Chapter 83 Disorders of Purine and Pyrimidine Metabolism
Purines are involved in all biologic processes; all cells require a balanced supply of purines for growth and survival. They provide the primary source of cellular energy through adenosine triphosphate (ATP) and, together with pyrimidines, provide the source for the RNA and DNA that stores, transcribes, and translates genetic information. Purines provide the basic coenzymes (NAD, NADH) for metabolic regulation and play a major role in signal transduction (GTP, cAMP, cGMP). Metabolically active nucleotides are formed from heterocyclic nitrogen-containing purine bases (guanine and adenine) and pyrimidine bases (cytosine, uridine, and thymine). The early steps in the biosynthesis of the purine ring are shown in Figure 83-1. Purines are primarily produced from endogenous sources and, in usual circumstances, dietary purines have a small role. The end product of purine metabolism in humans is uric acid (2,6,8-trioxypurine).
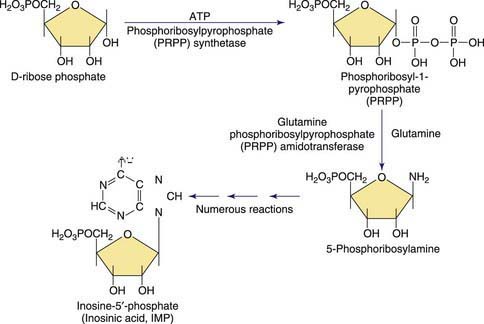
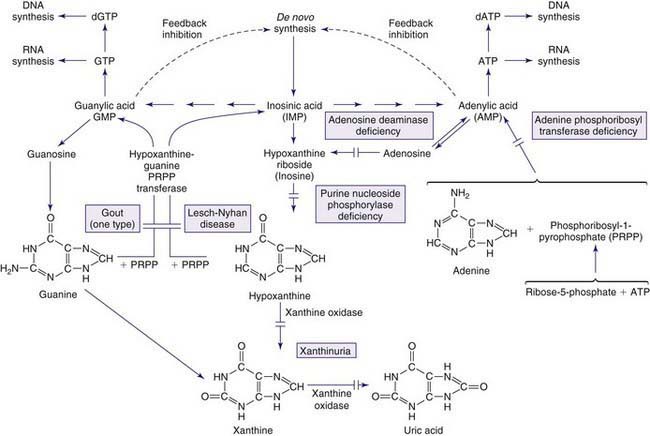
The metabolism of both purines and pyrimidines can be divided into 2 biosynthetic pathways and a catabolic pathway. The 1st, the de novo pathway, involves a multistep biosynthesis of phosphorylated ring structures from precursors such as CO2, glycine, and glutamine. Purine and pyrimidine nucleotides are produced from ribose-5-phosphate or carbamyl phosphate, respectively. The 2nd, a single-step salvage pathway, recovers purine and pyrimidine bases derived from either dietary intake or the catabolic pathway (Figs. 83-2 and 83-3; also see Fig. 83-1). In the de novo pathway, the nucleosides guanosine, adenosine, cytidine, uridine, and thymidine are formed by the addition of ribose-1-phosphate to the purine bases guanine or adenine, and to the pyrimidine bases cytosine, uracil, and thymine. The phosphorylation of these nucleosides produces monophosphate, diphosphate, and triphosphate nucleotides. Under usual circumstances, the salvage pathway predominates over the biosynthetic pathway. Synthesis is most active in tissues with high rates of cellular turnover, such as gut epithelium, skin, and bone marrow. The 3rd pathway is catabolism. The end product of the catabolic pathway of the purines is uric acid, whereas catabolism of pyrimidines produces citric acid cycle intermediates. Only a small fraction of the purines turned over each day are degraded and excreted.
Gout
Gout is associated with hereditary disorders in three different enzyme disorders that result in hyperuricemia. These include the severe form of HPRT deficiency (Lesch-Nyhan disease) and partial HPRT deficiency, superactivity of PP-ribose-P synthetase, and glycogen storage disease type I (glucose-6-phosphatase deficiency) (Chapter 81.1). In the 1st two, the basis of hyperuricemia is purine nucleotide and uric acid overproduction, whereas in the 3rd, it is both excessive uric acid production and diminished renal excretion of urate. Glycogen storage disease types III, V, and VII are associated with exercise-induced hyperuricemia, the consequence of rapid ATP utilization and failure to regenerate it effectively during exercise (Chapter 81.1). Autosomal dominant juvenile hyperuricemia, gouty arthritis, medullary cysts, and progressive renal insufficiency are features associated with familial juvenile hyperuricemic nephropathy (FJHN) and medullary cystic kidney disease type 1 (MCKD1) and type 2 (MCKD2). MCKD1 has been mapped to chromosome 1q21. FJHN and MCKD2 have been mapped to chromosome 16p11.2. FJHN and MCKD2 are proposed to be allelic and can result from uromodulin (UMOD) mutations; the term uromodulin-associated kidney disease (UAKD) has been proposed. Unlike the three inherited purine disorders that are X-linked and the recessively inherited glycogen storage disease, these are autosomal dominant conditions. Familial juvenile gout or familial juvenile hyperuricemic nephropathy is associated with severe renal hypoexcretion of uric acid. Although it most commonly presents from puberty up to the 3rd decade, it has been reported in infancy. It is characterized by early onset, hyperuricemia, gout, familial renal disease, and low urate clearance relative to glomerular filtration rate. It occurs in both males and females and is frequently associated with a rapid decline in renal function that may lead to death unless diagnosed and treated early. Once FJHN is recognized, presymptomatic detection is of critical importance to identify asymptomatic family members with hyperuricemia and to begin treatment, when indicated, to prevent nephropathy.
Abnormalities in Purine Salvage
Lesch-Nyhan Disease (LND)
Clinical Manifestations
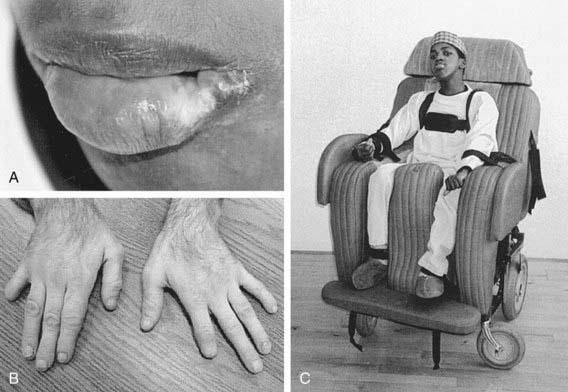
The age of onset of self-injury may be as early as 1 yr and, occasionally, as late as the teens. Self-injury occurs, although all sensory modalities, including pain, are intact. The self-injurious behavior usually begins with self-biting, although other patterns of self-injurious behavior emerge with time. Most characteristically, the fingers, mouth, and buccal mucosa are mutilated. Self-biting is intense and causes tissue damage and may result in the amputation of fingers and substantial loss of tissue around the lips (Fig. 83-4). Extraction of primary teeth may be required. The biting pattern can be asymmetric, with preferential mutilation of the left or right side of the body. The type of behavior is different from that seen in other intellectual disability syndromes involving self-injury; self-hitting and head banging are the most common initial presentations in other syndromes. The intensity of the self-injurious behavior generally requires that the patient be restrained. When restraints are removed, the individual with LND may appear terrified, and stereotypically place a finger in the mouth. The patient may ask for restraints to prevent elbow movement; when the restraints are placed or replaced, the patient may appear relaxed and better humored. Dysarthric speech may cause interpersonal communication problems; the higher-functioning children can express themselves fully and participate in verbal therapy.
Disorders Linked to Purine Nucleotide Synthesis
Phosphoribosylpyrophosphate (PRPP) Synthetase Superactivity
PRPP is a substrate involved in the synthesis of essentially all nucleotides and important in the regulation of the de novo pathways of purine and pyrimidine nucleotide synthesis. This enzyme produces PRPP from ribose-5-phosphate and ATP, as shown in Figures 83-1 and 83-2. PRPP is the 1st intermediary compound in the de novo synthesis of purine nucleotides that lead to the formation of inosine monophosphate. Superactivity of the enzyme results in an increased generation of PRPP. Because PRPP amidotransferase, the 1st enzyme of the de novo pathway, is not physiologically saturated by PRPP, the synthesis of purine nucleotides increases, and, consequently, the production of uric acid is increased. PRPP synthetase superactivity is 1 of the few hereditary disorders in which there is enhancement of the activity of an enzyme.
Disorders Resulting from Abnormalities in Purine Catabolism
Myoadenylate Deaminase Deficiency (Muscle Adenosine Monophosphate Deaminase Deficiency)
Pathogenesis
The inherited form of the disorder is an autosomal recessive trait. AMP-D1, the gene responsible for encoding muscle AMP deaminase, is located on the short arm of chromosome 1 (1p13-21). Population studies reveal that this mutant allele is found at high frequency in white populations. The disorder may be screened for by performing the forearm ischemic exercise test. The normal elevation of venous plasma ammonia after exercise that is seen in normal subjects is absent in AMP deaminase deficiency. Clinical manifestations are, most commonly, isolated muscle weakness, fatigue, myalgias after moderate-to-vigorous exercise, or cramps. Myalgias may be associated with an increased serum creatine kinase level and detectable electromyelographic abnormalities. Muscle wasting or histologic changes on biopsy are absent. The age of onset may be as early as 8 mo of life with ≈25% of cases recognized between 2 and 12 yr of age. The enzyme defect has been identified in asymptomatic family members. Secondary forms of muscle AMP deaminase deficiency have been identified in Werdnig-Hoffmann disease, Kugelberg-Welander syndrome, polyneuropathies, and amyotrophic lateral sclerosis (Chapter 604). The metabolic disorder involves the purine nucleotide cycle. The enzymes involved in this cycle are AMP deaminase, adenylosuccinate synthetase, and adenylosuccinase (see Fig. 83-2). It is proposed that muscle dysfunction in AMP deaminase deficiency results from impaired energy production during muscle contraction. It is unclear how individuals may carry the deficit and be asymptomatic. In addition to muscle dysfunction, a mutation of liver AMP deaminase has been proposed as a cause of primary gout, leading to overproduction of uric acid.
Disorders of Pyrimidine Metabolism
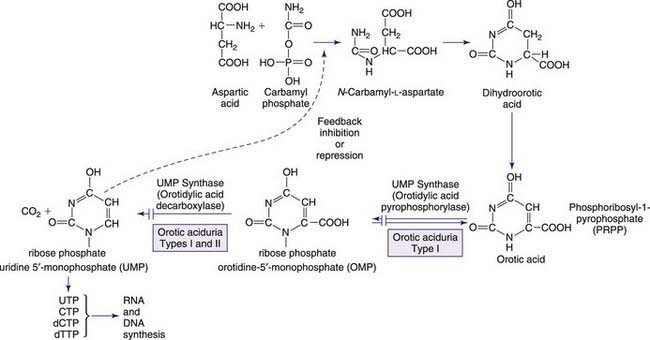
The pyrimidines are the building blocks of DNA and RNA and involved in the formation of coenzymes, as active intermediates in carbohydrate and phospholipid metabolism, glucuronidation in detoxification processes, and glycosylation of proteins and lipids. Pyrimidine synthesis differs from that of purines in that the single pyrimidine ring is 1st assembled and then linked to ribose phosphate to form uridine 5′-monophosphate (UMP). The pyrimidines uracil and thymine are degraded in 4 steps, as shown in Figure 83-3. Purine metabolism has an easily measurable end point in uric acid; however, there is no equivalent compound in pyrimidine metabolism. The 1st defect, hereditary orotic aciduria, is in the de novo synthetic pathway, whereas the other disorders involve either overactivity of or defects in the pyrimidine degradation pathway. Degradation disorders may present as anemia, neurologic disorders, or multisystem mitochondrial disorders. The 1st three steps of the degradation pathways for thymine and uracil make use of the same enzymes (DPD, DPH, and UP). These 3 steps result in the conversion into β-alanine from uracil. There is increasing evidence that pyrimidines play an important role in the regulation of the nervous system. Reduced production of the neurotransmitter function of β-alanine is hypothesized to produce clinical symptoms. These rare disorders may be overlooked because symptoms are not highly specific; they should be considered as possible causes of anemia and neurologic disease and are a contraindication for treatment of cancer patients with certain pyrimidine analogs, for example, 5-fluorouracil.
Hereditary Orotic Aciduria (Uridine Monophosphate Synthase Type 1 Deficiency)
Dihydropyrimidinase (DPH) Deficiency (Dihydropyrimidinuria)
N-Carbamyl-β-Amino Aciduria (Deficiency of β-Ureidopropionase)
Pathogenesis
Fluorescence in situ hybridization localized the human β-ureidopropionase gene to 22q11.2.
Uridine Monophosphate Hydrolase 1 Deficiency (Pyrimidine 5′-Nucleotidase Deficiency)
Uridine Monophosphate Hydrolase 1 Deficiency and Overactive Cytosolic 5′-Nucleotidase
Augoustides-Savvopoulou P, Papachristou F, Fairbanks LD, et al. Partial hypoxanthine-guanine phosphoribosyltransferase deficiency as the unsuspected cause of renal disease spanning three generations: a cautionary tale. Pediatrics. 2002;109:E17.
Cameron JS, Simmonds HA. Hereditary hyperuricemia and renal disease. Semin Nephrol. 2005;25:9-18.
Camici M, Micheli V, Ipata PL, et al. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem Int. 2010;56:367-378.
Ceballos-Picot I, Mockel L, Potier MC, et al. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Genet. 2009;18(13):2317-2327. Jul 1
Dabrowski E, Smathers SA, Ralstrom CS, et al. Botulinum toxin as a novel treatment for self-mutilation in Lesch-Nyhan syndrome. Dev Med Child Neurol. 2005;47:636-639.
Hartmann S, Okun JG, Schmidt C, et al. Comprehensive detection of disorders of purine and pyrimidine metabolism by HPLC with electrospray ionization tandem mass spectrometry. Clin Chem.. 2006;52:1127-1137.
Hladnik U, Nyhan WL, Bertelli M. Variable expression of HPRT deficiency in 5 members of a family with the same mutation. Arch Neurol. 2008;65:1240-1243.
Jinnah HA, De Gregorio L, Harris JC, et al. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res. 2000;463:309-326.
Jinnah HA, Visser JE, Harris JC, et al. Lesch-Nyhan Disease International Study Group. Delineation of the motor disorder of Lesch-Nyhan disease. Brain. 2006;129:1201-1217.
Loffler M, Fairbanks LD, Zameitat E, et al. Pyrimidine pathways in health and disease. Mol Med. 2005;11:430-437.
Marinaki AM, Escuredo E, Duley JA, et al. Genetic basis of hemolytic anemia caused by pyrimidine 5’ nucleotidase deficiency. Blood. 2001;97:3327-3332.
Mouchegh K, Zikánová M, Hoffmann GF, et al. Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: observation of 6 patients in 4 families. J Pediatr. 2007;150:57-61. e2
Nishino I, Spinazzola A, Hirano M. MNGIE: from nuclear DNA to mitochondrial DNA. Neuromuscul Disord. 2001;11:7-10.
Nyhan WL. Disorders of purine and pyrimidine metabolism. Mol Genet Metab. 2005;86:25-33.
Pralong E, Pollo C, Coubes P, et al. Electrophysiological characteristics of limbic and motor globus pallidus internus (GPI) neurons in two cases of Lesch-Nyhan syndrome. Neurophysiol Clin. 2005;35:168-173.
Pralong E, Pollo C, Villemure JG, et al. Opposite effects of internal globus pallidus stimulation on pallidal neurons activity. Mov Disord. 2007;22:1879-1884.
Race V, Marie S, Vincent MF, et al. Clinical, biochemical and molecular genetic correlations in adenylosuccinate lyase deficiency. Hum Mol Genet. 2000;9:2159-2165.
Schretlen DJ, Harris JC, Park K, et al. Neurocognitive functioning in Lesch-Nyhan disease and partial hypoxanthine-guanine phosphoribosyltransferase deficiency. J Int Neuropsychol Soc. 2001;7:805-812.
Sempere A, Arias A, Farré G, et al. Study of inborn errors of metabolism in urine from patients with unexplained mental retardation. J Inherit Metab Dis. 2010;33:1-7.
Teijeira S, San Millán B, Fernández JM, et al. Myoadenylate deaminase deficiency: clinico-pathological and molecular study of a series of 27 Spanish cases. Clin Neuropathol. 2009;28(2):136-142.
Torres RJ, Prior C, Puig JG. Efficacy and safety of allopurinol in patients with hypoxanthine-guanine phosphoribosyltransferase deficiency. Metabolism. 2007;56:1179-1186.





