[level-membership-for-pediatrics-category]
Drug-Induced Disorders of the Gastrointestinal Tract
Joseph Misdraji
Nonsteroidal Antiinflammatory Drugs
Sodium Polystyrene Sulfonate (Kayexalate)
Antacids, Sucralfate, and Gastric Mucosal Calcinosis
Chemical Colitis (Glutaraldehyde)
Introduction
Given the bewildering array of prescription and over-the-counter drugs and the fact that most are administered orally, it is not surprising that many drugs cause gastrointestinal (GI) pathology. Drug-related injury may be caused by direct toxic effects of the drug on the GI mucosa, toxic effects to the mesenchymal components of the gut including the enteric nervous system, systemic effects, or indirect damage (e.g., antibiotic-associated pseudomembranous colitis). The patterns of injury associated with drugs include virtually all types. Therefore, a complete medication list for each patient is essential to avoid misdiagnosing a drug reaction as another type of inflammatory disorder such as inflammatory bowel disease (IBD). Occasionally, a drug reaction may be suspected on the basis of specific findings within biopsy specimens, such as demonstration of certain crystals or pigments. More often, histology alone is not sufficient to implicate a specific drug as a cause of an inflammatory disorder of the gut.
In this chapter, a variety of common but nonspecific injury patterns, and the drugs associated with them, are described. Some of these patterns are organ specific, such as reactive gastropathy or pill esophagitis, whereas others are generalizable to the entire GI tract, such as ulcers, strictures, or vasculitis. The injury may be direct or indirect. Only the most common drug associations are discussed here. Also included is a discussion of specific drugs that are well known to cause GI damage, beginning with the most common ones, nonsteroidal antiinflammatory drugs (NSAIDs) and proton pump inhibitors (PPIs).
Nonspecific Injury Patterns
Organ-Specific
Esophagus
Pill Esophagitis
Pill esophagitis occurs secondary to caustic injury resulting from retention of a pill in the esophagus. This condition is often associated with failure to drink adequate amounts of liquid with the medication or consuming medications in the supine position before bedtime.1–5 The most common agents are antibiotics (particularly doxycycline, tetracycline, and clindamycin), NSAIDs, potassium chloride, iron supplements, ascorbic acid, quinidine, emepronium bromide, and alendronate.1,4–13 The mechanisms by which these drugs cause esophagitis vary. Tetracyclines, ascorbic acid, and ferrous sulfate produce acidic solutions when dissolved in water, suggesting that they produce an acid burn, whereas phenytoin produces an alkaline solution and possibly an alkaline burn.3,6 Production of local hyperosmolarity by potassium chloride and intracellular poisoning (after mucosal uptake) by doxycycline and NSAIDs may be the principal mechanisms of injury of these specific agents.3,6,14
Women and elderly patients are more often affected,1–4,6,8 although affected patients have a wide age range. Different types of drugs are consumed by different age groups. For instance, in a review of 650 reported cases, the average age of patients who had quinidine-related esophageal injury was 60 years, whereas that of patients injured by oral antibiotics was 30 years.13 Most patients have odynophagia, retrosternal pain, and dysphagia at presentation2–4,6,7,10 and do not reveal a history of preexisting esophageal dysmotility.1,3 Complications of pill esophagitis include esophageal strictures, hemorrhage, esophageal perforation, and even death.2,3,6,11,15
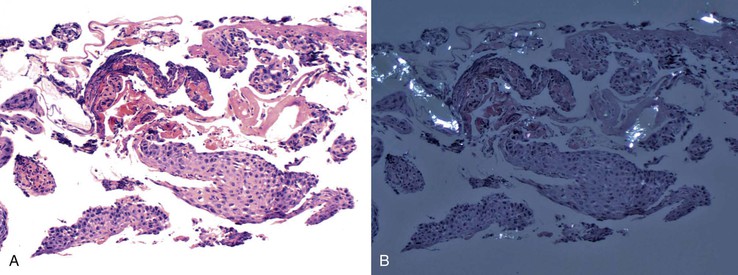
Endoscopic findings include erythema, mucosal denudation, discrete ulcers or erosions, and strictures.1,4,6,7,11,13,15,16 In some circumstances, the squamous epithelium exfoliates, forming an intraluminal cast, a condition known as esophagitis dissecans superficialis (see Chapter 16).9,17 Quinidine-induced esophageal injury occasionally manifests with an exuberant inflammatory exudate that mimics carcinoma.3,4 Remnants of the pill may also be seen in this condition.5 The most common anatomic sites of involvement are the midesophagus, typically at the level of the aortic arch (22 to 24 cm), and in patients with left atrial enlargement, the distal esophagus at 30 to 35 cm.2–5,10 However, distal involvement with stricture formation may occur, and this may be mistaken for reflux esophagitis.1,4 Histologic features are usually nonspecific. Acute inflammation, granulation tissue, erosions, and ulcers are common.7,10 When present, polarizable crystalline material may be an important clue to the diagnosis (Fig. 12.1), but it is not present in all cases.7 Multinucleation of squamous epithelial cells has been reported in cases of alendronate-associated esophageal injury.7,18 Esophageal strictures have been described in patients taking potassium chloride, doxycycline, tetracycline, acetylsalicylic acid, ascorbic acid, phenytoin, or quinidine.1
Stomach
Reactive Gastropathy
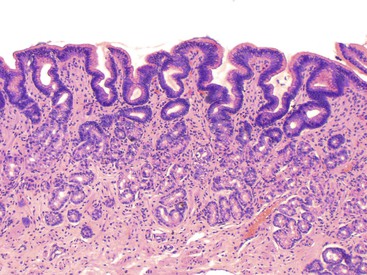
Reactive gastropathy was initially believed to be specifically related to gastric mucosal injury caused by reflux of duodenal contents into the stomach and was also known as alkaline gastritis or bile reflux gastritis.19,20 This distinctive histologic picture is now known to represent a nonspecific response to a variety of gastric irritants, of which bile is only one. NSAIDs and alcohol are common causes of reactive gastritis. The features of reactive gastropathy (also called chemical gastritis) include foveolar hyperplasia with a “corkscrew” appearance to the pits, surface epithelial degeneration with cuboidalization of the foveolar glandular cells, mucin depletion, edema, vascular congestion, a paucity of inflammatory cells in the lamina propria, and splaying of smooth muscle fibers in the lamina propria. The muscle fibers are oriented from the muscularis mucosae toward the mucosal lumen (Fig. 12.2).19,21 Occasional foci of atrophy with or without pseudopyloric or intestinal metaplasia are common and may reflect chronic injury resulting from the drug and subsequent repair.21
Small and Large Intestine
Colitis
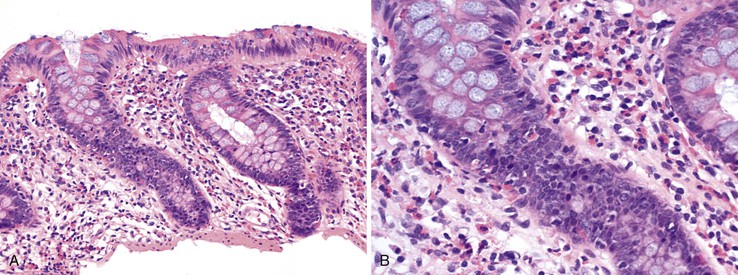
Many types of drugs cause colitis and in this capacity mimic other, more common forms of colitis, such as infectious colitis or IBD. The pattern of colitis may suggest particular agents (Table 12.1). For instance, eosinophilic colitis (Fig. 12.3) is associated with numerous drugs, including NSAIDs, gold compounds, and carbamazepine hypersensitivity.22–24 In one series of patients who underwent colonoscopy for probable drug-induced colitis, increased eosinophils were found in the left colon of patients taking NSAIDs, antiplatelet agents, or estroprogestinic agents, regardless of the colonoscopic findings.25 NSAIDs are associated with increased lymphocytes and scattered neutrophils in the lamina propria or, in some cases, an appearance similar to lymphocytic colitis.26 α-Methyldopa and NSAIDs may cause neutrophilic colitis, mimicking infection.27 Amoxicillin and levofloxacin have been implicated in pseudomembranous colitis.24 Chemical colitis from glutaraldehyde, alcohol, or hydrogen peroxide can mimic ischemic colitis.28–31 Therapy with antibody to cytotoxic T-lymphocyte–associated antigen 4 (anti-CTLA4) can induce an immune mediated enterocolitis that resembles ulcerative colitis.32,33
Table 12.1
Patterns of Colitis and Possible Drug-Related Etiologies
| Pattern of Colitis | Possible Drug |
| Eosinophilic colitis | NSAIDs, gold, carbamazepine, antiplatelet agents, estroprogestinic agents |
| Lymphocytic or collagenous colitis | NSAIDs, lansoprazole, ticlopidine, ranitidine, simvastatin, flutamide, carbamazepine, sertraline, penicillin V |
| Focal active colitis | NSAIDs, oral sodium phosphate |
| Ischemic colitis | NSAIDs, glutaraldehyde, antibiotics, chemotherapy, nasal decongestants, constipation-inducing medications, laxatives, vasopressor agents, cocaine, ergotamine, serotonin agonists/antagonists including sumatriptan, high-dose estrogen and progesterone, amphetamines, digitalis, diuretics, and immunomodulators such as interleukin 2 |
| Apoptotic colitis | Bowel preparation with oral sodium phosphate, laxatives, chemotherapeutic agents (especially 5-fluorouracil), NSAIDs, and cyclosporine A |
| Pseudomembranous colitis | NSAIDs, amoxicillin, levofloxacin, antibiotic-associated Clostridium difficile colitis |
| Immune mediated colitis | CTLA4 antibodies |
| Neutropenic colitis | Chemotherapy |
CTLA4, Cytotoxic T-lymphocyte–associated antigen 4; NSAIDs, nonsteroidal antiinflammatory drugs.
Ischemic Enteritis/Colitis
The clinical presentation of drug-induced ischemia varies depending on the offending agent, the specific mesenteric vessels involved, the interval between exposure to the drug and presentation, and the general status of the patient. Segmental involvement is typical. The histology is identical to ischemia from other causes (see Chapter 10) and may show ulceration, necrosis, edema, and fibrosis. Drugs associated with ischemic colitis include antibiotics, NSAIDs, chemotherapeutic agents such as taxanes, nasal decongestants, constipation-inducing medications, laxatives, vasopressor agents, cocaine, ergotamine, serotonin agonists/antagonists including sumatriptan, high-dose estrogen and progesterone, amphetamines, digitalis, diuretics, and immunomodulators such as interleukin 2.24,37
The mechanisms by which drugs cause ischemia vary. For instance, estrogens induce vascular thrombosis, ergotamine causes vascular spasm leading to proctitis with the formation of shallow ulcers, cocaine is a potent sympathomimetic mesenteric vasoconstrictor that produces severe intestinal ischemia, diuretics can cause extracellular fluid volume changes that favor peripheral circulation over mesenteric circulation, and antibiotics can cause hypersensitivity vasculitis. Chemical colitis, such as from glutaraldehyde or alcohol enemas, can also appear histologically indistinguishable from ischemic colitis.28–31
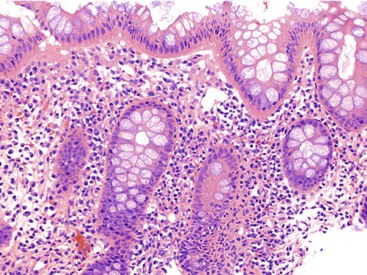
Focal Active Colitis
Focal active colitis (FAC) is defined as cryptitis that involves one or only a few crypts. It is associated with epithelial injury and occasionally with increased mononuclear inflammation in an otherwise unremarkable colonic biopsy (Fig. 12.4). Historically, FAC was believed to be strongly associated with Crohn’s disease, but more recent studies suggest that only a minority of patients with FAC either have or develop Crohn’s disease. For instance, in a 1997 study involving patients without a prior history of IBD, Greenson and colleagues38 found that most symptomatic patients with FAC had acute self-limited or infectious colitis, whereas FAC in asymptomatic patients carried no clinical significance. Crohn’s disease did not develop in any of the patients. However, 19 of 42 patients in this study were taking NSAIDs, leaving open the possibility that NSAIDs may cause FAC. Subsequently, FAC was reported by Driman and colleagues39 in patients given oral sodium phosphate as a bowel preparation regimen, and they suggested that the idiopathic cases in Greenson’s report may have been caused by such regimens. Other causes of FAC include Crohn’s disease and ischemic colitis.
Pseudo-obstruction (Ileus)
Drug-induced pseudo-obstruction, or paralytic ileus, rarely comes to the attention of surgical pathologists, because the treatment typically involves discontinuation of the offending agent. However, complications such as megacolon or perforation may ensue and may necessitate surgical intervention. Many drugs can damage the myenteric plexus, causing loss of neurons and schwannosis. For instance, narcotics, phenothiazines, tricyclic antidepressants, anthraquinone laxatives, anti-Parkinson drugs, clonidine, calcium channel blockers, and vincristine are associated with pseudo-obstruction.
Non–Organ-Specific
Ulcers (Solitary and Multiple)
Numerous drugs can lead to the formation of erosions or ulcers in the GI tract. These may be encountered as a single lesion with nonspecific features or as part of a more widespread ischemic, cytotoxic, or inflammatory process (Fig. 12.5). Potassium chloride was one of the earliest reported agents implicated in causing ulcers and strictures in the GI tract, including the stomach.40,41 Certainly, the most common type of drug implicated in gastric ulcers is NSAIDs.42 These drugs not only damage the mucosa but also retard ulcer healing.43 Other drugs associated with GI ulcers include alendronate, doxycycline, chemotherapeutic agents, corticosteroids, ferrous sulfate, Kayexalate, ergot, gold compounds, and colchicine.5,12,41,44–46
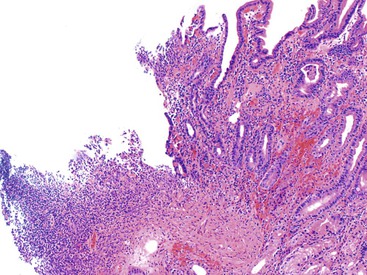
Perforation
Perforation caused by drugs is uncommon but may complicate severe ischemic, toxic, or inflammatory colitis. Intestinal perforation has been reported in association with immunosuppressive medications (e.g., steroids, azathioprine), NSAIDs, slow-release potassium chloride, flucytosine, and other medications. Corticosteroids, opioids, and NSAIDs are associated with an increased risk of diverticular perforation.47–49 GI perforation is strongly associated with NSAID use.50,51
Strictures
Esophageal strictures have been described in patients taking potassium chloride, doxycycline, tetracycline, acetylsalicylic acid, ascorbic acid, phenytoin, or quinidine.1 Several drugs have been implicated in intestinal strictures, including potassium chloride and pancreatic enzyme replacement. A dramatic example of drug-related intestinal and colonic strictures is diaphragm disease caused by NSAIDs (see below as well as Chapter 16 for details).
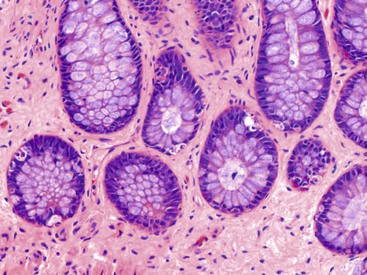
Apoptosis (Apoptotic Colopathy)
Normally, colonic epithelial cells migrate and ultimately mature toward the luminal surface of the mucosa. Eventually, senescent epithelial cells undergo apoptosis and then slough into the lumen or pass through the basement membrane into the lamina propria, where they are ingested by macrophages. Occasionally, increased apoptotic bodies are observed in the surface epithelium or immediately beneath the surface epithelium of otherwise normal-appearing mucosa. Sometimes the crypts may be involved as well (Fig. 12.6). There are a variety of potential causes, including medications used for bowel preparation (e.g., oral sodium phosphate). Laxatives may also cause an increase in the number of surface epithelial apoptotic bodies. Chronic use of laxatives may result in chronic degradation of increased numbers of apoptotic colonic epithelial cells and subsequent deposition of lipofuscin in macrophage lysosomes; this mechanism is presumed to lead to melanosis coli.52 Several other drugs have been associated with increased apoptotic bodies in the crypt epithelium, including chemotherapeutic agents (especially 5-fluorouracil), NSAIDs, and cyclosporine A.53 Other, non–drug-related causes of apoptotic colopathy include graft-versus-host disease, common variable immunodeficiency (CVID), cytomegalovirus, radiation injury, and human immunodeficiency virus (HIV) enteropathy.
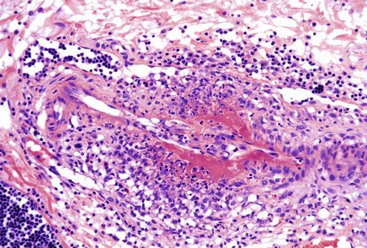
Vasculitis
Drug-induced vasculitis can affect the GI tract and result in ischemia. Quinidine, ranitidine, clarithromycin, angiotensin-converting enzyme inhibitors, acetylsalicylic acid, carbidopa/levodopa, ampicillin, chlorpromazine, and ciprofloxacin have been associated with Henoch-Schönlein purpura. GI involvement by Henoch-Schönlein purpura can cause diarrhea and vomiting and may be complicated by obstruction or perforation. GI involvement occurs most commonly in the second part of the duodenum, but Henoch-Schonlein purpura can also affect the esophagus, stomach, colon, and rectum. Discrete, coin like lesions that coalesce, as well as hemorrhagic and ecchymotic lesions, have been described. Biopsy specimens shows granulocytes in the wall of small arterioles or venules with necrosis of the vessel wall similar to leukocytoclastic vasculitis (Fig. 12.7).54
Another type of vasculitis possibly related to drugs is lymphocytic enterocolic phlebitis (see Chapter 10). This condition affects the right colon, small intestine, or sigmoid colon, often with ischemic consequences. In lymphocytic enterocolic phlebitis, veins show perivascular lymphocytic inflammation, with subendothelial aggregation, thickening of the vessel wall, and occasionally fibrinoid necrosis of the vessel (Fig. 12.8). In some cases, the inflammation is sparse and the myointimal hyperplasia is more prominent. Some cases reveal an associated lymphocytic infiltrate in the lamina propria and epithelium, analogous to lymphocytic colitis or collagenous colitis. Lymphocytic enterocolic phlebitis was initially described in three patients who had consumed rutoside, a phlebotonic drug commonly used in Europe to treat varicose veins.55 Although another reported case also occurred in a patient taking rutoside, subsequent cases have not corroborated the association with this drug. Some patients who had taken the antiandrogen drug flutamide have also been reported to develop this form of vasculitis.56,57 Therefore, although the exact cause of lymphocytic enterocolic phlebitis remains unknown, a hypersensitivity reaction to drugs is one possible etiology.
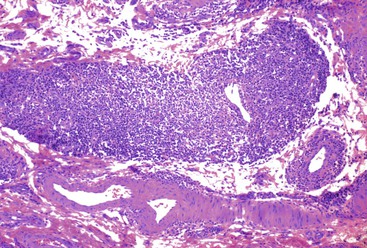
Indirect Injury
Infectious Colitis
Various drugs may lead to an increased risk or promote the development of certain infections of the GI tract. The association of antibiotics with pseudomembranous colitis caused by Clostridium difficile is the best-known example (see Chapters 4 and 17). Necrotizing enterocolitis, neutropenic enterocolitis, or other infections including GI candidiasis may develop in people who have received chemotherapy.58 Immunosuppressive agents predispose patients to opportunistic infectious organisms, such as cytomegalovirus.59,60
Patients with iron overload on deferoxamine therapy are predisposed to Yersinia infection, and both the iron overload and its treatment contribute to the risk of infection.61 Iron is an essential growth factor for bacteria, including Yersinia, but normally free iron in the host is too low to support the growth of virulent organisms. For bacterial pathogens, successful infection relies on invading cells, in part to acquire iron. Strains that have the Yersinia high-pathogenicity island are able to synthesize the siderophore yersiniabactin, enabling them to acquire iron from host proteins.62 Transfusion with its resultant iron load increases the amount of iron available to the organism, and iron overload impairs neutrophilic phagocytic activity.63 Furthermore, the addition of exogenous siderophores such as deferoxamine makes iron more available for the organism.62 The combination of iron overload and deferoxamine therapy increases the risk of Yersinia infection; in one study, invasive Yersinia infection was diagnosed in 14 patients with β-thalassemia at a frequency 5000-fold greater than in the general population, and all but 2 of these patients were taking deferoxamine at the time of diagnosis of infection.64
PPIs may also indirectly predispose to infection. Because gastric acid plays a role in killing bacteria, a reduction in gastric acid may reduce the ability to eliminate bacterial pathogens. An association between PPI use and bacterial gastroenteritis, primarily with Campylobacter and Salmonella, has been reported.65–67 Similarly, the reduced ability to kill bacterial spores may explain the reported association between PPI use and C. difficile infection.68
Specific Agents
Nonsteroidal Antiinflammatory Drugs
NSAIDs are the most widely prescribed drugs in the world. Not surprisingly, they are associated with numerous patterns of injury to the GI tract. NSAID-induced injury to the upper GI tract most commonly includes esophagitis and esophageal strictures; gastric, esophageal, and duodenal ulcers; GI bleeding; and perforation. Lower GI tract injury includes enteritis, virtually every pattern of colitis, ulceration, perforation, and a distinctive type of stricture disorder of the distal small intestine and colon termed diaphragm disease. NSAIDs can also exacerbate other preexisting diseases of the colon. The relative risk of serious GI complications in patients exposed to NSAIDs is 5 to 6 times higher than in nonexposed individuals.69 This risk seems to be declining, however, possibly because of a combination of factors such as the introduction of safer, more mucosal-friendly NSAIDs; widespread use of PPIs, which decrease the risk of NSAID injury to the stomach; and reduction in dosages being used.70
Pathogenesis
NSAIDs-induced GI injury occurs by means of various local and systemic mechanisms. Because NSAIDs are weak acids, they become un-ionized within the strong acid environment of the stomach and, as a result, may pass through cell membranes. Once inside the cytoplasm of cells, they become ionized and thus trapped within the cell (ion trapping) (Fig. 12.9).71,72 Within cells, NSAIDs uncouple oxidative phosphorylation, which depletes cells of adenosine triphosphate (ATP).71,72 The insult to epithelial cells results in increased mucosal permeability, which allows gastric acid, bacteria, or bile acids to further damage the mucosa. NSAIDs promote leukocyte-endothelial cell adhesion in the microvasculature, possibly resulting in ischemic mucosal damage. Most importantly, NSAIDs suppress prostaglandin synthesis via inhibition of cyclooxygenase (Fig. 12.10).72,73 Although some prostaglandins are responsible for inflammation, others, in particular prostaglandins E2 and I2, are involved in the regulation of mucosal mucin production, bicarbonate secretion, mucosal blood flow, epithelial cell proliferation, and epithelial restitution, all of which contribute to protection of the upper GI tract mucosa from acid.72 Suppression of these prostaglandins results in mucosal injury. Cyclooxygenase exists as two different isoforms, and the ability of NSAIDs to cause damage is related more to their ability to selectively inhibit COX-1 rather than COX-2.71 COX-1 is abundant in gastric mucosa; COX-2 is expressed at low levels in intact stomachs but is upregulated when COX-1 is inhibited or after injury.71 The introduction of selective COX-2 inhibitors was viewed as a promising development in the battle to find NSAIDs with fewer GI side effects. Two major studies, the Celecoxib Long-Term Arthritis Safety Study (CLASS)74 and the Vioxx Gastrointestinal Outcomes Research Trial (VIGOR)75 demonstrated that these agents are associated with a significantly lower incidence of upper GI events (e.g., perforations, ulcers, bleeding) compared with nonselective NSAIDs, although these claims are disputed among some authorities.76 Furthermore, because they selectively block prostacyclin production, COX-2 inhibitors leave thromboxane unopposed, which can lead to thrombotic events.
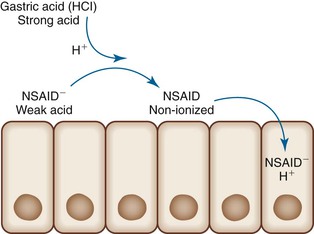
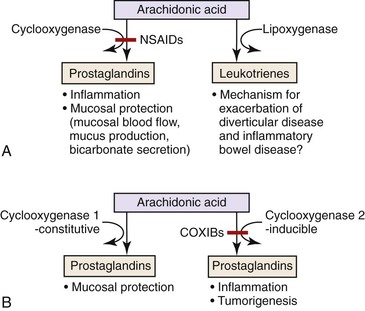
The mechanisms by which NSAIDs affect the lower GI tract may be similar to those that cause upper GI tract injury. Direct toxic effects on the mucosa, depletion of enterocyte ATP, and increased intestinal permeability have all been proposed as having a role in lower GI tract injury.77 Consistent with this theory is the fact that the ability of a NSAID to enter the enterohepatic circulation correlates with its ability to damage the intestinal tract, presumably by increasing exposure time of intestinal mucosa to high concentrations of the offending agent.77 However, parenteral administration of NSAIDs may also lead to lower GI tract injury, which suggests that there are also systemic mechanisms related to prostaglandin suppression, although oral preparations usually result in a greater degree of injury than parenteral formulations.77 Diversion of arachidonic acid to the lipoxygenase pathway results in the formation of leukotrienes and other inflammatory mediators, which helps explain the paradoxical exacerbation of (IBD) and diverticular disease experienced by some patients who consume NSAIDs.49 One other potential mechanism in which NSAIDs may damage the lower GI tract is by inducing ischemia. Intravenous indomethacin causes rapid splanchnic vasoconstriction in dogs; this may be sufficient to cause damage at “watershed” sites in the GI tract.
Risk Factors
Several factors help determine the likelihood of complications of NSAID use, including age, alcohol use, smoking, concomitant Helicobacter pylori infection, the type of NSAID, and the duration of use. Older age, alcohol use, and smoking are risk factors for complications related to NSAIDs, although the level of risk is difficult to quantify.51 The role of H. pylori is complex. Some evidence suggests that the risk of NSAID injury is paradoxically greater in H. pylori–negative patients, perhaps because H. pylori infection increases prostaglandin levels within gastric mucosa.43 Other studies suggest that H. pylori increases the risk of ulcer disease and bleeding in NSAID users and that eradication of H. pylori before NSAID therapy markedly reduces the incidence of ulcers or bleeding.78–80 H. pylori has numerous effects on the gastric mucosa, including enhancing prostaglandin production and increasing neutrophilic inflammation, and these may influence NSAID-induced gastric injury in a variety of ways.80 The duration of NSAID use may affect the types of injury sustained. Short-term use is more often seen in patients with gastric erosions or bleeding, whereas long-term use is associated with ileal and colonic strictures. The type of NSAID may also be relevant in the development of certain forms of injury. For example, the small intestine is exposed to higher concentrations of those NSAIDs that enter the enterohepatic circulation, increasing the likelihood of intestinal injury from those specific agents. Other factors that may help predict the risk of GI complications from NSAIDs include their COX-1 and COX-2 selectivity and plasma half-life.51
Pathology
Esophagitis and Esophageal Strictures
Pill esophagitis can be caused by various types of NSAIDs (see earlier discussion). Several studies have shown an association between NSAID use and the presence of erosive or necrotizing esophagitis, a greater severity of esophagitis, or poor improvement of esophagitis on follow-up.81–86 However, other studies have not shown an association between NSAID use and either the presence or the severity of esophagitis.87,88 NSAID use has also been associated with an increased risk of esophageal strictures.15,89,90
Erosions and Ulcers
NSAID-induced erosions usually occur in the gastric body and heal within a few days, regardless of whether the NSAID use has been continued. However, NSAID-induced ulcers are often large and multiple. They are more common in the gastric antrum than in the duodenum, and they are often painless.79,94 Lower GI ulcers most commonly involve the ileocecal region or proximal colon.95 However, studies with double-balloon endoscopy or capsule endoscopy have revealed mucosal breaks or ulcers in segments of the small intestine that are traditionally not amenable to endoscopic examination.96,97 Histologically, these ulcers are entirely nonspecific and are indistinguishable from other “idiopathic” benign colonic ulcers. Complications include stenoses or strictures, bleeding, and perforation.94
NSAID Colitis
Patients in whom NSAID-induced colitis develops usually have a history of NSAID intake for several months as therapy for chronic inflammatory conditions such as arthritis. Implicated most frequently are sustained-release NSAIDs, fenemate NSAIDs, and diclofenac preparations, although it is unclear whether this is a reflection of their common use in the general population. Women are affected more often than men. Presenting symptoms include bloody diarrhea, weight loss, iron deficiency anemia, and abdominal pain.98,99 Colonoscopic findings may be normal or may show only nonspecific inflammatory changes such as erythema, friability, small ulcers, or aphthous ulcers.99 Any segment of the colon may be affected, including segmental involvement or even pancolonic involvement (pancolitis).99,100 Cessation of NSAIDs causes resolution of the diarrhea and histologic inflammation.98 In protracted cases, some evidence suggests that some patients respond to steroids, antibiotics, or both.
Goldstein and Cinenza26 described the histologic features of 14 cases of NSAID-induced colitis in 1998; patchy colitis was present in 13 cases. Histologic examination showed an inflammatory infiltrate, mild in 9 cases and marked in 5, which was composed of a mixed infiltrate of lymphocytes, plasma cells, and neutrophils in 8 cases, predominantly neutrophilic inflammation in 4, and predominantly lymphoplasmacytic in 2 (Fig. 12.11). Seven cases showed surface erosion. Crypt “disarray” and mildly increased surface intraepithelial lymphocytes were observed in occasional cases, but none showed crypt distortion, granulomas, markedly increased surface intraepithelial lymphocytes, thickened subepithelial collagen table, or surface epithelial apoptotic bodies. Therefore, NSAID colitis is in the differential with causes of FAC such as Crohn’s disease and infectious colitis.
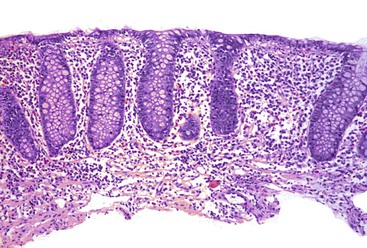
Additional studies have further defined the possible spectrum of NSAID colitis. In a case-control study of 31 patients with collagenous colitis, Riddell and co-workers34 demonstrated significantly more prevalent use of NSAIDs in patients with collagenous colitis compared with controls. In this report, NSAID use preceded the onset of diarrhea in all 19 patients using NSAIDs, the diarrhea improved in 3 patients after cessation of NSAIDs, and was recurrent in 1 on rechallenge. The authors suggested that the association between collagenous colitis and arthritis may, in fact, be caused by NSAIDs.
NSAIDs have also been implicated in lymphocytic colitis. In a study of 40 patients with lymphocytic colitis, half were using NSAIDs, and these patients had higher surface intraepithelial lymphocyte counts.36 In 2004, Goldstein and Bhanot101 described a paucicellular variant of lymphocytic colitis that had similar clinical associations with classic lymphocytic colitis, namely the female predominance, frequent normal appearance on endoscopy, and frequent presentation with watery stools. The frequency of NSAID use was similar in this group and the classic lymphocytic colitis group (21% and 24%, respectively). In a third group of 100 asymptomatic patients with descending colonic biopsy specimens obtained during screening colonoscopy, NSAID use was reported in 39% of those patients with morphologically normal colonic mucosa, in 50% of those with paucicellular lymphocytic colitis, and in 75% of those with classic lymphocytic colitis. The authors concluded that paucicellular lymphocytic colitis should be considered part of the spectrum of lymphocytic colitis and that NSAID use is associated with this pattern of injury. Other reports have also documented a link between NSAID use and either collagenous or lymphocytic colitis,99,102,103 although not all studies have been able to show an association.104
Miscellaneous Forms of NSAID-induced Colitis
Ischemic colitis has been reported in NSAID users. In a report of 11 cases of NSAID colitis, Puspok and colleagues99 described a pattern similar to ischemic colitis in 9 cases (Fig. 12.12). A patient with eosinophilic colitis and clinical features of hypersensitivity has also been reported.23 Two cases of “acute colitis” with neutrophilic cryptitis and mucin depletion were reported in patients taking etodolac.27 A case of pseudomembranous colitis was reported in a patient taking diclofenac.105 Increased apoptosis has also been described in patients using NSAIDs. Recently, Deshpande and associates106 reported frequent NSAID use among patients with unexplained chronic colitis, which raises the possibility that NSAIDs may, in some circumstances, cause colitis indistinguishable from IBD.
Diaphragm Disease
A rare but distinctive complication of long-term (usually >1 year) use of NSAIDs is diaphragm disease.107,108 At presentation, patients have subacute intestinal obstruction, iron deficiency anemia, or fecal occult blood.108–110 Other presenting symptoms include chronic diarrhea, change in bowel habits, abdominal pain, or weight loss. The disease was difficult to recognize before the advent of new techniques that help visualize remote segments of the small bowel, such as retrograde double-balloon enteroscopy and capsule endoscopy. Retention of the video capsule because of the strictures has been increasingly reported.108
Grossly, the characteristic diaphragm is a thin, concentric mucosal web of tissue (Fig. 12.13).110–112 Diaphragms commonly affect the ileum, ascending colon, or proximal transverse colon.99,109–111 They are often numerous (averaging approximately nine), cluster in the same general anatomic region, and are difficult to detect by inspection of only the external surface of the bowel.108,111,112 Histologically, submucosal fibrosis with fibers oriented perpendicular to the movement of fluids in the bowel lumen and mild mucosal inflammation are the key findings (Fig. 12.14).111 A “chaotic” arrangement of smooth muscle fibers, vascular elements, and neural elements resembling neuromuscular and vascular hamartomas has also been described.107,108,113 Mucosal erosions or ulcers may be seen at the apex of the stricture, along with marked regenerative changes of the epithilium.107,111 Other mucosal changes are variable and include villous blunting, cryptitis, crypt abscesses, increased eosinophils, or pseudopyloric metaplasia.107,111
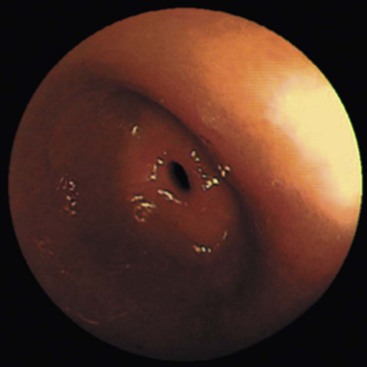
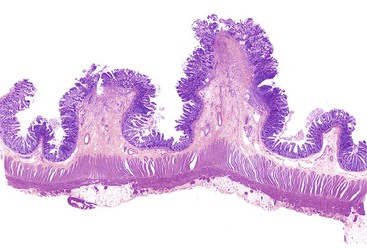
The presumed mechanism of injury involves the formation of a linear ulcer along the crests of haustral folds, with subsequent repair and fibrosis. The resulting band of fibrosis constricts the bowel lumen in a purse-string fashion to form a mucosal diaphragm. Reports of diaphragms in patients treated with suppositories114 and in bypassed ileal segments suggest that a systemic mechanism may be responsible for diaphragm disease.112 Endoscopic balloon dilatation has been reported to be successful in some cases as a form of initial management of diaphragms.115 Otherwise, resection is required. Recurrence has been reported.111
Perforation
Perforation of the GI tract is usually a complication of ulcer disease or diverticular disease, and NSAID use is associated with an increased risk of this complication.49,94,116 Perforations caused by NSAIDs carry a higher mortality rate than other forms of NSAID-induced GI disease. The risk of NSAID-related GI perforation is similar in the upper and lower GI tract.117 The type of NSAID and its plasma half-life influence the risk of perforation.117
Exacerbation of Diverticular Disease or Inflammatory Bowel Disease
NSAIDs may exacerbate preexisting inflammatory colonic diseases. Patients taking NSAIDs have an increased relative risk of symptomatic diverticular disease compared with patients who do not consume these agents.118 Diverticular disease–associated hemorrhage, perforation, and fistula formation have been reported with NSAID use.49,119–121 In patients with preexisting IBD, particularly ulcerative colitis, use of NSAIDs may precipitate relapse.122
Differential Diagnosis
NSAID-induced injury is difficult to distinguish from injury related to other agents. In the stomach, NSAID damage is indistinguishable from reactive gastritis related to other causes, such as bile or alcohol. NSAID colitis should be considered in any patient with diarrhea if a colonic biopsy specimen shows increased cellularity of the lamina propria and features of lymphocytic or collagenous colitis. Given the wide range of morphologic changes associated with NSAIDs, it is also important to consider NSAIDs in patients whose biopsy findings resemble ischemic colitis or pseudomembranous colitis and in patients with colonic perforation. There are no specific morphologic features that can help distinguish NSAIDs from the other causes of these disorders. Diaphragm disease may be difficult to distinguish from other stricturing disorders. A history of long-term NSAID use may be the most important and helpful piece of information in establishing a correct etiology. However, NSAID strictures tend to be more narrowly based than other types of strictures, multiple, and more often located in the ileum or right colon.
Proton Pump Inhibitors
The widespread and easy availability of PPIs has been a significant advance in the treatment of reflux esophagitis and peptic ulcer disease. These agents block gastric acid production by binding H+,K+-ATPase on the canalicular surface of parietal cell membranes, and they are very effective in reducing gastric acidity. However, evidence that they exacerbate corpus gastritis and atrophy, cause endocrine cell hyperplasia and gastric polyps, and may be associated with microscopic colitis has led to some concern regarding their long-term safety.
Exacerbation of Corpus Gastritis and Atrophy
One possible consequence of long-term use of PPIs is exacerbation of corpus gastritis and atrophy, both considered precursors of gastric cancer. Typically, H. pylori colonizes the gastric antrum more effectively than the corpus, and this is possibly related to acid production by the corpus.123 Suppression of gastric acidity presumably allows the organisms to colonize the corpus, enables better contact between the organisms and the corpus foveolar epithelium, and reduces buffering of the ammonia produced by the organism.123 The result may be increased and more severe corpus gastritis and atrophy in some patients.34,73,123–125 However, this is controversial, because some reports have found no association between PPI use and the development or progression of gastric atrophy in H. pylori–infected patients.126–130 Even without atrophy, corpus-predominant gastritis in H. pylori infection has been shown to increase the risk for the development of gastric cancer.131 Because treatment of H. pylori reverses corpus gastritis,132 patients should be evaluated for H. pylori and treated before beginning long-term therapy with PPIs.133
Endocrine Hyperplasia
Patients consuming antisecretory drugs show compensatory increased gastrin levels. The clinical relevance of hypergastrinemia has been explored in numerous studies. Patients treated with histamine H2 receptor antagonists have a twofold rise in serum gastrin levels, but endocrine cell hyperplasia has not been a clinically significant issue in this patient group.134 Long-term treatment with omeprazole is associated with a twofold to fourfold increase in serum gastrin levels in a subset of patients.134–138 Gastrin is trophic for fundic mucosa and enterochromaffin-like cells.139 Endocrine cell hyperplasia has been documented among patients receiving long-term PPI therapy.136–138,140 However, initial concerns that these patients would be at increased risk for the development of carcinoid tumors have not been realized.
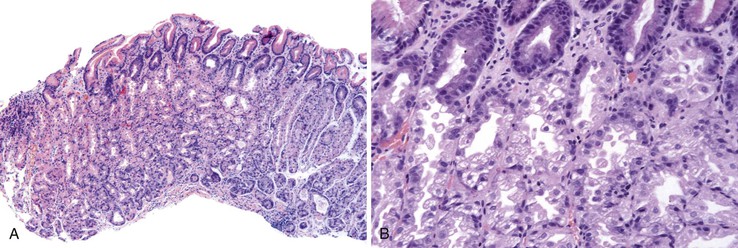
Parietal Cell Hyperplasia and Fundic Gland Polyps
Parietal cell hyperplasia characterized by enlarged and more numerous parietal cells that protrude into the gland lumen develops in many patients taking PPIs (Fig. 12.15).141–143 Fundic gland cysts may develop, presumably caused by obstruction of acid flow from the glands by protuberant parietal cells. This process results in changes that resemble fundic gland polyps.141,143 Several early reports described fundic gland polyps (sometimes multiple) in patients subjected to prolonged PPI use.124,144,145 In some of these patients, the polyps disappeared on discontinuation of the PPI and recurred with resumed use of the medication.144 Some authors have disputed these findings, however.146–148 The number of patients reported to have fundic gland polyps is small relative to the large number of patients taking PPIs. Moreover, this condition occurs in patients without H. pylori149 (possibly because the enzymatic degradation of gastric mucus by the organisms facilitates outflow from the gastric glands and protects against mucosal cyst formation), and the role of this infection as a confounding factor has been questioned. In a case-control study limited to patients without H. pylori gastritis, the incidence of fundic gland polyps among patients taking PPIs was similar to that in a control population.150 Since that report, however, several others have confirmed that prolonged PPI use is strongly associated with the development of polyps.151–153 In a prospective study, PPI use was the strongest risk factor for the development of fundic gland polyps, with an odds ratio of 9 in multiple logistic regression.153
Microscopic Colitis
Thomson and associates35 reported a series of 6 patients in whom either lymphocytic (5 cases) or collagenous (1 case) colitis developed secondary to lansoprazole. Colitis developed in all of them after switching from omeprazole to lansoprazole, and all cases of colitis resolved within 1 week after cessation of the drug.
Differential Diagnosis
Parietal cell hyperplasia secondary to PPI use can be a mimic of the gastric changes seen in Zollinger-Ellison syndrome. The latter often has more cystic dilatation of the glands. Also, PPI use is obviously many times more likely than Zollinger-Ellison syndrome. Clinical information can distinguish the two. Fundic gland polyps secondary to use of PPIs are indistinguishable from sporadic ones.
Sodium Polystyrene Sulfonate (Kayexalate)
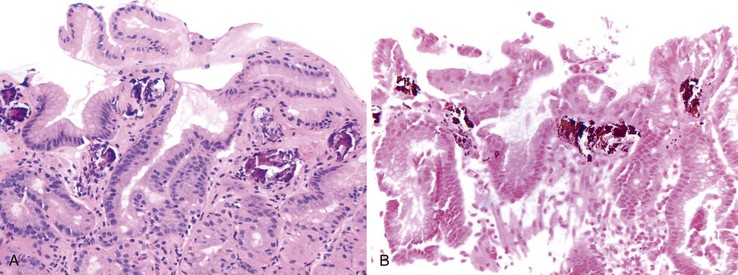
Hyperkalemia develops in patients with chronic renal insufficiency, and is often treated with sodium polystyrene sulfonate (Kayexalate), a cation-exchange resin administered either orally or as an enema. To reduce the risk of constipation and impaction, the resin is mixed with a hypertonic solution of sorbitol, a cathartic agent. Several cases have been reported of acute colonic necrosis in patients given sodium polystyrene sulfonate in sorbitol, with a mortality rate in one series of 36%.154–159 Most, but not all, cases occurred postoperatively, in patients with end-stage renal disease, or in patients with uremia.154–159
The extent of necrosis has been variable, ranging from involvement of the entire colon, rectum, and portions of the terminal ileum to involvement of smaller segments. Ulcers or pseudomembranes may develop, along with other, nonspecific features of colonic necrosis (e.g., dusky discoloration, edema). Histologically, lesions range from mucosal to transmural necrosis with perforation. The diagnostic finding is the presence of Kayexalate crystals overlying necrotic or ulcerated tissue. Kayexalate crystals are refractile and slightly basophilic, with a mosaic pattern. They stain red with either period acid-Schiff or acid-fast stain (Fig. 12.16).155,157
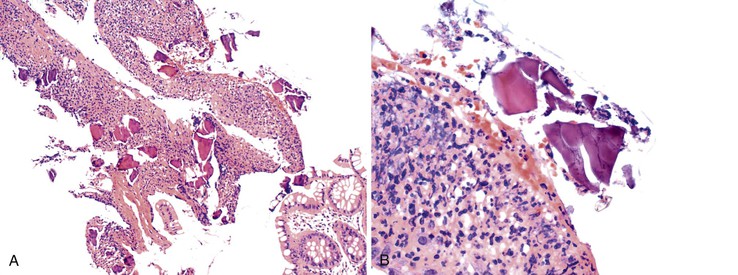
In the initial description of this condition, Lillemoe and co-workers160 demonstrated that the actual culprit is not sodium polystyrene sulfonate but the sorbitol with which it is administered. The mechanism by which this solution induces colonic necrosis is unknown, although uremia, changes in blood volume after dialysis, immunosuppressive therapy, peripheral vascular disease, and thrombocytopenia-related coagulation defects are all considered to be possible contributing factors. It is unclear whether renal failure predisposes patients to this complication or whether patients with renal failure are simply more likely to be hyperkalemic and thus more likely to be given sodium polystyrene sulfonate.
Upper GI tract injury caused by Kayexalate in sorbitol is uncommon. One case report described a patient with serpiginous ulcers in the cecum and gastric antrum associated with Kayexalate crystals.157 A subsequent series of 11 patients161 were found to have Kayexalate crystals in the esophagus (3 patients), stomach (2), duodenum (2), or both stomach and esophagus (4). The crystals were either adherent to intact mucosa or, in 8 patients (73%) with ulcers or erosions, admixed with exudates. GI bleeding was the most common indication for biopsy (36%). Unlike patients with lower GI tract injury, patients with upper GI tract injury do not normally require surgical intervention.
Differential Diagnosis
Kayexalate-induced necrosis must be distinguished from ischemic colitis or pseudomembranous colitis. The presence of Kayexalate crystals, clinical details, and a drug ingestion history distinguish Kayexalate-induced necrosis from ischemic colitis.
Iron Therapy
Patients taking iron tablets may sustain injury to the esophageal or gastric mucosa. Characteristic brown crystalline material may be detected in the lamina propria, in surface exudates, and, less often, in thrombosed vessels; the deposits are highlighted by Perl Prussian blue stain (Fig. 12.17).162–165 In most cases, the iron is associated with erosions or ulcers. A pattern of reactive gastropathy or chronic gastritis may be observed in some cases.165,166 In a study by Abraham and associates,166 half of the affected patients had underlying toxic, mechanical, or infectious conditions that might have predisposed them to mucosal injury, but other authorities have argued that iron causes the injury by itself.164 Iron therapy has also been associated with accumulation of iron within the cytoplasm of macrophages located in the small bowel lamina propria; this is termed pseudomelanosis (Fig. 12.18).164,167,168 The patterns of iron deposition that occur in association with iron medication should be distinguished from the presence of iron within gastric glandular cells, which is more often seen in patients with hereditary hemochromatosis.
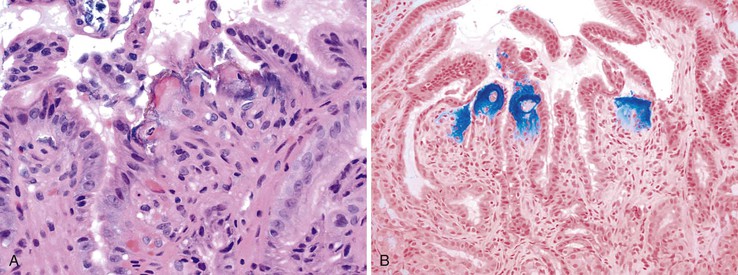
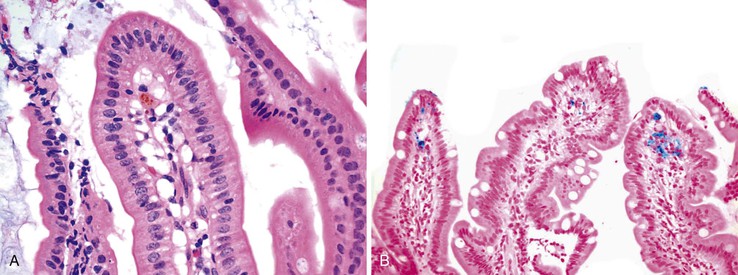
Differential Diagnosis
Iron-induced gastric injury is histologically similar to reactive gastritis from other causes. The only distinguishing feature is the presence of iron crystals in the subepithelial lamina propria, which are highlighted by iron stain.
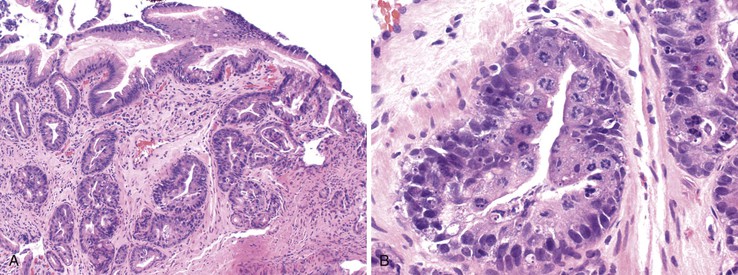
Antacids, Sucralfate, and Gastric Mucosal Calcinosis
The term gastric mucosal calcinosis refers to small calcifications within gastric mucosa, typically located beneath the surface epithelium, either in the antrum or in the body (Fig. 12.19). Calcinosis stains deep pink, or it may appear as partially calcified refractile material, some of which may be rimmed by histiocytes.169 The background gastric mucosa may be unremarkable or may show changes of reactive gastropathy. This condition is an example of so-called metastatic calcification that occurs in patients who have an imbalance of calcium and phosphate. Several conditions can lead to metastatic calcification, including drug use. In a report by Greenson and colleagues,169 all patients with this condition either had received an orthotopic transplant or had chronic renal failure, and all were taking either aluminum-containing antacids or sucralfate. The authors suggest that antacids play a role in the genesis of this condition. Elemental analysis of one case demonstrates that the deposits contained aluminum, phosphorus, calcium, and chlorine. Subsequently, Stroehlein and co-workers170 reported on six renal transplant recipients with gastric mucosal calcinosis, none of whom was taking sucralfate and only one of whom was taking low doses of an aluminum-containing antacid (Gelusil). Interestingly, many of these patients were taking H2 receptor antagonists or PPIs, which raises questions regarding the role of alkalization in precipitating calcium-phosphates in the stomach. Another case report describes gastric calcinosis in a nontransplantation patient who had dyspepsia and hypoparathyroidism and was taking various types of medications, including calcium acetate, calcitriol, and metoprolol (which contains small amounts of aluminum as an inactive ingredient).171
Colchicine
Colchicine is an alkaloid used to treat gout and other medical conditions. Its toxic effects are attributed to its ability to bind tubulin and inhibit its polymerization into microtubules, thereby interfering with neutrophil degranulation, chemotaxis, and mitosis. Colchicine toxicity may develop in patients with renal or hepatic failure, manifested by a cholera-like syndrome associated with dehydration, shock, bone marrow suppression, and acute renal failure.172 In the GI tract, colchicine toxicity is associated with a variable degree of mucosal injury in the esophagus, stomach, and small intestine. Reduced epithelial cell layers, nuclear swelling, and dyskeratosis have been described in the esophagus.172 In the duodenum, variable villous atrophy, nuclear pseudostratification, loss of polarity, and increased apoptotic bodies have been described (Fig. 12.20).173 One characteristic finding is the presence of numerous mitoses arrested in metaphase, particularly in neck cells.172,173 These may assume a characteristic “ring” pattern.173 These changes are not observed in patients taking colchicine who do not have clinical evidence of toxicity. Colchicine toxicity can mimic dysplasia, given the degree of nuclear pseudostratification, apoptotic debris, and increased mitotic figures that can develop with this drug. However, similar to other non-neoplastic conditions that mimic dysplasia, colchicine toxicity shows epithelial maturation, providing evidence of a reactive condition. The presence of ring mitoses is a key distinguishing feature. Clinical history and knowledge of drug ingestion are necessary.
Chemotherapy
Several types of GI complications related to chemotherapy have been described. Esophagitis with stricture formation may occur in patients who receive combination chemotherapy (often containing doxorubicin [Adriamycin]) and radiation therapy.5,58,174 Taxol toxicity is associated with mitotic arrest and the development of numerous ring mitoses, similar to colchicine toxicity, in any segment of the GI tract, but particularly in the esophagus.175,176
Most reports of chemotherapy-related injury to the GI tract describe gastroduodenal inflammation and ulceration in patients who have undergone hepatic arterial infusion chemotherapy (HAIC) for treatment of primary or metastatic carcinoma of the liver.45,46,177–179 The chemotherapeutic agents most often implicated are 5-fluoro-2-deoxyuridine (FUDR) and mitomycin C. Erosions and ulcers may develop in patients with upper GI complaints after HAIC, most often in the antropyloric region, and less often in the duodenum and esophagus.46,179 Histologic examination may show marked epithelial atypia in the region of the ulcer that can be mistaken for early carcinoma.45,177,178 Systemic chemotherapy and radiation therapy for esophageal and gastroesophageal junction carcinomas has also been reported to cause dysplasia-like atypia in the stomach.180 In one study, the prevalence of gastric dysplasia-like changes in an esophageal cancer population was 7.5%.181 The authors describe dysplasia-like changes in the foveolar epithelium and atrophic features with microcyst formation in the glands.
Atypia secondary to chemotherapy can mimic dysplasia or carcinoma. Features that assist in distinguishing these lesions from carcinoma and identifying chemotherapy effects include (1) preservation of mucosal architecture; (2) atypia limited to, or accentuated toward, the basilar gastric glands; (3) bizarre atypia exceeding that usually seen in carcinoma; (4) preservation of a low nucleus-to-cytoplasm (N : C) ratio; (5) prominent cytoplasmic eosinophilia, often with vacuolization; (6) few or no mitotic figures; (7) cytologic resemblance to radiation effect; (8) similar atypia within fibroblasts and endothelial cells; and (9) absence of intestinal metaplasia in adjacent gastric epithelium (Box 12.1).177 Dysplasia-like changes in the stomach can be distinguished from true dysplasia by a more common flat gross appearance, patchy distribution, lack of intestinal metaplasia, a combination of foveolar and gland involvement, evidence of retained surface maturation, cells with an open nuclear chromatin pattern with prominent nucleoli, retained nuclear polarity, mitoses confined to the lower two thirds of the pit epithelium without atypical mitoses, cytoplasmic hypereosinophilia or vacuolization or both, and gland microcystic changes in the former.180
Selective Internal Radiation
Selective internal radiation, in which injected radioactive yttrium 90 microspheres are used rather than traditional chemotherapeutic agents, can cause gastroduodenal ulceration and can lead to the appearance of black microspheres within mucosal capillaries (Fig. 12.21).12,181–183
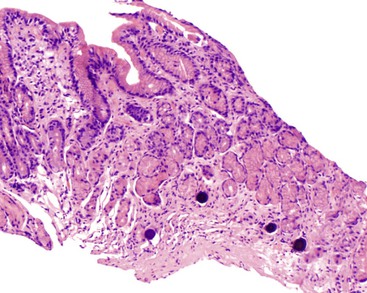
Anti-CTLA4 Antibody–Induced Colitis
CTLA4 is an inducible receptor the primary role of which is to downregulate the immune response, including innate immunity to cancer. By blocking this receptor, anti-CTLA4 antibodies disinhibit T cells and can cause tumor regression and necrosis. These monoclonal antibodies have been used to treat melanoma, prostate cancer, colorectal carcinoma, renal cell carcinoma, and non–small cell lung cancer. By disinhibiting T cells, these antibodies can cause a syndrome of autoimmune or autoinflammatory side effects, designated “immune-related adverse events.” The major toxicity is the induction of immune-mediated enterocolitis, which affects approximately 21% of patients32 and manifests with acute diarrhea 5 to 53 days after the last dose of medication.184,185 Diarrhea is described as watery, nonbloody, and only occasionally associated with fever, nausea, vomiting, or abdominal pain.32 Endoscopic features include edema, erythema, friability, ulceration, and fibrinopurulent exudates, usually in the distal colon.184,185 Histologic features mimic those of ulcerative colitis, such as lamina propria mixed inflammatory infiltrate, ulceration, loss of goblet cells, cryptitis, and crypt abscesses (Fig. 12.22).184,185 Occasional cases show increased eosinophils or granulomas.32 Although most cases affect the intestine, Beck and colleagues reported similar findings in the stomach and duodenum.32 Perforation of the colon may occur.32 Treatment involves high-dose corticosteroids and infliximab.32 The mortality rate is approximately 5%.32
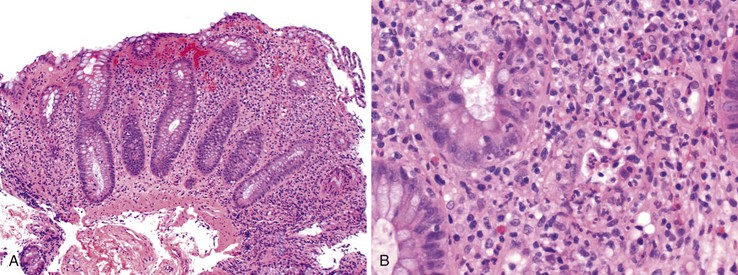
Neutropenic Colitis
Neutropenic colitis is a clinicopathologic syndrome characterized by an inflammatory or septic intraabdominal process in the setting of neutropenia; it is usually caused by chemotherapy for hematologic malignancies or solid tumors—or, less often, by other causes of neutropenia186–189 (see also Chapters 4 and 17). Chemotherapeutic agents traditionally associated with neutropenic colitis include cytosine arabinoside (ara-C), vincristine, doxorubicin, methotrexate, cyclophosphamide, etoposide (VP-16), daunomycin, and prednisone.190 More recently, it has been recognized in patients treated with vinorelbine, docetaxel, paclitaxel, carboplatin, gemcitabine, or 5-fluorouracil for ovarian, lung, colon, or breast cancer.190 Its predisposition to involve the ileal, appendiceal, and cecal mucosa accounts for the alternative terms necrotizing enteropathy, typhlitis, and ileocecal syndrome,191 but it can affect other segments of the intestine as well.190 It is characterized by damage to the mucosa with subsequent infection and systemic complications. The mucosal injury is likely drug induced in cases associated with chemotherapy; however, other factors may contribute, including local infection, necrosis of leukemic tumor deposits, or ischemia resulting from sepsis-related hypotension (Fig. 12.23).191 Regardless of the cause of the tissue damage, once the mucosal barrier has been breached, invasion by enteric and opportunistic organisms is facilitated by the neutropenic status of the host. Tissue necrosis because of infection may add to the insult.191 The number of organisms implicated in neutropenic colitis is large and includes Pseudomonas, Clostridium, Escherichia, Klebsiella, Enterobacter, and Candida. Also, cytomegalovirus can cause cecal ulcers in patients with malignancies, producing a similar syndrome.
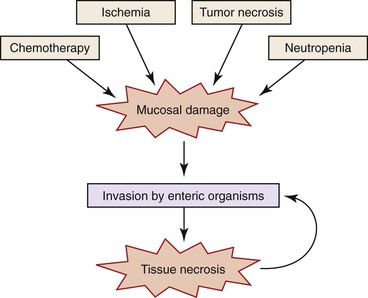
Classically, neutropenic colitis begins 7 to 10 days after chemotherapy treatment with fever, diarrhea (which may be bloody), abdominal pain, nausea, and vomiting.188,190,191 Blood cultures most often yield Clostridium septicum, C. difficile, Pseudomonas, Klebsiella, Enterobacter, and Escherichia coli.191 Stool cultures are usually negative.188 Computed tomographic findings of thickening of the bowel wall or pneumatosis intestinalis support the diagnosis but are not entirely specific.187,190 Recurrence may occur if the patient receives further chemotherapy.190
Grossly, the affected segment may appear dilated, edematous, and often hemorrhagic.190 Microscopic features of neutropenic colitis include edema, hemorrhage, ulceration, thromboses, pneumatosis, and mucosal or transmural necrosis with perforation. The epithelium may show changes compatible with chemotherapy effect. Fungal organisms may be seen in the superficial necrotic tissue, and bacteria may be found in deeper portions of the bowel wall.190 In patients with hematologic malignancies, tumor deposits may be found (Fig. 12.24).
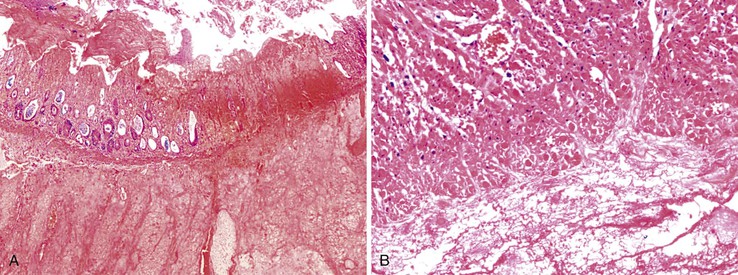
The treatment is challenging because of the difficulty in making a diagnosis coupled with the possibility of rapid progression to septic shock. Conservative management is typically used at onset, with surgical intervention reserved for patients who deteriorate or who have indications for surgery such as perforation, generalized peritonitis, or continuous bleeding despite correction of coagulopathy.191 In cases requiring surgery, right hemicolectomy is considered the operation of choice, but the surgical approach depends on the segment involved, with some cases requiring extensive small bowel resection.187 Recombinant granulocyte colony-stimulating factor may help reverse neutropenia.
Differential Diagnosis
The mortality rate for this disorder is almost 40% 190,191 The histology of neutropenic colitis overlaps with pseudomembranous colitis and ischemic colitis, but, in contrast to these other entities, neutrophils are absent or rare. The clinical scenario is also suggestive of neutropenic colitis.
Cathartics
Laxatives are divided into bulk, osmotic, and irritant (stimulant) types.
Bulk Laxatives
Bulk laxatives include derivatives of psyllium seeds (e.g., Metamucil). They are poorly absorbed by the colon and therefore cause water retention and loose stool. GI damage is not a complication of this group of laxatives.
Osmotic or Saline Laxatives
Osmotic or saline laxatives are either hyperosmotic (sodium phosphate, either oral or as a solution with sodium biphosphate used as an enema; magnesium sulfate; magnesium citrate) or isosmotic (polyethylene glycol). These agents act by increasing the osmolarity of stool, which leads to trapping of water and a consequent reduction in stool consistency; in the case of isosmotic solutions, the colon’s ability to absorb water is overwhelmed. Because these agents are usually well tolerated and effective, they are often used as bowel preparatory agents for endoscopic procedures.
In 1977, Meisel and colleagues192 reported that Fleet’s Enema (sodium biphosphate and sodium phosphate) and rectal bisacodyl caused endoscopically detectable mucosal abnormalities, including hyperemia, obliteration of the normal vascular pattern, and mucosal friability. Histologically, disruption of the surface epithelium was seen in patients treated with Fleet’s Enema, and loss of goblet cell mucin and alterations in the surface and crypt epithelium were observed in patients treated with rectal bisacodyl. Several studies have documented colonic pathology in association with oral sodium phosphate as well. In 1998, Driman and Preiksaitis39 reported aphthous ulcers (Fig. 12.25) in 2.6% of biopsy specimens from patients treated with oral sodium phosphate and focal cryptitis (FAC) in an additional 3.5%. Furthermore, they noted increased proliferation and increased apoptotic bodies in the crypts in these patients. Other reports have confirmed an association between FAC, erosions, or crypt apoptotic bodies and oral sodium phosphate.193–196 Another potential GI complication of hyperosmotic saline laxatives is ischemic colitis.197
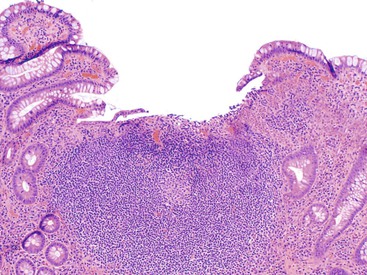
Stimulant or Irritant Laxatives
Stimulant or irritant laxatives promote intestinal motility or interfere with colonic mucosal transport of water and electrolytes. They fall into several groups: diphenylmethane derivatives (phenolphthalein, bisacodyl, and sodium picosulfate), anthraquinone derivatives (senna and cascara), ricinoleic acid (castor oil), and surface-acting agents (docusates). Anthraquinones are plant-derived compounds that undergo bacterial metabolism within the colon; this results in the production of the active forms, which induce fluid secretion and increase colonic motility. They are associated with melanosis coli, “cathartic colon,” and possibly neoplasia.
Melanosis coli refers to deposition of lipofuscin in macrophages within the lamina propria (Fig. 12.26). The proximal colon is more often affected than the left colon. The formation of lipofuscin is the result of breakdown of apoptotic colonic epithelial cells.52 In animal experiments,198 anthraquinone administration leads to a dose-related increase in apoptosis of the colonic surface epithelial cells. Most of the resulting apoptotic bodies are phagocytosed by macrophages and transported into the lamina propria, where they are transformed into lipofuscin within macrophage lysosomes. Patients with melanosis coli often have increased numbers of apoptotic bodies in the colonic surface epithelium. Although melanosis coli is often attributed to chronic anthraquinone laxative use, a history of laxative use is not always elicited in patients with this condition. An article by Lee53 describing increased apoptosis in drug-induced colitis (particularly that associated with 5-fluorouracil or NSAIDs) provided evidence that melanosis might also develop as a result of the use of other types of drugs. Therefore, melanosis coli may be viewed as a marker of increased colonic epithelial cell apoptosis, of which laxatives are one potential cause. In fact, the findings of moderate amounts of melanosis coli, increased crypt apoptosis, and scattered eosinophils may be a general sign of a drug reaction within the GI tract.
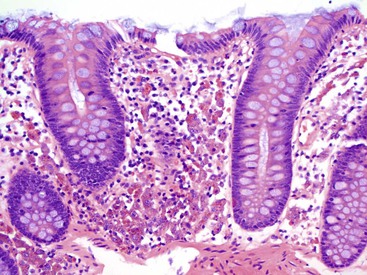
Evidence for enteric nervous system damage related to chronic laxative use is based on uncontrolled observations in only a few patients. In 1968, Smith reported the histologic findings in a colectomy specimen from a patient who had consumed laxatives on a long-term basis.199 This led to the long-held belief that chronic laxative use can cause damage to the enteric nervous system and could lead to loss of function of the colon, a state known as cathartic colon. In that initial report, Smith described reduced numbers of myenteric neurons and axons and an increased number of Schwann cells. The remaining neurons showed irregular, ill-defined margins and contained either one or two swollen processes. Axons were irregular in caliber and were scattered randomly, apparently undergoing regeneration. Soon afterward, Riemann and Schmidt described the ultrastructural changes in patients with cathartic colon.200 They found edematous distention or ballooning of axons, loss of structural elements such as neurosecretory granules and neurotubules, and increased lysosomes. Other reports described mucosal atrophy and fatty infiltration of the submucosa associated with fibrosis, but those authors were unable to confirm the type of neuronal changes described previously.201 Krishnamurthy and colleagues202 found abnormal neuronal changes, similar to those described in cathartic colon, in patients with severe idiopathic constipation, which raises the question of whether the histologic changes were caused by the laxatives or by the disorder that originally led to the patients’ consumption of laxatives. In 1996, Muller-Lissner203 questioned the existence of cathartic colon. He noted that despite increasing laxative use, the incidence of cathartic colon was decreasing. He speculated that a laxative that had fallen out of favor, podophyllin, may have been responsible for the cases of cathartic colon previously described. In sum, there is insufficient evidence to conclude that laxatives cause long-term neurologic damage to the colon.204,205
Mycophenolate Mofetil
Mycophenolate mofetil (MMF) is an immunosuppressive drug used mainly in the management of organ transplant rejection. MMF blocks the de novo pathway of purine synthesis. Enterocytes are partly dependent on this pathway, and GI toxicity is one of the main limitations of the drug. Diarrhea, nausea, vomiting, dysphagia, dyspepsia, melena, and hematemesis are common presenting symptoms and signs of MMF toxicity.206,207 Endoscopic findings may be relatively subtle; they range from normal to nonspecific mild changes such as erythema.207,208 Histologic features vary according to the segment of the GI tract involved but are reminiscent of graft-versus-host disease or Crohn’s disease. In the esophagus, erosions or esophagitis may be seen.206,207 In the stomach, reactive gastropathy, increased epithelial cell apoptosis, and granulomas are common.206,207 In the small intestine, villous atrophy, dilated damaged crypts, lamina propria edema, increased crypt apoptosis, and patchy neutrophilic inflammation are seen.206–209 Colonic biopsy specimens show crypt architectural disarray; dilated, damaged, and degenerated and atrophic crypts; lamina propria edema; mild inflammation; and increased crypt apoptosis (Fig. 12.27).207,210–213 Less often, the features in the small bowel and colon resemble Crohn’s disease, with increased lamina propria inflammation, granulomas, ulcers, and neutrophil cryptitis.207,212
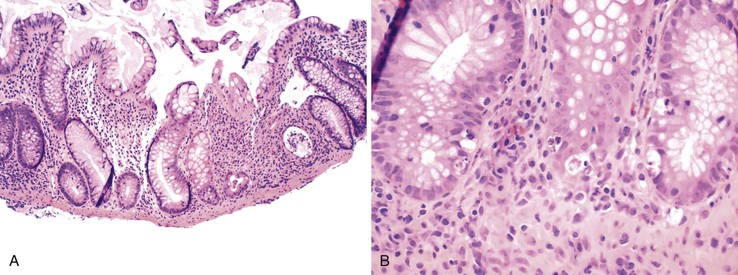
Chemical Colitis (Glutaraldehyde)
Chemical disinfectants are used to clean endoscopy instruments, and some of these contain compounds that are toxic to mucosae. Glutaraldehyde, at a concentration of 2%, is a common disinfectant and has been reported to cause chemical colitis, presumably when the agent is retained in the endoscope channel as a result of inadequate flushing after disinfection.31 Within a few hours to 2 or 3 days, patients experience abdominal pain, tenesmus, and bloody diarrhea.28,31 Endoscopic findings include sharply delineated patches of mucosal erythema, erosions, ulcerations, or mucosal necrosis, usually involving the rectum, sigmoid colon, and descending colon.31,214 Histologic features are indistinguishable from ischemic colitis or from injury caused by some toxin-producing bacteria. Features include mucin depletion and eosinophilia of the superficial lamina propria; in more injured areas, one may see breakdown of the epithelium beginning at the top of the mucosa and progressing toward the deep crypts, hemorrhage, fibrin deposition, and fibrin thrombi in capillaries within the lamina propria.31 Biopsy specimens obtained in later stages of disease show regenerative changes and migration of lymphocytes and plasma cells into the lamina propria.31
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]<
[/not-level-membership-for-pediatrics-category]
