[level-membership-for-basic-science-category]
Chapter 29 Drugs and haemostasis
• Coagulation system: the mode of action of drugs that promote coagulation and that prevent it (anticoagulants) and their uses.
• Fibrinolytic system: the mode of action of drugs that promote fibrinolysis (fibrinolytics) and their uses to lyse arterial and venous thrombi (thrombolysis).
• Platelets: the ways that drugs that inhibit platelet activity benefit arterial disease.
The coagulation system
Coagulation initiates with tissue factor (TF), a cell membrane protein that binds activated factor VII (indicated by adding the letter ‘a’, i.e. factor VIIa). Although there is a small fraction of circulating factor VII in the activated state, it has little or no enzymatic activity until it is bound to TF. Most non-vascular cells express TF in a constitutive1 fashion, whereas de novo TF synthesis can be induced in monocytes and damaged endothelial cells. Injury to the arterial or venous wall exposes extravascular TF-expressing cells to blood. Lipid-laden macrophages in the core of atherosclerotic plaques are particularly rich in TF, thereby explaining the propensity for thrombus formation at sites of plaque disruption. Once bound to TF, factor VIIa activates factor IX and factor X (to IXa and Xa, respectively), leading to thrombin generation and clot formation (Fig. 29.1).
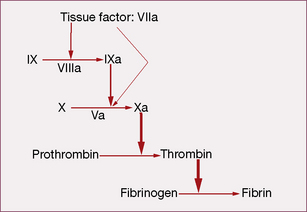
In the current model, blood coagulation starts with a transient release of tissue factor by damaged endothelium, resulting in the formation of sub-nanomolar amounts of thrombin via TF/VIIa-driven Xa formation (extrinsic-tenase). The initial thrombin activity is necessary to prime the system for a full thrombin explosion. Tissue factor pathway inhibitor (TFPI) rapidly shuts down this priming pathway and the full thrombin explosion is then dependent on factor IXa-driven Xa formation. Factor IXa-driven Xa formation (intrinsic-tenase) is amplified by the thrombin explosion itself, as thrombin forms a positive feedback loop by activating factor XIa (not shown in Fig. 29.1), which converts more IX to IXa.
• feedback activation of factor V and factor VIII
• activating platelet-bound factor XI, thereby leading to further factor Xa generation
• activating cells that provide the phospholipid surface required for assembly of the macromolecular enzymatic complexes.
Procoagulant drugs
Vitamin K
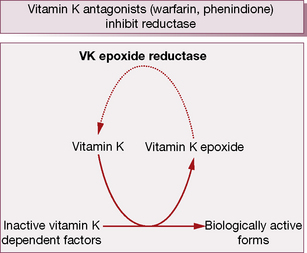
During γ-carboxylation of the proteins, the reduced and active form of vitamin KH2 converts to an epoxide, an oxidation product. Subsequently vitamin K epoxide reductase converts oxidised vitamin K back to the active vitamin K, i.e. there exists an interconversion cycle between vitamin K epoxide and reduced vitamin K (Fig. 29.2).
Vitamin K deficiency may arise from:
• bile failing to enter the intestine, e.g. obstructive jaundice or biliary fistula
• malabsorption syndromes, e.g. coeliac disease, or after extensive small intestinal resection
• reduced alimentary tract flora, e.g. in newborn infants and rarely after broad-spectrum antibiotics.
The following preparations of vitamin K are available:
Vitamin K is used to treat the following:
• Haemorrhage or threatened bleeding due to the coumarin or indanedione anticoagulants. Phytomenadione is preferred for its more rapid action; dosage regimens vary according to the degree of urgency and the original indication for anticoagulation.
• Haemorrhagic disease of the newborn, which develops usually between 2 and 7 days, and late haemorrhagic disease that presents at 6–7 months. Prophylaxis is recommended during the period of vulnerability with vitamin K (phytomenadione, as Konakion) 1 mg by single i.m. injection at birth. Alternatively, give vitamin K by mouth as two doses of a colloidal (mixed micelle) preparation of phytomenadione in the first week. Breast-fed babies should receive a further 2 mg at 1 month of age. Formula-fed babies do not need this last supplement as the formula contains vitamin K. Fears that intramuscular vitamin K might cause childhood cancer have been dispelled.
• Intestinal malabsorption syndromes; menadiol sodium phosphate should be used as it is water soluble.
Coagulation factor concentrates
Use of coagulation factor concentrates
• Superficial haemorrhage sometimes responds to local pressure.
• Minor bleeding can arrest with plasma factor concentrations of 0.25–0.30 units/mL, but severe bleeding requires at least 0.50 units/mL and surgical procedures or life-threatening haemorrhage require 0.75–1.00 units/mL by infusion of factor concentrate.
• In haemophilia A, factor VIII concentrate (t½ 8–12 h) is used for bleeding that is more than minor. Repeat dosing is necessary to maintain haemostatic levels.
• Factor IX (t½ 18–24 h) is used for bleeding that is more than minor in haemophilia B (Christmas disease).
• The speed of recovery of the affected joint or resolution of a haematoma determines the duration of therapy. After surgery, 7–14 days of replacement therapy is required to ensure adequate wound healing and to prevent secondary haemorrhage.
• Primary prophylaxis with factor concentrates two or three times weekly at doses sufficient to keep the factor above 0.01–0.02 units/mL reduces bleeding and hence the severity of chronic haemophilic arthropathy.
Desmopressin (DDAVP)
DDAVP is useful for treating patients with mild haemophilia A and von Willebrand’s disease, especially for short-term therapy. For dental extraction, a single injection of 0.3 micrograms/kg 1–2 h before surgery, combined with the oral antifibrinolytic drug, tranexamic acid, for 5–7 days after the procedure (see Antifibrinolytic drugs, p. 492), will produce normal haemostasis and prevent secondary haemorrhage.
DDAVP shortens the bleeding time in patients with renal or liver failure.
Anticoagulant drugs
• limiting thrombin generation, either as a result of inhibiting other proteases (clotting factors) involved in its generation or by reducing the activity of zymogens (the precursor inactive forms of the enzymes); or
• inhibiting (neutralising) thrombin activity, either directly or indirectly, depending on whether or not they activate the natural serpin-dependent anticoagulant pathway.2
Oral vitamin K antagonists (VKA)
Warfarin and other oral vitamin K antagonists (VKA) reduce the activity of zymogens.
During the γ-carboxylation of factors II (prothrombin), VII, IX and X (and also the natural anticoagulant proteins C and S), active vitamin K (KH2) is oxidised to an epoxide and must be reduced by the enzymes vitamin K epoxide reductase and vitamin K reductase to become active again (see the vitamin K cycle, p. 483). Coumarins3 are structurally similar to vitamin K and competitively inhibit vitamin K epoxide reductase and vitamin K reductase, so limiting availability of the active reduced form of the vitamin to form coagulant (and anticoagulant) proteins. The overall result is a shift in haemostatic balance in favour of anticoagulation because of the accumulation of clotting proteins with absent or decreased γ-carboxylation sites (PIVKAs).4
Uses of oral VKA
Oral vitamin K antagonist drugs are used to prevent and treat venous thrombosis and pulmonary embolus, and to prevent arterial thromboemboli in patients with atrial fibrillation or cardiac disease, including mechanical heart valves. The British Society for Haematology publishes recommended target INRs and duration of therapy for different thrombotic disorders, available at http://www.bcshguidelines.com. The following are general indications:
• Target INR 2.5 is appropriate for treatment of DVT; pulmonary embolism (PE); systemic embolism; prevention of venous thromboembolism in myocardial infarction; mitral stenosis with embolism; transient ischaemic attacks; atrial fibrillation; mechanical prosthetic aortic valves.
• Target INR 3.5 is preferred for recurrent DVT and PE when already on warfarin with target of 2.5, arterial disease and some mechanical prosthetic mitral valves.
• At least 6 weeks’ anticoagulation is recommended after calf vein thrombosis and at least 3 months after proximal DVT or PE. For patients with temporary risk factors and a low risk of recurrence, 3 months of treatment may be sufficient. For patients with idiopathic venous thromboembolism or permanent risk factors, at least 6 months’ anticoagulation is usual.
Parenteral anticoagulants
Heparin
After more tests and the preparation of other batches of heparophosphatide, I went one morning to the door of Dr. Howell’s office, and standing there (he was seated at his desk), I said ‘Dr. Howell, I have discovered antithrombin’. He was most skeptical. So I had the Deiner, John Schweinhant, bleed a cat. Into a small beaker full of its blood, I stirred all of a proven batch of heparophosphatides, and I placed this on Dr. Howell’s laboratory table and asked him to call me when it clotted. It never did clot. [It was heparin]5
Use of heparin
Treatment of established venous thromboembolism
Coincident with commencing heparin, patients usually start taking an oral vitamin K antagonist, typically warfarin in the UK. The INR is monitored and loading doses of VKA are given according to a validated loading protocol in order to minimise the risk of over-anticoagulation and bleeding. Ideally the INR should be measured daily during the first 4 days of loading with a VKA; guidance is available at http://www.bcshguidelines.com.
Fibrinolytic (thrombolytic) system
The system acts to remove intravascular fibrin, thereby restoring blood flow.
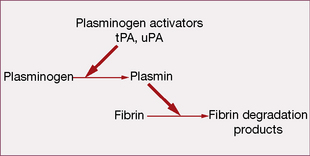
Drugs that promote fibrinolysis
Platelet function
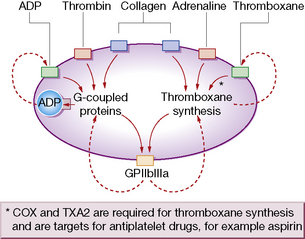
Platelets have rightly been termed ‘pharmacological packages’. To deliver the above functions, they must first undergo a process of activation that involves multiple agonists through numerous intracellular second-messenger pathways and complex networks (Fig. 29.3). These pathways converge on and activate the fibrinogen receptor, glycoprotein IIbIIIa (integrin αIIbβ3), inducing a conformational change that results in fibrinogen/fibrin binding. When fibrinogen occupies the receptor, outside-in signalling consolidates platelet activation by up-regulating second-messenger pathways, so providing a positive feedback loop.
Drugs that inhibit platelet activity (antiplatelet drugs)
(See also Myocardial infarction, p. 411.)
Aspirin prevents formation of both thromboxane A2 (TXA2) and prostacyclin (PGI2) (see Fig.16.1, p. 241). Therapeutic interest in the antithrombotic effect of aspirin has centred on separating these actions by using a low dose. In general, 75–100 mg/day by mouth will abolish synthesis of TXA2 without significant impairment of prostacyclin formation, i.e. amounts substantially below the 2.4 g/day used to control pain and inflammation. Laboratory testing of TXA2 production or TXA2-dependent platelet function can provide an assessment of the adequacy of aspirin dose. Among several causes of resistance to aspirin are genetic polymorphisms of COX-1 and other genes involved in thromboxane biosynthesis.6
Glycoprotein (GP) IIb–IIIa antagonists
The platelet glycoprotein IIb–IIIa complex is the predominant platelet integrin, a molecule restricted to megakaryocytes and platelets that mediates platelet aggregation by the binding of proteins such as fibrinogen and von Willebrand factor (vWF) (see Fig. 29.4). Hereditary absence of the GPIIb–IIIa complex (Glanzmann’s thrombasthenia) results in platelets that are incapable of aggregation by physiological agonists.
Uses of antiplatelet drugs
Antiplatelet therapy protects at-risk patients against stroke, myocardial infarction or death. A meta-analysis of 145 clinical trials of prolonged (> 1 month) antiplatelet therapy versus control, and trials between antiplatelet regimens, found that the chance of non-fatal myocardial infarction and non-fatal stroke fell by one-third, and that there was a one-sixth reduction in the risk of death from any vascular cause.7 Expressed in another way, in the first month after an acute myocardial infarction (a vulnerable period) aspirin prevents death, stroke or a further heart attack in about 4 of every 100 patients treated. Continuing treatment from the end of year 1 to year 3 conferred further benefit.
Aspirin is by far the most commonly used anti-platelet agent. The optimal dose is not certain, but one not exceeding aspirin 325 mg/day is acceptable, and 75–100 mg/day may be as effective and preferred where there is gastric intolerance. Aspirin alone (mainly) or aspirin plus dipyridamole greatly reduced the risk of occlusion where vascular grafts or arterial patency were studied systematically.8
Many patients who take aspirin for vascular disease may also require a NSAID, e.g. for joint disease. Given their common mode of action by inhibiting prostaglandin synthesis, this raises the issue that NSAIDs may block access of aspirin to active sites on platelets, with loss of cardioprotection. Retrospective cohort9 and case-control10 studies suggest no adverse interaction with ibuprofen, but the issue remains unresolved and in the meantime it seems prudent to take aspirin 2 h before a NSAID, e.g. at bedtime.
• Coagulation does not occur as a consequence of linear sequential enzyme activation pathways but by a network of simultaneous interactions, with regulation and modulation of these interactions during the thrombin generation process itself.
• Vitamin K is necessary for the final stage in the synthesis of coagulant factors II (prothrombin), VII, IX and X, and anticoagulant regulatory proteins, proteins C and S.
• Vitamin K is used to treat haemorrhage or threatened bleeding due to the coumarin or indanedione anticoagulants, haemorrhagic disease of the newborn and hypoprothrombinaemia due to intestinal malabsorption syndromes.
• Desmopressin increases the plasma concentration of factor VIII and von Willebrand factor, directly activates platelets, and is useful in patients with mild haemophilia A and von Willebrand’s disease.
• The predominant effect of anticoagulant drugs is to limit thrombin generation, or to neutralise thrombin.
• Warfarin and other oral vitamin K antagonists act by reducing the activity of vitamin K-dependent clotting factors (see above); they take 4–5 days to produce a therapeutic effect. Warfarin is the oral anticoagulant of choice, for it is reliably effective and has the lowest incidence of adverse effects.
• Oral VKA have a delayed pharmacodynamic effect relative to their pharmacokinetic profiles with both a slow on and off effect but the anticoagulant effect can be reversed with factor concentrate (II, VII, IX & X) and vitamin K.
• Oral direct thrombin and anti-Xa inhibitors have a fast pharmacodynamic effect in parallel with their pharmacokinetic profile. The anticoagulant effect cannot be reversed.
• Oral anticoagulant drugs are used to prevent and treat venous thrombosis and pulmonary embolus, and to prevent arterial thromboemboli in patients with atrial fibrillation or cardiac disease, including mechanical heart valves.
• Heparin depends for its anticoagulant action on the presence in plasma of antithrombin, a naturally occurring inhibitor of activated coagulation proteases that include thrombin, factor Xa and factor IXa.
• Patients with acute venous thromboembolism can be treated safely and effectively with low molecular weight heparin as outpatients.
• LMWHs and direct thrombin and Xa inhibitors are the preferred drugs for perioperative prophylaxis and are at least as effective as standard heparin for unstable angina.
• Fibrinolytic drugs dissolve thrombi in acutely occluded coronary arteries, thereby restoring blood flow to ischaemic myocardium and improving prognosis. The earlier thrombolysis is given the better the outcome. Thrombolysis is also effective for massive pulmonary emboli with cardiovascular compromise.
• Aspirin acetylates and thus inactivates cyclo-oxygenase (COX), the enzyme responsible for the first step in the formation of prostaglandins, and in low dose reduces platelet activity by preventing the formation of thromboxane.
• Clopidogrel inhibits ADP-dependent platelet aggregation; it reduces the risk of stroke, myocardial infarction or vascular death.
• GPIIb–IIIa antagonists block the final common pathway of platelet aggregation (the binding of fibrinogen or vWF to the GPIIb–IIIa complex) and are more complete inhibitors of platelets than either aspirin or clopidogrel.
• Antiplatelet therapy protects at-risk patients against stroke, myocardial infarction or death.
Baglin T., Barrowcliffe T.W., Cohen A., Greaves M. Guidelines on the use and monitoring of heparin. Br. J. Haematol.. 2006;133:19–34.
Baglin T.P., Keeling D.M., Watson H.G. Guidelines on oral anticoagulation (warfarin): third edition-2005 update. Br. J. Haematol.. 2006;132:277–285.
CRASH-2 trial collaborators, Shakur H., Roberts I., Bautista R., et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376:23–32.
De Meyer S.F., Vanhoorelbeke K., Broos K., et al. Antiplatelet drugs. Br. J. Haematol.. 2008;142:515–528.
Di Nisio M., Middeldorp S., Büller H.R., et al. Direct thrombin inhibitors. N. Engl. J. Med.. 2005;353:1028–1040.
Huntington J.A., Baglin T.P. Targeting thrombin: rational drug design from natural mechanisms. Trends Pharmacol. Sci.. 2003;24:589–595.
Patrono C., Garcia Rodriguez L.A., Landolfi R., Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med.. 2005;353:2373–2383.
Weitz J.I., Hirsh J., Samama M.M. New antithrombotic drugs: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133:234S–256S.
1 Genetically controlled by an active promoter and constantly produced rather than depending on the presence of an inducer.
2 Serpin: serine protease inhibitors. Antithrombin is the principal serpin involved in regulating coagulation.
3 Coumarins are present in many plants and are important in the perfume industry; the smell of new mown hay and grass is due to coumarins. Yellow sweet clover (King’s clover) is rich in coumarins and was used as a herbal medicine to reduce inflammation. It was a constituent of an ointment to ‘cool and dry and comfort the Membre’ of King Henry VIII of England, who enjoyed a particularly active sexual life (Cutler T 2003 College Commentary, May/June. Royal College of Physicians, London, p. 23). The discovery of coumarins as anticoagulants dates from investigation of an unexplained haemorrhagic disease of cattle that had eaten mouldy sweet clover. Subsequent research at the University of Wisconsin, USA, culminated in the isolation of the causative agent, dicoumarol (Stahmann M A, Huebner C F, Link K P 1941 Journal of Biological Chemistry 138:513–527).
4 Warfarin is 10 times more potent than dicoumarol and was originally used as a rodenticide. Its name is derived from the patent holder, Wisconsin Alumni Research Foundation, and the suffix comes from ‘coumarin’.
5 McLean gives a fascinating account of his struggles to pay his way through medical school, as well as his discovery of heparin in: McLean J 1959 Circulation XIX:75.
6 Hankey G J, Eikelboom J W 2006 Aspirin resistance. Lancet 367:606–617.
7 Antiplatelet Trialists’ Collaboration 1994 Collaborative overview of randomised trials of antiplatelet therapy – I: Prevention of death, myocardial infarction and stroke by prolonged antiplatelet therapy and various categories of patients. British Medical Journal 308:81–106.
8 Antiplatelet Trialists’ Collaboration 1994 Collaborative overview of randomised trials of antiplatelet therapy – II: Maintenance of vascular grafts or arterial patency by antiplatelet therapy. British Medical Journal 308:159–168.
9 García Rodríguez L A, Varas-Lorenzo C, Maguire A, González-Pérez A 2004 Nonsteroidal anti-inflammatory drugs and the risk of myocardial infarction in the general population. Circulation 109:3000–3006.
10 Patel T N, Goldberg K C 2004 Use of aspirin and ibuprofen compared with aspirin alone and the risk of myocardial infarction. Archives of Internal Medicine 164:852–856.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 29 Drugs and haemostasis
• Coagulation system: the mode of action of drugs that promote coagulation and that prevent it (anticoagulants) and their uses.
• Fibrinolytic system: the mode of action of drugs that promote fibrinolysis (fibrinolytics) and their uses to lyse arterial and venous thrombi (thrombolysis).
• Platelets: the ways that drugs that inhibit platelet activity benefit arterial disease.
The coagulation system
Coagulation initiates with tissue factor (TF), a cell membrane protein that binds activated factor VII (indicated by adding the letter ‘a’, i.e. factor VIIa). Although there is a small fraction of circulating factor VII in the activated state, it has little or no enzymatic activity until it is bound to TF. Most non-vascular cells express TF in a constitutive1 fashion, whereas de novo TF synthesis can be induced in monocytes and damaged endothelial cells. Injury to the arterial or venous wall exposes extravascular TF-expressing cells to blood. Lipid-laden macrophages in the core of atherosclerotic plaques are particularly rich in TF, thereby explaining the propensity for thrombus formation at sites of plaque disruption. Once bound to TF, factor VIIa activates factor IX and factor X (to IXa and Xa, respectively), leading to thrombin generation and clot formation (Fig. 29.1).
In the current model, blood coagulation starts with a transient release of tissue factor by damaged endothelium, resulting in the formation of sub-nanomolar amounts of thrombin via TF/VIIa-driven Xa formation (extrinsic-tenase). The initial thrombin activity is necessary to prime the system for a full thrombin explosion. Tissue factor pathway inhibitor (TFPI) rapidly shuts down this priming pathway and the full thrombin explosion is then dependent on factor IXa-driven Xa formation. Factor IXa-driven Xa formation (intrinsic-tenase) is amplified by the thrombin explosion itself, as thrombin forms a positive feedback loop by activating factor XIa (not shown in Fig. 29.1), which converts more IX to IXa.
• feedback activation of factor V and factor VIII
• activating platelet-bound factor XI, thereby leading to further factor Xa generation
• activating cells that provide the phospholipid surface required for assembly of the macromolecular enzymatic complexes.
Procoagulant drugs
Vitamin K
During γ-carboxylation of the proteins, the reduced and active form of vitamin KH2 converts to an epoxide, an oxidation product. Subsequently vitamin K epoxide reductase converts oxidised vitamin K back to the active vitamin K, i.e. there exists an interconversion cycle between vitamin K epoxide and reduced vitamin K (Fig. 29.2).
Vitamin K deficiency may arise from:
• bile failing to enter the intestine, e.g. obstructive jaundice or biliary fistula
• malabsorption syndromes, e.g. coeliac disease, or after extensive small intestinal resection
• reduced alimentary tract flora, e.g. in newborn infants and rarely after broad-spectrum antibiotics.
The following preparations of vitamin K are available:
Vitamin K is used to treat the following:
• Haemorrhage or threatened bleeding due to the coumarin or indanedione anticoagulants. Phytomenadione is preferred for its more rapid action; dosage regimens vary according to the degree of urgency and the original indication for anticoagulation.
• Haemorrhagic disease of the newborn, which develops usually between 2 and 7 days, and late haemorrhagic disease that presents at 6–7 months. Prophylaxis is recommended during the period of vulnerability with vitamin K (phytomenadione, as Konakion) 1 mg by single i.m. injection at birth. Alternatively, give vitamin K by mouth as two doses of a colloidal (mixed micelle) preparation of phytomenadione in the first week. Breast-fed babies should receive a further 2 mg at 1 month of age. Formula-fed babies do not need this last supplement as the formula contains vitamin K. Fears that intramuscular vitamin K might cause childhood cancer have been dispelled.
• Intestinal malabsorption syndromes; menadiol sodium phosphate should be used as it is water soluble.
Coagulation factor concentrates
Use of coagulation factor concentrates
• Superficial haemorrhage sometimes responds to local pressure.
• Minor bleeding can arrest with plasma factor concentrations of 0.25–0.30 units/mL, but severe bleeding requires at least 0.50 units/mL and surgical procedures or life-threatening haemorrhage require 0.75–1.00 units/mL by infusion of factor concentrate.
• In haemophilia A, factor VIII concentrate (t½ 8–12 h) is used for bleeding that is more than minor. Repeat dosing is necessary to maintain haemostatic levels.
• Factor IX (t½ 18–24 h) is used for bleeding that is more than minor in haemophilia B (Christmas disease).
• The speed of recovery of the affected joint or resolution of a haematoma determines the duration of therapy. After surgery, 7–14 days of replacement therapy is required to ensure adequate wound healing and to prevent secondary haemorrhage.
• Primary prophylaxis with factor concentrates two or three times weekly at doses sufficient to keep the factor above 0.01–0.02 units/mL reduces bleeding and hence the severity of chronic haemophilic arthropathy.
Desmopressin (DDAVP)
DDAVP is useful for treating patients with mild haemophilia A and von Willebrand’s disease, especially for short-term therapy. For dental extraction, a single injection of 0.3 micrograms/kg 1–2 h before surgery, combined with the oral antifibrinolytic drug, tranexamic acid, for 5–7 days after the procedure (see Antifibrinolytic drugs, p. 492), will produce normal haemostasis and prevent secondary haemorrhage.
DDAVP shortens the bleeding time in patients with renal or liver failure.
Anticoagulant drugs
• limiting thrombin generation, either as a result of inhibiting other proteases (clotting factors) involved in its generation or by reducing the activity of zymogens (the precursor inactive forms of the enzymes); or
• inhibiting (neutralising) thrombin activity, either directly or indirectly, depending on whether or not they activate the natural serpin-dependent anticoagulant pathway.2
Oral vitamin K antagonists (VKA)
Warfarin and other oral vitamin K antagonists (VKA) reduce the activity of zymogens.
During the γ-carboxylation of factors II (prothrombin), VII, IX and X (and also the natural anticoagulant proteins C and S), active vitamin K (KH2) is oxidised to an epoxide and must be reduced by the enzymes vitamin K epoxide reductase and vitamin K reductase to become active again (see the vitamin K cycle, p. 483). Coumarins3 are structurally similar to vitamin K and competitively inhibit vitamin K epoxide reductase and vitamin K reductase, so limiting availability of the active reduced form of the vitamin to form coagulant (and anticoagulant) proteins. The overall result is a shift in haemostatic balance in favour of anticoagulation because of the accumulation of clotting proteins with absent or decreased γ-carboxylation sites (PIVKAs).4
[/not-level-membership-for-basic-science-category]
