[level-membership-for-basic-science-category]
4 Drug interactions
Despite regulatory requirements to define the safety profile of new medicines including their potential for drug–drug interactions before marketing, the potential for adverse interactions is not always evident. This was illustrated by the worldwide withdrawal of the calcium channel blocker mibefradil, within months of launch, following reports of serious drug interactions (Li Wan Po and Zhang, 1998). In the past decade, a number of medicines have been either withdrawn from the market, for example, terfenadine, grepafloxacin and cisapride, or had their use restricted because of prolongation of the QT interval on the electrocardiogram, for example, thioridazine. Drug interactions are an important cause of QT prolongation which increases the risk of developing a life-threatening ventricular arrhythmia known as torsade de pointes (Roden, 2004).
Definition
An interaction is said to occur when the effects of one drug are altered by the co-administration of another drug, herbal medicine, food, drink or other environmental chemical agents (Baxter, 2010). The net effect of the combination may manifest as an additive or enhanced effect of one or more drugs, antagonism of the effect of one or more drugs, or any other alteration in the effect of one or more drugs.
Epidemiology
The reported incidence of drug–drug interactions in hospital admissions ranged from 0% to 2.8% in a review which included nine studies, all of which had some design flaws (Jankel and Fitterman, 1993). In the Harvard Medical Practice Study of adverse events, 20% of events in acute hospital inpatients were drug related. Of these, 8% were considered to be due to a drug interaction, suggesting that interactions are responsible for less than 2% of adverse events in this patient group (Leape et al., 1992).
In a 1-year prospective study of patients attending an Emergency Department, 3.8% resulted from a drug–drug interaction and most of these led to hospital admissions (Raschett et al., 1999). In a prospective UK study carried out on hospital inpatients, ADR were responsible for hospital admission in 6.5% of cases. Drug interactions were involved in 16.6% of adverse reactions, therefore being directly responsible for leading to hospital admission in approximately 1% of cases (Pirmohamed et al., 2004).
Susceptible patients
The risk of drug interactions increases with the number of drugs used. In a hospital study, the rate of ADR in patients taking 6–10 drugs was 7%, rising to 40% in those taking 16–20 drugs, with the exponential rise being largely attributable to drug interactions (Smith et al., 1969). In a high-risk group of emergency department patients, the risk of potential adverse drug interaction was 13% in patients taking 2 drugs and 82% in those taking 7 or more drugs (Goldberg et al., 1996).
Although polypharmacy is common and often unavoidable, it places certain patient groups at increased risk of drug interactions. Patients at particular risk include those with hepatic or renal disease, those on long-term therapy for chronic disease, for example, HIV infection, epilepsy, diabetes, patients in intensive care, transplant recipients, patients undergoing complicated surgical procedures and those with more than one prescriber. Critically ill and elderly patients are at increased risk not only because they take more medicines but also because of impaired homeostatic mechanisms that might otherwise counteract some of the unwanted effects. Interactions may occur in some individuals but not in others. The effects of interactions involving drug metabolism may vary greatly in individual patients because of differences in the rates of drug metabolism and in susceptibility to microsomal enzyme induction. Certain drugs are frequently implicated in drug interactions and require careful attention (Box 4.1).
Mechanisms of drug interactions
Pharmacokinetic interactions
Absorption
Induction or inhibition of drug transport proteins
The oral bioavailability of some drugs is limited by the action of drug transporter proteins, which eject drugs that have diffused across the gut lining back into the gut. At present, the most well-characterised drug transporter is P-glycoprotein. Digoxin is a substrate of P-glycoprotein and drugs that inhibit P-glycoprotein, such as verapamil, may increase digoxin bioavailability with the potential for digoxin toxicity (DuBuske, 2005).
Drug metabolism
CYP450 isoenzymes
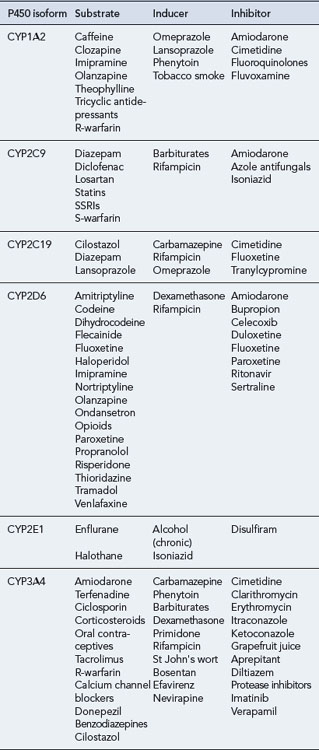
The CYP450 system comprises 57 isoenzymes, each derived from the expression of an individual gene. As there are many different isoforms of these enzymes, a classification for nomenclature has been developed, comprising a family number, a subfamily letter and a number for an individual enzyme within the subfamily (Wilkinson, 2005). Four main subfamilies of P450 isoenzymes are thought to be responsible for most (about 90%) of the metabolism of commonly used drugs in humans: CYP1, CYP2, CYP3 and CYP4. The most extensively studied isoenzyme is CYP2D6, also known as debrisoquine hydroxylase. Although there is overlap, each cytochrome 450 isoenzyme tends to metabolise a discrete range of substrates. Of the many isoenzymes, a few (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4) appear to be responsible for the human metabolism of most commonly used drugs.
The genes that encode specific cytochrome 450 isoenzymes can vary between individuals and, sometimes, ethnic groups. These variations (polymorphisms) may affect metabolism of substrate drugs. Interindividual variability in CYP2D6 activity is well recognised (see Chapter 5). It shows a polymodal distribution and people may be described according to their ability to metabolise debrisoquine. Poor metabolisers tend to have reduced first-pass metabolism, increased plasma levels and exaggerated pharmacological response to this drug, resulting in postural hypotension. By contrast, ultra-rapid metabolisers may require considerably higher doses for a standard effect. About 5–10% of white Caucasians and up to 2% of Asians and black people are poor metabolisers.
The effect of a cytochrome 450 isoenzyme on a particular substrate can be altered by interaction with other drugs. Drugs may be themselves substrates for a cytochrome 450 isoenzyme and/or may inhibit or induce the isoenzyme. In most instances, oxidation of a particular drug is brought about by several CYP isoenzymes and results in the production of several metabolites. So, inhibition or induction of a single isoenzyme would have little effect on plasma levels of the drug. However, if a drug is metabolised primarily by a single cytochrome 450 isoenzyme, inhibition or induction of this enzyme would have a major effect on the plasma concentrations of the drug. For example, if erythromycin (an inhibitor of CYP3A4) is taken by a patient being given carbamazepine (which is extensively metabolised by CYP3A4), this may lead to toxicity due to higher concentrations of carbamazepine. Table 4.1 gives examples of some drug substrates, inducers and inhibitors of the major cytochrome 450 isoenzymes.
Enzyme induction
Enzyme induction usually results in a decreased pharmacological effect of the affected drug. St John’s wort is now known to be a potent inducer of CYP3A (Mannel, 2004). Thus, when a patient receiving ciclosporin, tacrolimus, HIV-protease inhibitors, irinotecan or imatinib takes St John’s wort, there is a risk of therapeutic failure with the affected drug. However, if the affected drug has active metabolites, this may lead to an increased pharmacological effect. The effects of enzyme induction vary considerably between patients and are dependent upon age, genetic factors, concurrent drug treatment and disease state. Some examples of interactions due to enzyme induction are shown in Table 4.2.
| Drug affected | Inducing agent | Clinical outcome |
|---|---|---|
| Oral contraceptives | Rifampicin | Therapeutic failure of contraceptives |
| Rifabutin | Additional contraceptive precautions required | |
| Modafinil | Increased oestrogen dose required | |
| Ciclosporin | Phenytoin | Decreased ciclosporin levels with possibility of transplant rejection |
| Carbamazepine | ||
| St John’s wort | ||
| Paracetamol | Alcohol (chronic) | In overdose, hepatotoxicity may occur at lower doses |
| Corticosteroids | Phenytoin | Increased metabolism with possibility of therapeutic failure |
| Rifampicin |
Enzyme inhibition
Enzyme inhibition is responsible for many clinically significant interactions. Many drugs act as inhibitors of cytochrome 450 enzymes (see Box 4.2). A strong inhibitor is one that can cause ≥5-fold increase in the plasma area-under-the-curve (AUC) value or more than 80% decrease in clearance of CYP3A substrates. A moderate inhibitor is one that can cause ≥2- but <5-fold increase in the AUC value or 50–80% decrease in clearance of sensitive CYP3A substrates when the inhibitor is given at the highest approved dose and the shortest dosing interval. A weak inhibitor is one that can cause ≥1.25- but <2-fold increase in the AUC values or 20–50% decrease in clearance of sensitive CYP3A substrates when the inhibitor is given at the highest approved dose and the shortest dosing interval.
The clinical significance of this type of interaction depends on various factors, including dosage (of both drugs), alterations in pharmacokinetic properties of the affected drug such as half-life and patient characteristics such as disease state. Interactions of this type are again most likely to affect drugs with a narrow therapeutic range such as theophylline, ciclosporin, oral anticoagulants and phenytoin. For example, starting treatment with an enzyme inhibitor such as ritonavir in a patient taking sildenafil could result in a marked increase in sildenafil plasma concentrations. Some examples of interactions due to enzyme inhibition are shown in Table 4.3.
| Drug affected | Inhibiting agent | Clinical outcome |
|---|---|---|
| Anticoagulants (oral) | Ciprofloxacin | Anticoagulant effect increased and risk of bleeding |
| Clarithromycin | ||
| Azathioprine | Allopurinol | Enhancement of effect with increased toxicity |
| Clopidogrel | Lansoprazole | Reduced anti-platelet effect |
| Carbamazepine | Cimetidine | Antiepileptic levels increased with risk of toxicity |
| Phenytoin | ||
| Sodium valproate | ||
| Sildenafil | Ritonavir | Enhancement of sildenafil effect with risk of hypotension |
The isoenzyme CYP3A4, in particular, is present in the enterocytes. Thus, after oral administration of a drug, cytochrome 450 enzymes in the intestine and the liver may reduce the portion of a dose that reaches the systemic circulation, that is, the bioavailability of the drug. Drug interactions resulting in inhibition or induction of enzymes in the intestinal epithelium can have significant consequences. For example, by selectively inhibiting CYP3A4 in the enterocyte, grapefruit juice can markedly increase the bioavailability of some oral calcium channel blockers, including felodipine (Wilkinson, 2005). Such an interaction is usually considered to be a drug metabolism interaction, even though the mechanism involves an alteration in drug absorption. A single glass of grapefruit juice can cause CYP3A inhibition for 24–48 h and regular consumption may continuously inhibit enzyme activity. Consumption of grapefruit juice is therefore not recommended in patients receiving drugs that are extensively metabolised by CYP3A such as simvastatin, tacrolimus and vardenafil.
Elimination interactions
Changes in active renal tubule excretion
Drugs that use the same active transport system in the kidney tubules can compete with one another for excretion. Such competition between drugs can be used to therapeutic advantage. For example, probenecid may be given to increase the plasma concentration of penicillins by delaying renal excretion. With the increasing understanding of drug transporter proteins in the kidneys, it is now known that probenecid inhibits the renal secretion of many other anionic drugs via organic anion transporters (OATs; Lee and Kim, 2004). Increased methotrexate toxicity, sometimes life-threatening, has been seen in some patients concurrently treated with salicylates and some other non-steroidal anti-inflammatory drugs (NSAIDs). The development of toxicity is more likely in patients treated with high-dose methotrexate and those with impaired renal function. The mechanism of this interaction may be multifactorial but competitive inhibition of methotrexate’s renal tubular secretion is likely to be involved. If patients taking methotrexate are given salicylates or NSAIDs concomitantly, the dose of methotrexate should be closely monitored.
Drug transporter proteins
Drugs and endogenous substances are now known to cross biological membranes not just by passive diffusion but by carrier-mediated processes, often known as transporters. Significant advances in the identification of various transporters have been made and although their contribution to drug interactions is not yet clear, they are now thought to play a role in many interactions formerly attributed to cytochrome 450 enzymes (DuBuske, 2005).
P-glycoprotein (P-gp) is a large cell membrane protein that is responsible for the transport of many substrates, including drugs. It is a product of the ABCB1 gene (previously known as the multidrug resistance gene, MDR1) and a member of the adenosine triphosphate (ATP)-binding cassette family of transport proteins (ABC transporters). P-glycoprotein is found in high levels in various tissues including the renal proximal tubule, hepatocytes, intestinal mucosa, the pancreas and the blood–brain barrier. P-glycoprotein acts as an efflux pump, exporting substances into urine, bile and the intestinal lumen. Its activity in the blood–brain barrier limits drug accumulation in the central nervous system (CNS). Examples of some possible inhibitors and inducers of P-glycoprotein are shown in Table 4.4. The pumping actions of P-glycoprotein can be induced or inhibited by some drugs. For example, concomitant administration of digoxin and verapamil, a P-glycoprotein inhibitor, is associated with increased digoxin levels with the potential for digoxin toxicity. There is an overlap between CYP3A4 and P-glycoprotein inhibitors, inducers and substrates. Many drugs that are substrates for CYP3A4 are also substrates for P-glycoprotein. Therefore, both mechanisms may be involved in many of the drug interactions initially thought to be due to changes in CYP3A4. Digoxin is an example of the few drugs that are substrates for P-glycoprotein but not CYP3A4.
Table 4.4 Examples of inhibitors and inducers of P-glycoprotein
| Inhibitors | Atorvastatin |
| Ciclosporin | |
| Clarithromycin | |
| Dipyridamole | |
| Erythromycin | |
| Itraconazole | |
| Ketoconazole | |
| Propafenone | |
| Quinidine | |
| Ritonavir | |
| Valspodar | |
| Verapamil | |
| Inducers | Rifampicin |
| St John’s wort |
Pharmacodynamic interactions
Additive or synergistic interactions
If two drugs with similar pharmacological effects are given together, the effects can be additive (see Table 4.5). Although not strictly drug interactions, the mechanism frequently contributes to ADR. For example, the concurrent use of drugs with CNS-depressant effects such as antidepressants, hypnotics, antiepileptics and antihistamines may lead to excessive drowsiness, yet such combinations are frequently encountered. Combinations of drugs with arrhythmogenic potential such as antiarrhythmics, neuroleptics, tricyclic antidepressants and those producing electrolyte imbalance (e.g. diuretics) may lead to ventricular arrhythmias and should be avoided. Another example which has assumed greater importance of late is the risk of ventricular tachycardia and torsade de pointes associated with the concurrent use of more than one drug with the potential to prolong the QT interval on the electrocardiogram (Roden, 2004).
| Interacting drugs | Pharmacological effect |
|---|---|
| NSAID, warfarin, clopidogrel | Increased risk of bleeding |
| ACE inhibitors and K-sparing diuretic | Increased risk of hyperkalaemia |
| Verapamil and β-adrenergic antagonists | Bradycardia and asystole |
| Neuromuscular blockers and aminoglycosides | Increased neuromuscular blockade |
| Alcohol and benzodiazepines | Increased sedation |
| Pimozide and sotalol | Increased risk of QT interval prolongation |
| Clozapine and co-trimoxazole | Increased risk of bone marrow suppression |
Serotonin syndrome
Serotonin syndrome (SS) is associated with an excess of serotonin that results from therapeutic drug use, overdose or inadvertent interactions between drugs. Although severe cases are uncommon, it is becoming increasingly well recognised in patients receiving combinations of serotonergic drugs (Boyer and Shannon, 2005). It can occur when two or more drugs affecting serotonin are given at the same time or after one serotonergic drug is stopped and another started. The syndrome is characterised by symptoms including confusion, disorientation, abnormal movements, exaggerated reflexes, fever, sweating, diarrhoea and hypotension or hypertension. Diagnosis is made when three or more of these symptoms are present and no other cause can be found. Symptoms usually develop within hours of starting the second drug but occasionally they can occur later. Drug-induced SS is generally mild and resolves when the offending drugs are stopped. Severe cases occur infrequently and fatalities have been reported.
Drug–herb interactions
There has been a marked increase in the availability and use of herbal products in the UK over the past decade, which include Chinese herbal medicines and Ayurvedic medicines. Up to 24% of hospital patients report using herbal remedies (Constable et al., 2007). Such products often contain pharmacologically active ingredients which can give rise to clinically significant interactions when used inadvertently with other conventional drugs.
Conclusion
Whilst one should acknowledge the impossibility of memorising all potential drug interactions, health care workers need to be alert to the possibility of drug interactions and take appropriate steps to minimise their occurrence. Drug formularies and the Summary of Product Characteristics provide useful information about interactions. Other resources that may also be of use to prescribers include drug safety updates from regulators such as the Medicines and Health care products Regulatory Agency (available at http://www.mhra.gov.uk/index.htm), interaction alerts in prescribing software and the availability of websites which highlight interactions for specific drug classes, for example, HIV drugs (http://www.hiv-druginteractions.org/.).
Possible interventions to avoid or minimise the risk of a drug interaction include:
Answers
Answers
Baxter K., editor. Stockley’s Drug Interactions, ninth ed, London: Pharmaceutical Press, 2010.
Boyer E.W., Shannon M. Current concepts: the serotonin syndrome. N. Engl. J. Med.. 2005;352:1112-1120.
Constable S., Ham A., Pirmohamed M. Herbal medicines and acute medical emergency admissions to hospital. Br. J. Clin. Pharmacol.. 2007;63:247-248.
DuBuske L.M. The role of P-glycoprotein and organic anion-transporting polypeptides in drug interactions. Drug Saf.. 2005;28:789-801.
Goldberg R.M., Mabee J., Chan L., et al. Drug–drug and drug–disease interactions in the ED: analysis of a high-risk population. Am. J. Emerg. Med.. 1996;14:447-450.
Jankel C.A., Fitterman L.K. Epidemiology of drug–drug interactions as a cause of hospital admissions. Drug Saf.. 1993;9:55-59.
Leape L.L., Brennan T.A., Laird N., et al. The nature of adverse events in hospitalised patients: results of the Harvard Medical Practice Study II. N. Engl. J. Med.. 1992;324:377-384.
Lee W., Kim R.B. Transporters and renal drug elimination. Ann. Rev. Pharmacol. Toxicol.. 2004;44:137-166.
Li Wan Po A., Zhang W.Y. What lessons can be learnt from withdrawal of mibefradil from the market? Lancet. 1998;351:1829-1830.
Mannel M. Drug interactions with St John’s wort: mechanisms and clinical implications. Drug Saf.. 2004;27:773-797.
Pirmohamed M., James S., Meakin S., et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18820 patients. Br. Med. J.. 2004;329:15-19.
Raschett R., Morgutti M., Mennitti-Ippolito F., et al. Suspected adverse drug events requiring emergency department visits or hospital admissions. Eur. J. Clin. Pharmacol.. 1999;54:959-963.
Roden D.M. Drug-induced prolongation of the QT interval. N. Engl. J. Med.. 2004;350:1013-1022.
Smith J.W., Seidl L.G., Cluff L.E. Studies on the epidemiology of adverse drug reactions v. clinical factors influencing susceptibility. Ann. Intern. Med.. 1969;65:629.
Wilkinson G.R. Drug therapy: drug metabolism and variability among patients in drug response. N. Engl. J. Med.. 2005;352:2211-2221.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
4 Drug interactions
Despite regulatory requirements to define the safety profile of new medicines including their potential for drug–drug interactions before marketing, the potential for adverse interactions is not always evident. This was illustrated by the worldwide withdrawal of the calcium channel blocker mibefradil, within months of launch, following reports of serious drug interactions (Li Wan Po and Zhang, 1998). In the past decade, a number of medicines have been either withdrawn from the market, for example, terfenadine, grepafloxacin and cisapride, or had their use restricted because of prolongation of the QT interval on the electrocardiogram, for example, thioridazine. Drug interactions are an important cause of QT prolongation which increases the risk of developing a life-threatening ventricular arrhythmia known as torsade de pointes (Roden, 2004).
Definition
An interaction is said to occur when the effects of one drug are altered by the co-administration of another drug, herbal medicine, food, drink or other environmental chemical agents (Baxter, 2010). The net effect of the combination may manifest as an additive or enhanced effect of one or more drugs, antagonism of the effect of one or more drugs, or any other alteration in the effect of one or more drugs.
Epidemiology
The reported incidence of drug–drug interactions in hospital admissions ranged from 0% to 2.8% in a review which included nine studies, all of which had some design flaws (Jankel and Fitterman, 1993). In the Harvard Medical Practice Study of adverse events, 20% of events in acute hospital inpatients were drug related. Of these, 8% were considered to be due to a drug interaction, suggesting that interactions are responsible for less than 2% of adverse events in this patient group (Leape et al., 1992).
In a 1-year prospective study of patients attending an Emergency Department, 3.8% resulted from a drug–drug interaction and most of these led to hospital admissions (Raschett et al., 1999). In a prospective UK study carried out on hospital inpatients, ADR were responsible for hospital admission in 6.5% of cases. Drug interactions were involved in 16.6% of adverse reactions, therefore being directly responsible for leading to hospital admission in approximately 1% of cases (Pirmohamed et al., 2004).
Susceptible patients
The risk of drug interactions increases with the number of drugs used. In a hospital study, the rate of ADR in patients taking 6–10 drugs was 7%, rising to 40% in those taking 16–20 drugs, with the exponential rise being largely attributable to drug interactions (Smith et al., 1969). In a high-risk group of emergency department patients, the risk of potential adverse drug interaction was 13% in patients taking 2 drugs and 82% in those taking 7 or more drugs (Goldberg et al., 1996).
Although polypharmacy is common and often unavoidable, it places certain patient groups at increased risk of drug interactions. Patients at particular risk include those with hepatic or renal disease, those on long-term therapy for chronic disease, for example, HIV infection, epilepsy, diabetes, patients in intensive care, transplant recipients, patients undergoing complicated surgical procedures and those with more than one prescriber. Critically ill and elderly patients are at increased risk not only because they take more medicines but also because of impaired homeostatic mechanisms that might otherwise counteract some of the unwanted effects. Interactions may occur in some individuals but not in others. The effects of interactions involving drug metabolism may vary greatly in individual patients because of differences in the rates of drug metabolism and in susceptibility to microsomal enzyme induction. Certain drugs are frequently implicated in drug interactions and require careful attention (Box 4.1).
Mechanisms of drug interactions
Pharmacokinetic interactions
Absorption
Induction or inhibition of drug transport proteins
The oral bioavailability of some drugs is limited by the action of drug transporter proteins, which eject drugs that have diffused across the gut lining back into the gut. At present, the most well-characterised drug transporter is P-glycoprotein. Digoxin is a substrate of P-glycoprotein and drugs that inhibit P-glycoprotein, such as verapamil, may increase digoxin bioavailability with the potential for digoxin toxicity (DuBuske, 2005).
Drug metabolism
CYP450 isoenzymes
The CYP450 system comprises 57 isoenzymes, each derived from the expression of an individual gene. As there are many different isoforms of these enzymes, a classification for nomenclature has been developed, comprising a family number, a subfamily letter and a number for an individual enzyme within the subfamily (Wilkinson, 2005). Four main subfamilies of P450 isoenzymes are thought to be responsible for most (about 90%) of the metabolism of commonly used drugs in humans: CYP1, CYP2, CYP3 and CYP4. The most extensively studied isoenzyme is CYP2D6, also known as debrisoquine hydroxylase. Although there is overlap, each cytochrome 450 isoenzyme tends to metabolise a discrete range of substrates. Of the many isoenzymes, a few (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4) appear to be responsible for the human metabolism of most commonly used drugs.
The genes that encode specific cytochrome 450 isoenzymes can vary between individuals and, sometimes, ethnic groups. These variations (polymorphisms) may affect metabolism of substrate drugs. Interindividual variability in CYP2D6 activity is well recognised (see Chapter 5). It shows a polymodal distribution and people may be described according to their ability to metabolise debrisoquine. Poor metabolisers tend to have reduced first-pass metabolism, increased plasma levels and exaggerated pharmacological response to this drug, resulting in postural hypotension. By contrast, ultra-rapid metabolisers may require considerably higher doses for a standard effect. About 5–10% of white Caucasians and up to 2% of Asians and black people are poor metabolisers.
[/not-level-membership-for-basic-science-category]
