Chapter 582 Disorders of Sex Development
Sexual Differentiation (Chapter 576)
In normal differentiation, the final form of all sexual structures is consistent with normal sex chromosomes (either XX or XY). A 46,XX complement of chromosomes as well as genetic factors such as DAX1 and the signaling molecule WNT-4 are necessary for the development of normal ovaries. Development of the male phenotype is even more complex. It requires a Y chromosome and, specifically, an intact SRY gene, which, in association with other genes such as SOX9, SF1, and WT1 and others (Chapter 576), directs the undifferentiated gonad to become a testis. Aberrant recombinations may result in X chromosomes carrying SRY, resulting in XX males, or Y chromosomes that have lost SRY, resulting in XY females.
Chromosomal aberrations may result in ambiguity of the external genitalia. Conditions of aberrant sex differentiation may also occur with the XX or XY genotype. The appropriate term for what was previously called intersex is disorders of sex development (DSD). This term defines a condition “in which development of chromosomal, gonadal or anatomical sex is atypical.” It is becoming more preferable to use the term “atypical genitalia” rather than “ambiguous genitalia.” Comparison with the previous terms and a new etiologic classification are seen in Tables 582-1 and 582-2. Some of the genes involved in disorders of sex development are listed in Table 576-1.
| PREVIOUS | CURRENTLY ACCEPTED |
|---|---|
| Intersex | Disorders of sex development (DSD) |
| Male pseudohermaphrodite | 46,XY DSD |
| Undervirilization of an XY male | 46,XY DSD |
| Undermasculinization of an XY male | 46,XY DSD |
| 46,XY intersex | 46,XY DSD |
| Female pseudohermaphrodite | 46,XX DSD |
| Overvirilization of an XX female | 46,XX DSD |
| Masculinization of an XX female | 46,XX DSD |
| 46,XX intersex | 46,XX DSD |
| True hermaphrodite | Ovotesticular DSD |
| Gonadal intersex | Ovotesticular DSD |
| XX male or XX sex reversal | 46,XX testicular DSD |
| XY sex reversal | 46,XY complete gonadal dysgenesis |
From Lee PA, Houk CP, Ahmed SF, et al: Consensus statement on management of intersex disorders, Pediatrics 118:e488–e500, 2006.
Table 582-2 ETIOLOGIC CLASSIFICATION OF DISORDERS OF SEX DEVELOPMENT (DSD)
46,XX-DSD
Androgen Exposure
Disorder of Ovarian Development
Undetermined Origin
Associated with genitourinary and gastrointestinal tract defects
46,XY DSD
Defects in Testicular Development
Deficiency of Testicular Hormones
Defect in Androgen Action
Ovotesticular DSD
Sex Chromosome DSD
From Lee PA, Houk CP, Ahmed SF, et al: Consensus statement on management of intersex disorders, Pediatrics 118:e488–e500, 2006.
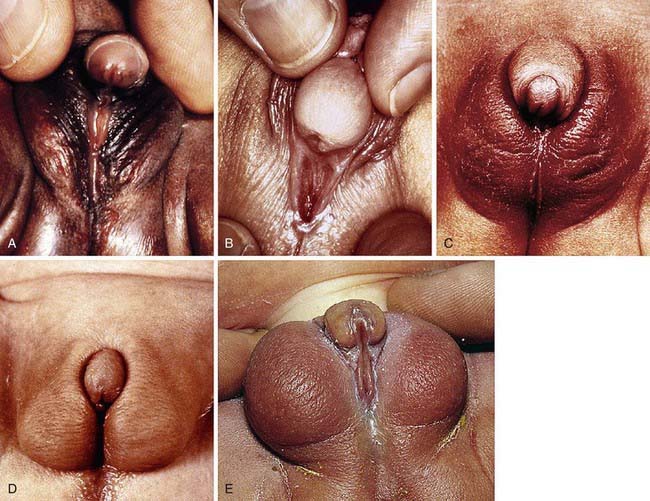
Development of the external genitalia begins with the potential to be either male or female (Fig. 582-1). Virilization of a female, the most common form of DSD, results in varying phenotypes (Fig. 582-2), which start from the basic genital appearances of the embryo (see Fig. 582-1).
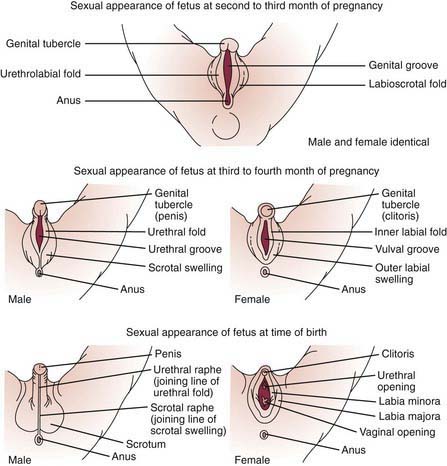
Figure 582-1 Schematic demonstration of differentiation of normal male and female genitalia during embryogenesis.
(From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby, p 328.)
Diagnostic Approach to the Patient with Atypical or Ambiguous Genitalia
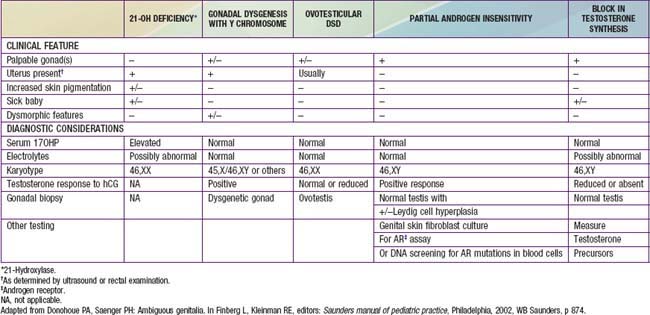
After a complete history and physical exam, the common diagnostic approach includes multiple steps, described in the following outline. These steps are usually performed simultaneously rather than waiting for results of 1 test prior to performing another, due to the sensitive and sometimes urgent nature of the condition. Careful attention to the presence of physical features other than the genitalia is crucial, to determine if a diagnosis of a particular multisystem syndrome is possible. These are described in more detail in Chapters 582.1, 582.2, and 582.3. A summary of many features of commonly encountered causes of DSD is provided in Table 582-3.
Diagnostic tests include the following:
582.1 46,XX DSD
Congenital Adrenal Hyperplasia (Chapter 570.1)
This is the most common cause of genital ambiguity and of 46,XX DSD. Females with the 21-hydroxylase and 11-hydroxylase defects are the most highly virilized, although minimal virilization also occurs with the type II 3β-hydroxysteroid dehydrogenase defect (see Fig. 582-1). Salt losers tend to have greater degrees of virilization than do non–salt-losing patients. Masculinization may be so intense that a complete penile urethra results, and the condition may mimic a male with bilateral cryptorchidism.
Aromatase Deficiency
In genotypic females, the rare condition of aromatase deficiency during fetal life leads to 46,XX DSD and results in hypergonadotropic hypogonadism at puberty because of ovarian failure to synthesize estrogen (see Fig. 568-1).
Bose HS, Pescovitz OH, Miller WL. Spontaneous feminization in a 46,XX female patient with congenital lipoid adrenal hyperplasia due to a homozygous frameshift mutation in the steroidogenic acute regulatory protein. J Clin Endocrinol Metab. 1997;82:1511-1515.
Diamond M, Sigmundson K. Sex reassignment at birth. Arch Pediatr Adolesc Med. 1997;151:298-304.
Diamond DA, Mitchell C, Lamb K, et al. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: attitudes and practices of pediatric urologists. J Pediatr. 2006;148:445-449.
Frade Costa EM, Bilharinho Mendonca B, Inacio M, et al. Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertil Steril. 1997;67:229-232.
Mendonca BB, Leite MV, DeCastro M, et al. Female pseudohermaphroditism caused by a novel homozygous mutation of the GR gene. J Clin Endocrinol Metab. 2002;87:1805-1809.
Moisan AM, Ricketts ML, Tardy V, et al. New insight into the molecular basis of 3β-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-4425.
Morishima A, Grumbach MM, Simpson ER, et al. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689-3698.
Mullis PE, Yoshimura N, Kuhlmann B, et al. Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997;82:1739-1745.
Parisi MA, Ramsdell LA, Burns MW, et al. A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med. 2007;9:348-357.
Saenger P. New developments in congenital lipoid adrenal hyperplasia and steroidogenic acute regulatory protein. Pediatr Clin North Am. 1997;44:397-421.
Wallien MS, Cohen-Kettenis PT. Psychosexual outcome of gender-dysphoric children. J Am Acad Child Adolesc Psychiatry. 2008;47:1413-1423.
582.2 46,XY DSD
Defects in Testicular Differentiation
XY Pure Gonadal Dysgenesis (Swyer Syndrome)
Pure gonadal dysgenesis also occurs in XX individuals (Chapter 580).
Defects in Testicular Hormones
Five genetic defects have been delineated in the enzymatic synthesis of testosterone by fetal testis, and a defect in Leydig cell differentiation has been described. These defects produce 46,XY males with inadequate masculinization (see Fig. 576-1). Because levels of testosterone are normally low before puberty, an hCG stimulation test may be needed in children to assess the ability of the testes to synthesize testosterone.
Leydig Cell Aplasia
High serum LH and low follicle-stimulating hormone (FSH) were noted in 1 male with hypogonadism owing to a mutation in the gene for the β subunit of FSH (see Table 577-1).
3β-Hydroxysteroid Dehydrogenase Deficiency
Males with this form of congenital adrenal hyperplasia (Chapter 570) have various degrees of hypospadias, with or without bifid scrotum and cryptorchidism and, rarely, a complete female phenotype. Affected infants usually develop salt-losing manifestations shortly after birth. Incomplete defects, occasionally seen in boys with premature pubarche, as well as late-onset nonclassic forms have been reported. These children have point mutations of the gene for type II 3β-hydroxysteroid enzyme, resulting in impairment of steroidogenesis in the adrenals and gonads; the impairment may be unequal between adrenals and gonads. Normal pubertal changes in some boys could be explained by the normally present type I 3β-hydroxysteroid dehydrogenase present in many peripheral tissues. Infertility is frequent. There is no correlation between degree of salt wasting and degree of phenotypic abnormality.
Deficiency of 17-Hydroxylase/17,20 Lyase
A single enzyme (P450C17 or CYP17) encoded by a single gene on chromosome 10q24.3 has both 17-hydroxylase and 17,20 lyase activities in adrenal and gonadal tissues (Chapter 570). Many different genetic lesions have been reported. Genetic males usually have a complete female phenotype or, less often, various degrees of undervirilization from labioscrotal fusion to perineal hypospadias and cryptorchidism. Pubertal development fails to occur in both genetic sexes.
Defects in Androgen Action
5α-Reductase Deficiency
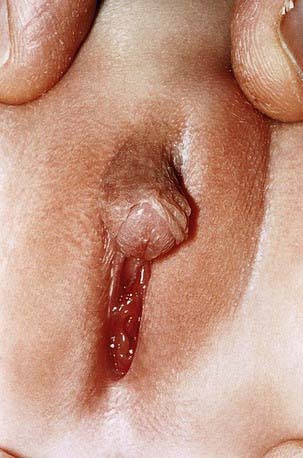
The phenotype most commonly associated with this condition results in boys who have a small phallus, bifid scrotum, urogenital sinus with perineal hypospadias, and a blind vaginal pouch (Fig. 582-3). Testes are in the inguinal canals or labioscrotal folds and are normal histologically. There are no müllerian structures. Wolffian structures—the vas deferens, epididymis, and seminal vesicles—are present. Most affected patients have been identified as females. At puberty, virilization occurs; the phallus enlarges, the testes descend and grow normally, and spermatogenesis occurs. There is no gynecomastia. Beard growth is scanty, acne is absent, the prostate is small, and recession of the temporal hairline fails to occur. Virilization of the Wolffian duct is caused by the action of testosterone itself, although masculinization of the urogenital sinus and external genitals depends on the action of DHT during the critical period of fetal masculinization. Growth of facial hair and of the prostate also appears to be DHT dependent.
Androgen Insensitivity Syndromes
Clinical Manifestations
The clinical spectrum of patients with AISs, all of whom have a 46,XY chromosomal complement, range from phenotypic females (in complete AIS) to males with various forms of ambiguous genitalia and undervirilization (partial AIS, or clinical syndromes such as Reifenstein syndrome) to phenotypically normal-appearing males with infertility. In addition to normal 46,XY chromosomes, the presence of testes and normal or elevated testosterone and LH levels are common to all such children (Figs. 582-4 and 582-5).
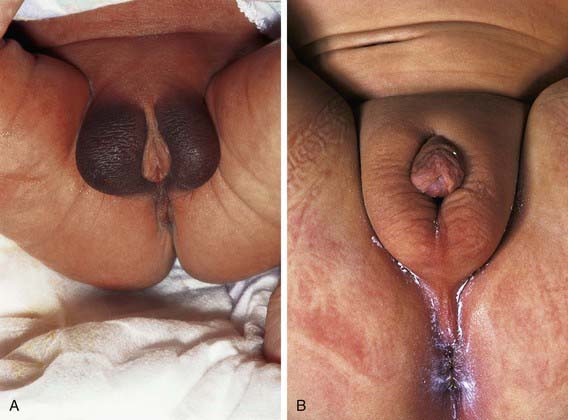
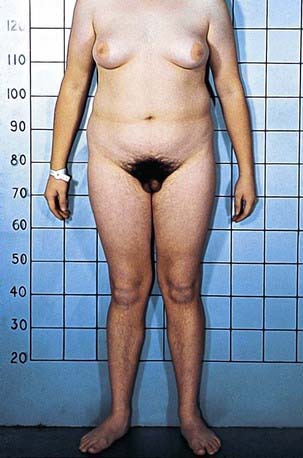
Prepubertal children with this disorder are often detected when inguinal masses prove to be testes or when a testis is unexpectedly found during herniorrhaphy in a phenotypic female. About 1-2% of girls with an inguinal hernia prove to have this disorder. In infants, elevated gonadotropin levels should suggest the diagnosis. In adults, amenorrhea is the usual presenting symptom. In prepubertal children, the condition must be differentiated from other types of XY under virilized males in which there is complete feminization. These include XY gonadal dysgenesis (Swyer syndrome), true agonadism, Leydig cell aplasia including LH receptor defects, and 17-ketosteroid reductase deficiency; all these conditions, unlike complete AIS, are characterized by low levels of testosterone as neonates and during adult life and by failure to respond to hCG during the prepubertal years. Although patients with complete AIS have unambiguously female external genitals at birth, those with partial AIS have a wide variety of phenotypic presentations ranging from perineoscrotal hypospadias, bifid scrotum, and cryptorchidism to extreme under virilization appearing as clitoromegaly and labial fusion. Some forms of partial AIS have been known as specific syndromes. Patients with Reifenstein syndrome have incomplete virilization characterized by hypogonadism, severe hypospadias, and gynecomastia (see Fig. 582-5). Gilbert-Dreyfus and Lubs are additional syndromes classified as partial AIS. In all cases, abnormalities in the androgen receptor gene have been identified.
Undetermined Causes
Other XY undervirilized males display great variability of the external and internal genitalia and various degrees of phallic and müllerian development. Testes may be histologically normal or rudimentary, or there may only be 1. Even the newer techniques may find no recognized cause in a up to 50% of children with 46,XY DSD. Some ambiguity of the genitalia is associated with a wide variety of chromosomal aberrations, which must always be considered in the differential diagnosis, the most common being the 45,X/46,XY syndrome (Chapter 580.1). It may be necessary to karyotype several tissues to establish mosaicism. Other complex genetic syndromes, many resulting from single gene mutations, are associated with varying degrees of ambiguity of the genitalia, particularly in the male. These entities must be identified on the basis of the associated extragenital malformations.
Smith-Lemli-Opitz syndrome is an autosomal recessive disorder caused by mutations in the sterol Δ7-reductase gene located on chromosome 11q12-q13. It is characterized by prenatal and postnatal growth retardation, microcephaly, ptosis, anteverted nares, broad alveolar ridges, syndactyly of the 2nd-3rd toes, and severe mental retardation (Chapter 80.3). Its incidence is 1/20,000 to 1/60,000; 70% are male. Genotypic males usually have genital ambiguity and, occasionally, partial sex reversal with female genital ambiguity or complete sex reversal with female external genitals. Müllerian duct derivatives are usually absent. Affected 46,XX patients have normal genitalia. Two types of Smith-Lemli-Opitz syndrome have been recognized. The classical form (type I) described earlier and the acrodysgenital syndrome, which is usually lethal within 1 yr and is associated with severe malformations, postaxial polydactyly, and extremely abnormal external genitalia (type II). Pyloric stenosis is associated with Smith-Lemli-Opitz syndrome type I and Hirschsprung disease with type II. Cleft palate, skeletal abnormalities, and 1 case of a lipoma of the pituitary gland have been seen in type II cases. Some authors believe in a spectrum of disease severity rather than in the above classification. Low plasma cholesterol with elevated 7-dehydrocholesterol, its precursor, are found in types 1 and 2, and the levels do not correlate with severity. Maternal apolipoprotein E values do seem to correlate with severity. The most common prenatal expression of Smith-Lemli-Opitz syndrome is intrauterine growth retardation (see Chapter 80.3 for treatment).
Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105-1099.
Canto P, Vilchis F, Chavez B, et al. Mutations of the 5α-reductase type 2 gene in eight Mexican patients from six different pedigrees with 5α-reductase-2 deficiency. Clin Endocrinol. 1997;46:155-160.
Chu J, Zhang R, Zhao Z, et al. Male fertility is compatible with an Arg840 Cys substitution in the AR in a large Chinese family affected with divergent phenotypes of AR insensitivity syndrome. J Clin Endocrinol Metab. 2002;87:347-351.
Diamond DA, Mitchell C, Lamb K, et al. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: attitudes and practices of pediatric urologists. J Pediatr. 2006;148:445-449.
Diamond M, Sigmundson K. Sex reassignment at birth. Arch Pediatr Adolesc Med. 1997;151:298-304.
Frade Costa EM, Bilharinho Mendonca B, Inacio M, et al. Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertil Steril. 1997;67:229-232.
Geissler WM, Davis DL, Wu L, et al. Male pseudohermaphroditism caused by mutation of testicular 17β-hydroxysteroid dehydrogenase 3. Nat Genet. 1994;7:34-39.
Gul D. Third case of WAGR syndrome with severe obesity and constitutional deletion of chromosome (11)(p12p14). Am J Med Genet. 2002;107:70-71.
Hiort O, Sinnecker GHG, Holterbus PM, et al. Inherited and de novo androgen receptor gene mutations: investigation of single-case families. J Pediatr. 1998;132:939-943.
Hochberg Z, Chayen R, Reiss N, et al. Clinical, biochemical and genetic findings in a large pedigree of male and female patients with 5α-reductase 2 deficiency. J Clin Endocrinol Metab. 1996;81:2821-2827.
Kremer H, Karaaij R, Toledo SPA, et al. Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat Genet. 1995;9:160-164.
McPherson EW, Clemens MM, Gibbons RJ, et al. X-linked α-thalassemia/mental retardation (ATR-X) syndrome: a new kindred with severe genital anomalies and mild hematologic expression. Am J Med Genet. 1995;55:302-306.
Mebarki F, Sanchez R, Rheaumes E, et al. Non-salt-losing male pseudohermaphroditism due to the novel homozygous N100S mutation in the type II 3 β-hydroxysteroid dehydrogenase gene. J Clin Endocrinol Metab. 1995;80:2127-2134.
Mendonca BB, Inacio M, Costa EMF, et al. Male pseudohermaphroditism due to steroid 5α-reductase 2 deficiency. Medicine. 1996;75:64-76.
Moisan AM, Ricketts ML, Tardy V, et al. New insight into the molecular basis of 3β-hydroxysteroid dehydrogenase deficiency: identification of eight mutations in the HSD3 gene in eleven patients from seven new families and comparison of the functional properties of twenty-five mutant enzymes. J Clin Endocrinol Metab. 1999;84:4410-4425.
Mongan NP, Jaaskelainen J, Green K, et al. Two de novo mutations in the AR gene cause the complete androgen insensitivity syndrome in a pair of monozygotic twins. J Clin Endocrinol Metab. 2002;87:1057-1061.
Mueller RF. The Denys-Drash syndrome. J Med Genet. 1994;31:471-477.
Parisi MA, Ramsdell LA, Burns MW, et al. A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med. 2007;9:348-357.
Quigley CA, French FS. Androgen insensitivity syndromes. Curr Ther Endocrinol Metab. 1994;5:342-351.
Reardon W, Gibbons RJ, Winter RM, et al. Male pseudohermaphroditism in sibs with the α-thalassemia/mental retardation (ATR-X) syndrome. Am J Med Genet. 1995;55:285-287.
Saenger P. New developments in congenital lipoid adrenal hyperplasia and steroidogenic acute regulatory protein. Pediatr Clin North Am. 1997;44:397-421.
Sarafoglou K, Ostrer H. Clinical Review 111—familial sex reversal: a review. J Clin Endocrinol Metab. 2000;85:483-493.
Tajima T, Fujieda K, Kouda N, et al. Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency. J Clin Endocrinol Metab. 2001;86:3820-3825.
Wallien MS, Cohen-Kettenis PT. Psychosexual outcome of gender-dysphoric children. J Am Acad Child Adolesc Psychiatry. 2008;47:1413-1423.
Weidemann W, Peters B, Romalo G, et al. Response to androgen treatment in a patient with partial androgen insensitivity and a mutation in the deoxyribonucleic acid-binding domain of the androgen receptor. J Clin Endocrinol Metab. 1998;83:1173-1176.
582.3 Ovotesticular DSD
In ovotesticular DSD, both ovarian and testicular tissues are present, either in the same or in opposite gonads. Affected patients have ambiguous genitalia, varying from normal female with only slight enlargement of the clitoris to almost normal male external genitalia (see Fig. 582-2A).
Damiani D, Fellous M, McElreavey K, et al. True hermaphroditism: clinical aspects and molecular studies in 16 cases. Eur J Endocrinol. 1997;136:201-204.
Diamond DA, Mitchell C, Lamb K, et al. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: attitudes and practices of pediatric urologists. J Pediatr. 2006;148:445-449.
Diamond M, Sigmundson K. Sex reassignment at birth. Arch Pediatr Adolesc Med. 1997;151:298-304.
Frade Costa EM, Bilharinho Mendonca B, Inacio M, et al. Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertil Steril. 1997;67:229-232.
Krob G, Braun A, Kuhnle U. True hermaphroditism: geographical distribution, clinical findings, chromosomes and gonadal histology. Eur J Pediatr. 1994;153:2-10.
Parisi MA, Ramsdell LA, Burns MW, et al. A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med. 2007;9:348-357.
Cameron FJ, Montalto J, Byrt E, et al. Gonadal dysgenesis: associations between clinical features and sex of rearing. Endocr J. 1997;44:95-104.
Creighton S, Minto C. Managing intersex. Br Med J. 2001;323:1264-1265.
Daaboul J, Frader J. Ethics and the management of the patient with intersex: a middle way. J Pediatr Endocrinol Metab. 2001;14:1575-1583.
Diamond DA, Mitchell C, Lamb K, et al. Sex assignment for newborns with ambiguous genitalia and exposure to fetal testosterone: attitudes and practices of pediatric urologists. J Pediatr. 2006;148:445-449.
Diamond M, Sigmundson K. Sex reassignment at birth. Arch Pediatr Adolesc Med. 1997;151:298-304.
Frade Costa EM, Bilharinho Mendonca B, Inacio M, et al. Management of ambiguous genitalia in pseudohermaphrodites: new perspectives on vaginal dilation. Fertil Steril. 1997;67:229-232.
Houk CP, Hughes IA, Ahmed SF, et al. Summary of consensus statement on intersex disorders and their management. Pediatrics. 2006;118:753-757.
Lee PA, Houk CP, Ahmed SF, et al. Consensus statement on management of intersex disorders. Pediatrics. 2006;118:e488-e500.
MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med. 2004;350:367-378.
Parisi MA, Ramsdell LA, Burns MW, et al. A gender assessment team: experience with 250 patients over a period of 25 years. Genet Med. 2007;9:348-357.
Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056-1061.



