Disorders of sexual differentiation
1. Describe the first level of sexual differentiation.
The first level of sexual differentiation is the establishment of chromosomal sex. Most infants are 46,XX females or 46,XY males. Genetic sex determines gonadal sex. Gonadal structures differentiate from the “bipotential,” or primordial, gonadal ridge. The Y chromosome contains an area known as the sex-determining region, or SRY. The SRY gene product initiates the differentiation of the bipotential gonad into a testis. In its absence, the gonad becomes an ovary.
2. What is the next level of sex determination?
The next level of sex determination involves the genital duct structures. The genital duct structures are initially identical in the male and female. In the normal male, testicular Leydig cells produce testosterone, which is necessary to maintain ipsilateral wolffian duct structures (e.g., vas deferens, epididymis, seminal vesicles). The Sertoli cells of the testis produce müllerian-inhibiting factor (MIF), which acts ipsilaterally to cause regression of müllerian duct structures (fallopian tubes, uterus, upper third of the vagina). In the absence of testosterone and MIF, müllerian duct structures are preserved, and wolffian duct structures regress.
3. Discuss the development of the external genitalia.
Male and female external genitalia arise from the same embryologic structures. In the absence of androgen stimulation, these structures remain in the female pattern, whereas the presence of androgens causes male differentiation (virilization). For complete virilization, testosterone must be converted to dihydrotestosterone (DHT) by the enzyme 5-alpha-reductase, and androgen receptors must be functional. Excessive androgens virilize a female. Inadequate androgen production, inability to convert testosterone to DHT, or inability to respond to androgens, as in androgen receptor defects, results in undervirilization of a male.
4. What is testis-determining factor (TDF)?
The TDF promotes differentiation of the bipotential gonad into a testis; SRY was eventually characterized as the TDF. SRY belongs to a family of DNA binding proteins. Specific manipulations have shown that the introduction of SRY results in sex reversal of XX mice, and site-directed mutagenesis of the SRY gene in XY mice yields an XY female. The activation of SRY is influenced by the Wilms’ tumor suppressor gene, WT1. Other genes that play a role downstream of SRY include SOX9, SF-1, DAX1,WNT4, DMRT1, ATRX, DHH, and GATA4.
5. Describe the Lyon hypothesis. In which cells are two X chromosomes necessary for normal development?
Dr. Mary Lyon addressed the question of the extra X chromosomal material in females. Simply put, if two X chromosomes are necessary in each cell, how can males be developmentally normal? Lyon suggested that in each cell, one of the two X chromosomes is inactive, and in any given cell line, which X is active is randomly determined. In fact, the inactive X may be identified in many cells as a clump of chromatin at the nuclear membrane (Barr body). The important exception is in the ovary, where two functional X chromosomes are necessary for normal sustained ovarian development. Without two X chromosomes per cell (as in 45 XO Turner syndrome), the ovary involutes and leaves only fibrous tissue.
6. Discuss normal male sexual differentiation.
The fetus is sexually bipotential. Figure 42-1 shows schematically how male development is accomplished. The undifferentiated gonad is derived from coelomic epithelium, mesenchyme, and germ cells, which, in the presence of SRY, give rise to Leydig cells, Sertoli cells, seminiferous tubules, and spermatogonia. Testes are formed at 7 weeks. Testicular production of testosterone (Leydig cells) leads to wolffian duct development, whereas MIF (Sertoli cells) leads to müllerian duct regression. Masculinization of the external genitalia is mediated by DHT, which is produced from testosterone by the action of the enzyme 5-alpha-reductase.
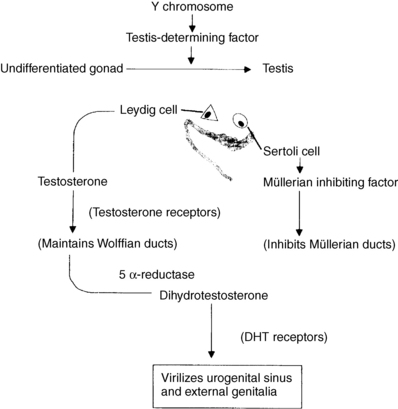
Figure 42-1. Normal male development. DHT, Dihydrotestosterone.
7. Describe normal female sexual differentiation.
In the absence of SRY, the undifferentiated gonad gives rise to follicles, granulosa cells, theca cells, and ova. Ovarian development occurs in the thirteenth to sixteenth week of gestation. Lack of testosterone and MIF allows regression of the wolffian ducts and maintenance of the müllerian ducts, respectively. Lack of DHT results in the maintenance of female external genitalia.
8. How is external genital development determined?
The external genitalia arise from the urogenital tubercle, urogenital swelling, and urogenital folds. In females, these become the clitoris, labia majora, and labia minora, respectively. In males, under the influence of DHT, the genital tubercle becomes the glans of the penis, the urogenital folds elongate and fuse to form the shaft of the penis, and the genital swellings fuse to form the scrotum. Fusion is completed by 70 days of gestation, and penile growth continues to term.
Female differentiation does not require ovaries or hormonal influence, whereas normal development of male genitalia requires normal testosterone synthesis, conversion to DHT by 5-alpha-reductase, and normal androgen receptors (Fig. 42-2).
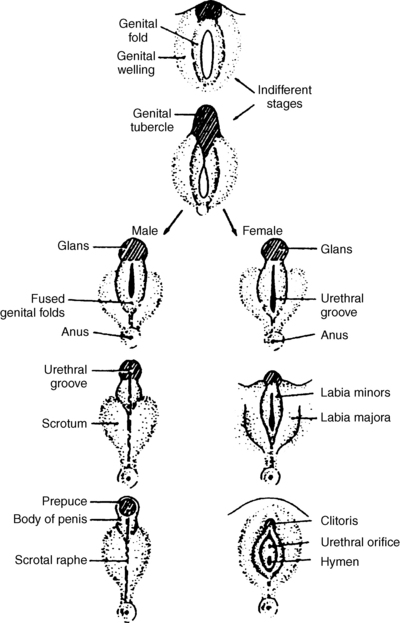
9. The differential diagnosis of disorders of sexual differentiation (DSD) is complex, but it may be simplified by an approach based on an understanding of the process of sexual differentiation. Can you devise such a classification?
 KEY POINTS 1: DISORDERS OF SEXUAL DIFFERENTIATION
KEY POINTS 1: DISORDERS OF SEXUAL DIFFERENTIATION
1. Sexual ambiguity in a newborn must be seen as a medical, social, and psychological emergency requiring a multidisciplinary team approach to assign a sex of rearing. Members of the team include the pediatric endocrinologist, urologist, geneticist, pediatrician, appropriate counselors, and an ethicist.
2. Evaluation of ambiguity must consider the four major categories of children presenting with this problem: virilized 46,XX females, undervirilized 46,XY males, disorders of gonadal differentiation, and unclassified forms (cryptorchidism, hypospadias, developmental anomalies).
3. The most common cause of sexual ambiguity in newborns is congenital adrenal hyperplasia secondary to 21-hydroxylase deficiency.
4. As a general rule, gonadal tissue containing Y chromosomal material is at higher risk for development of malignancy. Consideration must be given to surgical removal of such gonads at some point.




10. What is a virilized female?
A virilized female (previously called female pseudohermaphroditism) is characterized by a 46,XX karyotype, ovaries, normal müllerian duct structures, absent wolffian duct structures, and virilized genitalia resulting from exposure to androgens during the first trimester. See Table 42-2.
TABLE 42-2.
PRADER CLASSIFICATION: DEGREE OF VIRILIZATION OF EXTERNAL GENITALIA
| Type 1 | Clitoral hypertrophy |
| Type 2 | Clitoral hypertrophy, urethral and vaginal orifices present, but very near |
| Type 3 | Clitoral hypertrophy, single urogenital orifice, posterior fusion of the labia majora |
| Type 4 | Penile clitoris, perineoscrotal hypospadias, complete fusion of the labia majora |
| Type 5 | Complete masculinization (normal-looking male genitalia) but no palpable testes |
11. What is the most common cause of a virilized female?
The most common cause is congenital adrenal hyperplasia (CAH) resulting from 21-hydroxylase deficiency. In fact, this disorder is the single most common cause of sexual ambiguity. In this condition, the gene responsible for encoding the 21-hydroxylase enzyme is inactive. This enzyme blockage occurs along the pathway to cortisol and aldosterone. Because of low or absent levels of cortisol, the feedback mechanism produces increased adrenocorticotropic hormone (ACTH), which drives the pathway further and results in accumulation of precursor hormones, the measurement of which is useful for making a diagnosis. Increased ACTH also drives the production of excess adrenal androgens, which result in virilization. Virilization may also be caused by maternal ingestion of androgens or synthetic progesterones during the first trimester of pregnancy.
12. How do virilized female infants present?
Affected infants may present with a wide spectrum of ambiguity, ranging from clitoromegaly alone to complete fusion of the labial swellings to form a scrotum and large phallus. (Beware the infant with bilaterally undescended testes.) Even in the most virilized girls, a penile urethra is rare.
13. What is an undervirilized male?
An undervirilized male (previously called male pseudohermaphroditism) refers to a 46,XY male who has ambiguous or female external genitalia. The abnormality may range from hypospadias to a completely female phenotype. Such disorders result from deficient androgen stimulation of genital development and most often are secondary to Leydig cell agenesis, testosterone biosynthetic defects, 5-alpha-reductase deficiency, and partial or total androgen resistance (androgen receptor defects).
14. Which boys with hypospadias should be evaluated for sexual ambiguity?
First-degree (coronal or glandular) hypospadias as the sole presenting genital abnormality has no apparent endocrine basis and need not be evaluated. The incidence of this anomaly is between 1 and 8 in 1000 births. In contrast, perineoscrotal hypospadias is a feature of many causes of sexual ambiguity, and a child with this finding should be fully evaluated as sexually ambiguous.
15. What is gonadal dysgenesis?
Patients with Y-related chromosomal or genetic disorders that cause maldevelopment of one or both testes are said to have gonadal dysgenesis. They present with ambiguous genitalia and may have hypoplasia of wolffian duct structures and inadequate virilization. MIF may be absent, thus allowing müllerian duct structures to persist. Duct asymmetry is therefore common. The Y-containing dysgenetic testes are at risk for developing gonadoblastomas and must be removed.
16. An infant is born with ambiguous genitalia, and the sex of the infant is uncertain. How do you proceed?
Honesty and diplomacy are essential. Explain that the genitalia are not yet fully developed and that further testing is necessary to determine the infant’s sex. Reference to more commonly understood birth defects may be useful. Explain that while several days may be necessary to complete the testing and that a team will participate to make an accurate diagnosis and a considered recommendation, completion of the birth certificate should not be postponed, and sex assignment should not be delayed.
17. What history is necessary to evaluate the infant?
Maternal history is particularly important and should include illnesses, drug ingestion, alcohol intake, and ingestion of hormones during pregnancy. Was progestational therapy used for threatened abortion or androgens for endometriosis? Does the mother have signs of excessive androgen? Explore family history for occurrence of ambiguity, neonatal deaths, consanguinity, or infertility.
18. How should you direct the physical examination?
The diagnosis of the origin of sexual ambiguity can rarely be made by examination alone, but physical findings can help to direct further evaluation. Look for the following:





19. What other areas should be evaluated?
Certain forms of CAH may cause dehydration, hypertension, or areolar or genital hyperpigmentation. Turner’s stigmata may be present, including webbed neck, low hairline, and edema of hands and feet. Other associated congenital anomalies may indicate a complex that includes sexual ambiguity.
20. Explain which radiographic studies are necessary.
Structural studies are needed to address the presence of gonads and müllerian structures. Pelvic ultrasound examination by qualified and experienced personnel should be performed as soon as possible to look for a uterus. The presence of gonads, fallopian tubes, and a vaginal vault may also be determined. If necessary, a genitogram may be performed by inserting contrast material into the urogenital orifice (or vaginal orifice) to define vaginal size, presence of a cervix, and any fistulas.
21. Explain the role of karyotyping.
A karyotype is essential and must be obtained expeditiously. Buccal smears are absolutely contraindicated because they are inaccurate. In many laboratories, a karyotype can be completed within 48 to 72 hours. Some laboratories can also perform rapid fluorescence in situ hybridization analysis for the presence of the SRY gene.
22. What laboratory test is very helpful in almost all cases?
Because 21-hydroxylase deficiency is a common cause of sexual ambiguity, the level of 17-hydroxyprogesterone (17-OHP) should be assessed in all such infants who do not have palpable gonads.
23. How is further evaluation directed?
Further evaluation must be directed by information provided through the history, examination, and initial studies. Determining the presence or absence of palpable gonads (presumably testes), the presence or absence of a uterus, and the karyotype allows classification of the infant as a virilized female, an undervirilized male, having a disorder of gonadal differentiation, or having one of the unclassified forms.
24. The infant has no palpable gonads and has fused labioscrotal folds and a prominent phallus. The ultrasound scan reveals a uterus and tubes with possible ovaries. The karyotype is 46,XX. How do you proceed now?
The infant is a virilized female. If there is no history of maternal androgen ingestion or virilization, the infant has one of three forms of CAH. Of these, 21-hydroxylase deficiency is most common and is confirmed by finding an elevated serum level of 17-OHP. In 11-beta-hydroxylase deficiency, 11-deoxycortisol is elevated, whereas 17-hydroxypregnenolone and dehydroepiandrosterone (DHEA) are elevated in 3-beta-hydroxysteroid dehydrogenase deficiency. The baseline levels are usually diagnostic but can be confirmed by an ACTH stimulation test. The electrolyte disturbances seen with such disorders do not usually occur until 8 to 14 days of life; however, plasma renin activity is elevated earlier and should be measured as a marker of salt wasting. Screening of newborns for CAH with measurement of a 17-OHP level is now mandated in all 50 of the United States and in many countries throughout the world.
25. An undervirilized male represents a more complex diagnostic dilemma. In an infant with palpable gonads, no müllerian structures, and a 46,XY karyotype, how do you proceed?
Defects in testosterone synthesis include three enzyme blocks common to the adrenal and testicular pathways (StAR defect, 3-beta-hydroxysteroid dehydrogenase deficiency, and 17-alpha-hydroxylase deficiency). Enzyme blocks are diagnosed with ACTH stimulation testing and measurement of steroid precursors. Infants with StAR defects have no measurable precursors but show high levels of ACTH and a low cortisol response. Infants with 3-beta-hydroxysteroid dehydrogenase deficiency have elevated levels of 17-hydroxypregnenolone and DHEA. Patients with 17-alpha-hydroxylase deficiency have elevated levels of progesterone, desoxycorticosterone, and corticosterone, with associated hypertension (Fig. 42-3).
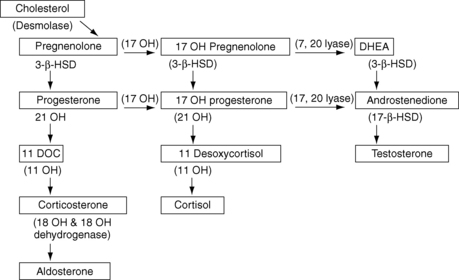
26. Discuss the two remaining defects that involve deficiencies of testicular, but not adrenal, enzymes.
The two remaining defects in testosterone synthesis involve deficiencies of specific testicular rather than adrenal enzymes: 17,20-lyase and 17-beta-hydroxysteroid dehydrogenase. Thus they are not associated with elevations of ACTH or electrolyte disturbances. Both deficiencies are diagnosed by measuring the precursor response to administration of human chorionic gonadotropin (hCG). Infants with 17,20-lyase deficiency have elevated levels of 17-hydroxypregnenolone and 17-OHP, whereas infants with 17-beta-hydroxysteroid dehydrogenase deficiency have elevated levels of DHEA and androstenedione.
27. What other possibilities should be investigated?
Infants with Leydig cell hypoplasia have low levels of testosterone before and after hCG stimulation but normal adrenal function. Testicular biopsy reveals normal seminiferous tubules and Sertoli cells but absent or few Leydig cells.
Stimulation with hCG also allows measurement of the testosterone-to-DHT ratio. If the ratio is elevated, 5-alpha-reductase deficiency should be suspected and may be confirmed by cultures of genital skin fibroblasts.
Finally, normal testosterone levels with no abnormalities in ACTH and hCG test results lead to the diagnosis of partial androgen insensitivity (androgen receptor defects). The diagnosis is made by demonstrating abnormal androgen binding in cultures of genital skin fibroblasts in a research laboratory or by molecular analysis.
28. What is complete androgen insensitivity?
The androgen receptor, encoded on the X chromosome, binds testosterone and, more avidly, DHT. Androgen insensitivity results from abnormalities of the androgen receptor. Complete androgen resistance occurs with a frequency of 1 in 20,000 to 1 in 64,000 XY individuals.
29. How do infants with complete androgen insensitivity present?
Complete androgen insensitivity (testicular feminization) rarely manifests as ambiguity in the newborn period or early childhood. Unless the testes have descended and are palpable in the labia majora, affected infants appear as phenotypically normal females.
Affected children grow as normal females until puberty. They feminize with normal breast development at puberty because high levels of testosterone are aromatized to estrogen, but they have no pubic or axillary hair and no menses. Because they produce MIF, they lack müllerian duct structures. Wolffian duct structures are also rudimentary or absent because these patients lack normal testosterone receptors. Gender identity is usually female. Patients come to medical attention because of primary amenorrhea. The diagnosis is therefore frequently made when patients are in their middle to late teens.
30. When should intraabdominal testicular tissue be removed?
The intraabdominal testes of androgen insensitivity or XY gonadal dysgenesis are at risk for malignancy (up to 20% in some series), particularly after the onset of puberty. Timing of gonadectomy is debated. Because the risk of malignancy is low until puberty, some clinicians prefer to leave the gonads intact until spontaneous pubertal development; however, because carcinoma in situ has been found in prepubertal patients, other practitioners recommend early removal. If the testes are removed before puberty, estrogen therapy is necessary for normal pubertal progression. Because the upper section of the vagina is müllerian in origin, affected individuals may have shortened vaginas and require plastic surgical repair.
31. Summarize the physiologic results of 5-alpha-reductase deficiency.
Deficiency of 5-alpha-reductase impairs the conversion of testosterone to DHT and leads to incomplete virilization and differentiation of the external genitalia, which are dependent on the action of DHT. The disorder is particularly well documented in large kindreds in the Dominican Republic and Gaza, in whom it is inherited as an autosomal recessive condition.
32. Describe the clinical picture in children with 5-alpha-reductase deficiency.
Male infants with 5-alpha-reductase deficiency are born with sexual ambiguity. External genitalia range from a penis with simple hypospadias to a blind vaginal pouch and clitoris-like phallus. The most common presentation is a urogenital sinus with a blind vaginal pouch. During puberty, affected boys undergo virilization; affected females are normal.
Traditionally, infants with 5-alpha-reductase deficiency were raised as females until puberty, then continued life as males, and, in some cases, achieved fertility. More recently, however, the condition has been recognized early in life, and affected males are now raised from infancy as boys.
33. What is an ovotesticular DSD “true hermaphrodite”?
Ovotesticular DSD, previously known as true hermaphroditism, is a disorder of gonadal differentiation in which individuals have both ovarian and testicular elements. Affected children may have bilateral ovotestes, an ovary or testis on one side with an ovotestis on the other, or an ovary on one side and testis on the other. Because the effects of MIF and testosterone on duct structures are ipsilateral and localized, internal duct development is often asymmetric. Thus, a fallopian tube and unicornuate uterus, with absent or vestigial male duct structures, may develop on the side without testicular elements, whereas an epididymis, vas deferens, and seminal vesicles without müllerian structures may develop on the side with testicular elements. The genitalia may be male, female, or ambiguous, depending on the amount of functioning testicular tissue.
34. Why is a multidisciplinary team necessary in approaching an infant with sexual ambiguity?
Sexual ambiguity is a complex issue. An accurate diagnosis is essential and may take some time. Sex of assignment must be based not only on the underlying diagnosis and karyotype but also on the potential for adult sexual function, fertility, and psychological health. For these reasons, input from several specialties, including endocrinology, genetics, neonatology, psychology, urology, and an ethicist, is important. All members of the team must communicate adequately with each other. Parents must fully understand the medical recommendation for sex assignment and required therapy. They must wholeheartedly agree and support the assigned sex to avoid ambivalence, which can lead to gender confusion and psychological trauma for the child.
35. How is the decision about sex assignment made?
Exogenous and endogenous hormones are clearly important, as is the appearance of the genitalia. The decision about sex assignment must be carefully made, taking into consideration each “level” of sex determination. Sex assignment also depends on fetal sex hormone exposure, the potential for adult sexual function, and psychological and cultural considerations. It is vital that parents completely understand and support the decision because ambivalence about sex of rearing may result in gender confusion and psychological trauma.
36. After the cause of sexual ambiguity has been determined in an infant, what factors should be considered in assigning a sex of rearing?
Arriving at a precise diagnosis provides the treating team an understanding of potential risks and benefits of either sex assignment. For example, in poorly virilized males, the difference in outcomes among children with defects in testosterone synthesis, complete androgen insensitivity, and 5-alpha-reductase deficiency is enormous. A child with defective synthesis of testosterone may be raised male or female, depending on other factors; a child with complete androgen insensitivity should be raised female; and a boy with 5-alpha-reductase deficiency usually is raised male. Yet children affected by any of the three conditions have 46,XY karyotypes.
37. What other factors must be considered?
 What is the potential for unambiguous genital appearance?
What is the potential for unambiguous genital appearance?
 What is the potential for normal sexual function?
What is the potential for normal sexual function?
 Is there a potential for fertility?
Is there a potential for fertility?

 What are the factors likely to affect gender identity and psychological health?
What are the factors likely to affect gender identity and psychological health?


38. To which gender are virilized females usually assigned?
Virilized females are usually assigned a female sex. They have normal ovaries as well as müllerian structures and, with surgical correction and steroid replacement, can have normal sexual function and achieve fertility. However, severely virilized females should be assigned a male sex.
39. How is sex assignment determined in undervirilized males?
Undervirilized males are often infertile, and sex assignment has usually been based on phallic size. Because a stretched penile length of 2.5 cm is 2.5 SD below the mean, an infant with a phallus smaller than 2.5 cm may be assigned a female sex of rearing. However, phallic size (penis or clitoris) has been challenged as a major factor in decisions of gender assignment. Adult social and fulfilling sexual function should be the primary goals of gender assignment. If male sex assignment is contemplated, a trial of depot testosterone (25 mg every 3-4 weeks) for 1 to 3 months indicates whether phallic growth is possible.
40. Summarize the factors that determine sex assignment in patients with gonadal dysgenesis.
In patients with gonadal dysgenesis and Y chromosomal material, gonadectomy is necessary, and fertility is not possible. Internal duct structure is also frequently deranged. Small phallic size usually leads to a female sex assignment.
41. How is sex assignment determined in ovotesticular DSD?
True hermaphrodites who have a unilateral ovary and müllerian structures may have spontaneous puberty and normal fertility and may be raised as females. External genital size and structure may allow male assignment, but more commonly, external genitalia are poorly virilized, and affected infants are assigned a female sex.
42. What principles should be kept in mind when sex assignments are made?
We have much to learn about gender identity and must consider which decisions may be made later than previously thought (e.g., surgery). Some surgical interventions are cosmetic, and some affected patients have expressed the wish to make the decisions in adolescence or adulthood. This field challenges many of our perceptions of sex and gender and our role as physicians. Although the infant with genital ambiguity presents a medical and social emergency, decisions should be made carefully, cautiously, and with all necessary biochemical and anatomic information available. Most important, the multidisciplinary team approach must involve the parents in an open and honest discussion of the options. In the end, it is the parents who come first in decision making on sex assignment.
Brown, J, Warne, G. Practical management of the intersex infant. J Pediatr Endocrinol Metab. 2005;18:3–23.
Douglas, G, Axelrad, ME, Brandt, ML, et al. Guidelines for evaluating and managing children born with disorders of sexual development. Pediatr Ann. 2012;41:4.
Eugenides, J. Middlesex [novel]. New York: Picador; 2003.
Goodall, J. Helping a child to understand her own testicular feminization. Lancet. 1991;337:33–35.
Houk, CP, Lee, PA, Intersexed states. diagnosis and management. Endocrinol Metab Clin North Am 2005;34:791–810.
Jasso, N, Boussin, L, Knebelmann, B, et al. Anti-müllerian hormone and intersex states. Trends Endocrinol Metab. 1991;2:227–233.
Kaplan, S. Clinical Pediatric Endocrinology. Philadelphia: Saunders; 1990.
Lee, PA, Houk, CP, Ahmed, SF, Hughes, IA. Consensus statement on management of intersex disorders. Pediatrics. 2006;118:e488–e500.
Low, Y, Hutson, JM, Murdoch Children’s Research Institute Sex Study Group. rules for clinical diagnosis in babies with ambiguous genitalia. J Paediatr Child Health 2003;39:406–413.
McGillivray, BC. The newborn with ambiguous genitalia. Semin Perinatol. 1992;16:365–368.
Mieszczak, J, Houk, CP, Lee, PA, et al. Assignment of the sex of rearing in the neonate with a disorder of sex development. Curr Opin Pediatr. 2009;21:541–547.
Meyers-Seifer, CH, Charest, NJ. Diagnosis and management of patients with ambiguous genitalia. Semin Perinatol. 1992;16:332–339.
Mulaikal, RM, Migeon, CJ, Rock, JA, et al. Fertility rates in female patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med. 1987;316:178–182.
Pagona, R. Diagnostic approach to the newborn with ambiguous genitalia. Pediatr Clin North Am. 1987;34:1019–1031.
Penny, R. Ambiguous genitalia. Am J Dis Child. 1990;144:753.
Rangecroft, L, British Association of Paediatric Surgeons working party on the surgical management of children born with ambiguous genitalia. surgical management of ambiguous genitalia. Arch Dis Child 2003;88:799–801.
Thigpen, AE, Davis, DL, Gautier, T, et al, Brief report. the molecular basis of steroid 5 alpha-reductase deficiency in a large Dominican kindred. N Engl J Med 1992;327:1216–1219.
Warne, GL, Kanumakala, S. Molecular endocrinology of sex differentiation. Semin Reprod Med. 2002;20:169–180.
Zucker, KJ, et al. Psychosexual development of women with congenital adrenal hyperplasia. Horm Behav. 1996;30:300–311.
