inducer.
3 Silverstein F E, Faich G, Goldstein J L et al 2000 Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. Journal of the American Medical Association 284:1247–1255.
4 Farkouh M E, Kirshner H, Harrington R A et al 2004 Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet 364:675–684.
5 Bresalier R S, Sandler R S, Quan H et al 2005 Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New England Journal of Medicine 352:1092–1102.
6 Cannon C P, Curtis S P, FitzGerald G A et al 2006 Cardiovascular outcomes with etoricoxib and diclofenac in patients with osteoarthritis and rheumatoid arthritis in the Multinational Etoricoxib and Diclofenac Arthritis Long-term (MEDAL) programme: a randomised comparison. Lancet 368:1771–1781.
7 Fosbøl E L, Folke F, Jacobsen S et al 2010 Cause-specific cardiovascular risk associated with nonsteroidal antiinflammatory drugs among healthy individuals. Circulation, Cardiovascular Quality and Outcomes 3:395–405.
8 Aronoff D M, Oates J A, Boutaud O et al 2006 New insights into the mechanism of action of acetaminophen: its clinical pharmacological characteristics reflect inhibition of the two prostaglandin H2 synthases. Clinical Pharmacology and Therapeutics 79:9–19.
9 A 73-year-old woman was taking paracetamol for pain relief but added a paracetamol-containing proprietary preparation for cold relief, in effect nearly doubling the dose. She died of paracetamol poisoning. Her husband said that his wife ‘knew that too much paracetamol was dangerous but she did not realise there was paracetamol in [the proprietary preparation]’ which she bought at a supermarket that did not have a dispensary counter where she could have received advice. Report 2011 British Medical Journal 342:971.
10 Chan E S, Cronstein B N 2010 Methotrexate – how does it really work? Nature Reviews Rheumatology 6:175–178.
11 Anonymous 2000 Leflunomide for rheumatoid arthritis. Drug and Therapeutics Bulletin 38:52–54.
12 Fielder A, Graham E, Jones S et al 1998 Royal College of Ophthalmologists guidelines: ocular toxicity and hydroxychloroquine. Eye 12:907–909.
13 Costedoat-Chalumeau N, Amoura Z, du Huong L T et al 2005 Safety of hydroxychloroquine in pregnant patients with connective tissue diseases. Review of the literature. Autoimmunity Reviews 4:111–115.
14 www.ivig.nhs.uk (accessed November 2011)
15 British Thoracic Society Standards of Care Committee 2005 BTS recommendations for assessing risk and for managing Mycobacterium tuberculosis infection and disease in patients due to start anti-TNF-α treatment. Thorax 60:800–805.
16 BMJ Group 2008 Rituximab and abatacept for rheumatoid arthritis. Drug and Therapeutics Bulletin 46:57–61.
17 [No authors listed] 2010 Tocilizumab for rheumatoid arthritis. Drug and Therapeutics Bulletin 48(1):9–12.
18 Pincus T, Koch G G, Sokka T et al 2001 A randomized, double-blind, crossover clinical trial of diclofenac plus misoprostol versus acetaminophen in patients with osteoarthritis of the hip or knee. Arthritis and Rheumatism 44:1587–1598.
19 European League Against Rheumatism.
20 Sundy J S, Baraf H S, Becker M A et al 2008 Efficacy and safety of intravenous (IV) pegloticase (PGL) in subjects with treatment failure gout (TFG): phase 3 results from GOUT1 and GOUT2 [Abstract]. Arthritis and Rheumatism 58(Suppl.):S400.
21 Kennedy T, McCabe C, Struthers G et al 2005 BSR guidelines on standards of care for persons with rheumatoid arthritis. Rheumatology (Oxford) 44:553–556.
22 McCarey D W, McInnes I B, Madhok R et al 2004 Trial of atorvastatin in rheumatoid arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet 363:2015–2021.
23 Bone and Tooth Society, National Osteoporosis Society and Royal College of Physicians. Glucocorticoid-induced osteoporosis – guidelines for prevention and treatment. 2002. Available online at:http://www.rcplondon.ac.uk/pubs/books/glucocorticoid/
24 The T score is the number of standard deviations above or below the mean bone mineral density of a 25-year-old (sex-matched) individual. A T score below − 2.5 indicates osteoporosis.
25 Kyle S, Chandler D, Griffiths C E M et al 2005 Guideline for anti-TNF-alpha therapy in psoriatic arthritis. Rheumatology (Oxford) 44:390–397.
26 Keat A, Barkham N, Bhalla A et al 2005 BSR guidelines for prescribing TNF-α blockers in adults with ankylosing spondylitis. Report of a working party of the British Society for Rheumatology. Rheumatology (Oxford) 44:939–947.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 16 Drugs for inflammation and joint disease
Inflammation
Several drugs in current use act on the various stages of this inflammatory process. Antagonists of TNFα, IL-1 and IL-6 are available (see Biologic agents, p. 253). Colchicine, used in the treatment of gout, interferes with neutrophil chemotaxis, thus inhibiting their recruitment to the site of inflammation.
Many leucocytes, including mast cells and macrophages, as well as endothelial cells, synthesise pro-inflammatory eicosanoids and platelet-activating factor (PAF) (Fig. 16.1). These are 20-carbon unsaturated fatty acids derived from phospholipid substrates in the plasma membrane by the enzymes phospholipase A2, cyclo-oxygenase (COX) and lipo-oxygenase (which are induced by IL-1). The prostaglandins, thromboxanes and leukotrienes have diverse pro-inflammatory roles. Leukotrienes promote the activation and accumulation of leucocytes at sites of inflammation. Prostaglandins induce vasodilatation of the microcirculation and are important in pain signalling from locally inflamed tissue. Platelet-activating factor and thromboxane A2 affect the coagulation and fibrinolytic cascades. Non-steroidal anti-inflammatory drugs (NSAIDs), including aspirin, inhibit COX and hence prostaglandin and thromboxane synthesis. Glucocorticoids act by inducing the synthesis of lipocortin-1, a polypeptide that inhibits phospholipase A2, and thereby exert a broad anti-inflammatory effect. The leukotriene receptor antagonists montelukast and zafirlukast cause bronchodilatation and are used to treat asthma.
The adaptive immune response
An adaptive immune response is initiated when a helper T cell recognises a peptide antigen presented on the surface of an antigen-presenting cell (APC) and is activated (Fig. 16.2). The activated helper T cell is then able to activate other types of T cell and B cells. This results in the proliferation of adaptive cellular effectors, the generation and release of antibodies by plasma cells and the production of a range of cytokines by the participating leucocytes. On occasion, amplification loops may become self-perpetuating, leading to chronic autoimmune disease.
Pharmacological manipulation of inflammatory mediators
Mode of action
GCs, being lipophilic, diffuse across the cell membrane and bind the cytosolic glucocorticoid receptor (GR) (Fig. 16.3). Receptor polymorphisms influence the strength of the receptor interaction, and represent one source of variation in sensitivity to exogenous steroids. Once bound, the GC-GR complex translocates to the nucleus where it acts in at least two ways to alter gene transcription:
1. The GC-GR complex binds to the glucocorticoid response element within target gene promoters, increasing transcription of various anti-inflammatory genes. These include I-κB, which inhibits the activation of nuclear factor (NF)-κB, and the cytokines IL-4, IL-10, IL-13 and transforming growth factor (TGF)β, which have immunosuppressive and anti-inflammatory activity.
2. The GC-GR complex interferes with the binding of the transcription factors activating protein (AP)-1 and NF-κB to their response elements. This action decreases the transcription of a range of pro-inflammatory mediators. These include IL-1β and TNFα; IL-2, which stimulates T-cell proliferation; a range of chemokines and cellular adhesion molecules; metalloproteinases; COX-2; and inducible nitric oxide synthase.
3. GCs increase synthesis of the polypeptide lipocortin-1, which inhibits phospholipase A2 and thereby the synthesis of prostaglandins, thromboxane A2 and PAF (see Fig. 16.1).
Non-steroidal anti-inflammatory drugs (NSAIDs)
Recently concern has arisen over the effect of traditional NSAIDs and COX-2 inhibitors on the cardiovascular system, with analysis of the VIGOR1 study showing that rofecoxib in particular increases the risk of myocardial infarction (rofecoxib has since been withdrawn from use). While they retain an important role in the treatment of acute gout, inflammatory arthritis, ankylosing spondylitis and dysmenorrhea, long-term prescription should only be undertaken following a full discussion with the patient regarding the balance of risks and benefits.
Mode of action
NSAIDs inhibit cyclo-oxygenase (COX), which catalyses the synthesis of prostaglandins and thromboxane from arachidonic acid (see Fig. 16.1). There are two isoforms of COX:
• COX-1 is constitutively2 expressed in most cell types.
• COX-2 is induced when inflammatory cells (fibroblasts, endothelial cells and macrophages) are activated by cytokines such as IL-1β and TNFα. It is also often upregulated in cancer cells, and is constitutively expressed in the kidney and brain.
NSAIDs can be categorised according to their COX specificity as:
Pharmacokinetics
NSAIDs are absorbed almost completely from the gastrointestinal tract, tend not to undergo first-pass elimination (see p. 87), are highly protein bound and have small volumes of distribution. Their t½ values in plasma tend to group into short (1–5 h) or long (10–60 h). Differences in t½ are not necessarily reflected proportionately in duration of effect, as peak and trough drug concentrations at their intended site of action following steady-state dosing are much less than those in plasma. The vast majority of NSAIDs are weak organic acids and localise preferentially in the synovial tissue of inflamed joints (see pH partition hypothesis, p. 80).
Adverse effects
Gastrointestinal
NSAID-associated gastrointestinal disease appears to result from the inhibition of COX–1-mediated production of cytoprotective mucosal prostaglandins, especially PGI2 and PGE2, which inhibit acid secretion in the stomach, promote mucus production and enhance mucosal perfusion. Several large randomised controlled trials have investigated the incidence of gastrointestinal adverse effects in traditional NSAIDs compared with coxibs. The VIGOR (rofecoxib versus naproxen), CLASS3 (celecoxib versus ibuprofen and diclofenac) and TARGET4 (lumiracoxib versus ibuprofen and naproxen) studies all indicate that coxib use leads to an approximately 50% reduction of upper gastrointestinal adverse events.
Cardiovascular
The VIGOR and APPROVE5 trials reported increased thrombotic cardiovascular events in patients treated with rofecoxib, leading to concerns about a class effect of the coxibs. It was suggested that COX-2 selectivity resulted in an imbalance between prostacyclin and thromboxane production, an effect which would not be seen with traditional NSAIDs which inhibited the synthesis of both equally. Subsequent data from the prospective MEDAL6 and TARGET trials have not supported a class effect based on COX-2 selectivity. These studies suggest that treatment with either a coxib or an NSAID results in a small increase in cardiovascular risk. The risk is dose-related and rofecoxib, particularly at doses exceeding 50 mg per day, confers the highest cardiovascular risk in the majority of studies. A recent study of more than 1 million patients quantified cardiovascular risk as a composite of coronary death, non-fatal myocardial infarction and fatal and non-fatal stroke, and reported that diclofenac and rofecoxib were associated with the highest cardiovascular risk, while naproxen and perhaps celecoxib at doses ≤ 200 mg per day were the least likely to cause a cardiovascular event.7 NICE guidelines recommend that patients with pro-thrombotic risk, coronary artery or cerebrovascular disease should not be prescribed NSAIDs or a coxib. For other patients, treatment decisions should be made on an individual patient basis taking into account both cardiovascular and gastrointestinal risk factors. The medication should be prescribed for the shortest possible time and regularly reviewed.
Principal interactions
• Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers: there is a risk of renal impairment and hyperkalaemia.
• Quinolone antimicrobials: convulsions may occur if NSAIDs are co-administered.
• Anticoagulant (warfarin) and antiplatelet agents (dypiridamole, clopidogrel): increased risk of gastrointestinal bleeding with NSAIDs.
• Antihypertensives: their effect is lessened due to sodium retention by inhibition of renal prostaglandin formation.
• Ciclosporin and tacrolimus: nephrotoxic effect is exacerbated by NSAIDs.
• Cytotoxics: renal tubular excretion of methotrexate is reduced by competition with NSAIDs, with risk of methotrexate toxicity (low-dose methotrexate given weekly avoids this hazard).
• Diuretics: NSAIDs cause sodium retention and reduce diuretic efficacy and there is a risk of hyperkalaemia with potassium-sparing diuretics.
• Lithium: NSAIDs delay the excretion of lithium by the kidney and may cause lithium toxicity.
Paracetamol (acetaminophen)
Mode of action and uses
Paracetamol is an effective treatment for mild-moderate pain and for relieving fever. It does not affect platelet function or disrupt the GI mucosal barrier. Paracetamol has analgesic efficacy equivalent to aspirin, but in therapeutic doses it has only weak anti-inflammatory effects, a functional separation that reflects its differential inhibition of enzymes responsible for prostaglandin synthesis.8 For this reason, some would not class paracetamol as an NSAID.
Acute overdose
Severe hepatocellular damage and renal tubular necrosis can result from taking 150 mg/kg body-weight (about 10 or 20 tablets) in one dose.9 Patients at particular risk include:
Aspirin (acetylsalicylic acid)
Mode of action
• An antiplatelet effect due to permanent inactivation, by acetylation, of COX-1 in platelets, preventing synthesis of thromboxane A2. Platelets cannot regenerate the enzyme and the resumption of thromboxane A2 production is dependent on the entry of new platelets into the circulation (platelet lifespan is 7 days). Thus a continuous antiplatelet effect is achieved with low doses.
• Respiratory stimulation is a characteristic of aspirin intoxication and occurs both directly by stimulation of the respiratory centre and indirectly through increased carbon dioxide production.
• Although aspirin in high dose reduces renal tubular reabsorption of uric acid so increasing its elimination, other treatments for hyperuricaemia are preferred. Indeed aspirin should be avoided in gout as low doses inhibit uric acid secretion and on balance its effects on uric acid elimination are adverse.
