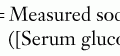119 Disorders of Sexual Development
Normal Sexual Development
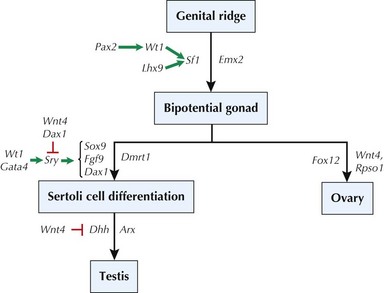
The normal development of the reproductive organs in humans begins with the differentiation of primordial structures and ends with the completion of secondary sexual characteristics that begins during puberty (see Chapter 67 for a discussion on puberty). Around the fifth week of gestation, the genital ridge develops into the appropriate gonads based on the chromosomal sex (XX or XY) established at conception. A complex series of genetic events must occur to establish the appropriate differentiation to an ovary or testis. It was clear that genes on the Y chromosome were involved in this differentiation because the presence of a Y chromosome resulted in male sexual characteristics, such as in XY or XXY individuals. Before these genes were known, they were referred to as “testis-determining factor” (TDF) because their presence led to testis formation. Ovaries and other female-specific reproductive organs were believed to develop by “default” because of the absence of Y chromosome material or TDF. Accordingly, research in mice and humans with X-Y translocations determined that the SRY gene (sex-determining region, Y) encoded for a transcription factor that is necessary for male reproductive organ development. Additional genes have also been identified for testis formation (Figure 119-1), and mutations in any of these genes (e.g., SOX9, DMRT1) have been associated with sex-reversal in XY patients. It is also important to note that more recent data have demonstrated an active role of several genes (FOXL2, WNT4, RSPO1) in ovary development in XX individuals, contradicting the older “default” female development notion.
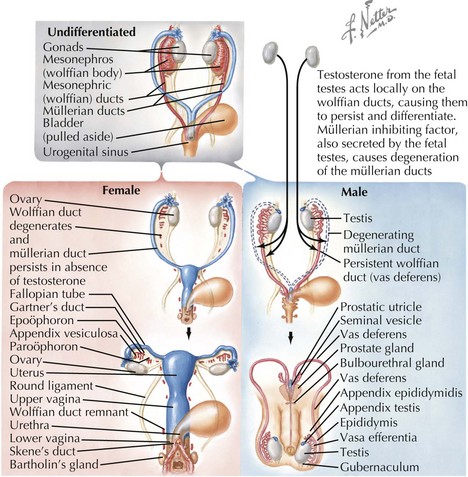
After the development of the gonads, the genital ducts must differentiate into the appropriate internal structures necessary for reproduction (Figure 119-2). In the undifferentiated stage, both mesonephric (Wolffian) and paramesonephric (Müllerian) ducts are present. In males, the newly formed testes secrete anti-Müllerian hormone (AMH), inhibin B, and testosterone between weeks 7 and 11, causing the Müllerian ducts to regress and the Wolffian ducts to persist, eventually becoming the seminal vesicles, vas deferentia, and epididymis. The opposite occurs in females, in whom the absence of testicular secretions causes the mesonephric ducts to regress and the Müllerian ducts to persist, forming the fallopian tubes, uterus, and upper third of the vagina
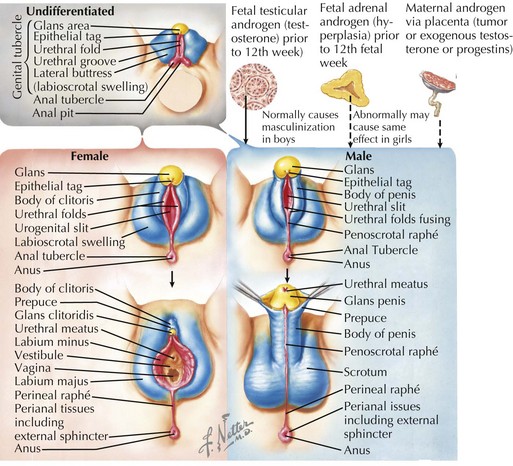
The undifferentiated genital tubercle, labioscrotal swellings, and urethral folds are present around gestational week 4, and the external genitalia begin to become more distinguishable during weeks 9 to 13. In males, the presence of testosterone and the testosterone derivative dihydrotestosterone (converted by 5-αreductase) causes the genital tubercle to form the glans penis, stimulating the urethral folds to fuse ventrally and form the spongy urethra and resulting in part of the penile shaft and labioscrotal swellings to fuse and form the scrotum (Figure 119-3). The absence of testosterone and dihydrotestosterone in females causes the genital tubercle to form the glans clitoris and the urethral folds and labioscrotal swelling to remain unfused (except posteriorly) to form the labia minora and labia majora, respectively. In males, testicular descent begins during the twelfth week of gestation and finishes during the third trimester.
Etiology, Pathogenesis, and Clinical Presentation
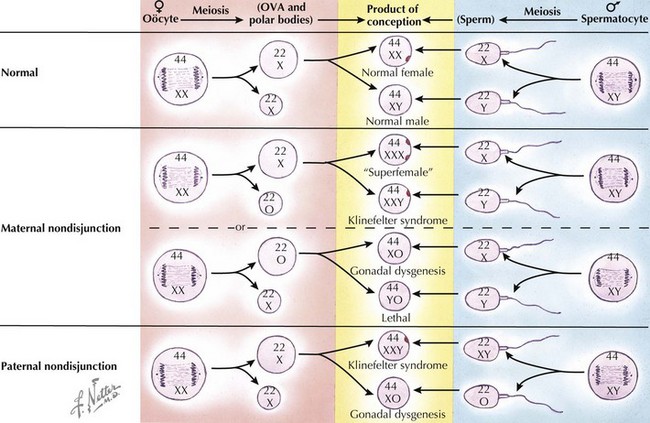
Because of the myriad genes associated with urogenital development, many genetic abnormalities may alter the structure and function of the reproductive organs. As noted above, these DSDs have been recently subtyped into sex chromosome DSDs, 46,XY DSDs, and 46,XX DSDs (Table 119-1). Sex chromosome DSDs are caused by various mechanisms (Figure 119-4) and include Klinefelter syndrome (47,XXY) and variants, Turner syndrome (45,X) and variants, and other types of XX/XY mosaicism, as discussed in Chapter 118, and typically result in largely normal prenatal sexual development but reduced function of the gonads during and after puberty. Most other genetic abnormalities may include point mutations, frameshift mutations, or deletions of specific genes that cause the protein product to be nonfunctional and thereby affect gonad differentiation, leading to a disparity between the chromosomal sex (XX or XY) and gonad development (ovary or testis).
Table 119-1 Classification of Disorders of Sexual Differentiation
| Sex Chromosome DSDs | 46,XY DSDs | 46,XX DSDs |
|---|---|---|
| A. 47,XXY | A. Disorders of testicular development | A. Disorders of ovarian development |
| B. 45,X | 1. Complete or partial gonadal dysgenesis (e.g., SRY, SOX9, SF1, WT1, DHH) | 1. Gonadal dysgenesis |
| C. 45,X/46,XY | 2. Ovotesticular DSD | 2. Ovotesticular DSD |
| D. 46,XX/46,XY mosaicism | 3. Testis regression | 3. Testicular DSD (SRY+, dup SOX9) |
| B. Disorders of androgen synthesis | B. Androgen excess | |
| 1. LH receptor mutations | 1. Fetal (3-β hydroxysteroid dehydrogenase 2), 21-hydroxylase (CYP21A2), P450 oxidoreductase (POR), 11-β hydroxylase (CYP11B1), glucocorticoid receptor mutations | |
| 2. Smith-Lemli-Opitz (cholesterol biosynthesis defect) | 2. Fetoplacental (aromatase [CYP19], oxidoreductase [POR]) | |
| 3. Cholesterol side-chain cleavage (CYP11A1) | 3. Maternal (virilizing tumors [luteoma], androgenic drugs) | |
| 4. 17-α hydroxylase (CYP17) | ||
| 5. P450 oxidoreductase (POR) | ||
| 6. 5-α reductase 2 | ||
| C. Disorders of androgen action | C. Other | |
| 1. Androgen insensitivity syndrome | 1. Syndromic associations (cloacal anomalies) | |
| 2. Drugs and environmental modulators | 2. Müllerian agenesis or hypoplasia (MURCS) | |
| 3. Uterine abnormalities (MODY5) | ||
| D. Other | 4. Vaginal atresia (McKusick-Kaufman) | |
| 1. Syndromic associations of male genital development (cloacal anomalies, Robinow, Aarskog, hand-foot-genital) | 5. Labial adhesions | |
| 2. Persistent Müllerian duct syndrome | ||
| 3. Vanishing testis syndrome | ||
| 4. Isolated hypospadias | ||
| 5. Congenital hypogonatropic hypogonadism | ||
| 6. Cryptorchidism (INSL3, GREAT) | ||
| 7. Environmental influences |
DSD, disorders of sexual development; LH, luteinizing hormone; MURCS, Müllerian duct aplasia, renal agenesis or ectopia, and cervical somite dysplasia; MODY5, maturity-onset diabetes of the young.
Adapted from Hughes IA: Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab 22:119-134, 2008.
46,XY Disorders of Sexual Development
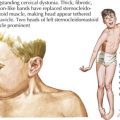
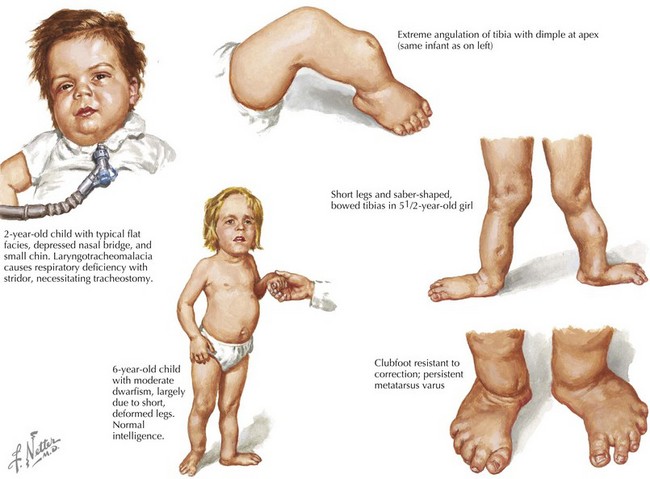
Other genes that may be associated with various 46,XY DSDs are listed in Table 119-1. SOX9 is a transcription factor that along with SF1 regulates transcription of AMH. SOX9 is also involved in chondrocyte differentiation, and mutations of this gene cause a skeletal disorder called camptomelic dysplasia (Figure 119-5), where about 67% of XY patients develop as phenotypic females with abnormalities that include gonadal dysgenesis; the presence of Müllerian structures such as a uterus, fallopian tubes, and vagina; and external female genitalia. WT1 is another transcription factor involved in testis formation and synergizes with SF1 to induce AMH. Mutations of WT1 are seen in patients with Denys-Drash syndrome, in which XY patients have various urogenital anomalies, including ambiguous genitalia. In addition, larger deletions that include WT1 cause WAGR syndrome (Wilms’ tumor, aniridia, genitourinary anomalies, and mental retardation). DHH is an important signaling molecule involved in various areas of morphogenesis, including male gonadal development, and mutations in DHH are associated with XY gonadal dysgenesis.
Evaluation and Management
Diagnosis
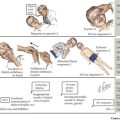
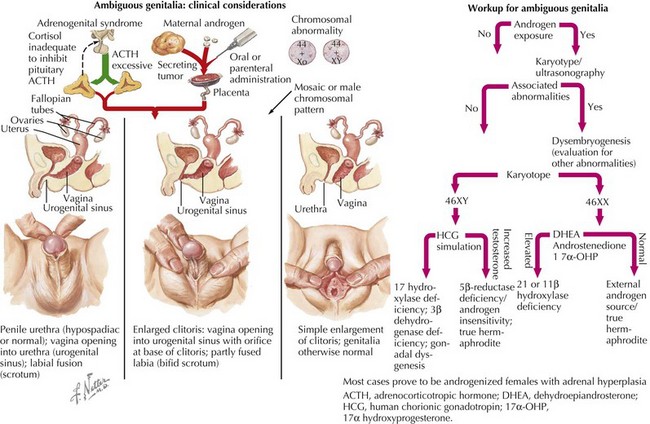
Laboratory and radiologic studies vary for each patient and are based on the historical and physical findings (Figure 119-6). The genetic investigation focuses on determining the number of X and Y chromosomes by karyotype, fluorescence in situ hybridization, or microarray analysis to determine the chromosomal sex (e.g., 46,XY, 46,XX, 45,X, 46,XX/46,XY) and any evidence of mosaicism. Specific genes can also be analyzed for mutations based on other associated findings on the physical examination or serum chemistries. Serum electrolytes are necessary to evaluate sodium concentration in cases of suspected CAH. Other CAH studies include the levels of 17-OH-progesterone (elevated in CAH) and 11-deoxycortisol (high in 11-β hydroxylase deficiency; low in 21-hydroxylase deficiency). If 17-OH-progesterone is normal, testosterone and dihydrotestosterone levels (after human chorionic gonadotropin [hCG] stimulation) may help elucidate the cause of some 46,XY DSDs (e.g., high testosterone to low dihydrotestosterone is seen in 5-α reductase 2 deficiency). hCG stimulation may also determine anorchia if there is no significant change in androgen levels combined with elevated luteinizing hormone and follicle-stimulating hormone levels. An ultrasound should be the first radiologic procedure performed because it may determine the Müllerian anatomy and possibly the presence and location of the gonads. Computed tomography or magnetic resonance imaging may also assist in clarifying these structures further. Finally, infants with intraabdominal or nonpalpable gonads may require exploratory laparoscopy with biopsy to determine if dysgenetic gonads, ovotestes, or streak testes are present.
Achermann JC, Hughes IA. Disorders of sexual development. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, editors. Williams Textbook of Endocrinology. ed 11. Philadelphia: Saunders Elsevier; 2008:783-848.
Burke LW. Female genital system. New York, Oxford Press. Stevenson RE, Hall JG, editors. Human Malformations and Related Anomalies. ed 2. 2006:1279-1306.
Hughes IA. Disorders of sex development: a new definition and classification. Best Pract Res Clin Endocrinol Metab. 2008;22:119-134.
Kolon TF. Disorders of sexual development. Curr Urol Rep. 2008;9:172-177.
Martin RA. Male genital system. New York, Oxford Press. Stevenson RE, Hall JG, editors. Human Malformations and Related Anomalies. ed 2. 2006:1251-1278.
Moore KL, Persuad TVN. editors: The urogenital system. In The Developing Human, ed 8, Philadelphia: Saunders Elsevier; 2008:243-284.