[level-membership-for-pediatrics-category]Chapter 125
Disorders of Sex Differentiation
Embryology, Sex Differentiation, and Gonad Differentiation
Overview: The three important precursor components needed for genital system development are the genital ridge and the two sets of internal sex ducts, the müllerian-paramesonephric and the wolffian-mesonephric. Around 7 weeks’ gestation, the embryologic genital ridge becomes either an ovary or a testis. The development of the male genital system is an “active” process requiring testes and müllerian inhibiting substance (MIS). The sex-determining region (SRY gene) on the Y chromosome’s short arm encodes a testis-determining factor. Under this factor’s influence (and in the presence of H-Y antigens found in the cell membranes of normal XY males) there is normal testis development with germ cells in the genital ridge differentiating into Sertoli cells and Leydig cells. Sertoli cells secrete MIS, which causes complete müllerian duct system involution. Leydig cells produce testosterone. The enzyme 5a-reductase converts testosterone intracellularly within the target tissues into the powerful androgen dihydrotestosterone (DHT). DHT allows the wolffian duct system to develop into the epididymis, vas deferens, ejaculatory duct, and seminal vesicles. If no Y chromosome exists or if abnormal encoding of testes determining factor is present, the gonad will passively differentiate into an ovary between 11 weeks to 17 weeks’ gestation in the presence of two X chromosomes. Absence of two X chromosomes may lead to abnormal or streak ovaries. The ovaries and their hormones, however, are thought to have no apparent role in sex differentiation of the female genital tract. The absence of MIS leads to persistence of müllerian structures, which develop into the fallopian tubes, uterus, cervix, and upper vagina. The wolffian ducts involute in the absence of testosterone. The undifferentiated external genitalia include the urogenital tubercle, urogenital swelling, and urogenital folds. DHT stimulation in males causes these structures to develop into the glans penis, scrotum, and penile shaft, respectively. In females, they develop into the clitoris, labia majora, and labia minora, respectively. The prostate gland develops from the urogenital sinus.1–6
Disorders of Sex Differentiation Without Ambiguity of External Genitalia (Box 125-1)
Turner Syndrome (XO Gonadal Dysgenesis)
Overview: Classic Turner syndrome (isochromatous 45,XO karyotype pattern) is the most common gonadal dysgenesis associated with an abnormal karyotype in girls and is nonfamilial. A single X chromosome is the probable cause for the presence of streak ovaries (streaks or ridges of connective tissue in the mesosalpinges parallel to the fallopian tubes), rather than normal ovaries. Some functional ovarian elements are present in a few cases. Fallopian tubes, a uterus, and a vagina are present, and no wolffian duct derivatives are found.1,5,6
Patients with classic Turner syndrome have several somatic findings. Affected patients are short in stature, with a distinctive facies that includes low-set ears, a low hairline, and a high, arched palate. They have a short, broad, and webbed neck, widely spaced nipples, and a shield chest. Skeletal abnormalities are common—classically short fourth metacarpal, fifth metacarpal, or both. Cystic hygromas of the neck area are noted in fetal life; and the webbed neck is thought to be a residuum. Other variably seen findings include large aortic roots, coarctation of the aorta, renal anomalies (horseshoe kidneys), duplication anomalies, ureteropelvic junction obstruction, and Hashimoto thyroiditis. Patients with Turner syndrome have a history of delayed onset of puberty, no breast development or vaginal mucosal estrogenization (but presence of pubic and axillary hair), infantile internal and external genitalia, and primary amenorrhea.1,7,8
Mosaic Turner Syndrome
One fourth of patients with 45,XO Turner syndrome have a so-called chromatin-positive pattern. Their karyotype is a mosaic consisting most often of a mixture of 45,XO and 46,XX chromosomes. Less often, other mosaic patterns (e.g., XO/XXX or XO/XX/XXX) or 46,XX and an abnormal X chromosome are present. In such cases, the gonads may consist of a streak ovary on one side and a hypoplastic or normal ovary on the other side, bilateral hypoplastic ovaries, or essentially normal ovaries. External and internal genitalia are entirely female, without wolffian duct remnants. These patients usually do not have the somatic abnormalities typically attributed to classic Turner syndrome, but many are short.1 Patients with mosaic Turner syndrome may develop secondary sex characteristics at puberty (found to occur in about 50%), and some may menstruate regularly.1,9
Amenorrhea Caused by Hypergonadotropic Hypogonadism
Primary amenorrhea (defined as a lack of menses by age 16 years), with high levels of circulating follicle-stimulating hormone and luteinizing hormone by serum assays, occurs because of ovarian failure; gonadal tissues fail to respond to endogenous gonadotropins. Patients included in this category have classic Turner syndrome, gonadal dysgenesis (including 46,XY and familial 46,XX), and secondary ovarian failure as a result of radiation or chemotherapy or on an autoimmune basis (autoimmune oophoritis).1,5,6,8
46,XY Gonadal Dysgenesis
Patients with 46,XY gonadal (X-linked recessive or sex-limited autosomal dominant) dysgenesis are phenotypically female, with streak gonads and infantile internal and external female genitalia. Patients are usually first diagnosed as having an abnormality in adolescence. As with other forms of gonadal dysgenesis, in such patients, the sex chromosome may not be absent but is abnormal. A deletion of the small arm of the Y chromosome (testis-determining factor or MIS) may be present. Mosaicism may lead to the development of ovaries. If a Y chromosome is a component of the karyotype of a patient with gonadal dysgenesis and ovaries, the patient has an increased risk of developing a gonadoblastoma within a dysgenetic ovary. Seminomas also occur in these patients.1,2
Familial 46,XX Gonadal Dysgenesis
Patients with familial 46,XX gonadal (sporadic or autosomal recessive) dysgenesis have gonads that consist of bilateral streaks in some cases, whereas in others hypoplastic ovaries or a hypoplastic ovary may be present on one side and a streak gonad on the other. The internal and external genitalia are entirely female, without wolffian duct derivatives. Incomplete puberty may be observed in patients with residual ovarian tissue. Sexual infantilism and primary amenorrhea are typical findings in those patients with bilateral streak gonads.1,2
Phenotypic Males
Klinefelter Syndrome (47,XXY Seminiferous Tubular Dysgenesis)
Seminiferous tubular dysgenesis (Klinefelter syndrome) is the most common aberration of the human sex chromosome. The typical 47,XXY karyotype is found in phenotypic males with primary hypogonadism. It is nonfamilial, occurring in 1 in every 750 to 1000 males. Variants have been described with less common chromosomal abnormalities, including XX/XXY or XY/XXY mosaicism, as well as XXXY, XXXXY, XXYY, or XXXYY sex chromosomal karyotypes. The external genitalia, especially the testes, are small. The testes are usually less than 3 cm in length and are firm. Cryptorchidism and hypospadias are common, with future development of azoospermia and sterility in most patients. The diagnosis is usually not made until after puberty. Gynecomastia develops in almost half of the older patients, and affected patients (particularly those with the classic 47,XXY karyotype) are at an increased risk for breast cancer. Rare cases of testicular and extragonadal germ cell neoplasms have been reported.1,5,6
Persistent Müllerian Duct Syndrome
Persistent müllerian duct syndrome is a rare type of sexual differentiation abnormality in males caused by a deficiency of müllerian inhibiting factor (MIF). These patients usually have a normal 46,XY karyotype and are phenotypically male, but they have a small uterus and fallopian tubes and a small vagina connected to the posterior urethra at the level of the verumontanum. Unilateral or bilateral cryptorchidism is common, as are unilateral or bilateral inguinal hernias. A uterus (uteri hernia syndrome), fallopian tubes, or sometimes a testis may be found in the hernia. The disorder, typically sporadic, has been reported in siblings.1,2,10,11
Disorders of Sex Differentiation with Ambiguous External Genitalia (Box 125-2)
Pseudohermaphroditism (Intersex)
Pseudohermaphroditism or intersex problems are abnormalities in which nonaccord of chromosomal, gonadal, and genital sex is present. Unlike true hermaphroditism, in which two types of gonadal tissue are present, pseudohermaphroditism has nonaccord, but only one gender’s gonads are present. By definition, male intersex patients have testes and female intersex patients have ovaries or ovarian tissue.1
Female Intersex
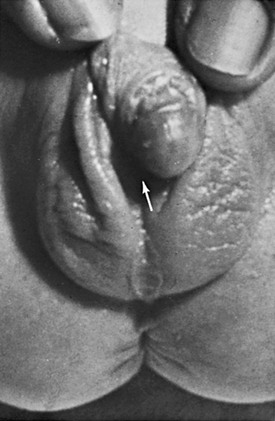
Overview: Female intersex is usually diagnosed in neonatal life in chromosomally normal females (46,XX) with masculinized external genitalia. The cause is usually increased fetal adrenal androgen production, most commonly from congenital adrenal hyperplasia or adrenogenital syndrome. Congenital adrenal hyperplasia (Fig. 125-2) is, by far, the most common cause of abnormal sex differentiation in females, occurring in 1 in 15,000 live births worldwide. It is caused by an inherited deficiency of enzymes involved in adrenocortical hormone biosynthesis. Affected patients have normal ovaries, a uterus, and fallopian tubes and no testicular tissue or internal wolffian duct derivatives. In most cases, the external genitalia are ambiguous (Fig. 125-3), with a prominent penis or partially fused labial scrotal folds.
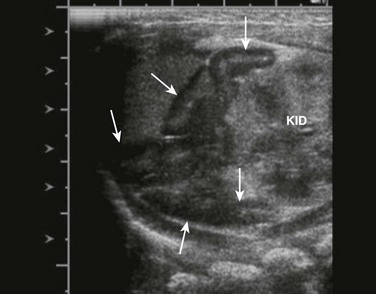
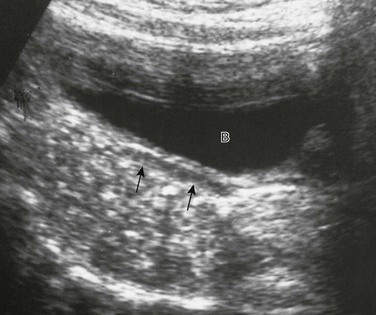
Figure 125-2 Adrenal ultrasound.
Sagittal view of adrenal and upper left kidney. A very enlarged adrenal (arrows) is seen superior to the upper portion of the kidney. The patient was a newborn with ambiguous genitalia and had three times the normal level of 17 hydroxyprogesterone. Her karyotype proved to be 46,XX. She was diagnosed with salt-wasting 21 hydroxylase deficiency as the cause of her congenital adrenal hyperplasia.
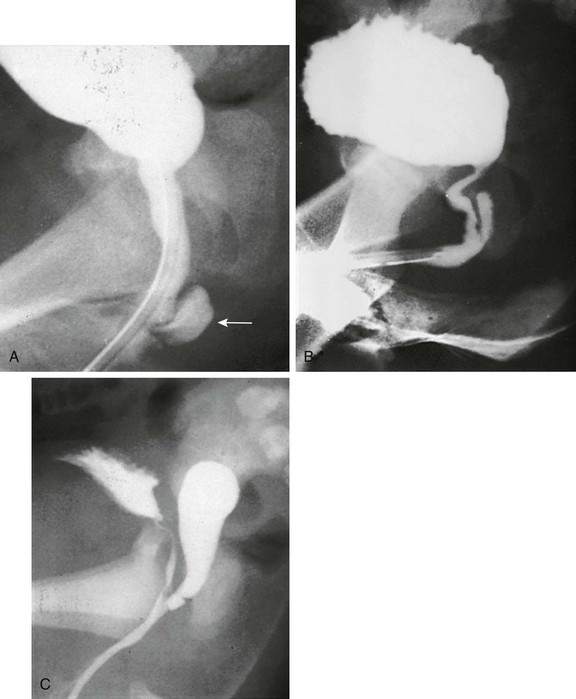
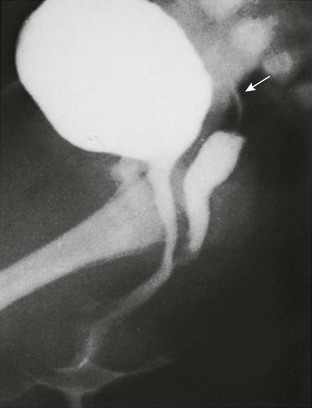
Imaging: A variously sized vagina is connected with the posterior urethra, forming a urogenital sinus, which commonly empties at the base of the penis. No gonads can be palpated in the labioscrotal folds or in the inguinal canal of such patients because they are located within the pelvis. Voiding cystourethrography (VCUG) often shows a male-type elongated urethra (Fig. 125-4). These patients are potentially fertile with external genital reconstruction and correct sex assignment.1,2
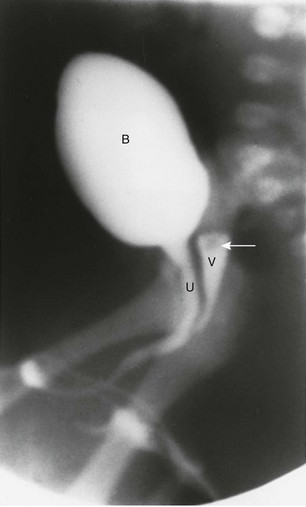
Figure 125-4 Congenital adrenal hyperplasia.
Lateral voiding cystourethrography image shows a bladder (B) with an elongated male-type urethra (U). Posterior to it is a vagina (V) with a subtle impression (arrow) of a cervix superiorly. The patient was a 46,XX neonate from an area in Puerto Rico, where congenital adrenal hyperplasia is common. (From Cohen HL, Haller J. Pediatric and adolescent genital abnormalities. Clin Diagn Ultrasound. 1988;24:187-216. With permission. Copyright Elsevier Science USA.)
Male Intersex
Male intersex patients are true males with a normal 46,XY male karyotype, present H-Y antigens, normal or mildly defective (and usually undescended) testes, but incomplete masculinization or frank ambiguity of their external genitalia. Decreased testosterone production and a lack of MIF production results in a karyotypically normal male with a female phenotype (except for partial masculinization of the external genitalia), and incomplete inhibition of the development of müllerian elements such as the uterus, vagina, and fallopian tubes (Fig. 125-5 and e-Fig. 125-6). Such patients usually have no secondary sexual development at puberty and may have an infantile uterus on ultrasonography. If the production of MIF by the testes is not affected, no internal müllerian system structures (uterus and fallopian tubes) will develop. The biochemical defect may be decreased androgen synthesis, decreased DHT production as a result of deficiency of 5a-reductase, or a defect in the androgen receptors. In many cases, the exact etiology remains unknown.1,2,5,6,12,13
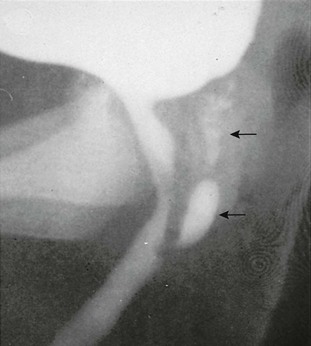
Figure 125-5 A male infant with severe hypospadias (perineal and scrotoperineal), bilateral cryptorchidism, small penis, and unfused scrotal folds.
The image shows a utricle or blind vaginal pouch that is very short and extending off the distal aspect of the posterior urethra. Arrows show spermatic ducts emptying into the utricle. The male urethra is somewhat short. See e-Figure 125-6.
e-Figure 125-6 Male pseudohermaphroditism.
A, Incomplete testicular feminization syndrome in a patient with a predominantly female phenotype. Voiding cystourethrography shows a mildly elongated urethra and retrograde opacification of a short, blind-ending vaginal pouch (arrow) in the perineum behind the urethra. Seminal ducts may end in a vaginal pouch such as this. A similar configuration can be seen in patients with complete testicular feminization syndrome and in patients with 5a-reductase deficiency. B, Three-year-old child with ambiguous external genitalia and 5a-reductase deficiency. A retrograde urethrogram shows a urogenital sinus and a small vagina without a cervical imprint. A more common pattern in 5a-reductase deficiency is that shown in A. C, Infant with severe hypospadias. A retrograde urethrogram shows a somewhat longer utricle connected with the distal end of the posterior urethra. The male urethra is somewhat short.
Testicular Feminization Syndrome (Androgen Receptor Defect)
Testicular feminization syndrome (X-linked recessive) is a form of intersex in which patients with 46,XY karyotype have well-formed testes (usually undescended within the abdomen or inguinal region) that produce androgens and MIF. However, lack of end-organ response to androgens is caused by a defect in a specific cytoplasmic receptor protein (cytosol receptor) that normally binds DHT to the plasma membrane and transports it to the nuclear chromatin. Müllerian system development is inhibited, and patients do not develop a uterus, fallopian tubes, or the upper two thirds of the vagina. They do develop secondary female sexual characteristics via circulating estrogens (produced from the breakdown of testosterone and adrenal steroids as well as from direct production by the testes). Patients with the complete form of the abnormality appear as phenotypically normal females, although they may have inguinal or labial masses resulting from the undescended testes (Fig. 125-7). They have normal breast development and may or may not have a short, blind-ending vagina behind the urethral opening. They frequently present with amenorrhea and respond to substitutional estrogen therapy. In the incomplete form (10% to 20% of cases) of testicular feminization (incomplete androgen receptor defect), patients present earlier in life compared with those with the complete form and have ambiguous genitalia. Affected patients may have a predominantly female phenotype (incomplete testicular feminization) or a predominantly male phenotype (Reifenstein syndrome).1,2
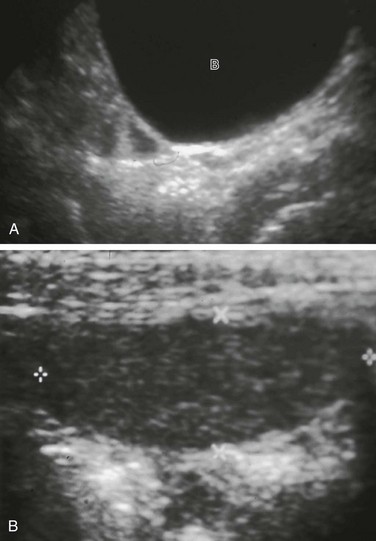
Figure 125-7 Testicular feminization syndrome.
A, A phenotypically normal 16-year-old girl, who presented with primary amenorrhea. Longitudinal midline sonogram showed no uterus posterior to the bladder (B). Ovaries were not seen. Karyotyping showed 46,XY, and the patient was proven to have testicular feminization syndrome. B, On examination for primary amenorrhea, this patient was noted to have bilateral inguinal masses. An oval structure (cursors) without contained cysts was found in the patient’s left inguinal region on ultrasonography and proved to be an undescended testis. (Courtesy of Joseph Yee, MD.)
Gonadal Dysgenesis
Mixed Gonadal Dysgenesis (Dysgenetic Male Pseudohermaphroditism)
Mixed or asymmetric gonadal dysgenesis (also known as XO/XY gonadal dysgenesis) is a relatively common form of abnormal sexual differentiation, usually occurring sporadically. Affected patient karyotypes are most often a mosaic of 45,XO and 46,XX. At times, XO/XYY or other mosaicisms are seen. These patients often have a streak gonad similar to that seen in Turner syndrome on one side and a usually dysgenetic (but occasionally normal) testis on the other. A fallopian tube is often present on the side of the streak gonad, and a vas deferens may be present on the side of the testis. The testis is usually intraabdominal but can be partially or completely descended. The external genitalia cover a wide spectrum of appearances from that of an almost normal female to that of an essentially normal male with hypospadias. Most patients have ambiguous external genitalia (as seen in other intersex situations), with a penis or clitoris of variable size, unfused labioscrotal folds, and a variously sized vagina connected with the urethra (urogenital sinus) that commonly empties at the base of the penis (Fig. 125-8). A uterus, said to be present in all cases, is usually small or rudimentary. Most patients are raised as females, but at puberty, some virilization may take place (usually without gynecomastia).1,2
Familial 46,XY Gonadal Dysgenesis
Familial 46,XY gonadal dysgenesis is an X-linked autosomal dominant trait associated with camptomelic dwarfism. The gonads are variable and may be bilateral streaks, bilateral dysgenetic testes, or a streak on one side and a dysgenetic testis on the other (mixed gonadal dysgenesis). Patients with bilateral streak gonads have a female phenotype with normal fallopian tubes, uterus, and vagina, usually clitoromegaly, and absence of wolffian duct derivatives. Sexual infantilism and amenorrhea are expected at puberty. Patients with bilateral dysgenetic testes or with a mixed form of gonadal dysgenesis typically have ambiguous or incompletely masculinized external genitalia. Müllerian and wolffian duct derivatives are present but may be hypoplastic or rudimentary.1,5,6
Drash Syndrome—Gonadal Dysgenesis, Nephropathy, and Wilms Tumor
Drash syndrome is an uncommon form of gonadal dysgenesis and male intersex. Patients most often have a 46,XY karyotype, bilateral gonadal dysgenesis with a variable histologic pattern, ambiguous external genitalia, and intraabdominal testes. They have chronic glomerulonephritis with histologic features similar to those of congenital nephrosis. They develop end-stage renal disease in early life. More than half of these patients develop a Wilms tumor at a young age, with the tumor purportedly occurring only in those patients with a female phenotype. In patients with Drash syndrome, 20% to 30% develop gonadal neoplasia.14,15
46,XY Gonadal Agenesis (Vanishing Testes Syndrome)
Patients with this condition are males with a 46,XY karyotype. No gonads are present, and male sex differentiation is absent or incomplete as a result of testicular resorption of unknown cause in early fetal life (13 to 14 weeks’ gestation). The external genitalia are ambiguous, and usually, both müllerian and wolffian duct derivatives are completely absent. Vanishing testes syndrome is differentiated from cases of bilateral anorchia (congenital absence of the testes), which are presumably caused by resorption of the testes beyond 13 to 14 weeks’ gestational age. Patients with bilateral anorchia have normal male sex development and no residual müllerian structures.1,16
XX Maleness
XX male syndrome (de la Chapelle syndrome) is a very unusual cause of ambiguous genitalia occurring in 4 to 5 cases per 100,000 individuals. Because of errors in meiosis, an unequal interchange of sex chromosomes occurs such that individuals may have one or two X chromosomes containing the male SRY gene. Patients are sterile but usually have two small testes, usually descended. Gynecomastia may be present, but more often, the individuals are phenotypically male and have no intraabdominal müllerian tissue despite being genetically female.1,17
True Hermaphroditism
True hermaphroditism is a rare condition. It is a sporadic disorder in which the affected patient has both testicular and ovarian tissues in the same or contralateral gonads. More than half such patients have the 46,XX karyotype. Mosaic karyotype patterns with at least one line with a Y chromosome do exist, including XO/XY, XX/XXY, or XX/XY chimerism (30%). Fifteen percent of patients have the 46,XY karyotype. All true hermaphrodites are H-Y antigen positive regardless of karyotype. In patients with the 45,XX karyotype, undetected Y chromosomal material is probably present and transferred to another chromosome. The testes or ovotestes may be intraabdominal, in the inguinal region, in the scrotal area, or in the labia majora. The ovaries of hermaphrodites are almost always intraabdominal. Internal gonadal ducts are usually consistent with the ipsilateral gonad (i.e., a vas on the side of a testis and a fallopian tube on the side of an ovary). In the case of ovotestes, the associated internal gonadal duct is usually a fallopian tube. A uterus is found in almost all cases but is most often hypoplastic and may be bicornuate.1
A wide spectrum of external genitalia ranges from normal male to ambiguous to female. Cryptorchidism is common. Inguinal hernia is common as well and, as with normal patients, can contain a gonad with its internal gonadal duct or even a uterus. About 75% of hermaphrodites are brought up as males. At puberty, usually some virilization as well as gynecomastia occurs.1,2
Gonadal Neoplasia of Patients with Disorders of Sex Differentiation
The gonads in several intersex disorders (with or without ambiguous genitalia) are at an increased risk for developing neoplasms. Patients with the highest risk of developing gonadal neoplasia are those with XO/XY mixed gonadal dysgenesis and those with 46,XY gonadal dysgenesis. The incidence of a gonadal tumor in both these conditions increases from 3% to 4% by age 10 years to 10% to 20% within the second decade of life and to 70% or more in older patients. Neoplasms are almost always germ cell in type, including seminoma or dysgerminoma and gonadoblastoma. Less commonly, patients may develop a gonadal teratoma, teratocarcinoma, yolk sac tumor, embryonal carcinoma of the adult type, or choriocarcinoma. These neoplasms are rare in patients who do not have a Y chromosome as part of their karyotype. The risk is apparently related to the H-Y antigen.1,2
Diagnostic Evaluation of Patients with Ambiguous Genitalia
Overview: The discovery of anomalous or ambiguous genitalia in a newborn has been described as an emergency from a social perspective and, hence to many, from a clinical perspective as well. The identification of the uterus, vagina, or urogenital sinus, by using ultrasonography, contrast fistulogram, or vaginogram, is paramount in the decision on how to rear the child. These findings can then be correlated and sexual identification aided by karyotyping, including fluorescent studies for Y chromosomes, specific analysis of the Y chromosome for the testis-determining gene, culture of genital skin fibroblasts for androgen receptor binding, and tests for androgen responsiveness. The appearance of the external genitalia is seldom diagnostic of a specific intersex disorder, but palpable gonads in the inguinal canal, labioscrotal folds, or scrotum can exclude female pseudohermaphroditism in most cases. Sometimes, the definitive anatomic diagnosis is made only at laparoscopy or laparotomy and on the basis of gonadal biopsy. The main role of ultrasonography is identification of the uterus, a relatively easy task in the female newborn.1,2
Imaging: A detailed radiographic study of the lower genitourinary tract (genitography) is important for diagnosis and as a guide in surgical reconstructive procedures. Patients usually have a urogenital sinus, a common terminal channel for the anterior urethra and posterior vaginal pouch. This sinus usually empties at the base of the penis. VCUG, particularly on lateral view, may outline the entire anatomy needed for evaluation. If the urethral catheter can be advanced only to the vagina, an injection with the catheter in that position may opacify the vagina, the urogenital sinus, and often the proximal urethra. If confusion about the anatomy persists during the VCUG examination, a retrograde injection of contrast material may be attempted through a catheter with the tip placed just inside the “urethral” meatus. At times, a coude catheter (with a curved tip) manipulated under fluoroscopic control into the urethra and bladder or into the vagina may improve contrast study of the area. An effort should be made on the VCUG or vaginogram to determine if a uterine cervix is present. Often, if a uterus (especially if normal) is present, a mass impression of the cervix on the contrast medium–filled vagina (see Fig. 125-4) is evident. A cervical imprint, however, may not be apparent if the vagina is not sufficiently distended or if the uterus is hypoplastic. A uterine cervix is present in all female intersex patients and is a common finding among many patients with mixed gonadal dysgenesis or true hermaphroditism as well. All members of these groups may have a hypoplastic or rudimentary uterus that may not be noted radiographically. Cervical imprints are not seen in male pseudohermaphroditism or male hypospadias.1,2
In some patients evaluated by VCUG or contrast vaginogram, the urogenital sinus is quite short and joined by the vagina very close to the perineal surface (Fig. 125-9). In other patients, it is a much longer channel, joined by the vagina at a much higher level, occasionally near the bladder neck. A very high insertion of the vagina in the urogenital sinus may pose a problem at the time of vaginal reconstruction because of the danger of injuring the external urethral sphincter. In patients evaluated for disorders of sex differentiation or ambiguous genitalia, the vagina may vary in size from a small cavity to an organ of normal size for the patient’s age. Occasionally, its distal end is stenosed or is completely obliterated, resulting in hydrocolpos at birth or hematocolpos at puberty (Fig. 125-10).1
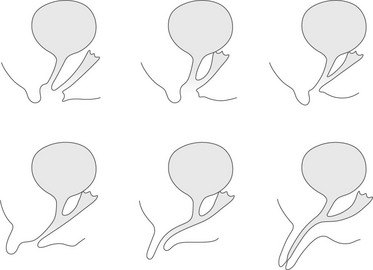
Figure 125-9 Different types of urogenital sinus are arranged in order of increasing masculinization, from an almost normal female pattern to a penile urethra.
The urogenital sinus is of variable length, and the vagina may enter the urogenital sinus at various levels.
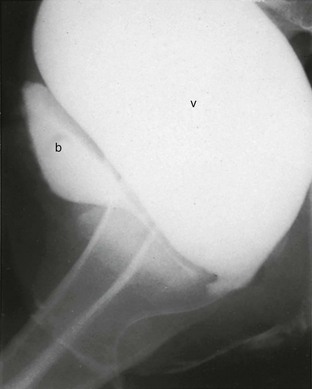
Figure 125-10 Hydrocolpos in adrenogenital syndrome.
Lateral view from a retrograde vaginogram of a 14-year-old girl with adrenogenital syndrome, and the urogenital sinus shows a catheter within a large obstructed vagina. She had presented with abdominal pain and a pelvic mass. v, vagina; b, bladder.
Pelvic ultrasonography is valuable in evaluating infants with ambiguous genitalia. Ultrasonography is an excellent imaging tool for identification of uterine tissue, although it may be difficult with hypoplastic uteri. The proximal vagina may also be demonstrated, particularly if it contains urine, but the urethra and urogenital sinus cannot be studied by this method. Ultrasonography does not replace genitography, but it is of particular value when genitography is unsuccessful. Ovaries are often seen, but fallopian tubes, unless obstructed, are more difficult to image. Ultrasonography is an excellent tool for the identification of the adrenal gland in the newborn and in the older child. Pelvic magnetic resonance imaging is of great value in evaluating patients with complex anatomy, particularly those with complex müllerian duct system abnormalities, in whom ultrasonography and genitography do not provide sufficient information (Fig. 125-11 and e-Fig. 125-12).1,18–20
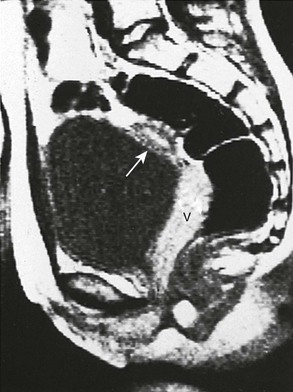
Figure 125-11 Anatomic evaluation of a 13-year-old with known adrenogenital syndrome being evaluated before surgical vaginoplasty.
Sagittal T1-weighted magnetic resonance image shows the vagina (V) throughout its course posterior to the bladder and anterior to rectum. Uterine tissue (arrow) is seen anterosuperior to it.
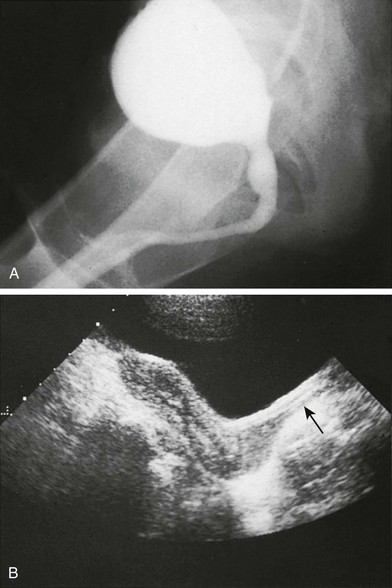
e-Figure 125-12 Anatomic evaluation of a 13-year-old with known adrenogenital syndrome being evaluated before surgical vaginoplasty (same patient as shown in Fig. 125-11).
A, Voiding cystourethrography shows a phallic urethra. A vagina is not seen. B, A sagittal sonogram shows a normal adult uterus. The proximal two thirds of the vagina (arrow) are noted and appear normal.
 What the Clinician Needs to Know
What the Clinician Needs to Know
Ahmed, SF, Rodie, M. Investigation and initial management of ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24(2):197–218.
Barthold, JS. Disorders of sex differentiation: a pediatric urologist’s perspective of new terminology and recommendations. J Urol. 2011;185(2):393–400.
Chavhan, GB, Parra, DA, Oudjhane, K, et al. Imaging of ambiguous genitalia: classification and diagnostic approach. Radiographics. 2008;28(7):1891–1904.
Choi, HK, Cho, KS, Lee, HW, et al. MR imaging of intersexuality. Radiographics. 1998;18(1):83–96.
Cohen-Kettenis, PT. Psychosocial and psychosexual aspects of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2010;24(2):325–334.
Gillam, LH, Hewitt, JK, Warne, GL. Ethical principles for the management of infants with disorders of sex development. Horm Res Paediatr. 2010;74(6):412–418.
Lambert, SM, Vilain, EJ, Kolon, TF. A practical approach to ambiguous genitalia in the newborn period. Urol Clin North Am. 2010;37(2):195–205.
Vidal, I, Gorduza, DB, Haraux, E, et al. Surgical options in disorders of sex development (DSD) with ambiguous genitalia. Best Pract Res Clin Endocrinol Metab. 2010;24(2):311–324.
References
1. Cohen, HL. Anomalies of sex differentiation. In: Slovis T, ed. Caffey’s pediatric diagnostic imaging. 11th ed. Philadelphia, PA: Mosby (Elsvier); 2008:2394–2405.
2. Currarino, G, Wood, B, Majd, M. Abnormalities of the genital tract. In: Silverman F, Kuhn J, eds. Caffey’s pediatric x-ray diagnosis. 9th ed. St. Louis: Mosby–Year Book; 1993:1375–1401.
3. Rey, RA, Grinspon, RP. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2011;25:221–238.
4. Kucinskas, L, Just, W. Human male sex determination and sexual differentiation: pathways, molecular interaction and genetic disorders. Medicina (Kaunas). 2005;41:633–640.
5. Purcell, K, Lingappa, V, Taylor, R. Disorders of the female reproductive tract. In: McPhee S, Ganong W, eds. Pathophysiology of disease. An introduction to clinical medicine. 5th ed. New York: McGraw-Hill; 2006:624–651.
6. McPhee, S. Disorders of the male reproductive tract. In: McPhee S, Ganong W, eds. Pathophysiology of disease. An introduction to clinical medicine. 5th ed. New York: McGraw-Hill; 2006:652–675.
7. Boechat, MI, Westra, SJ, Lippe, B. Normal US appearance of ovaries and uterus in four patients with Turner’s syndrome and 45,X karyotype. Pediatr Radiol. 1996;26:37–39.
8. Nidecker, A, Cohen, HL, Zinn, HL. Amenorrhea in the adolescent or young adult. In: Bluth E, Benson C, Ralls P, et al, eds. Ultrasound. A practical approach to clinical problems. 2nd ed. New York: Thieme; 2008:513–527.
9. Hall, JG, Gilchrist, DM. Turner syndrome and its variants. Pediatr Clin North Am. 1990;37:1421–1440.
10. Fernandes, E, Fernandes, E, Hollabaugh, S, et al. Persistent müllerian duct syndrome. Urology. 1990;36:516–518.
11. Halbertsma, F, Otten, B, Wijnen, R, et al. A baby boy with cryptorchism, inguinal hernia and internal female genitalia: the persistent Müllerian duct syndrome. Ned Tijdschr Geneeskd. 2004;148:484–487.
12. Sivit, CJ, Hung, W, Taylor, G, et al. Sonography in neonatal congenital adrenal hyperplasia. AJR Am J Roentgenol. 1991;156:141–143.
13. Speiser, P. Prenatal treatment of congenital adrenal hyperplasia. J Urol. 1999;162:534–536.
14. Koziell, A, Grundy, R. Frasier and Denys-Drash syndromes: different disorders or part of a spectrum? Arch Dis Child. 1999;81(4):365–369.
15. Coppes, MJ, Huff, V, Pelletier, J. Denys-Drash syndrome: relating a clinical disorder to genetic alterations in the tumor suppressor gene WT1. J Pediatr. 1993;123(5):673–678.
16. Marcantonio, SM, Fechner, PY, Migeon, CJ, et al. Embryonic testicular regression sequence: a part of the clinical spectrum of 46,XY gonadal dysgenesis. Am J Med Genet. 1994;49:1–5.
17. de la Chapelle, A. The etiology of maleness in XX men. Hum Genet. 1981;58:105–116.
18. Cohen, HL, Bober, S, Bow, S. Imaging the pediatric pelvis: the normal and abnormal genital tract and simulators of its diseases. Urol Radiol. 1992;14:273–283.
19. Ratani, R, Cohen, HL, Fiore, E. Pediatric gynecologic ultrasound. Ultrasound Q. 2004;20:127–139.
20. Cohen, HL, Lemond, T. Gynecologic imaging of the pediatric patient. In: Fielding J, Brown D, Thurmond A, eds. Gynecologic imaging. Philadelphia: Elsevier Saunders; 2011:547–564.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 125
Disorders of Sex Differentiation
Embryology, Sex Differentiation, and Gonad Differentiation
Overview: The three important precursor components needed for genital system development are the genital ridge and the two sets of internal sex ducts, the müllerian-paramesonephric and the wolffian-mesonephric. Around 7 weeks’ gestation, the embryologic genital ridge becomes either an ovary or a testis. The development of the male genital system is an “active” process requiring testes and müllerian inhibiting substance (MIS). The sex-determining region (SRY gene) on the Y chromosome’s short arm encodes a testis-determining factor. Under this factor’s influence (and in the presence of H-Y antigens found in the cell membranes of normal XY males) there is normal testis development with germ cells in the genital ridge differentiating into Sertoli cells and Leydig cells. Sertoli cells secrete MIS, which causes complete müllerian duct system involution. Leydig cells produce testosterone. The enzyme 5a-reductase converts testosterone intracellularly within the target tissues into the powerful androgen dihydrotestosterone (DHT). DHT allows the wolffian duct system to develop into the epididymis, vas deferens, ejaculatory duct, and seminal vesicles. If no Y chromosome exists or if abnormal encoding of testes determining factor is present, the gonad will passively differentiate into an ovary between 11 weeks to 17 weeks’ gestation in the presence of two X chromosomes. Absence of two X chromosomes may lead to abnormal or streak ovaries. The ovaries and their hormones, however, are thought to have no apparent role in sex differentiation of the female genital tract. The absence of MIS leads to persistence of müllerian structures, which develop into the fallopian tubes, uterus, cervix, and upper vagina. The wolffian ducts involute in the absence of testosterone. The undifferentiated external genitalia include the urogenital tubercle, urogenital swelling, and urogenital folds. DHT stimulation in males causes these structures to develop into the glans penis, scrotum, and penile shaft, respectively. In females, they develop into the clitoris, labia majora, and labia minora, respectively. The prostate gland develops from the urogenital sinus.1–6
Disorders of Sex Differentiation Without Ambiguity of External Genitalia (Box 125-1)
Turner Syndrome (XO Gonadal Dysgenesis)
Overview: Classic Turner syndrome (isochromatous 45,XO karyotype pattern) is the most common gonadal dysgenesis associated with an abnormal karyotype in girls and is nonfamilial. A single X chromosome is the probable cause for the presence of streak ovaries (streaks or ridges of connective tissue in the mesosalpinges parallel to the fallopian tubes), rather than normal ovaries. Some functional ovarian elements are present in a few cases. Fallopian tubes, a uterus, and a vagina are present, and no wolffian duct derivatives are found.1,5,6
Patients with classic Turner syndrome have several somatic findings. Affected patients are short in stature, with a distinctive facies that includes low-set ears, a low hairline, and a high, arched palate. They have a short, broad, and webbed neck, widely spaced nipples, and a shield chest. Skeletal abnormalities are common—classically short fourth metacarpal, fifth metacarpal, or both. Cystic hygromas of the neck area are noted in fetal life; and the webbed neck is thought to be a residuum. Other variably seen findings include large aortic roots, coarctation of the aorta, renal anomalies (horseshoe kidneys), duplication anomalies, ureteropelvic junction obstruction, and Hashimoto thyroiditis. Patients with Turner syndrome have a history of delayed onset of puberty, no breast development or vaginal mucosal estrogenization (but presence of pubic and axillary hair), infantile internal and external genitalia, and primary amenorrhea.1,7,8
Mosaic Turner Syndrome
One fourth of patients with 45,XO Turner syndrome have a so-called chromatin-positive pattern. Their karyotype is a mosaic consisting most often of a mixture of 45,XO and 46,XX chromosomes. Less often, other mosaic patterns (e.g., XO/XXX or XO/XX/XXX) or 46,XX and an abnormal X chromosome are present. In such cases, the gonads may consist of a streak ovary on one side and a hypoplastic or normal ovary on the other side, bilateral hypoplastic ovaries, or essentially normal ovaries. External and internal genitalia are entirely female, without wolffian duct remnants. These patients usually do not have the somatic abnormalities typically attributed to classic Turner syndrome, but many are short.1 Patients with mosaic Turner syndrome may develop secondary sex characteristics at puberty (found to occur in about 50%), and some may menstruate regularly.1,9
Amenorrhea Caused by Hypergonadotropic Hypogonadism
Primary amenorrhea (defined as a lack of menses by age 16 years), with high levels of circulating follicle-stimulating hormone and luteinizing hormone by serum assays, occurs because of ovarian failure; gonadal tissues fail to respond to endogenous gonadotropins. Patients included in this category have classic Turner syndrome, gonadal dysgenesis (including 46,XY and familial 46,XX), and secondary ovarian failure as a result of radiation or chemotherapy or on an autoimmune basis (autoimmune oophoritis).1,5,6,8
46,XY Gonadal Dysgenesis
Patients with 46,XY gonadal (X-linked recessive or sex-limited autosomal dominant) dysgenesis are phenotypically female, with streak gonads and infantile internal and external female genitalia. Patients are usually first diagnosed as having an abnormality in adolescence. As with other forms of gonadal dysgenesis, in such patients, the sex chromosome may not be absent but is abnormal. A deletion of the small arm of the Y chromosome (testis-determining factor or MIS) may be present. Mosaicism may lead to the development of ovaries. If a Y chromosome is a component of the karyotype of a patient with gonadal dysgenesis and ovaries, the patient has an increased risk of developing a gonadoblastoma within a dysgenetic ovary. Seminomas also occur in these patients.1,2
Familial 46,XX Gonadal Dysgenesis
Patients with familial 46,XX gonadal (sporadic or autosomal recessive) dysgenesis have gonads that consist of bilateral streaks in some cases, whereas in others hypoplastic ovaries or a hypoplastic ovary may be present on one side and a streak gonad on the other. The internal and external genitalia are entirely female, without wolffian duct derivatives. Incomplete puberty may be observed in patients with residual ovarian tissue. Sexual infantilism and primary amenorrhea are typical findings in those patients with bilateral streak gonads.1,2
Phenotypic Males
Klinefelter Syndrome (47,XXY Seminiferous Tubular Dysgenesis)
Seminiferous tubular dysgenesis (Klinefelter syndrome) is the most common aberration of the human sex chromosome. The typical 47,XXY karyotype is found in phenotypic males with primary hypogonadism. It is nonfamilial, occurring in 1 in every 750 to 1000 males. Variants have been described with less common chromosomal abnormalities, including XX/XXY or XY/XXY mosaicism, as well as XXXY, XXXXY, XXYY, or XXXYY sex chromosomal karyotypes. The external genitalia, especially the testes, are small. The testes are usually less than 3 cm in length and are firm. Cryptorchidism and hypospadias are common, with future development of azoospermia and sterility in most patients. The diagnosis is usually not made until after puberty. Gynecomastia develops in almost half of the older patients, and affected patients (particularly those with the classic 47,XXY karyotype) are at an increased risk for breast cancer. Rare cases of testicular and extragonadal germ cell neoplasms have been reported.1,5,6
Persistent Müllerian Duct Syndrome
Persistent müllerian duct syndrome is a rare type of sexual differentiation abnormality in males caused by a deficiency of müllerian inhibiting factor (MIF). These patients usually have a normal 46,XY karyotype and are phenotypically male, but they have a small uterus and fallopian tubes and a small vagina connected to the posterior urethra at the level of the verumontanum. Unilateral or bilateral cryptorchidism is common, as are unilateral or bilateral inguinal hernias. A uterus (uteri hernia syndrome), fallopian tubes, or sometimes a testis may be found in the hernia. The disorder, typically sporadic, has been reported in siblings.1,2,10,11
Disorders of Sex Differentiation with Ambiguous External Genitalia (Box 125-2)
Pseudohermaphroditism (Intersex)
Pseudohermaphroditism or intersex problems are abnormalities in which nonaccord of chromosomal, gonadal, and genital sex is present. Unlike true hermaphroditism, in which two types of gonadal tissue are present, pseudohermaphroditism has nonaccord, but only one gender’s gonads are present. By definition, male intersex patients have testes and female intersex patients have ovaries or ovarian tissue.1
Female Intersex
Overview: Female intersex is usually diagnosed in neonatal life in chromosomally normal females (46,XX) with masculinized external genitalia. The cause is usually increased fetal adrenal androgen production, most commonly from congenital adrenal hyperplasia or adrenogenital syndrome. Congenital adrenal hyperplasia (Fig. 125-2) is, by far, the most common cause of abnormal sex differentiation in females, occurring in 1 in 15,000 live births worldwide. It is caused by an inherited deficiency of enzymes involved in adrenocortical hormone biosynthesis. Affected patients have normal ovaries, a uterus, and fallopian tubes and no testicular tissue or internal wolffian duct derivatives. In most cases, the external genitalia are ambiguous (Fig. 125-3), with a prominent penis or partially fused labial scrotal folds.
Figure 125-2 Adrenal ultrasound.
Sagittal view of adrenal and upper left kidney. A very enlarged adrenal (arrows) is seen superior to the upper portion of the kidney. The patient was a newborn with ambiguous genitalia and had three times the normal level of 17 hydroxyprogesterone. Her karyotype proved to be 46,XX. She was diagnosed with salt-wasting 21 hydroxylase deficiency as the cause of her congenital adrenal hyperplasia.
Imaging: A variously sized vagina is connected with the posterior urethra, forming a urogenital sinus, which commonly empties at the base of the penis. No gonads can be palpated in the labioscrotal folds or in the inguinal canal of such patients because they are located within the pelvis. Voiding cystourethrography (VCUG) often shows a male-type elongated urethra (Fig. 125-4
[/not-level-membership-for-pediatrics-category]
