[level-membership-for-pediatrics-category]
Chapter 622 Disorders of the Retina and Vitreous
Retinopathy of Prematurity
Classification
The phases and severity of the disease process are classified into 5 stages. Stage 1 is characterized by a demarcation line that separates vascularized from avascular retina. This line lies within the plane of the retina and appears relatively flat and white. Often noted is abnormal branching or arcading of the retinal vessels that lead into the line. Stage 2 is characterized by a ridge; the demarcation line has grown, acquiring height, width, and volume and extending up and out of the plane of the retina. Stage 3 is characterized by the presence of a ridge and by the development of extraretinal fibrovascular tissue (Fig. 622-1A). Stage 4 is characterized by subtotal retinal detachment caused by traction from the proliferating tissue in the vitreous or on the retina. Stage 4 is subdivided into 2 phases: (a) subtotal retinal detachment not involving the macula and (b) subtotal retinal detachment involving the macula. Stage 5 is total retinal detachment.
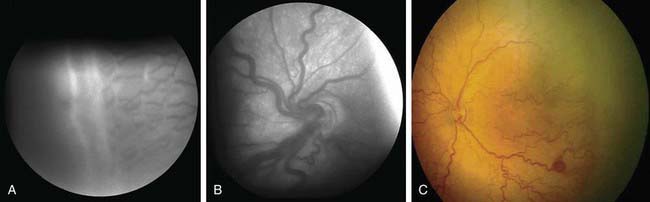
When signs of posterior retinal vascular changes accompany the active stages of ROP, the term plus disease is used (see Fig. 622-1B,C). Patients reaching the point of dilatation and tortuosity of the retinal vessels also often demonstrate the associated findings of engorgement of the iris, pupillary rigidity, and vitreous haze.
Diagnosis
Systematic serial ophthalmologic examinations of infants at risk are recommended. In 2006 the American Academy of Pediatrics (AAP) published new screening guidelines for ROP. Infants with a birth weight of <1,500 g or gestational age of ≤32 wk and selected infants with a birth weight between 1,500 and 2,000 g or gestational age of >32 wk with an unstable clinical course, including those requiring cardiorespiratory support and who are believed by their attending pediatrician or neonatologist to be at high risk, should have retinal screening examinations. The timing of the initial screening exam is based on the infant’s age. Table 622-1 was developed from an evidence-based analysis of the Mutlicenter Trial of Cryotherapy for ROP. The examination can be stressful to fragile preterm infants, and the dilating drops can have untoward side effects. Infants must be carefully monitored during and after the examination. Some neonatologists and ophthalmologists advocate the use of topical tetracaine and/or oral sucrose to reduce the discomfort and stress to the infant. Follow-up is based on the initial findings and risk factors but is usually at 2 wk or less.
Table 622-1 TIMING OF FIRST EYE EXAMINATION BASED ON GESTATIONAL AGE AT BIRTH
| GESTATIONAL AGE AT BIRTH (wk) | AGE AT INITIAL EXAMINATION (wk) | |
|---|---|---|
| Postmenstrual | Chronologic | |
| 22 | 31 | 9 |
| 23 | 31 | 8 |
| 24 | 31 | 7 |
| 25 | 31 | 6 |
| 26 | 31 | 5 |
| 27 | 31 | 4 |
| 28 | 32 | 4 |
| 29 | 33 | 4 |
| 30 | 34 | 4 |
| 31 | 35 | 4 |
| 32 | 36 | 4 |
Prevention
Prevention of ROP ultimately depends on prevention of premature birth and its attendant problems. The association between ROP and oxygen saturation has been studied for decades. More-recent research has focused on keeping severely premature infants at lower oxygen saturation (85-92%) at age <34 wk and maintaining them at higher oxygen saturation (92-97%) at age >34 wk. This reduction in oxygen saturation early in the infant’s life effectively reduces the phase I hyperoxia and can stimulate the retina to develop normally. The reversal of the hypoxic phase II by elevating the oxygen saturation might ultimately decrease the incidence of severe ROP by down-regulating the secretion of VEGF. Most likely, a multicenter, prospective, randomized study will need to be performed to answer this question. Some investigators have suggested supplemental vitamin E for its antioxidant properties in infants at risk for ROP. Its efficacy has not been proved; at certain dosage levels, it can produce untoward side effects (Chapter 91.2).
Retinoblastoma
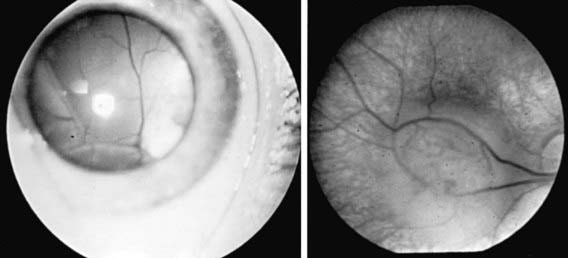
Retinoblastoma (Fig. 622-2, Chapter 496) is the most common primary malignant intraocular tumor of childhood. It occurs in approximately 1/15,000 live births; 250-300 new cases are diagnosed in the United States annually. Hereditary and nonhereditary patterns of transmission occur; there is no gender or race predilection. The hereditary form is usually bilateral and multifocal, whereas the nonhereditary form is generally unilateral and unifocal. About 15% of unilateral cases are hereditary. Bilateral cases often manifest earlier than unilateral cases. Unilateral tumors are often large by the time they are discovered. The average age at diagnosis is 15 mo for bilateral cases, compared with 25 mo for unilateral cases. It is unusual for a child to present with a retinoblastoma after 3 yr of age. Rarely, the tumor is discovered at birth, during adolescence, or even in early adulthood.
Retinitis Pigmentosa
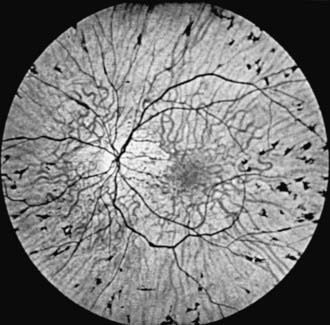
Retinitis pigmentosa (RP) is a progressive retinal degeneration characterized by pigmentary changes, arteriolar attenuation, usually some degree of optic atrophy, and progressive impairment of visual function. Dispersion and aggregation of the retinal pigment produce various ophthalmoscopically visible changes, ranging from granularity or mottling of the retinal pigment pattern to distinctive focal pigment aggregates with the configuration of bone spicules (Fig. 622-3). Other ocular findings include subcapsular cataract, glaucoma, and keratoconus.
Clinically similar secondary pigmentary retinal degenerations that need to be differentiated from RP occur in a wide variety of metabolic diseases, neurodegenerative processes, and multifaceted syndromes. Examples include the progressive retinal changes of the mucopolysaccharidoses (particularly Hurler, Hunter, Scheie, and Sanfilippo syndromes) and certain of the late-onset gangliosidoses (Batten-Mayou, Spielmeyer-Vogt, and Jansky-Bielschowsky diseases), the progressive retinal degeneration that is associated with progressive external ophthalmoplegia (Kearns-Sayre syndrome), and the RP-like changes in the Laurence-Moon and Bardet-Biedl syndromes. The retinal manifestations of abetalipoproteinemia (Bassen-Kornzweig syndrome) and Refsum disease are also similar to those found in RP. The diagnosis of these latter two disorders in a patient with presumed RP is important because treatment is possible. There is also an association of RP and congenital hearing loss, as in Usher syndrome (Chapters 80 and 82).
Cherry Red Spot
Because of the special histologic features of the macula, certain pathologic processes affecting the retina produce an ophthalmoscopically visible sign referred to as a cherry red spot, a bright to dull red spot at the center of the macula surrounded and accentuated by a grayish-white or yellowish halo. The halo is a result of a loss of transparency of the retinal ganglion cell layer secondary to edema, lipid accumulation, or both. Because ganglion cells are not present in the fovea, the retina surrounding the fovea is opacified but the fovea transmits the normal underlying choroidal color (red), accounting for the presence of the cherry red spot. A cherry red spot typically occurs in certain sphingolipidoses, principally in Tay-Sachs disease (GM2 type 1), in the Sandhoff variant (GM2 type 2), and in generalized gangliosidosis (GM1 type 1). Similar but less-distinctive macular changes occur in some cases of metachromatic leukodystrophy (sulfatide lipidosis), in some forms of neuronopathic Niemann-Pick disease, and in certain mucolipidoses (Chapters 80.4 and 80.5). The cherry red spot that characteristically occurs as a result of retinal ischemia secondary to vasospasm, ocular contusion, or occlusion of the central retinal artery must be differentiated from the cherry red spot of neurodegenerative disease.
Phakomas
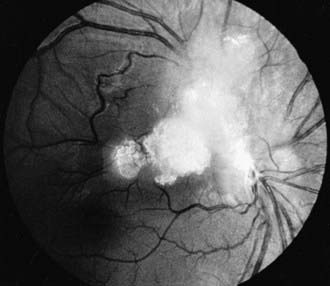
Phakomas (Chapter 589) are the herald lesions of the hamartomatous disorders. In Bourneville disease (tuberous sclerosis), the distinctive ocular lesion is a refractile, yellowish, multinodular cystic lesion arising from the disc or retina; the appearance of this typical lesion is often compared with that of an unripe mulberry (Fig. 622-4). Equally characteristic and more common in tuberous sclerosis are flatter, yellow to whitish retinal lesions, varying in size from minute dots to large lesions approaching the size of the disc. These lesions are benign astrocytic proliferations. Rarely, similar retinal phakomas occur in von Recklinghausen disease (neurofibromatosis). In von Hippel-Lindau disease (angiomatosis of the retina and cerebellum), the distinctive fundus lesion is a hemangioblastoma; this vascular lesion usually appears as a reddish globular mass with large paired arteries and veins passing to and from the lesion. In Sturge-Weber syndrome (encephalofacial angiomatosis), the fundus abnormality is a choroidal hemangioma; the hemangioma can impart a dark color to the affected area of the fundus, but the lesion is best seen with fluorescein angiography.
Coats Disease
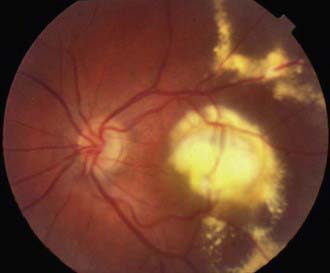
Coats disease is an exudative retinopathy of unknown cause characterized by telangiectasia of retinal vessels with leakage of plasma to form intraretinal and subretinal exudates and by retinal hemorrhages and detachment (Fig. 622-5). The condition is usually unilateral. It predominantly affects boys, usually appearing in the 1st decade. The condition is nonfamilial and for the most part occurs in otherwise healthy children. The most common presenting signs are blurring of vision, leukocoria, and strabismus. Rubeosis of the iris, glaucoma, and cataract can develop. Treatment with photocoagulation or cryotherapy may be helpful.
Hypertensive Retinopathy
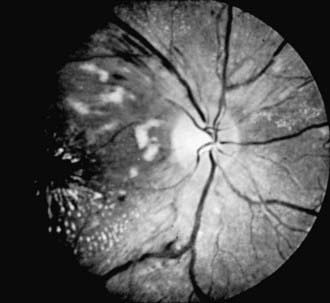
In the early stages of hypertension, no retinal changes may be observable. Generalized constriction and irregular narrowing of the arterioles are usually the first signs in the fundus. Other alterations include retinal edema, flame-shaped hemorrhages, cotton-wool spots (retinal nerve fiber layer infarcts), and papilledema (Fig. 622-6). These changes are reversible if the hypertension can be controlled in the early stages, but in long-standing hypertension, changes may be irreversible. Thickening of the vessel wall can produce a silver- or copper-wire appearance. Hypertensive retinal changes in a child should alert the physician to renal disease, pheochromocytoma, collagen disease, and cardiovascular disorders, particularly coarctation of the aorta.
Diabetic Retinopathy
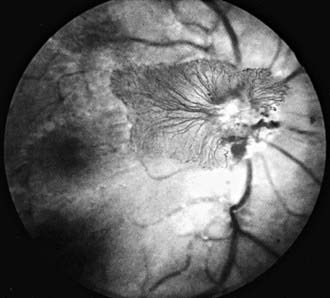
Proliferative retinopathy, the more serious form, is characterized by neovascularization and proliferation of fibrovascular tissue on the retina, extending into the vitreous. Neovascularization can occur on the optic disc (NVD), elsewhere on the retina (NVE), or on the iris and in the anterior chamber angle (NVI, or rubeosis irides) (Fig. 622-7). Traction on these new vessels leads to hemorrhage and eventually scarring. The vision-threatening complications of proliferative diabetic retinopathy are retinal and vitreous hemorrhages, cicatrization, traction, and retinal detachment. Neovascularization of the iris can lead to secondary glaucoma if not treated promptly.
Treatment
Vitrectomy and other intraocular surgery may be necessary in patients with nonresolving vitreous hemorrhage or traction retinal detachment. The value of technologic advances, such as insulin infusion pumps and pancreatic transplants, in preventing ocular complications is under investigation (Chapter 583).
Trauma-Related Retinopathy
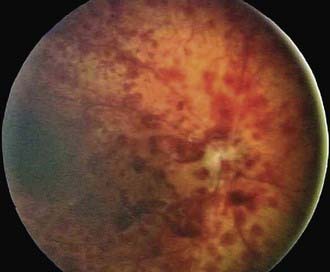
Retinal changes can occur in patients who suffer trauma to other parts of the body. The occurrence of retinal hemorrhages in infants who have been physically abused is well documented (Fig. 622-8; Chapter 37). Retinal, subretinal, subhyaloid, and vitreous hemorrhages have been described in infants and young children with inflicted neurotrauma. Often there are no signs of direct trauma to the eye, periocular region, or head. Such cases can result from violent shaking of an infant, and permanent retinal damage can result.
Abramson DH, Frank CM, Susman M, et al. Presenting signs of retinoblastoma. J Pediatr. 1998;132:505-508.
American Academy of Pediatrics. Screening examinations of premature infants for retinopathy of prematurity. Pediatrics. 2006;117:572-576.
Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231-2239.
Chang M, McLean IW, Merritt JC. Coats’ disease: a study of 62 histologically confirmed cases. J Pediatr Ophthalmol Strabismus. 1984;21:163-168.
Chiang MF, Wang L, Busuioc M, et al. Telemedical retinopathy of prematurity diagnosis. Arch Ophthamol. 2007;125:1531-1538.
Cremers FPM, Collin RWJ. Promised and challenges of genetic therapy for blindness. Lancet. 2009;374:1569-1570.
Cryotherapy for Retinopathy of Prematurity Cooperative Group. 15-year outcomes following threshold retinopathy of prematurity. Arch Ophthalmol. 2005;123:311-318.
Dass AB, Trese MT. Surgical results of persistent hyperplastic primary vitreous. Ophthalmology. 1999;106:280-284.
Early Treatment Diabetic Retinopathy Research Study Group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report 1. Arch Ophthalmol. 1985;103:1796-1806.
Early Treatment for Retinopathy of Prematurity Cooperative Group. Revised indications for the treatment of retinopathy of prematurity: results of the Early Treatment for Retinopathy of Prematurity randomized trial. Arch Ophthalmol. 2003;121:1684-1696.
El-Aziz MM, Barragan I, O’Driscoll CA, et al. EYS, encoding an ortholog of Drosophila, Spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genetics. 2008;40:1285-1287.
Gallie B. Canadian guidelines for retinoblastoma care. Can J Ophthalmol. 2009;44:639-642.
George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthmol. 1996;114:274-280.
Good WV. Retinopathy of prematurity and the peripheral retina. J Pediatr. 2008;153:591-592.
Hardwig P, Robertson DM. Von Hippel-Lindau disease: a familial, often lethal, multi-system phakomatosis. Ophthalmology. 1984;91:263-270.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795-1809.
Hellström A, Ley D, Hansen-Pupp I, et al. New insights into the development of retinopathy of prematurity—importance of early weight gain. Acta Paediatr. 2009;99:502-508.
Hollands H, Johnson D, Brox AC, et al. Acute-onset floaters and flashes. JAMA. 2009;302:2243-2249.
Kim JW, Abramson DH, Dunkel IJ. Current management strategies for intraocular retinoblastoma. Drugs. 2007;67(15):2173-2185.
Koenekoop RK. Successful RPE65 gene replacement and improved visual function in humans. Ophthalm Genet. 2008;29(3):89-91.
Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597-1604.
Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240-2248.
Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364(7):603-614.
Miyakulo H, Hashimoto K, Miyakulo S. Retinal vascular pattern in familial exudative vitreoretinopathy. Ophthalmology. 1984;91:1524-1530.
Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88:688-692.
Noble KG, Carr RE. Leber’s congenital amaurosis: a retrospective study of 33 cases and a histopathological study of one case. Arch Ophthalmol. 1978;96:818-821.
Pawlik D, Lauterbach R, Turyk E. Fish-oil fat emulsion supplementation may reduce the risk of severe retinopathy in VLBW infants. Pediatrics. 2011;127(2):223-228.
Richter GM, Williams SL, Starren J, et al. Telemedicine for retinopathy of prematurity diagnosis: evaluation and challenges. Surv Ophthalmol. 2009;54:671-685.
Sage J. Hope in sight for retinoblastoma. Nat Med. 2007;13:30-31.
Sears JE, Pietz J, et al. A change in oxygen supplementation can decrease the incidence of ROP. Ophthalmology. 2009;116(3):513-518.
Shalev B, Farr A, Repka MX. Randomized comparison of diode laser photocoagulation versus cryotherapy for threshold retinopathy of prematurity: seven year outcome. Am J Ophthalmol. 2001;132:76-80.
Shields CL, Gorry T, Shields JA. Outcome of eyes with unilateral sporadic retinoblastoma based on the initial external findings by the family and the pediatrician. J Pediatr Ophthalmol Strabismus. 2004;41:143-149.
Shields CL, Palamar M, Sharma P, et al. Retinoblastoma regression patterns following chemoreduction and adjuvant therapy in 557 tumors. Arch Ophthalmol. 2009;127:282-290.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 622 Disorders of the Retina and Vitreous
Retinopathy of Prematurity
Classification
The phases and severity of the disease process are classified into 5 stages. Stage 1 is characterized by a demarcation line that separates vascularized from avascular retina. This line lies within the plane of the retina and appears relatively flat and white. Often noted is abnormal branching or arcading of the retinal vessels that lead into the line. Stage 2 is characterized by a ridge; the demarcation line has grown, acquiring height, width, and volume and extending up and out of the plane of the retina. Stage 3 is characterized by the presence of a ridge and by the development of extraretinal fibrovascular tissue (Fig. 622-1A). Stage 4 is characterized by subtotal retinal detachment caused by traction from the proliferating tissue in the vitreous or on the retina. Stage 4 is subdivided into 2 phases: (a) subtotal retinal detachment not involving the macula and (b) subtotal retinal detachment involving the macula. Stage 5 is total retinal detachment.
When signs of posterior retinal vascular changes accompany the active stages of ROP, the term plus disease is used (see Fig. 622-1B,C). Patients reaching the point of dilatation and tortuosity of the retinal vessels also often demonstrate the associated findings of engorgement of the iris, pupillary rigidity, and vitreous haze.
Diagnosis
Systematic serial ophthalmologic examinations of infants at risk are recommended. In 2006 the American Academy of Pediatrics (AAP) published new screening guidelines for ROP. Infants with a birth weight of <1,500 g or gestational age of ≤32 wk and selected infants with a birth weight between 1,500 and 2,000 g or gestational age of >32 wk with an unstable clinical course, including those requiring cardiorespiratory support and who are believed by their attending pediatrician or neonatologist to be at high risk, should have retinal screening examinations. The timing of the initial screening exam is based on the infant’s age. Table 622-1 was developed from an evidence-based analysis of the Mutlicenter Trial of Cryotherapy for ROP. The examination can be stressful to fragile preterm infants, and the dilating drops can have untoward side effects. Infants must be carefully monitored during and after the examination. Some neonatologists and ophthalmologists advocate the use of topical tetracaine and/or oral sucrose to reduce the discomfort and stress to the infant. Follow-up is based on the initial findings and risk factors but is usually at 2 wk or less.
Table 622-1 TIMING OF FIRST EYE EXAMINATION BASED ON GESTATIONAL AGE AT BIRTH
| GESTATIONAL AGE AT BIRTH (wk) | AGE AT INITIAL EXAMINATION (wk) | |
|---|---|---|
| Postmenstrual | Chronologic | |
| 22 | 31 | 9 |
| 23 | 31 | 8 |
| 24 | 31 | 7 |
| 25 | 31 | 6 |
| 26 | 31 | 5 |
| 27 | 31 | 4 |
| 28 | 32 | 4 |
| 29 | 33 | 4 |
| 30 | 34 | 4 |
| 31 | 35 | 4 |
| 32 | 36 | 4 |
Prevention
Prevention of ROP ultimately depends on prevention of premature birth and its attendant problems. The association between ROP and oxygen saturation has been studied for decades. More-recent research has focused on keeping severely premature infants at lower oxygen saturation (85-92%) at age <34 wk and maintaining them at higher oxygen saturation (92-97%) at age >34 wk. This reduction in oxygen saturation early in the infant’s life effectively reduces the phase I hyperoxia and can stimulate the retina to develop normally. The reversal of the hypoxic phase II by elevating the oxygen saturation might ultimately decrease the incidence of severe ROP by down-regulating the secretion of VEGF. Most likely, a multicenter, prospective, randomized study will need to be performed to answer this question. Some investigators have suggested supplemental vitamin E for its antioxidant properties in infants at risk for ROP. Its efficacy has not been proved; at certain dosage levels, it can produce untoward side effects (Chapter 91.2).
Retinoblastoma
Retinoblastoma (Fig. 622-2, Chapter 496) is the most common primary malignant intraocular tumor of childhood. It occurs in approximately 1/15,000 live births; 250-300 new cases are diagnosed in the United States annually. Hereditary and nonhereditary patterns of transmission occur; there is no gender or race predilection. The hereditary form is usually bilateral and multifocal, whereas the nonhereditary form is generally unilateral and unifocal. About 15% of unilateral cases are hereditary. Bilateral cases often manifest earlier than unilateral cases. Unilateral tumors are often large by the time they are discovered. The average age at diagnosis is 15 mo for bilateral cases, compared with 25 mo for unilateral cases. It is unusual for a child to present with a retinoblastoma after 3 yr of age. Rarely, the tumor is discovered at birth, during adolescence, or even in early adulthood.
