[level-membership-for-pulmolory-and-respiratory-category]CHAPTER 22
Drugs affecting circulation: antihypertensives, antianginals, antithrombotics
After reading this chapter, the reader will be able to:
1. Define terms that pertain to drugs affecting circulation: antihypertensives, antianginals, and antithrombotics
2. Categorize the stages of normal to high blood pressure
3. Define a hypertensive crisis, and differentiate between hypertensive emergency and hypertensive urgency.
4. Design an algorithm for the pharmacotherapy of hypertension.
5. Compare and contrast the clinical pharmacology of the agents used for hypertensive pharmacotherapy
6. Describe the chronotherapeutic effect of blood pressure, and design a pharmacotherapy regimen based on this principle
7. Describe the mechanism of action of angiotensin-converting enzyme inhibitors, calcium channel blockers, and β blockers
8. Compare and contrast the clinical pharmacology of spironolactone and eplerenone
9. List drug-drug interactions relevant to antihypertensives and plausible mechanisms
10. Describe the formation and elimination of an acute coronary thrombus
11. Describe the pathophysiology of angina and the drugs used to treat angina
12. List the agents in each of the following antithrombotic classes: anticoagulants, antiplatelets, and thrombolytics
13. Describe the mechanism of action of heparin
14. Compare and contrast the clinical pharmacology of heparin and low-molecular-weight heparin (LMWH)
15. List the laboratory parameters that may be used to monitor for the effect of heparin, LMWH, and direct thrombin inhibitors
16. Describe the mechanism of heparin-induced and warfarin-induced paradoxical thrombosis
17. Compare and contrast the clinical pharmacology of aspirin, clopidogrel, ticlopidine, and dipyridamole
18. Describe the role of genetic polymorphism in the antiplatelet activity of clopidogrel and anticoagulant effect of warfarin
19. Describe the indication and mechanism of action of glycoprotein IIb/IIIa inhibitors
20. List the indications and contraindication of thrombolytic agents
Drug that prevents or breaks up blood clots in conditions such as thrombosis or embolism; antithrombotics include anticoagulants, antiplatelets, and thrombolytics.
Arterial blood pressure (blood pressure)
Defined hemodynamically as the product of systemic vascular resistance and cardiac output (heart rate × stroke volume).
Damage to the heart and the blood vessels or circulation, including to the brain, kidney, and the eye.
Influencing the rate of rhythmic movements (heartbeat).
Human biologic variations of rhythm within a 24-hour cycle.
Measurement of the renal clearance of endogenous creatinine per unit time; approximates glomerular filtration rate (GFR) but overestimates GFR by 10% to 15%; used for drug-dosing guidelines.
Covalently cross-linked degradation fragments of the cross-linked fibrin polymer during plasmin-mediated fibrinolysis; level increases after the onset of fibrinolysis and allows for identification of the presence of fibrinolysis.
Maximum dose of a drug, beyond which it no longer exerts a therapeutic effect; however, its toxic effect increases.
Fibrin split or fibrinogen degradation products (FDPs)
Small peptides that result following the action of plasmin on fibrinogen and fibrin in the fibrinolytic process. FDPs are anticoagulant substances that can cause bleeding if fibrinolysis becomes uncontrolled and excessive.
Glomerular filtration rate (GFR)
Volume of water filtered from the plasma by the kidney via the glomerular capillary walls into Bowman capsules per unit time; considered to be 90% of creatinine clearance and equivalent to insulin clearance.
Blood pressure greater than 180/120 mm Hg, with the elevation of blood pressure accompanied by acute, progressing target organ injury.
Blood pressure greater than 180/120 mm Hg without signs or symptoms of acute target organ complications.
Drug influencing the contractility of a muscle (heart).
Intrinsic sympathomimetic activity (ISA)
Having the ability to activate and block adrenergic receptors, producing a net stimulatory effect on the sympathetic nervous system.
Treatment of disease by drug therapy.
Enzyme also known as angiotensinogenase, released by the kidney in response to a lack of renal blood flow and responsible for converting angiotensinogen into angiotensin I.
Neurotransmitter or hormone replacements that may be weaker or inert.
The circulatory system comprises an integral functional part of the cardiopulmonary system. Drug therapy affecting the circulation is seen in the acute critical care, outpatient care, and home care environments. This chapter presents three classes of drug therapy, all targeted at the circulatory system. After a brief review of the epidemiology, etiology, and pathophysiology of hypertension, the multiple drug groups used as antihypertensives are described. Drugs used to treat angina pectoris are the second group of drugs described. The third group of agents affecting circulation, antithrombotics, comprises several classes of drugs used to regulate clotting mechanisms.
Hypertension
Epidemiology and etiology
More than 1 billion people worldwide and 1 in every 4 Americans has high blood pressure (≥140/90 mm Hg). Hypertension adversely affects numerous body organs, including the heart, brain, kidney, and eye. Damage to these organ systems resulting from hypertension is termed cardiovascular disease (CVD). Uncontrolled hypertension increases CVD morbidity and mortality by increasing the risk of developing left ventricular hypertrophy, angina, myocardial infarction (MI), heart failure, stroke, peripheral arterial disease, retinopathy, and kidney disease. One of eight deaths can be attributed to hypertension, and the World Health Organization reports that suboptimal blood pressure (systolic blood pressure above 115 mm Hg) is responsible for 62% of cerebrovascular disease and 49% of ischemic heart disease. Blood pressure increases with age, and hypertension is more prevalent in adults older than 65 years. This fact is of great concern because it is estimated that by 2040, 25% of the American population will be older than 65. Hypertension occurs more frequently in men than in women and occurs in more blacks than whites. Evidence suggests that individuals who are normotensive have a greater than 90% lifetime risk for developing hypertension by age 55 (Table 22-1).1
TABLE 22-1
JNC-VII Classification of Blood Pressure for Adults
| CATEGORY | SYSTOLIC (mm Hg) | DIASTOLIC (mm Hg) |
| Normal | <120 | <80 |
| Prehypertension | 120-139 | 80-89 |
| Hypertension | ||
| Stage 1 | 140-159 | 90-99 |
| Stage 2 | ≥160 | ≥100 |
Hypertension is diagnosed by the mean of two or more separate seated blood pressure determinations on different days. The Seventh Report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC-VII), published in 2003, is the most up-to-date guideline compared with other hypertension treatment recommendations.2 The American Heart Association (AHA) released a statement regarding the treatment of hypertension as it relates to the prevention and management of ischemic heart disease, which differed from the JNC-VII recommendations.3 Significant differences between JNC-VII and the AHA statement include expanding the category of the high-risk hypertensive to include patients with known coronary artery disease (CAD) or CAD risk equivalents (e.g., carotid artery disease, peripheral arterial disease, or abdominal aortic aneurysm or a 10-year Framingham risk score of more than 10%).
In almost all cases, the etiology of hypertension is unknown, and it is termed either primary hypertension or essential hypertension. The prevalence of secondary hypertension is less than 10%; secondary hypertension includes many disease-induced and drug-induced etiologies. Disease-induced causes of hypertension include Cushing syndrome, hyperparathyroidism, hyperthyroidism, pheochromocytoma, primary aldosteronism, and kidney disease. Drug-induced causes of hypertension include amphetamines, corticosteroids, cyclosporine, erythropoietin, estrogens, nonsteroidal antiinflammatory drugs (NSAIDs) including cyclooxygenase-1 inhibitors (e.g., ibuprofen and naproxen) and cyclooxygenase-2 inhibitors (e.g., celecoxib), pseudoephedrine, sibutramine, tacrolimus, venlafaxine, high sodium–containing over-the-counter (OTC) products (e.g., Alka-Seltzer effervescent antacid tablets), OTC weight loss products (e.g., ephedrine-containing diet pills), and chronic alcohol ingestion.4,5
Hypertensive crisis
A patient with blood pressure greater than 180/120 mm Hg is considered to be in a hypertensive crisis. A hypertensive crisis represents either a hypertensive urgency or a hypertensive emergency. Hypertensive urgencies usually signify high blood pressures without signs or symptoms of acute target organ complications; however, patients may present with severe headaches, shortness of breath, nosebleeds, or severe anxiety. In these situations, improvement in blood pressure control can be accomplished over a period of 24 to 48 hours.2 Overaggressive use of intravenous drugs and oral medications can cause too rapid a decrease in blood pressure. Rapid decrease in blood pressure can result in hypoperfusion of organs such as the brain, kidneys, and heart. Oral antihypertensive agents such as captopril, clonidine, and labetalol are routinely used to manage hypertensive urgencies, followed by close observation for several hours. Patients can benefit from antihypertensive medication adjustments if they are found to be noncompliant with taking their medications.
< ?xml:namespace prefix = "mml" />
Hypertension pharmacotherapy
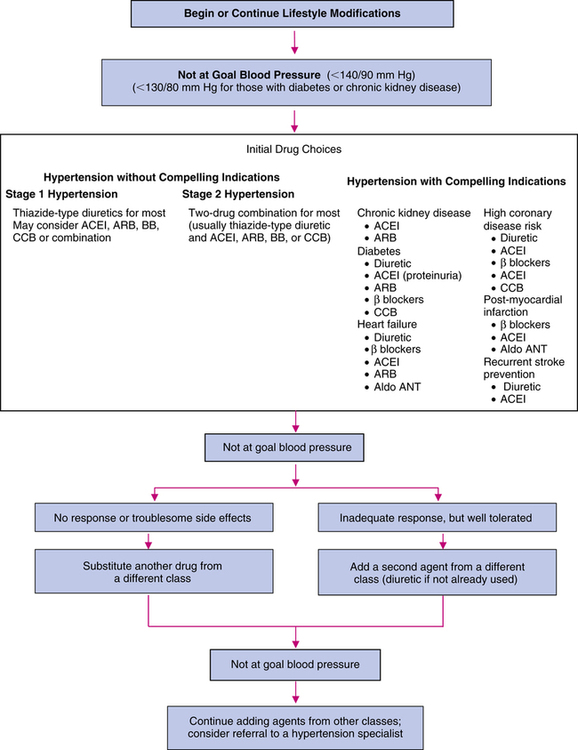
First-line agents for the treatment of uncomplicated hypertension are thiazide-type diuretics, including ACEIs, ARBs, β blockers, and CCBs.2 Vasodilators, α-blocking agents, α2 agonists, and antiadrenergic agents are considered second-line antihypertensives.2 It is unknown if direct renin inhibitors (DRIs), a new class of antihypertensives, should be considered as first-line agents because long-term morbidity and mortality data are currently unavailable. For stage 1 hypertension, pharmacotherapy should be initiated for most patients with a low dose of a once-daily agent, usually a thiazide-type diuretic, and titrated upward until blood pressure control is achieved or intolerable adverse effects occur. Clinicians should be cognizant that monotherapy achieves effective blood pressure control in only 60% to 70% of patients. Thiazide diuretics profoundly decrease CVD morbidity and mortality, enhance the antihypertensive effects of the other antihypertensives, and are very useful in achieving blood pressure control. For stage 2 hypertension, because a higher response rate may be achieved by initiating low-dose combination antihypertensives, usually a thiazide-type diuretic plus an alternative first-line agent is used. The low-dose combination method may minimize adverse effects and may maximize efficacy and compliance.6,7 Figure 22-1 presents an algorithm for the management of hypertension.2,3
Angiotensin-converting enzyme inhibitors
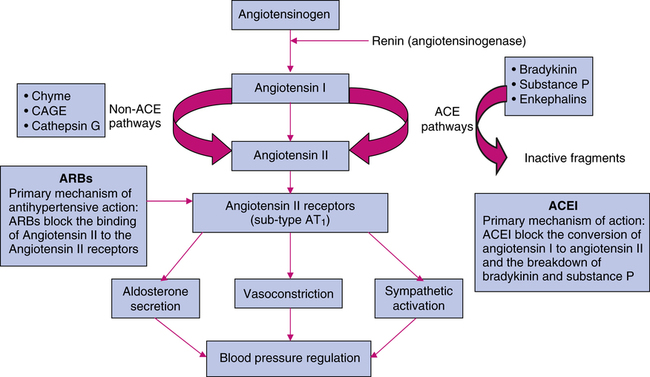
ACEIs act primarily through suppression of the renin-angiotensin-aldosterone system (RAAS). Because of a lack of renal blood flow, renin is released into the circulation, where it acts on angiotensinogen to produce angiotensin I. In the pulmonary vasculature, angiotensin I is converted by angiotensin-converting enzyme (ACE) to angiotensin II. Angiotensin II is a highly potent endogenous vasoconstrictor that also stimulates aldosterone secretion from the zona glomerulosa cells of the adrenal cortex, contributing to sodium and water retention.8 Angiotensin II also stimulates the release of catecholamines from the adrenergic nerve endings and mediates the release of central sympathetic outflow. ACE is abundant in the endothelial cells of blood vessels and to a lesser extent in the kidneys.
ACEIs block the conversion of angiotensin I to angiotensin II by competing with the physiologic substrate angiotensin I for the active site of ACE (Figure 22-2). The affinity of ACEIs for ACE is approximately 30,000 times greater than for angiotensin I. ACEIs also inhibit kininase which is responsible for the degradation of bradykinin and other vasodilating substances, including prostaglandin E2 (PGE2) and prostacyclin (PGI2), which enhances the antihypertensive effects of these drugs. Because ACEIs are potent antihypertensives in patients with low-renin hypertension, the effects on bradykinin may have an integral role in the mechanism of action of these agents. The hemodynamic effects of ACEIs are a reduction of peripheral arterial resistance, an increase in cardiac output, little or no change in heart rate, an increase in renal blood flow, and unchanged glomerular filtration rate (GFR). ACEIs have mild antihyperlipidemic effects.
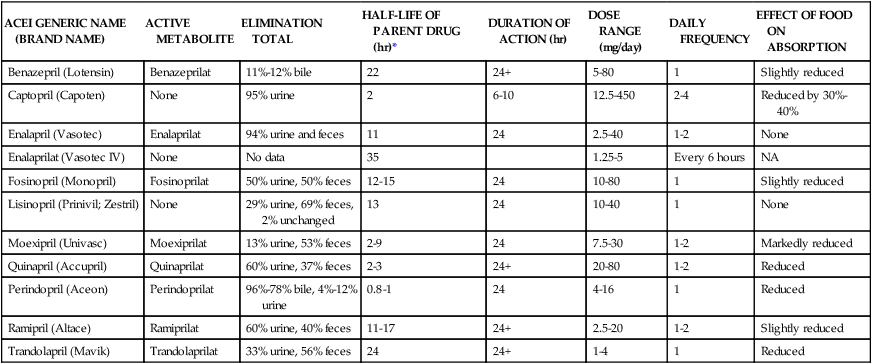
ACEIs rival diuretics as the most effective first-line antihypertensives to decrease CVD morbidity and mortality in various settings. In contrast to β blockers and thiazide diuretics, ACEIs do not induce glucose intolerance, hyperlipidemia, or hyperuricemia. ACEIs are homogeneous, which means there is very little variability among ACEIs in terms of efficacy and toxicity. With the exception of captopril, all ACEIs are generally administered once or twice daily. Enalaprilat is the only available parenteral ACEI. Table 22-2 presents the pharmacokinetics and dosing guidelines for ACEIs.4,5
TABLE 22-2
Pharmacokinetics and Dosing Guidelines for Angiotensin-Converting Enzyme Inhibitors (ACEIs)
| ACEI GENERIC NAME (BRAND NAME) | ACTIVE METABOLITE | ELIMINATION TOTAL | HALF-LIFE OF PARENT DRUG (hr)* | DURATION OF ACTION (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY | EFFECT OF FOOD ON ABSORPTION |
| Benazepril (Lotensin) | Benazeprilat | 11%-12% bile | 22 | 24+ | 5-80 | 1 | Slightly reduced |
| Captopril (Capoten) | None | 95% urine | 2 | 6-10 | 12.5-450 | 2-4 | Reduced by 30%-40% |
| Enalapril (Vasotec) | Enalaprilat | 94% urine and feces | 11 | 24 | 2.5-40 | 1-2 | None |
| Enalaprilat (Vasotec IV) | None | No data | 35 | 1.25-5 | Every 6 hours | NA | |
| Fosinopril (Monopril) | Fosinoprilat | 50% urine, 50% feces | 12-15 | 24 | 10-80 | 1 | Slightly reduced |
| Lisinopril (Prinivil; Zestril) | None | 29% urine, 69% feces, 2% unchanged | 13 | 24 | 10-40 | 1 | None |
| Moexipril (Univasc) | Moexiprilat | 13% urine, 53% feces | 2-9 | 24 | 7.5-30 | 1-2 | Markedly reduced |
| Quinapril (Accupril) | Quinaprilat | 60% urine, 37% feces | 2-3 | 24+ | 20-80 | 1-2 | Reduced |
| Perindopril (Aceon) | Perindoprilat | 96%-78% bile, 4%-12% urine | 0.8-1 | 24 | 4-16 | 1 | Reduced |
| Ramipril (Altace) | Ramiprilat | 60% urine, 40% feces | 11-17 | 24+ | 2.5-20 | 1-2 | Slightly reduced |
| Trandolapril (Mavik) | Trandolaprilat | 33% urine, 56% feces | 24 | 24+ | 1-4 | 1 | Reduced |
Angiotensin II receptor blockers
Several nonrenin and non-ACE pathways are used for the production of angiotensin II (see Figure 22-2). Nonrenin pathways generate angiotensin II from angiotensinogen via tissue plasminogen activator, cathepsin G, and tonin. Non-ACE enzymes that generate angiotensin II from angiotensin I are cathepsin G, chymostatin-sensitive angiotensin II–generating enzyme, and chymase. ACEIs incompletely block the synthesis of angiotensin II. ARBs are angiotensin II type 1 (AT1) receptor antagonists. AT1 receptors are found in many tissues, such as vascular smooth muscle, myocardial tissue, brain, kidney, liver, uterus, and adrenal glands (cortex and medulla). Many tissues also have an AT2 receptor; however, it is not known to have effects on myocardial hemostasis. ARBs have 1000-fold greater affinity for AT1 receptors than AT2 receptors and generally do not block the AT2 receptor. Because ARBs do not inhibit ACE, they do not interfere with the concentrations of bradykinins and substance P. This kinin-sparing effect may explain why ARBs have a low incidence of inducing cough or angioedema. However, the beneficial effects of kinins, including blood pressure–lowering potency, may be sacrificed.
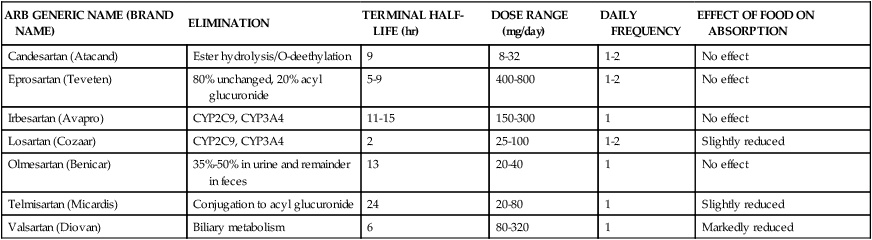
Compared with ACEIs, ARBs are considered as potent or slightly weaker antihypertensive agents. The inhibition of bradykinin by ACEIs may account for its augmented antihypertensive effect. Angiotensin II receptor blockers arguably are considered second-line agents to ACEIs for hypertension and heart failure and are indicated when ACEI-induced cough or other adverse effects are intolerable. However, ARBs may be considered superior to ACEIs in patients with type 2 diabetic nephropathy. ARBs are administered once or twice daily. Using the combination of an ACEI and an ARB has not been well studied; however, its beneficial effects have been observed in patients with heart failure and nephrotic syndrome. Table 22-3 presents the pharmacokinetics and dosing guidelines for ARBs.4,5,8
TABLE 22-3
Pharmacokinetics and Dosing Guidelines for Angiotensin II Receptor Blockers (ARBs)
| ARB GENERIC NAME (BRAND NAME) | ELIMINATION | TERMINAL HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY | EFFECT OF FOOD ON ABSORPTION |
| Candesartan (Atacand) | Ester hydrolysis/O-deethylation | 9 | 8-32 | 1-2 | No effect |
| Eprosartan (Teveten) | 80% unchanged, 20% acyl glucuronide | 5-9 | 400-800 | 1-2 | No effect |
| Irbesartan (Avapro) | CYP2C9, CYP3A4 | 11-15 | 150-300 | 1 | No effect |
| Losartan (Cozaar) | CYP2C9, CYP3A4 | 2 | 25-100 | 1-2 | Slightly reduced |
| Olmesartan (Benicar) | 35%-50% in urine and remainder in feces | 13 | 20-40 | 1 | No effect |
| Telmisartan (Micardis) | Conjugation to acyl glucuronide | 24 | 20-80 | 1 | Slightly reduced |
| Valsartan (Diovan) | Biliary metabolism | 6 | 80-320 | 1 | Markedly reduced |
Direct renin inhibitors
DRIs act by inhibiting renin, the enzyme that is the first step of the RAAS (see Figure 22-2). Renin is responsible for the conversion of angiotensinogen to angiotensin I, which is the rate-limiting step in RAAS. Renin inhibition also leads to decreased formation of angiotensin II and aldosterone. However, all agents that inhibit the RAAS, such as ACEIs, have the potential to inhibit feedback inhibition of renin leading to increases in renin and its activity. This effect can be blocked with the use of a renin inhibitor. DRIs can be used alone or in combination with other antihypertensive agents.
Aliskiren is administered once daily at a dose of 150 to 300 mg. It has very poor oral bioavailability; only about 2.5% is absorbed. Absorption of aliskiren is substantially decreased by high fatty meals; patients should always take it the same way: either with or without food. It undergoes minimal hepatic metabolism by CYP3A4. Cyclosporine and itraconazole, which are potent inhibitors of CYP3A4, were shown to increase aliskiren levels significantly and should not be used concomitantly. Other CYP3A4 inhibitors were also shown to increase aliskiren levels, but the clinical significance of their interaction is unknown. Aliskiren has also been shown to reduce the effectiveness of furosemide by 30% to 50%. The effectiveness of furosemide should be monitored when these two agents are used concomitantly. Approximately 25% of the absorbed dose is excreted unchanged in the urine. Most of the unabsorbed drug is excreted in the feces. No dosage adjustments are recommended at this time in patients with renal or hepatic impairment.9,10
Calcium channel blockers
Dihydropyridine CCBs are potent vasodilators; these agents include amlodipine, felodipine, isradipine, nicardipine, nifedipine, and nisoldipine. With the exception of nifedipine, dihydropyridines have negligible chronotropic effects. Immediate-release nifedipine, especially when administered as a liquid (pseudosublingual), causes a potent reflex tachycardia that increases coronary oxygen demand and has been implicated with an increased risk of MI and stroke. Only sustained-release dosage forms of nifedipine are indicated for hypertension.11 Amlodipine and plausibly felodipine may be used in patients with heart failure because these agents do not decrease cardiac contractility. CCBs are very effective antihypertensive agents in both elderly and black patients. Table 22-4 presents the pharmacokinetics and dosing guidelines for calcium antagonists.2–5
TABLE 22-4
Pharmacokinetics and Dosing Guidelines for Calcium Channel Blockers
| CALCIUM ANTAGONIST GENERIC NAME (BRAND NAME) | ONSET OF ACTION OF ORAL DOSAGE FORMS (hr) | HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Nondihydropyridines | ||||
| Verapamil (Calan, Isoptin) | 0.5 | 3-7 | 180-480 | 3-4 |
| Verapamil SR (Calan SR, Isoptin SR) | 0.5 | 3-7 | 120-480 | 1-2 |
| Verapamil ER (Covera-HS) | 4-5 | 2.8-7.4 | 180-420 | Once at bedtime |
| Verapamil chronotherapeutic oral drug absorption (Verelan PM) | 4-5 | 3-7 | 100-400 | Once at bedtime |
| Diltiazem (Cardizem) | 0.5 | 3.5 | 90-360 | 3-4 |
| Diltiazem ER capsules (Cardizem CD, Cartia XT, Dilacor XR, Diltia XT, Tiazac, Taztia XT) | 1 | 5 | 90-540 | 1-2 |
| Diltiazem ER tablets (Cardizem LA) | 3-4 | 6-9 | 120-540 | Once daily (morning or evening) |
| Dihydropyridines | ||||
| Amlodipine (Norvasc) | 6-12 | 30-50 | 2.5-10 | 1 |
| Felodipine (Plendil) | 2-5 | 11-16 | 5-20 | 1 |
| Isradipine (DynaCirc) | 2 | 8 | 2.5-10 | 2 |
| Isradipine CR (DynaCirc CR) | 2 | 8 | 2.5-10 | 1 |
| Nicardipine (Cardene) | 20 minutes | 2-4 | 60-120 | 3 |
| Nicardipine SR (Cardene SR) | 20 minutes | 2-4 | 60-120 | 2 |
| Nifedipine (Adalat, Procardia)* | 20 minutes | 2-5 | 30-120 | 3-4 |
| Nifedipine LA (Adalat CC, Procardia XL) | 20 minutes | 7 | 30-120 | 1 |
| Nimodipine (Nimotop)† | ND | 1-2 | 360 | Every 4 hours for 21 days |
| Nisoldipine (Sular) | ND | 7-12 | 20-60 | 1 |
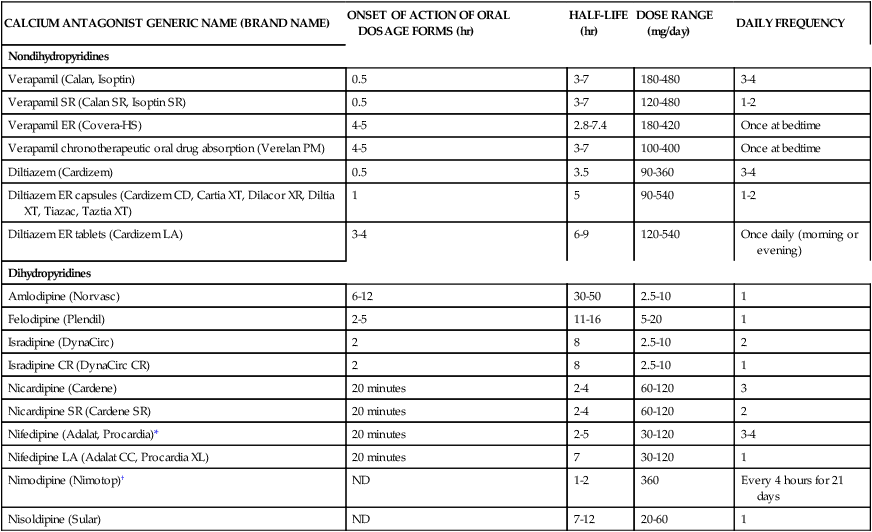
*Nifedipine (prompt release) is not indicated for hypertension.
β blockers
The antihypertensive effects of β blockers have multiple mechanisms of action and are as follows:
• Blockade of the β receptors on the renal juxtaglomerular cells, leading to renin blockade and decreased angiotensin II concentrations
• Blockade of myocardial β receptors, leading to decreased cardiac contractility and heart rate, diminishing cardiac output
• Blockade of central nervous system (CNS) β receptors, leading to decreased sympathetic output from the CNS and plausibly blockade of peripheral β receptors, decreasing norepinephrine concentrations
β blockers are indicated for hypertension, angina pectoris, cardiac dysrhythmias, secondary prevention of MI, chronic heart failure, and pheochromocytoma. β blockers are no longer considered first-line agents in treatment of essential hypertension but should be reserved as add-on therapy to other antihypertensive agents. β blockers are also used for migraine prophylaxis, hypertrophic subaortic stenosis, tremors, alcohol withdrawal syndrome, prophylaxis of esophageal variceal rebleeding, anxiety, symptoms of thyrotoxicosis, and in combination with α blockers for pheochromocytoma. Table 22-5 presents the pharmacokinetics and dosing guidelines for β blockers.2,3,5,9
TABLE 22-5
Pharmacokinetics and Dosing Guidelines for β Blockers
| β BLOCKER GENERIC NAME (BRAND NAME) | α BLOCKADE | β1 SELECTIVITY | ISA | LIPID SOLUBILITY | HALF-LIFE (hr) | DOSE RANGE (mg) | DAILY FREQUENCY |
| Acebutolol (Sectral) | 0 | + | + | Low | 3-4 | 200-200 | 2 |
| Atenolol (Tenormin) | 0 | + | 0 | Low | 6-9 | 25-100 | 1 |
| Betaxolol (Kerlone) | 0 | + | 0 | Low | 14-24 | 5-20 | 1 |
| Bisoprolol (Zebeta) | 0 | ++ | + | Low | 9-12 | 25-200 | 1 |
| Carteolol (Cartrol) | 0 | 0 | + | Low | 6 | 2.5-10 | 1 |
| Carvedilol (Coreg) | + | 0 | 0 | High | 7-10 | 6.25-50 | 2 |
| Labetalol (Trandate, Normodyne) | + | 0 | 0 | Moderate | 3-5 | 100-2400 | 2 |
| Metoprolol (Lopressor) | 0 | + | 0 | Moderate | 3-5 | 50-200 | 1-2 |
| Metoprolol ER (Toprol-XL) | 0 | + | 0 | Moderate | 3-7 | 25-200 | 1 |
| Nadolol (Corgard) | 0 | 0 | 0 | Low | 14-24 | 20-240 | 1 |
| Nebivolol (Bystolic) | 0 | +++ | ? | Low | 11-30 | 5-40 | 1 |
| Penbutolol (Levatol) | 0 | 0 | + | High | 5 | 20-80 | 1 |
| Pindolol (Visken) | 0 | 0 | +++ | Moderate | 3-4 | 10-60 | 2 |
| Propranolol (Inderal) | 0 | 0 | 0 | High | 4-6 | 40-240 | 2 |
| Propranolol LA (Inderal, InnoPran XL) | 0 | 0 | 0 | High | 8-10 | 80-640 | 1 |
| Timolol (Blocadren) | 0 | 0 | 0 | Low | 3-4 | 20-40 | 2 |

Diuretics
Diuretics are divided into the following five classes:
1. Thiazides and thiazide-like agents
4. Carbonic anhydrase inhibitors (CAIs) (e.g., acetazolamide [Diamox])
Thiazide and thiazide-like diuretics
Because thiazides cause hypercalcemia, they may be a useful adjunct in the management and prevention of osteoporosis. Although chlorthalidone, indapamide, and metolazone do not possess the benzothiadiazine structure, pharmacologically they act like thiazide diuretics—they are thiazide-like in structure and activity. Thiazide diuretics lose their antihypertensive potency in patients with a creatinine clearance less than 30 mL/min. Indapamide retains its potency, however, in patients with a creatinine clearance greater than 15 mL/min. Metolazone is the only thiazide-like diuretic that retains potency in patients with a creatinine clearance less than 15 mL/min. Despite the thiazide-like structure of metolazone, its pharmacologic effects are similar to those of loop diuretics. Metolazone is often added to a loop diuretic in patients with diuretic resistance, achieving a synergistic diuretic effect. Mykrox tablets are a formulation of metolazone with a higher bioavailability than conventional metolazone, resulting in a more rapid diuretic effect; Mykrox is not therapeutically equivalent to Zaroxolyn. Table 22-6 presents the pharmacokinetics and dosing guidelines for thiazide and thiazidelike diuretics.2,4,5
TABLE 22-6
Pharmacokinetics and Dosing Guidelines for Thiazides and Thiazide-Like Diuretics
| THIAZIDE/THIAZIDE-LIKE DIURETIC GENERIC NAME (BRAND NAME) | BIOAVAILABILITY | PEAK EFFECT (hr) | DURATION OF DIURESIS (hr) | HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Chlorothiazide (Diuril) | 10-20 | 2 (PO), 0.5 (IV) | 6-12 (PO), 2 (IV) | 1-2 | 500-2000 | 1-2 |
| Chlorthalidone (Hygroton) | 65 | 2 | 24-72 | 35-55 | 15-200 | 1 |
| Hydrochlorothiazide (Esidrix, HydroDIURIL, Oretic, Microzide) | 65-75 | 4-6 | 6-12 | 2.5-4.5 | 25-100 | 1-3 |
| Indapamide (Lozol) | 95 | 2 | 24-36 | 14-18 | 1.25-5 | 1 |
| Metolazone (Zaroxolyn) | 65 | 2 | 12-24 | 6-20 | 5-20 | 1 |
| Metolazone (Mykrox) | 2-4 | 12-24 | 14 | 0.5-1 mg | 1 |

Loop diuretics
Loop diuretics are indicated for chronic heart failure, ascites with or without hepatic cirrhosis, renal failure, pulmonary edema, hypercalcemia, hypermagnesemia, and syndrome of inappropriate antidiuretic hormone. Loop diuretics are second-line diuretics in the management of hypertension; however, they are superior to thiazide diuretics in diuresis and decreasing blood pressure for patients with renal insufficiency. Table 22-7 presents the pharmacokinetics and dosing guidelines for oral loop diuretics.2,4,5
TABLE 22-7
Pharmacokinetics and Dosing Guidelines for Oral Loop Diuretics
| LOOP DIURETIC GENERIC NAME (BRAND NAME) | BIOAVAILABILITY (%) | ONSET (hr) | DURATION (hr) | HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Bumetanide (Bumex) | 70-95 | 0.5-1 | 5-6 | 0.8 ± 0.2 | 0.5-10 | 1 |
| Ethacrynic acid (Edecrin) | 100 | 0.5 | 6-8 | 2-4 | 50-200 | 1-2 |
| Furosemide (Lasix) | 60 | 0.5-1 | 6-8 | 0.5-1.1 | 40-240 | 1-2 |
| Torsemide (Demadex) | 80 | 0.5-1 | 1 | 2-4 | 5-200 | 1 |

Aldosterone antagonists
Spironolactone (Aldactone) and eplerenone (Inspra) are aldosterone antagonists that exert their effect on the late distal tubule and collecting duct. Spironolactone, a weak diuretic, is used primarily for its aldosterone antagonist effects. Spironolactone is indicated for hypertension, management of hepatic cirrhosis (diuretic of choice), primary hyperaldosteronism, hypokalemia, and heart failure. For hypertension, spironolactone is used in combination with other antihypertensives or to spare potassium when administered with diuretics. The chemical structure of spironolactone resembles the structure of the corticosteroids and may explain its sexual adverse effects, such as impotence, decreased libido, gynecomastia, deepening of the voice, menstrual irregularities, and hirsutism. Other spironolactone-induced adverse effects include diarrhea, gastritis, skin rashes, drowsiness, lethargy, ataxia, headaches, and confusion. Similar to the other potassium-sparing diuretics, spironolactone may cause hyperkalemia. Table 22-8 presents the pharmacokinetics and dosing guidelines for the aldosterone antagonists.2,4,5
TABLE 22-8
Pharmacokinetics and Dosing Guidelines for Aldosterone Antagonists
| ALDOSTERONE ANTAGONIST GENERIC NAME (BRAND NAME) | ACTIVE METABOLITE | ELIMINATION TOTAL | ONSET OF ACTION (hr) | PEAK RESPONSE | DURATION OF ACTION (hr) | HALF-LIFE OF PARENT DRUG (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY | EFFECT OF FOOD ON ABSORPTION |
| Spironolactone (Aldactone) | Canrenone | 47%-57% renal, 35%-41% fecal | 2-4 | 6-8 hours | 16-24 | 1.4 | 25-400 | 1-2 | Increased |
| Eplerenone (Inspra) | None | 67% renal, 32% fecal | 1-2 | 4 weeks | 24 | 3.5-6 | 50-100 | 1-2 | No effect |

Centrally acting adrenergic agents
The most effective and least toxic α2 agonist is the clonidine transdermal therapeutic system (Catapres-TTS), which achieves sustained levels of clonidine for 7 days. The sustained clonidine levels avoid the peaks and troughs associated with the prompt release dosage form, and treatment is relatively devoid of the troublesome anticholinergic and CNS side effects. The clonidine patch is applied to a hairless area of intact skin on the upper torso. On the initial application, the clonidine patch takes 2 to 3 days to achieve target blood levels and a therapeutic effect. It is recommended to coadminister clonidine oral tablets along with the patch for the first 2 to 3 days of therapy. The most common adverse effects of the patch are local skin rashes and irritation. Table 22-9 presents the pharmacokinetics and dosing guidelines for α2 agonists.2,4,5
TABLE 22-9
Pharmacokinetics and Dosing Guidelines for Centrally Acting Adrenergic Agents (α2 Agonists)
| α2 AGONIST GENERIC NAME (BRAND NAME) | ONSET OF ACTION (hr) | PEAK EFFECT (hr) | DURATION OF ACTION (hr) | HALF-LIFE (hr) | ELIMINATION | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Methyldopa (Aldomet) | 4-6 | 6-9 | 24-48 | 1.25 | Renal (biphasic) | 500-2000 | 2-3 |
| Clonidine (Catapres) | 0.5-1 | 3-5 | 24 | 6-20 | Renal (40%-60%) | 0.1-2.4 | 2-4 |
| Guanfacine (Tenex) | 2.5 | 6 | 24 | 17 | Renal (50%) | 1-3 | Once at bedtime |
| Guanabenz (Wytensin) | 1 | 2-5 | 6-8 | 7-10 | Renal (70%-80%) | 4-32 | 2 |

α1-adrenergic antagonists
α1-adrenergic receptor antagonists selectively block postsynaptic α1 receptors. Total peripheral resistance is reduced through arterial and venous dilation; these agents decrease both preload and afterload and cause a potent first-dose sympathetic reflex increase in heart rate and renin activity.12 α1-adrenergic antagonists cause a first-dose phenomenon that manifests with orthostatic hypotension, tachycardia, palpitations, dizziness, headaches, and syncope. After several doses, despite persistent vasodilation, tolerance to the first-dose phenomenon develops, and heart rate, renin, and cardiac output return to normal. To minimize the first-dose phenomenon, initial doses of α1-adrenergic antagonists should be low and administered at bedtime.
α1-adrenergic antagonists are indicated for hypertension, benign prostatic hyperplasia (BPH), heart failure, and Raynaud vasospasm except for uroselective α1-adrenergic antagonists (tamsulosin and alfuzosin), which are indicated only for BPH. In contrast to other antihypertensives, α1-adrenergic antagonists have favorable effects on the lipoprotein profile and may decrease triglycerides and low-density lipoproteins and increase high-density lipoproteins by 5% to 10%, making them an ideal drug of choice. However, the ALLHAT (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial) study compared doxazosin with other antihypertensives (chlorthalidone) and revealed a 25% higher incidence of combined cardiovascular morbidity in patients receiving doxazosin.13 A higher incidence of doxazosin-induced stroke, heart failure, angina, and coronary revascularization was reported. On the basis of the results of this study, α1-adrenergic antagonists are considered second-line antihypertensive therapy. Table 22-10 presents the pharmacokinetics and dosing guidelines for α1-adrenergic antagonists.2,4,5
TABLE 22-10
Pharmacokinetics and Dosing Guidelines for α1-Adrenergic Receptor Antagonists
| α1 ANTAGONIST GENERIC NAME (BRAND NAME) | ELIMINATION ROUTES | PEAK (hr) | DURATION (hr) | HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Doxazosin (Cardura) | 63% feces, 9% urine | 6 | 18-36 | 11 | 1 to 16 | 1 |
| Prazosin (Minipress) | 90% feces, 10% urine | 1.5 | 8-10 | 2 | 3-40 | 2-3 |
| Terazosin (Hytrin) | 60% feces, 40% urine | 2 | 24 | 14 | 120 | 1-2 |

Angina
Epidemiology, etiology, and pathophysiology
Ischemic heart disease can manifest as many clinical variants such as stable exertional angina; unstable (rest, preinfarction, crescendo) angina; coronary vasomotion; vasospasm associated with atypical, variant, or Prinzmetal angina; silent myocardial ischemia; or MI. Angina pectoris (chest pain) is a symptom or marker of myocardial ischemia. Ischemia is defined as a lack of oxygen and decreased or no blood flow to the myocardium. From 2002-2006, greater than 10 million Americans older than 20 years were diagnosed with angina pectoris.14 Women often initially present with angina, whereas men present with MI. CAD, when present, tends to be less severe in women than men.
Pharmacotherapy
Pharmacotherapy for angina pectoris includes nitrates, β blockers, CCBs, and ranolazine (Ranexa). Ranolazine was approved by the U.S. Food and Drug Administration (FDA) in 2006 for the treatment of chronic stable angina in combination with amlodipine, β blockers, or nitrates.15 For the management of vasospastic and chronic stable angina, diltiazem, verapamil, amlodipine, and nifedipine are indicated. For the management of angina, β blockers are usually dosed to achieve a resting heart rate of 50 to 60 beats/min and a maximal exercise heart rate of 100 beats/min. Goal blood pressure for patients with documented CAD (i.e., chronic stable angina, unstable angina, non–ST segment elevation MI, ST segment elevation MI) is less than 130/80 mm Hg because of increased risk for mortality. All patients with angina should receive daily aspirin (75 to 100 mg/day) to prevent MI.16–19
Nitrates
Nitroglycerin reduces myocardial oxygen demand by causing venodilation of coronary arteries and collaterals, resulting in decreased end-diastolic pressures. Venous effects predominate; however, nitroglycerin can affect arteries at high doses. The cellular mechanism of action of nitrates is depicted in Figure 22-3. Nitrates are indicated for acute treatment or prophylaxis of angina, acute MI, acute heart failure, low-output syndromes, and hypertension (intravenous). Nitrates may be administered by various routes and are readily available in multiple preparations, including oral, intravenous, ointment, transdermal, translingual, and sublingual tablets. Sublingual nitroglycerin is indicated for acute anginal relief. Sublingual nitroglycerin has an onset of action of minutes and duration of action of 30 minutes. Sublingual nitroglycerin should be administered every 5 minutes until relief is obtained. If pain relief is not achieved after three doses in 15 minutes, emergency care should be sought. This algorithm is recommended for patients who were previously prescribed nitroglycerin. For patients who were never prescribed nitroglycerin, it is recommended that patients should seek emergency care if symptoms worsen or persist 5 minutes after taking the first sublingual nitroglycerin.19 Sublingual tablets must always be stored in their original glass container, and any unused tablets should be discarded 6 months after the original container is opened because of loss of potency. Other forms of nitroglycerin are isosorbide dinitrate (Isordil) and isosorbide mononitrate (Imdur, Ismo, and Monoket). Table 22-11 presents the pharmacokinetics and dosing guidelines of nitrates.
TABLE 22-11
Pharmacokinetics and Dosing Guidelines for Nitrates
| NAME | DOSAGE FORMS | ONSET OF ACTION (min) | DURATION OF ACTION (hr) | INITIAL DOSE |
| Nitroglycerin |
• Oral capsule, ER: 2.5-9 mg every 12 hours; may increase to every 8 hours if needed and if tolerated
• Topical ointment: 7.5-30 mg applied twice daily to a 36-square-inch area of truncal skin
• Transdermal patch: 0.2-0.4 mg/hr
• Sublingual tablet: 0.3-0.6 mg every 5 minutes, 3 times
• Sublingual spray: 1-2 metered sprays onto or under the tongue; may repeat in 3-5 minutes, with no more than 3 metered sprays in 15 minutes

ER, Extended release; IV, intravenous; PVC, polyvinyl chloride.

Ranolazine
Ranolazine is indicated for the treatment of patients with chronic angina who have not achieved an adequate response with other antianginal drugs. In 2007, the American College of Cardiology Foundation (ACCF) and the American Heart Association (AHA) suggested that ranolazine may be safely administered for symptomatic relief after unstable angina or non–ST segment elevation MI, but it does not seem to reduce significantly cardiovascular death, MI, or recurrent ischemia. Ranolazine provides antiischemic effects that complement the benefits of CCBs, β blockers, and nitrates. Although the exact mechanism of how ranolazine exerts its antianginal and antiischemic effects is unknown, it is speculated that it selectively inhibits the late phase of the inward sodium channel in ischemic myocytes resulting in decreased myocardial oxygen consumption.20 Ranolazine increases exercise tolerance, which reduces angina frequency and the need for emergent nitroglycerin interventions. In contrast to standard antianginal medications, ranolazine does not alter blood pressure or heart rate. The initial adult dose of ranolazine extended release tablets is 500 mg twice daily, with a maximal dose of 1 g twice daily.
Antithrombotic agents
Antithrombotics may be defined as agents that prevent or break up blood clots in conditions such as thrombosis or embolism. Three categories of antithrombotic agents are currently available in the United States: anticoagulants, antiplatelets, and thrombolytics. Anticoagulant agents work by preventing the formation of the fibrin clot and preventing further clot formation in already existing thrombi. Antiplatelet agents inhibit the action of platelets in the initial stage of the clotting process. Thrombolytics break up thrombi by degrading fibrin. Box 22-1 lists currently available antithrombotic agents.21
Formation and elimination of acute coronary thrombus
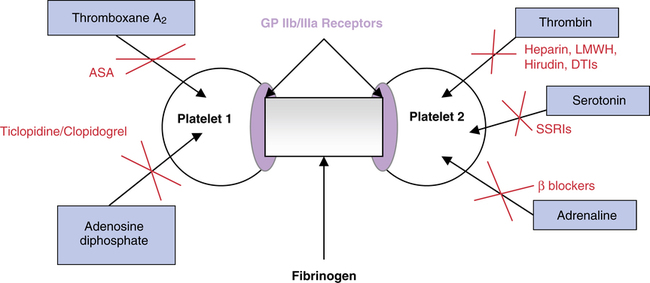
Under normal conditions, the body maintains an equilibrium state between clot formation (thrombosis) and clot breakdown (fibrinolysis).22 Thromboses are initiated by an injury to the endothelial wall of a coronary vessel. When injury occurs, the anticoagulated endothelial surface is disrupted, and the highly procoagulant subendothelial surface is exposed. Instantaneously, platelets aggregate in response to the release of chemotactic substances, such as thromboxane A2, followed by platelet adhesion to the subendothelial vessel surface, representing the initial step in clot formation. Platelet adhesion is mediated mainly by von Willebrand factor. von Willebrand factor is present in the subendothelium and is actively recruited when the subendothelium is injured. Adhered platelets are exposed to many subendothelial proteins, such as collagen and thrombin. Collagen and thrombin also promote platelet activation. Activated platelets release platelet agonists such as adenosine diphosphate (ADP), norepinephrine, serotonin, and arachidonic metabolites, mitigating and amplifying platelet aggregation and forming an unstable thrombus or platelet plug.
The most important consequence of platelet activation is the expression of platelet receptor glycoprotein (GP) IIb/IIIa on the platelet’s surface, allowing binding to fibrinogen. Fibrinogen binds to the two GP IIb/IIIa molecules, causing a cross-linking of receptors on adjacent platelets and initiating platelet aggregation. Triggers affecting platelet aggregation and their antagonists are depicted in Figure 22-4. Fibrinogen is converted into fibrin monomers by the action of thrombin; this is the final step in clot formation. Homeostasis is complete when the fibrin clot becomes insoluble within the vessel. This stable fibrin clot is the end result of the coagulation cascade. Under normal conditions, multiple inhibitors and control mechanisms keep these reactions localized to the site of the injury.
The fibrin clot ultimately must be removed for hemostasis to be maintained. Activation of the fibrinolytic system by tissue plasminogen activators (t-PAs), which are present in most body fluids and tissues, results in the conversion of plasminogen to plasmin, initiating the dissolution of fibrin and fibrinogen. The breakdown of fibrinogen and fibrin results in polypeptides termed fibrin split or fibrinogen degradation products (FDPs). FDPs are anticoagulant substances that can cause bleeding if fibrinolysis becomes uncontrolled and excessive. D-dimers are fragments of plasmin-digested, cross-linked fibrin that increase in concentration after the onset of fibrinolysis. Blood testing for D-dimer fragments may assist in the diagnosis of pathogenic venous thromboembolism. The extrinsic and intrinsic pathways of the coagulation system are depicted and described in Figure 22-5.2,3,18

Anticoagulant agents
Heparins: unfractionated heparin and low-molecular-weight heparin
Heparin is extracted from porcine intestinal mucosa or bovine lungs; however, because of the high propensity of thrombocytopenia with the bovine lung derivative, only the porcine derivative is routinely employed in practice. Heparin serves as a catalyst that accelerates the rate of the thrombin-to-antithrombin III reaction by at least 1000-fold by serving as a catalytic template to which both bind, resulting in a ternary complex (heparin, thrombin, and antithrombin). Antithrombin III is a large protein (58,000 Da) that is synthesized in the liver and is known as a suicide substrate. Heparin is a high-molecular-weight complex mucopolysaccharide containing specific pentasaccharide units and approximately 45 monosaccharide side chains with a mean molecular mass of 12,000 Da (range 5000 to 30,000 Da).23
High-molecular-weight heparin (UFH) is cleared faster and requires more frequent dosing or an intravenous continuous infusion. The half-life of UFH is approximately 30 to 60 minutes, whereas the half-life of LMWH is 4 to 5 hours, allowing for once-daily or twice-daily LMWH administration. The onset of action of heparin is within 6 hours of initiation of a continuous infusion. LMWH time to peak anti–factor Xa activity is 2 to 5 hours. Each commercially available LMWH is synthesized by different mechanisms, possesses moderately different pharmacokinetic and pharmacodynamic characteristics, and has different FDA-approved indications and dosing regimens; these agents are not interchangeable. Table 22-12 presents the pharmacokinetic properties and dosing parameters for all heparins.
TABLE 22-12
Pharmacokinetic Properties and Dosing Guidelines of Heparins
| DOSING | |||||
| HEPARIN FORMULATION | MOLECULAR WEIGHT (Da) | ANTI-Xa/ANTI-IIa RATIO | HALF-LIFE (min) | PROPHYLAXIS | TREATMENT |
| Dalteparin (Fragmin) | 4000-6000 | 2:1 | 119-139 |
General surgery: 5000 U every 24 hours Postoperative start: 2500 U 4-8 hours after surgery, then 5000 U every 24 hours Preoperative start, day of surgery: 2500 U within 2 hours before surgery, 2500 U 4-8 hours after surgery, then 5000 U every 24 hours Perioperative start, evening before surgery: 5000 U until 14 hours before surgery, 5000 U 4-8 hours after surgery, then 5000 U every 24 hours |
200 U/kg every 24 hours or 100 U/kg every 12 hours
Kidney impairment: No specific dosage adjustment recommended
Extended VTE treatment in patients with cancer:
Days 1-30: 200 U/kg every 24 hours
Months 2-6: 150 U/kg every 24 hours
Unstable angina or non–Q wave MI: 120 U/kg (maximum of 10,000 U) every 12 hours with concurrent aspirin therapy until clinically stable
General surgery: 40 mg every 24 hours; for CrCl <30 mL/min use 30 every 24 hours
Hip/knee orthopedic surgery: 30 mg every 12 hours (TKR/THR); 40 mg every 24 hours (THR only); if CrCl <30 mL/min, use 30 mg every 24 hours
Acute medical illness: 40 mg every 24 hours; if CrCl <30 mL/min use 30 mg every 24 hours
IV: 80 U/kg (or 5000 U) IV push followed by continuous infusion of 18 U/kg/hr (or 1300 U/hr)
Monitored dosing: initial 17,500 U or 250 U/kg then 250 U every 12 hours
Unmonitored dosing: Initial: 333 U/kg then 250 U/kg every 12 hours
STEMI: Bolus of 60 U/kg (maximum: 400 U), then 12 U/kg/hr (maximum: 1000 U/hr) as continuous infusion, with target aPTT of 1.5-2 times upper limit of control
NSTEMI: Initial bolus of 60 U/kg (maximum of 4000 U) followed by 12 U/kg/hr (maximum of 1000 U/hr). Dose adjustment to correspond to therapeutic range equivalent to heparin levels of 0.3-0.7 U/mL by anti–factor Xa determinations

*Half-life of UFH has saturable binding, and half-life increases with doses more than 400 U/kg.
Activated partial thromboplastin time (aPTT) is used to monitor the effects of heparin because it is sensitive to the inhibitory effects of thrombin, factor Xa, and factor IXa and correlates with heparin levels. When the concentration of plasma heparin is 0.1 to 1 U/mL, aPTT and thrombin time are prolonged. The goal of heparin therapy is to prevent unwanted clotting without an increased risk of hemorrhage. This goal may be accomplished by maintaining aPTT between 2 and 2.5 times the upper limit of the control value. aPTT should not be used to monitor LMWHs. The effect of LMWHs may be monitored on the basis of anti–factor Xa levels; however, because the relationship between anti–factor Xa levels and clinical outcomes is tenuous, routine measurement is not indicated and should be reserved for special populations, such as patients with renal disease, obese patients, and underweight patients.24
More recently, there has been a major change in the United States Pharmacopeia (USP) monograph of UFH. As a result of the heparin contamination problem encountered from 2007-2009, a new reference standard for heparin and a new test to determine potency were established by the FDA. These changes have resulted in an estimated 10% reduction in anticoagulant activity of UFH, which was validated. Because of this decrease in potency, the intravenous dose of UFH may need to be increased to achieve target aPTT, and more frequent or intensive aPTT monitoring may be required. No dose adjustment is needed for subcutaneous administration of UFH.25
Adverse effects induced by UFH and LMWHs include bleeding, hematoma, early thrombocytopenia, delayed thrombocytopenia with or without white clot syndrome, hyperkalemia, osteoporosis (with prolonged use), and increase in liver enzyme tests (LETs). An increase in LETs may occur in 10% to 30% of patients receiving LMWHs or high molecular weight heparins. However, the increase in LETs seems benign and has not been associated with any cases of hepatic sequelae. Early-onset heparin-induced thrombocytopenia type 1 (HIT-1) manifests with a decrease in platelets of approximately 50,000/mm3. The decrease in platelets is transient and inconsequential. Delayed-onset heparin-induced thrombocytopenia type 2 (HIT-2) is due to the formation of antiplatelet antibodies between days 6 and 12.
If a patient has heparin-dependent antibodies present in plasma from previous heparin exposure, HIT-2 may occur at any time. HIT-2 is dependent on platelet factor-4 binding. These platelet antibodies aggregate and form the basis for the paradoxical heparin-induced white clot syndrome. The white clot syndrome is a medical emergency that may manifest as pulmonary embolism, MI, stroke, renal or hepatic thrombosis, or skin necrosis and gangrene. The diagnosis of HIT-2 is clinical and may be confirmed by several laboratory tests. Clinical diagnosis of HIT-2 includes a significant reduction in the platelet count of greater than 50% or a decrease in the platelet count to less than 100,000/mm3 or both. Ostensibly, the risk of thrombocytopenia is greatest with UFH and lowest with LMWHs; however, LMWHs cannot be administered as an alternative to heparin because of greater than 95% cross-reactivity.
The antidote for heparin is protamine sulfate. Protamine sulfate is derived from the sperm of mature testes of salmon and related species. Protamine is electropositive and rapidly binds to the electronegative heparin to form salts that have no anticoagulant effect. Protamine also causes a dissociation of heparin–antithrombin III complexes in favor of a heparin-protamine complex. The recommended neutralizing dose of protamine is 1 mg for every 100 U of heparin, up to a total of protamine 50 mg per dose. Protamine should be administered by slow intravenous infusion over at least 1 to 3 minutes to prevent hypotension, bradycardia, or dyspnea. Patients who have previously received protamine-containing insulin, have undergone a vasectomy, or have a known sensitivity to fish or medications derived from fish (calcitonin-salmon, cold water fish oils containing omega-3 fatty acids, and oyster shell–derived calcium supplements) are at an increased risk for experiencing allergic reactions such as anaphylaxis and developing antiprotamine antibodies.24 Excessive protamine may act as an anticoagulant, resulting in bleeding complications; a careful underdosing strategy is suggested. There is no proven method for neutralizing LMWHs. Protamine seems to neutralize approximately 60% of the anti–factor Xa activity of LMWHs. UFH may be the preferred parenteral anticoagulant in patients who are at risk for clinically significant bleeding, such as patients with end-stage renal disease receiving hemodialysis treatments.
Direct thrombin inhibitors
Natural hirudin is produced in trace amounts by the salivary glands of the leech Hirudo medicinalis. Lepirudin is a recombinant hirudin derived from yeast cells. FDA-approved dosing recommendations include an initial bolus dose of 0.4 mg/kg body weight followed by 0.15 mg/kg/hr continuous infusion. Current ACCP guidelines have a slightly different recommendation. The ACCP recommends a starting dose of no more than 0.10 mg/kg/hr and a lower bolus dose of 0.2 mg/kg to be administered only in the setting of life-threatening or limb-threatening thrombosis. These new recommendations were introduced because of the high rates of bleeding associated with the FDA-approved dosing.26
Warfarin (coumadin)
The initial dose of warfarin should be the expected maintenance dose. Loading doses of warfarin are no longer recommended. A study found that when hospitalized patients were given an average warfarin maintenance dose of 5 mg, the INR was usually greater than or equal to 2 within 4 or 5 days and was associated with less excessive anticoagulation compared with a 10-mg dose.27 Warfarin starting doses of less than 5 mg may be most appropriate in elderly patients. Warfarin interferes with the hepatic synthesis of vitamin K–dependent clotting factors II, VII, IX, and X and endogenous anticoagulant proteins C and S. The time to complete anticoagulation with warfarin is not immediate. Inhibition of coagulation factors begins 12 to 24 hours after administration; however, the complete antithrombotic effects of warfarin may not occur until 5 to 7 days after initiation of therapy.
Many factors, such as diet, disease states, and drugs, can alter the pharmacologic characteristics and effects of warfarin.28 Patients should be counseled to eat a healthy and consistent diet. An increased intake of vitamin K–containing supplements and foods, such as green leafy vegetables, may result in a reduced anticoagulant response, decreased INR, and subsequently treatment failure such as an embolism. Conversely, abrupt decreases in vitamin K dietary intake may result in an increased anticoagulant response with an increased INR and subsequent risk of hemorrhage. Hepatic disease and, to a lesser extent, renal disease may decrease elimination of warfarin and increase the effects of warfarin. A plethora of drugs can increase or decrease the effects of warfarin. It is prudent to measure the INR frequently when factors that interact with warfarin are added to a patient’s regimen. Table 22-13 lists selected significant warfarin drug interactions.

NSAIDs, Nonsteroidal antiinflammatory drugs; TMP-SMX, trimethoprim-sulfamethoxazole.
The role of genetic polymorphism in the management of warfarin has been the focus of interest in more recent studies. CYP2C9, which plays an integral role in the metabolism of the S-isomer of warfarin, and VKORC1 (VKOR complex subunit 1), the gene that determines the activity of vitamin K epoxide reductase, both undergo genetic polymorphism leading to variances in warfarin responses between patients. Studies have shown that testing for a patient’s genetic type can lead to decreased major bleeding or thromboembolic events resulting in hospital admission.28 Genetic test kits (e.g., GeneMedRx) are available for purchase but are not covered by insurance plans and can be expensive. Genotyping is still considered a novelty and not routinely used in practice.
Antiplatelet agents
Aspirin
Dipyridamole
The second European Stroke Prevention Study (ESPS-2) showed that dipyridamole modified-release formulation, 200 mg given twice daily, is effective in the secondary prevention of stroke and TIA compared with placebo and that coadministration with aspirin, 25 mg twice daily, provides additional benefit.28 ESPS-2 showed that the relative risk reduction for stroke with aspirin administration compared with placebo was 18.1% (P = .013); for modified-release dipyridamole, the relative risk reduction was 16.3% (P = .039); and with the combination, it was 37% (P < .001). Aggrenox must not be substituted by prompt-release dipyridamole and aspirin. The recommended dosage of Aggrenox is one capsule twice daily, swallowed whole. Because of increased risk of headache, it may be advisable to start with one capsule daily at bedtime up to 1 week until tolerated before increasing the dose to twice daily.
Clopidogrel (plavix)
Clopidogrel (Plavix) is a prodrug thienopyridine derivative platelet aggregation inhibitor that interferes with platelet membrane function by inhibiting ADP-induced platelet-fibrinogen binding and subsequent platelet-platelet interactions. Indications for clopidogrel include the reduction of atherosclerotic events in patients with a history of MI, stroke, or peripheral arteriolar disease and acute coronary syndrome (ACS) regardless of whether a patient is managed medically, by percutaneous coronary intervention (PCI), or by coronary artery bypass grafting. Clopidogrel is slightly more effective than aspirin in reducing the combined risk of ischemic stroke, MI, or vascular death in patients with atherosclerotic vascular disease.29 For patients with ACS, clopidogrel plus aspirin was found to be superior to aspirin alone in reducing composite endpoints of MI, stroke, and cardiovascular death. Clopidogrel has not shown superiority to aspirin for stroke prophylaxis except in patients with peripheral vascular disease.
Statins or 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitors have also been an area of interest with regard to clopidogrel drug interactions. Similar to clopidogrel, statins are CYP3A4 substrates. In vivo studies that examined the degree of platelet inhibition by clopidogrel in patients taking a statin have shown that there is a significant decline in platelet inhibition with the use of statins. However, no large clinical trials have shown that this interaction can lead to negative clinical outcomes. Despite the lack of clinical trials evaluating the interaction between statins and clopidogrel, caution is advised when combining these two medications. If statin therapy is required, pravastatin (Pravachol) or rosuvastatin (Crestor) should be considered because these drugs do not undergo CYP3A4 metabolism and potentially are devoid of any significant drug interactions.30
Testing for genetic polymorphism has also received considerable emphasis in the FDA boxed warning for clopidogrel. Although genetic polymorphism for CYP2C19 has been shown in several studies to reduce antiplatelet activity and increase major adverse cardiac events (MACEs), prospective studies show clinical efficacy of personalizing antiplatelet therapy based on genotype analysis. The recommendation issued by the FDA to clinicians is to consider alternative treatment strategies, such as combining clopidogrel with cilostazol or using high-dose clopidogrel (600 mg loading dose followed by 150 mg daily for ACS) in patients identified as poor CYP2C19 metabolizers. The efficacy and safety of these approaches remain uncertain.31 Alternatively, one may consider switching to another antiplatelet agent, such as prasugrel, which does not undergo CYP2C19 metabolism.
Ticlopidine (ticlid)
Ticlopidine (Ticlid), a thienopyridine, is a platelet aggregation inhibitor that interferes with platelet membrane function by inhibiting ADP-induced platelet-fibrinogen binding and subsequent platelet-platelet interactions. The effect of ticlopidine on platelet function is irreversible and lasts for the life of the platelet. Ticlopidine is indicated for stroke. In the Ticlopidine Aspirin Stroke Study (TASS), the ticlopidine group had a 21% greater relative risk reduction for stroke compared with the aspirin group and a 9% greater reduction in stroke, MI, or vascular death at 3 years.31 The Canadian-American Ticlopidine Study (CATS) showed that ticlopidine reduced the relative risk of stroke, MI, or vascular death by 30% compared with placebo (10.8%; P = .006).32 The half-life of ticlopidine is 14 hours; however, with repeat dosing it approaches 4 to 5 days. Steady state is achieved within 14 to 21 days. Platelet aggregation is inhibited 50% within 4 days and 60% to 70% within 10 days. Platelet aggregation and bleeding time return to normal within 14 days after ticlopidine discontinuation. Ticlopidine is extensively metabolized by the liver; active metabolites have not been elucidated.
Prasugrel (effient)
Prasugrel (Effient) is a prodrug thienopyridine derivative platelet aggregation inhibitor that interferes with platelet membrane function by inhibiting ADP-induced platelet-fibrinogen binding and subsequent platelet-platelet interactions. When prasugrel is compared with other agents in the same class, it has a very limited scope; it is indicated only for the prevention of thrombosis in patients with ACS undergoing PCI. Prasugrel in combination with aspirin decreases nonfatal MI slightly more than clopidogrel in combination with aspirin but with an increased risk of bleeding. Prasugrel is contraindicated in patients who have had history of a TIA or stroke. In patients older than 75 years or in patients who weigh less than 60 kg, prasugrel was shown to have a greater risk of bleeding, which outweighs its benefit.33 Prasugrel is extensively metabolized by hydrolysis in the liver followed by CYP3A4 and CYP2D6; its metabolites are eliminated via the kidneys and feces. The half-life of the active metabolite of prasugrel is 7 to 8 hours. The onset of action of prasugrel can be seen in 30 minutes. The average platelet inhibition observed with prasugrel is 50% to 80%. Platelet aggregation and bleeding time return to normal approximately 5 to 9 days after prasugrel discontinuation. The dose of prasugrel in ACS is a 60-mg loading dose followed by 10 mg once daily (plus aspirin). In patients who weigh less than 60 kg, a decreased dose of 5 mg is recommended, although there have not been any clinical trials to support this practice.
The most common adverse effect of prasugrel is hemorrhage, which is greater than that observed with clopidogrel. Other adverse effects are similar to clopidogrel. A higher incidence of colonic neoplasm was seen during the trial for its approval.34,35
Glycoprotein iib/iiia inhibitors
GP IIb/IIIa inhibitors are indicated for the treatment of patients with ACS—unstable angina or non–ST segment elevation acute MI—and patients who are medically managed and patients undergoing PCI. Abciximab (ReoPro) is the GP IIb/IIIa inhibitor of choice for PCI. The management of unstable angina or non–ST segment elevation acute MI includes the use of aspirin, heparin, and a GP IIb/IIIa inhibitor. This combination has led to a decrease in the composite endpoints of new MI or death.36 In patients who are being managed medically for a non–ST segment elevation ACS, the use of GP IIb/IIIa inhibitors has been marginalized to patients with moderate to high risk based on a risk assessment score and continued ischemia or patients with diabetes. GP IIb/IIIa inhibitors are administered via continuous intravenous infusions. Oral formulations of GP IIb/IIIa inhibitors failed to display efficacy in clinical trials and are unavailable.
The most common adverse effect reported during therapy was bleeding. The incidence of major bleeding manifesting as gastrointestinal, genitourinary, or intracranial hemorrhage with the three-drug combination was only slightly greater than with aspirin and heparin alone, illustrating the safety of the GP IIb/IIIa inhibitors. Although minor bleeding with GP IIb/IIIa inhibitors is common (10%), it is generally inconsequential. Because these agents may cause thrombocytopenia, daily platelet, hemoglobin, and hematocrit monitoring is required. Abciximab has been implicated as a cause of immune-mediated thrombocytopenia in 5% of patients. Table 22-14 presents the pharmacologic characteristics of GP IIb/IIIa inhibitors.
TABLE 22-14
Characteristics of Glycoprotein (GP) IIb/IIIa Inhibitors
| ABCIXIMAB (REOPRO) | TIROFIBAN (AGGRASTAT) | EPTIFIBATIDE (INTEGRILIN) | |
| Common uses | Adjunct to PCI | Management of ACS, medically or with PCI | Management of ACS, medically or with PCI; adjunct to PCI |
| Pharmacology | Chimeric human-murine monoclonal antibody Fab fragment GP IIb/IIIa inhibitor | Nonpeptide GP IIb/IIIa inhibitor | Cyclic heptapeptide GP IIb/IIIa inhibitor |
| Origin | Antibodies from immunized mice | Chemically derived | Active component of snake venom peptides |
| Binding to platelets | Irreversible | Reversible | Reversible |
| Elimination half-life | 30 minutes | 2 hours | 2.5 hours |
| Platelet function recovery | Approximately 48 hours | Approximately 4 hours | Approximately 4 hours |
| Elimination | Renal, lymphatic system | 65% renal, 25% biliary | 50% renal, 30% metabolized in plasma into amino acids |

ACS, Acute coronary syndrome; PCI, percutaneous coronary intervention.
Thrombolytic agents
For the treatment of ST segment elevation ACS manifesting with at least 1 mm of ST segment elevation in two or more contiguous ECG leads, all available thrombolytics (streptokinase, alteplase, reteplase, and tenecteplase) are indicated; however, streptokinase is considered a second-line agent because of its lack of fibrin specificity. Eligible patients should receive thrombolytic therapy within 12 hours of symptom onset; however, benefit can be realized for 24 hours. Thrombolytics are preferred to primary PCI when patients present within 3 hours of symptom onset and the door to primary PCI time would be greater than 90 minutes. For acute massive pulmonary embolism, alteplase is the only thrombolytic indicated. Alteplase is reserved for patients with acute massive pulmonary embolism who present with symptoms within 2 weeks but optimally within 5 days. Alteplase is the only thrombolytic indicated for the management of acute ischemic stroke for patients who present within 3 hours of symptom onset and no later than 6 hours after onset.37 The use of thrombolytics is often precluded because of their extensive list of contraindications. The absolute and relative contraindications for the use of thrombolytics are listed in Box 22-2.
The most common adverse effect associated with these agents is major and minor bleeding. Major bleeding includes gastrointestinal, genitourinary, respiratory tract, retroperitoneal, and intracranial hemorrhage. Minor bleeding often manifests as superficial or surface bleeding as a result of arterial punctures and surgical intervention. Thrombolytic-induced hemorrhagic stroke in patients older than 75 years occurs more often with alteplase than with streptokinase. Patients older than 75 years should receive streptokinase rather than alteplase. Alteplase and tenecteplase are known to be fibrin specific because they promote the conversion of plasminogen into plasmin in the presence of clot-bound fibrin only, with limited systemic proteolysis. The increased fibrin specificity is believed to induce less extensive systemic depletion of clotting factors such as fibrinogen and plasminogen. The clinical relevance of thrombolytic fibrin specificity has not been elucidated. Thrombolytics have rarely been associated with cholesterol embolization manifesting as purple toe syndrome, livedo reticularis, acute renal failure, gangrene, MI, bowel infarction, stroke, and rhabdomyolysis. When thrombolytics are used for ACS, they can cause reperfusion arrhythmias manifesting as bradycardia or ventricular tachyarrhythmias. Table 22-15 presents the pharmacologic properties of thrombolytic agents.38
TABLE 22-15
Pharmacologic Properties of Thrombolytic Agents
| STREPTOKINASE | ALTEPLASE (rtPA) | RETEPLASE (rPA) | TENECTEPLASE (TNK-tPA) | |
| Brand name | Streptase | Activase | Retavase | TNKase |
| Source | Streptococcal culture | Recombinant DNA technology using heterologous mammalian tissue culture | Recombinant DNA technology using Escherichia coli | Recombinant DNA technology using Chinese hamster ovary cells |
| Common uses | Pulmonary embolism, deep vein thrombosis, peripheral arterial occlusion, clearance of occluded central venous access devices, ST segment elevation | Pulmonary embolism, stroke, clearance of occluded central venous access device, ST segment elevation | Myocardial infarction, ST segment elevation | Myocardial infarction, ST segment elevation |
| Type of agent | Bacterial proactivator | Tissue plasminogen activator | Tissue plasminogen activator | Tissue plasminogen activator |
| Plasma half-life (min) | 12-18 | 2-6 | 13-16 | 90-130 |
| Fibrinolytic activation | Systemic | Systemic | Systemic | Systemic |
| Antigenic | Yes | No | No | No |
| Fibrin specific | + | +++ | ++ | ++++ |
| Systemic bleeding risk | +++ | ++ | ++ | + |
| ICH risk | + | ++ | ++ | ++ |

 SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
Answers can be found in Appendix A.
1. List adverse effects associated with angiotensin-converting enzyme inhibitors (ACEIs).
2. Which of the β blockers possess intrinsic sympathomimetic activity (ISA)?
3. Which of the β blockers possess selective β1-blocker activity?
4. List adverse effects associated with α1-adrenergic antagonists.
5. What are the most common side effects of nitrates?
6. List metabolic effects associated with thiazide diuretics.
7. Name five medications that may cause drug-induced increases in blood pressure.
8. Which calcium channel blocker is most likely to cause constipation?
9. Identify the best available parameter to monitor the effects of warfarin.
10. What is the antidote for heparin?
11. What is the mechanism of action of warfarin?
12. Name the pharmacologic class responsible for inhibiting the final pathway in platelet aggregation.
13. Which thrombolytic is recommended for patients older than 75 years who present with ST segment elevation myocardial infarction?
14. Name the only ACEI that is available in a parenteral dosage form.
15. Identify the best available parameter to monitor the effects of heparin.
16. Is clopidogrel or ticlopidine superior to aspirin for stroke prevention?
 CLINICAL SCENARIO
CLINICAL SCENARIO[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]CHAPTER 22
Drugs affecting circulation: antihypertensives, antianginals, antithrombotics
After reading this chapter, the reader will be able to:
1. Define terms that pertain to drugs affecting circulation: antihypertensives, antianginals, and antithrombotics
2. Categorize the stages of normal to high blood pressure
3. Define a hypertensive crisis, and differentiate between hypertensive emergency and hypertensive urgency.
4. Design an algorithm for the pharmacotherapy of hypertension.
5. Compare and contrast the clinical pharmacology of the agents used for hypertensive pharmacotherapy
6. Describe the chronotherapeutic effect of blood pressure, and design a pharmacotherapy regimen based on this principle
7. Describe the mechanism of action of angiotensin-converting enzyme inhibitors, calcium channel blockers, and β blockers
8. Compare and contrast the clinical pharmacology of spironolactone and eplerenone
9. List drug-drug interactions relevant to antihypertensives and plausible mechanisms
10. Describe the formation and elimination of an acute coronary thrombus
11. Describe the pathophysiology of angina and the drugs used to treat angina
12. List the agents in each of the following antithrombotic classes: anticoagulants, antiplatelets, and thrombolytics
13. Describe the mechanism of action of heparin
14. Compare and contrast the clinical pharmacology of heparin and low-molecular-weight heparin (LMWH)
15. List the laboratory parameters that may be used to monitor for the effect of heparin, LMWH, and direct thrombin inhibitors
16. Describe the mechanism of heparin-induced and warfarin-induced paradoxical thrombosis
17. Compare and contrast the clinical pharmacology of aspirin, clopidogrel, ticlopidine, and dipyridamole
18. Describe the role of genetic polymorphism in the antiplatelet activity of clopidogrel and anticoagulant effect of warfarin
19. Describe the indication and mechanism of action of glycoprotein IIb/IIIa inhibitors
20. List the indications and contraindication of thrombolytic agents
Drug that prevents or breaks up blood clots in conditions such as thrombosis or embolism; antithrombotics include anticoagulants, antiplatelets, and thrombolytics.
Arterial blood pressure (blood pressure)
Defined hemodynamically as the product of systemic vascular resistance and cardiac output (heart rate × stroke volume).
Damage to the heart and the blood vessels or circulation, including to the brain, kidney, and the eye.
Influencing the rate of rhythmic movements (heartbeat).
Human biologic variations of rhythm within a 24-hour cycle.
Measurement of the renal clearance of endogenous creatinine per unit time; approximates glomerular filtration rate (GFR) but overestimates GFR by 10% to 15%; used for drug-dosing guidelines.
Covalently cross-linked degradation fragments of the cross-linked fibrin polymer during plasmin-mediated fibrinolysis; level increases after the onset of fibrinolysis and allows for identification of the presence of fibrinolysis.
Maximum dose of a drug, beyond which it no longer exerts a therapeutic effect; however, its toxic effect increases.
Fibrin split or fibrinogen degradation products (FDPs)
Small peptides that result following the action of plasmin on fibrinogen and fibrin in the fibrinolytic process. FDPs are anticoagulant substances that can cause bleeding if fibrinolysis becomes uncontrolled and excessive.
Glomerular filtration rate (GFR)
Volume of water filtered from the plasma by the kidney via the glomerular capillary walls into Bowman capsules per unit time; considered to be 90% of creatinine clearance and equivalent to insulin clearance.
Blood pressure greater than 180/120 mm Hg, with the elevation of blood pressure accompanied by acute, progressing target organ injury.
Blood pressure greater than 180/120 mm Hg without signs or symptoms of acute target organ complications.
Drug influencing the contractility of a muscle (heart).
Intrinsic sympathomimetic activity (ISA)
Having the ability to activate and block adrenergic receptors, producing a net stimulatory effect on the sympathetic nervous system.
Treatment of disease by drug therapy.
Enzyme also known as angiotensinogenase, released by the kidney in response to a lack of renal blood flow and responsible for converting angiotensinogen into angiotensin I.
Neurotransmitter or hormone replacements that may be weaker or inert.
The circulatory system comprises an integral functional part of the cardiopulmonary system. Drug therapy affecting the circulation is seen in the acute critical care, outpatient care, and home care environments. This chapter presents three classes of drug therapy, all targeted at the circulatory system. After a brief review of the epidemiology, etiology, and pathophysiology of hypertension, the multiple drug groups used as antihypertensives are described. Drugs used to treat angina pectoris are the second group of drugs described. The third group of agents affecting circulation, antithrombotics, comprises several classes of drugs used to regulate clotting mechanisms.
Hypertension
Epidemiology and etiology
More than 1 billion people worldwide and 1 in every 4 Americans has high blood pressure (≥140/90 mm Hg). Hypertension adversely affects numerous body organs, including the heart, brain, kidney, and eye. Damage to these organ systems resulting from hypertension is termed cardiovascular disease (CVD). Uncontrolled hypertension increases CVD morbidity and mortality by increasing the risk of developing left ventricular hypertrophy, angina, myocardial infarction (MI), heart failure, stroke, peripheral arterial disease, retinopathy, and kidney disease. One of eight deaths can be attributed to hypertension, and the World Health Organization reports that suboptimal blood pressure (systolic blood pressure above 115 mm Hg) is responsible for 62% of cerebrovascular disease and 49% of ischemic heart disease. Blood pressure increases with age, and hypertension is more prevalent in adults older than 65 years. This fact is of great concern because it is estimated that by 2040, 25% of the American population will be older than 65. Hypertension occurs more frequently in men than in women and occurs in more blacks than whites. Evidence suggests that individuals who are normotensive have a greater than 90% lifetime risk for developing hypertension by age 55 (Table 22-1).1
TABLE 22-1
JNC-VII Classification of Blood Pressure for Adults
| CATEGORY | SYSTOLIC (mm Hg) | DIASTOLIC (mm Hg) |
| Normal | <120 | <80 |
| Prehypertension | 120-139 | 80-89 |
| Hypertension | ||
| Stage 1 | 140-159 | 90-99 |
| Stage 2 | ≥160 | ≥100 |
Hypertension is diagnosed by the mean of two or more separate seated blood pressure determinations on different days. The Seventh Report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC-VII), published in 2003, is the most up-to-date guideline compared with other hypertension treatment recommendations.2 The American Heart Association (AHA) released a statement regarding the treatment of hypertension as it relates to the prevention and management of ischemic heart disease, which differed from the JNC-VII recommendations.3 Significant differences between JNC-VII and the AHA statement include expanding the category of the high-risk hypertensive to include patients with known coronary artery disease (CAD) or CAD risk equivalents (e.g., carotid artery disease, peripheral arterial disease, or abdominal aortic aneurysm or a 10-year Framingham risk score of more than 10%).
In almost all cases, the etiology of hypertension is unknown, and it is termed either primary hypertension or essential hypertension. The prevalence of secondary hypertension is less than 10%; secondary hypertension includes many disease-induced and drug-induced etiologies. Disease-induced causes of hypertension include Cushing syndrome, hyperparathyroidism, hyperthyroidism, pheochromocytoma, primary aldosteronism, and kidney disease. Drug-induced causes of hypertension include amphetamines, corticosteroids, cyclosporine, erythropoietin, estrogens, nonsteroidal antiinflammatory drugs (NSAIDs) including cyclooxygenase-1 inhibitors (e.g., ibuprofen and naproxen) and cyclooxygenase-2 inhibitors (e.g., celecoxib), pseudoephedrine, sibutramine, tacrolimus, venlafaxine, high sodium–containing over-the-counter (OTC) products (e.g., Alka-Seltzer effervescent antacid tablets), OTC weight loss products (e.g., ephedrine-containing diet pills), and chronic alcohol ingestion.4,5
Hypertensive crisis
A patient with blood pressure greater than 180/120 mm Hg is considered to be in a hypertensive crisis. A hypertensive crisis represents either a hypertensive urgency or a hypertensive emergency. Hypertensive urgencies usually signify high blood pressures without signs or symptoms of acute target organ complications; however, patients may present with severe headaches, shortness of breath, nosebleeds, or severe anxiety. In these situations, improvement in blood pressure control can be accomplished over a period of 24 to 48 hours.2 Overaggressive use of intravenous drugs and oral medications can cause too rapid a decrease in blood pressure. Rapid decrease in blood pressure can result in hypoperfusion of organs such as the brain, kidneys, and heart. Oral antihypertensive agents such as captopril, clonidine, and labetalol are routinely used to manage hypertensive urgencies, followed by close observation for several hours. Patients can benefit from antihypertensive medication adjustments if they are found to be noncompliant with taking their medications.
< ?xml:namespace prefix = "mml" />
Hypertension pharmacotherapy
First-line agents for the treatment of uncomplicated hypertension are thiazide-type diuretics, including ACEIs, ARBs, β blockers, and CCBs.2 Vasodilators, α-blocking agents, α2 agonists, and antiadrenergic agents are considered second-line antihypertensives.2 It is unknown if direct renin inhibitors (DRIs), a new class of antihypertensives, should be considered as first-line agents because long-term morbidity and mortality data are currently unavailable. For stage 1 hypertension, pharmacotherapy should be initiated for most patients with a low dose of a once-daily agent, usually a thiazide-type diuretic, and titrated upward until blood pressure control is achieved or intolerable adverse effects occur. Clinicians should be cognizant that monotherapy achieves effective blood pressure control in only 60% to 70% of patients. Thiazide diuretics profoundly decrease CVD morbidity and mortality, enhance the antihypertensive effects of the other antihypertensives, and are very useful in achieving blood pressure control. For stage 2 hypertension, because a higher response rate may be achieved by initiating low-dose combination antihypertensives, usually a thiazide-type diuretic plus an alternative first-line agent is used. The low-dose combination method may minimize adverse effects and may maximize efficacy and compliance.6,7 Figure 22-1 presents an algorithm for the management of hypertension.2,3
Angiotensin-converting enzyme inhibitors
ACEIs act primarily through suppression of the renin-angiotensin-aldosterone system (RAAS). Because of a lack of renal blood flow, renin is released into the circulation, where it acts on angiotensinogen to produce angiotensin I. In the pulmonary vasculature, angiotensin I is converted by angiotensin-converting enzyme (ACE) to angiotensin II. Angiotensin II is a highly potent endogenous vasoconstrictor that also stimulates aldosterone secretion from the zona glomerulosa cells of the adrenal cortex, contributing to sodium and water retention.8 Angiotensin II also stimulates the release of catecholamines from the adrenergic nerve endings and mediates the release of central sympathetic outflow. ACE is abundant in the endothelial cells of blood vessels and to a lesser extent in the kidneys.
ACEIs block the conversion of angiotensin I to angiotensin II by competing with the physiologic substrate angiotensin I for the active site of ACE (Figure 22-2). The affinity of ACEIs for ACE is approximately 30,000 times greater than for angiotensin I. ACEIs also inhibit kininase which is responsible for the degradation of bradykinin and other vasodilating substances, including prostaglandin E2 (PGE2) and prostacyclin (PGI2), which enhances the antihypertensive effects of these drugs. Because ACEIs are potent antihypertensives in patients with low-renin hypertension, the effects on bradykinin may have an integral role in the mechanism of action of these agents. The hemodynamic effects of ACEIs are a reduction of peripheral arterial resistance, an increase in cardiac output, little or no change in heart rate, an increase in renal blood flow, and unchanged glomerular filtration rate (GFR). ACEIs have mild antihyperlipidemic effects.
ACEIs rival diuretics as the most effective first-line antihypertensives to decrease CVD morbidity and mortality in various settings. In contrast to β blockers and thiazide diuretics, ACEIs do not induce glucose intolerance, hyperlipidemia, or hyperuricemia. ACEIs are homogeneous, which means there is very little variability among ACEIs in terms of efficacy and toxicity. With the exception of captopril, all ACEIs are generally administered once or twice daily. Enalaprilat is the only available parenteral ACEI. Table 22-2 presents the pharmacokinetics and dosing guidelines for ACEIs.4,5
TABLE 22-2
Pharmacokinetics and Dosing Guidelines for Angiotensin-Converting Enzyme Inhibitors (ACEIs)
| ACEI GENERIC NAME (BRAND NAME) | ACTIVE METABOLITE | ELIMINATION TOTAL | HALF-LIFE OF PARENT DRUG (hr)* | DURATION OF ACTION (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY | EFFECT OF FOOD ON ABSORPTION |
| Benazepril (Lotensin) | Benazeprilat | 11%-12% bile | 22 | 24+ | 5-80 | 1 | Slightly reduced |
| Captopril (Capoten) | None | 95% urine | 2 | 6-10 | 12.5-450 | 2-4 | Reduced by 30%-40% |
| Enalapril (Vasotec) | Enalaprilat | 94% urine and feces | 11 | 24 | 2.5-40 | 1-2 | None |
| Enalaprilat (Vasotec IV) | None | No data | 35 | 1.25-5 | Every 6 hours | NA | |
| Fosinopril (Monopril) | Fosinoprilat | 50% urine, 50% feces | 12-15 | 24 | 10-80 | 1 | Slightly reduced |
| Lisinopril (Prinivil; Zestril) | None | 29% urine, 69% feces, 2% unchanged | 13 | 24 | 10-40 | 1 | None |
| Moexipril (Univasc) | Moexiprilat | 13% urine, 53% feces | 2-9 | 24 | 7.5-30 | 1-2 | Markedly reduced |
| Quinapril (Accupril) | Quinaprilat | 60% urine, 37% feces | 2-3 | 24+ | 20-80 | 1-2 | Reduced |
| Perindopril (Aceon) | Perindoprilat | 96%-78% bile, 4%-12% urine | 0.8-1 | 24 | 4-16 | 1 | Reduced |
| Ramipril (Altace) | Ramiprilat | 60% urine, 40% feces | 11-17 | 24+ | 2.5-20 | 1-2 | Slightly reduced |
| Trandolapril (Mavik) | Trandolaprilat | 33% urine, 56% feces | 24 | 24+ | 1-4 | 1 | Reduced |
Angiotensin II receptor blockers
Several nonrenin and non-ACE pathways are used for the production of angiotensin II (see Figure 22-2). Nonrenin pathways generate angiotensin II from angiotensinogen via tissue plasminogen activator, cathepsin G, and tonin. Non-ACE enzymes that generate angiotensin II from angiotensin I are cathepsin G, chymostatin-sensitive angiotensin II–generating enzyme, and chymase. ACEIs incompletely block the synthesis of angiotensin II. ARBs are angiotensin II type 1 (AT1) receptor antagonists. AT1 receptors are found in many tissues, such as vascular smooth muscle, myocardial tissue, brain, kidney, liver, uterus, and adrenal glands (cortex and medulla). Many tissues also have an AT2 receptor; however, it is not known to have effects on myocardial hemostasis. ARBs have 1000-fold greater affinity for AT1 receptors than AT2 receptors and generally do not block the AT2 receptor. Because ARBs do not inhibit ACE, they do not interfere with the concentrations of bradykinins and substance P. This kinin-sparing effect may explain why ARBs have a low incidence of inducing cough or angioedema. However, the beneficial effects of kinins, including blood pressure–lowering potency, may be sacrificed.
Compared with ACEIs, ARBs are considered as potent or slightly weaker antihypertensive agents. The inhibition of bradykinin by ACEIs may account for its augmented antihypertensive effect. Angiotensin II receptor blockers arguably are considered second-line agents to ACEIs for hypertension and heart failure and are indicated when ACEI-induced cough or other adverse effects are intolerable. However, ARBs may be considered superior to ACEIs in patients with type 2 diabetic nephropathy. ARBs are administered once or twice daily. Using the combination of an ACEI and an ARB has not been well studied; however, its beneficial effects have been observed in patients with heart failure and nephrotic syndrome. Table 22-3 presents the pharmacokinetics and dosing guidelines for ARBs.4,5,8
TABLE 22-3
Pharmacokinetics and Dosing Guidelines for Angiotensin II Receptor Blockers (ARBs)
| ARB GENERIC NAME (BRAND NAME) | ELIMINATION | TERMINAL HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY | EFFECT OF FOOD ON ABSORPTION |
| Candesartan (Atacand) | Ester hydrolysis/O-deethylation | 9 | 8-32 | 1-2 | No effect |
| Eprosartan (Teveten) | 80% unchanged, 20% acyl glucuronide | 5-9 | 400-800 | 1-2 | No effect |
| Irbesartan (Avapro) | CYP2C9, CYP3A4 | 11-15 | 150-300 | 1 | No effect |
| Losartan (Cozaar) | CYP2C9, CYP3A4 | 2 | 25-100 | 1-2 | Slightly reduced |
| Olmesartan (Benicar) | 35%-50% in urine and remainder in feces | 13 | 20-40 | 1 | No effect |
| Telmisartan (Micardis) | Conjugation to acyl glucuronide | 24 | 20-80 | 1 | Slightly reduced |
| Valsartan (Diovan) | Biliary metabolism | 6 | 80-320 | 1 | Markedly reduced |
Direct renin inhibitors
DRIs act by inhibiting renin, the enzyme that is the first step of the RAAS (see Figure 22-2). Renin is responsible for the conversion of angiotensinogen to angiotensin I, which is the rate-limiting step in RAAS. Renin inhibition also leads to decreased formation of angiotensin II and aldosterone. However, all agents that inhibit the RAAS, such as ACEIs, have the potential to inhibit feedback inhibition of renin leading to increases in renin and its activity. This effect can be blocked with the use of a renin inhibitor. DRIs can be used alone or in combination with other antihypertensive agents.
Aliskiren is administered once daily at a dose of 150 to 300 mg. It has very poor oral bioavailability; only about 2.5% is absorbed. Absorption of aliskiren is substantially decreased by high fatty meals; patients should always take it the same way: either with or without food. It undergoes minimal hepatic metabolism by CYP3A4. Cyclosporine and itraconazole, which are potent inhibitors of CYP3A4, were shown to increase aliskiren levels significantly and should not be used concomitantly. Other CYP3A4 inhibitors were also shown to increase aliskiren levels, but the clinical significance of their interaction is unknown. Aliskiren has also been shown to reduce the effectiveness of furosemide by 30% to 50%. The effectiveness of furosemide should be monitored when these two agents are used concomitantly. Approximately 25% of the absorbed dose is excreted unchanged in the urine. Most of the unabsorbed drug is excreted in the feces. No dosage adjustments are recommended at this time in patients with renal or hepatic impairment.9,10
Calcium channel blockers
Dihydropyridine CCBs are potent vasodilators; these agents include amlodipine, felodipine, isradipine, nicardipine, nifedipine, and nisoldipine. With the exception of nifedipine, dihydropyridines have negligible chronotropic effects. Immediate-release nifedipine, especially when administered as a liquid (pseudosublingual), causes a potent reflex tachycardia that increases coronary oxygen demand and has been implicated with an increased risk of MI and stroke. Only sustained-release dosage forms of nifedipine are indicated for hypertension.11 Amlodipine and plausibly felodipine may be used in patients with heart failure because these agents do not decrease cardiac contractility. CCBs are very effective antihypertensive agents in both elderly and black patients. Table 22-4 presents the pharmacokinetics and dosing guidelines for calcium antagonists.2–5
TABLE 22-4
Pharmacokinetics and Dosing Guidelines for Calcium Channel Blockers
| CALCIUM ANTAGONIST GENERIC NAME (BRAND NAME) | ONSET OF ACTION OF ORAL DOSAGE FORMS (hr) | HALF-LIFE (hr) | DOSE RANGE (mg/day) | DAILY FREQUENCY |
| Nondihydropyridines | ||||
| Verapamil (Calan, Isoptin) | 0.5 | 3-7 | 180-480 | 3-4 |
| Verapamil SR (Calan SR, Isoptin SR) | 0.5 | 3-7 | 120-480 | 1-2 |
| Verapamil ER (Covera-HS) | 4-5 | 2.8-7.4 | 180-420 | Once at bedtime |
| Verapamil chronotherapeutic oral drug absorption (Verelan PM) | 4-5 | 3-7 | 100-400 | Once at bedtime |
| Diltiazem (Cardizem) | 0.5 | 3.5 | 90-360 | 3-4 |
| Diltiazem ER capsules (Cardizem CD, Cartia XT, Dilacor XR, Diltia XT, Tiazac, Taztia XT) | 1 | 5 | 90-540 | 1-2 |
| Diltiazem ER tablets (Cardizem LA) | 3-4 | 6-9 | 120-540 | Once daily (morning or evening) |
| Dihydropyridines | ||||
| Amlodipine (Norvasc) | 6-12 | 30-50 | 2.5-10 | 1 |
| Felodipine (Plendil) | 2-5 | 11-16 | 5-20 | 1 |
| Isradipine (DynaCirc) | 2 | 8 | 2.5-10 | 2 |
| Isradipine CR (DynaCirc CR) | 2 | 8 | 2.5-10 | 1 |
| Nicardipine (Cardene) | 20 minutes | 2-4 | 60-120 | 3 |
| Nicardipine SR (Cardene SR) | 20 minutes | 2-4 | 60-120 | 2 |
| Nifedipine (Adalat, Procardia)* | 20 minutes | 2-5 | 30-120 | 3-4 |
| Nifedipine LA (Adalat CC, Procardia XL) | 20 minutes | 7 | 30-120 | 1 |
| Nimodipine (Nimotop)† | ND | 1-2 | 360 | Every 4 hours for 21 days |
| Nisoldipine (Sular) | ND | 7-12 | 20-60 | 1 |
*Nifedipine (prompt release) is not indicated for hypertension.
β blockers
The antihypertensive effects of β blockers have multiple mechanisms of action and are as follows:
• Blockade of the β receptors on the renal juxtaglomerular cells, leading to renin blockade and decreased angiotensin II concentrations
• Blockade of myocardial β receptors, leading to decreased cardiac contractility and heart rate, diminishing cardiac output
• Blockade of central nervous system (CNS) β receptors, leading to decreased sympathetic output from the CNS and plausibly blockade of peripheral β receptors, decreasing norepinephrine concentrations
β blockers are indicated for hypertension, angina pectoris, cardiac dysrhythmias, secondary prevention of MI, chronic heart failure, and pheochromocytoma. β blockers are no longer considered first-line agents in treatment of essential hypertension but should be reserved as add-on therapy to other antihypertensive agents. β blockers are also used for migraine prophylaxis, hypertrophic subaortic stenosis, tremors, alcohol withdrawal syndrome, prophylaxis of esophageal variceal rebleeding, anxiety, symptoms of thyrotoxicosis, and in combination with α blockers for pheochromocytoma. Table 22-5 presents the pharmacokinetics and dosing guidelines for β blockers.2,3,5,9
TABLE 22-5
Pharmacokinetics and Dosing Guidelines for β Blockers
| β BLOCKER GENERIC NAME (BRAND NAME) | α BLOCKADE | β1 SELECTIVITY | ISA | LIPID SOLUBILITY | HALF-LIFE (hr) | DOSE RANGE (mg) | DAILY FREQUENCY |
| Acebutolol (Sectral) | 0 | + | + | Low | 3-4 | 200-200 | 2 |
| Atenolol (Tenormin) | 0 | + | 0 | Low | 6-9 | 25-100 | 1 |
| Betaxolol (Kerlone) | 0 | + | 0 | Low | 14-24 | 5-20 | 1 |
| Bisoprolol (Zebeta) | 0 | ++ | + | Low | 9-12 | 25-200 | 1 |
| Carteolol (Cartrol) | 0 | 0 | + | Low | 6 | 2.5-10 | 1 |
| Carvedilol (Coreg) | + | 0 | 0 | High | 7-10 | 6.25-50 | 2 |
| Labetalol (Trandate, Normodyne) | + | 0 | 0 | Moderate | 3-5 | 100-2400 | 2 |
| Metoprolol (Lopressor) | 0 | + | 0 | Moderate | 3-5 | 50-200 | 1-2 |
| Metoprolol ER (Toprol-XL) | 0 | + | 0 | Moderate | 3-7 | 25-200 | 1 |
| Nadolol (Corgard) | 0 | 0 | 0 | Low | 14-24 | 20-240 | 1 |
| Nebivolol (Bystolic) | 0 |
