Chapter 128 Disorders of the Complement System
128.2 Genetic Deficiencies of Complement Components
Congenital deficiencies of all 11 components of the classical-membrane attack pathway and of factor D and properdin of the alternative pathway are described (Table 128-1). All of the components of the classical and alternative pathways except properdin are inherited as autosomal recessive co-dominant traits. Each parent transmits a gene that codes for synthesis of half the serum level of the component. Deficiency results from inheritance of 1 null gene from each parent; the hemizygous parents typically have CH50 levels that are low normal. Properdin deficiency is transmitted as an X-linked trait.
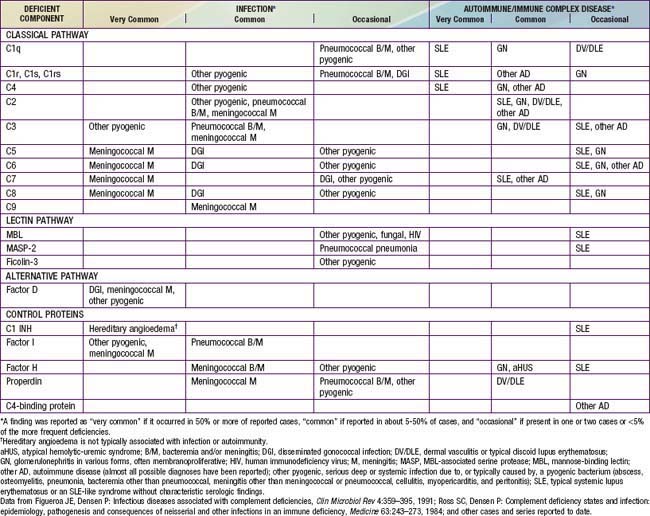
Most patients with primary C1q deficiency have systemic lupus erythematosus (SLE), an SLE-like syndrome without typical SLE serology, a chronic rash that has shown an underlying vasculitis on biopsy, or membranoproliferative glomerulonephritis (MPGN). Some C1q-deficient children have serious infections, including septicemia and meningitis. Individuals with C1r, C1s, combined C1r/C1s, C4, C2, or C3 deficiency also have a high incidence of autoimmune syndromes (see Table 128-1), especially SLE or an SLE-like syndrome in which antinuclear antibody level is not elevated.
Casanova JL, Abel L. Human mannose-binding lectin in immunity: friend, foe, or both? J Exp Med. 2004;110:1295-1299.
Fijen CAP, Kuijper EJ, te Bulte MT, et al. Assessment of complement deficiency in patients with meningococcal disease in the Netherlands. Clin Infect Dis. 1999;28:98-105.
Jonsson G, Truedsson L, Sturfelt G, et al. Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Medicine. 2005;84:23-34.
Munthe-Fog L, Hummelshoj T, Honore C, et al. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. N Engl J Med. 2009;360:2637-2644.
Tebruegge M, Curtis N. Epidemiology, etiology, pathogenesis, and diagnosis of recurrent bacterial meningitis. Clin Microbiol Rev. 2008;21:519-537.
128.3 Deficiencies of Plasma, Membrane, or Serosal Complement Control Proteins
Congenital deficiencies of 5 plasma complement control proteins have been described (see Table 128-1). Factor I deficiency was reported originally as a deficiency of C3 resulting from hypercatabolism. The 1st patient described had suffered a series of severe pyogenic infections similar to those associated with agammaglobulinemia or congenital deficiency of C3. Factor I is an essential regulator of both pathways. Its deficiency permits prolonged existence of C3b in the C3 convertase of the alternative pathway, C3bBb, resulting in constant activation of the alternative pathway and cleavage of more C3 to C3b, in circular fashion. Intravenous infusion of plasma or purified factor I induced a prompt rise in serum C3 concentration in the patient and a return to normal of in vitro C3-dependent functions such as opsonization.
The effects of factor H deficiency are like those of factor I deficiency because factor H assists in dismantling the alternative pathway C3 convertase. Levels of C3, factor B, total hemolytic activity, and alternative pathway activity have been low or undetectable in these patients. Patients have sustained systemic infections due to pyogenic bacteria, particularly N. meningitidis. Many have had glomerulonephritis or atypical HUS (aHUS) (Chapter 512). Mutations in genes encoding membrane cofactor protein (MCP), factors I or B, C3, or the endothelial anti-inflammatory protein thrombomodulin, or autoantibodies to factor H, have also been associated with aHUS. The few patients thus far reported as having C4-binding protein deficiency have about 25% of the normal levels of the protein and no typical disease presentation, although one had angioedema and Behçet disease.
Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
The Medical Letter. Three new drugs for hereditary angioedema. Med Lett. 2010;52(1345):66-67.
Morgan BP. Hereditary angioedema—therapies old and new. N Engl J Med. 2010;363(6):581-583.
Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676-1687.
Zuraw BL. Hereditary angioedema. N Engl J Med. 2008;359:1027-1036.
Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363(6):513-522.
128.4 Secondary Disorders of Complement
Eichenfield LF, Johnston RBJr. Secondary disorders of the complement system. Am J Dis Child. 1989;143:595-602.
Wang RH, Phillips GJr, Medof ME, et al. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. J Clin Invest. 1993;92:1326-1355.
Wurzner R. Evasion of pathogens by avoiding recognition or eradication by complement, in part via molecular mimicry. Mol Immunol. 1999;36:249-260.




