Chapter 556 Disorders of Pubertal Development
Precocious puberty is defined by the onset of secondary sexual characters before the age of 8 years in girls and 9 years in boys. The variation in the age of the onset of puberty in normal children, particularly of different ethnicities, makes this definition somewhat arbitrary. It remains in use by most clinicians.
Depending on the primary source of the hormonal production, precocious puberty may be classified as central (also known as gonadotropin dependent, or true) or peripheral (also known as gonadotropin independent or precocious pseudopuberty) (Table 556-1). Central precocious puberty is always isosexual and stems from hypothalamic-pituitary-gonadal activation with ensuing sex hormone secretion and progressive sexual maturation. In peripheral precocious puberty, some of the secondary sex characters appear, but there is no activation of the normal hypothalamic-pituitary-gonadal interplay. In this latter group, the sex characteristics may be isosexual or heterosexual (contrasexual; Chapters 577–582).
Table 556-1 CONDITIONS CAUSING PRECOCIOUS PUBERTY
CENTRAL (GONADOTROPIN DEPENDENT, TRUE PRECOCIOUS PUBERTY)
COMBINED PERIPHERAL AND CENTRAL
PERIPHERAL (GONADOTROPIN INDEPENDENT, PRECOCIOUS PSEUDOPUBERTY)
Girls
Boys
INCOMPLETE (PARTIAL) PRECOCIOUS PUBERTY
hCG, human chorionic gonadotropic; SCTAT, sex-cord tumor with annular tubules.
Peripheral precocious puberty can induce maturation of the hypothalamic-pituitary-gonadal axis and trigger the onset of central puberty. This mixed type of precocious puberty occurs commonly in conditions such as congenital adrenal hyperplasia, McCune-Albright syndrome, and familial male-limited precocious puberty, when the bone age reaches the pubertal range (10.5-12.5 yr).
556.1 Central Precocious Puberty
Central precocious puberty is defined by the onset of breast development before the age of 8 years in girls and by the onset of testicular development (volume ≥ 4 mL) before the age of 9 years in boys, as a result of the early activation of the hypothalamic-pituitary-gonadal axis. It occurs 5- to 10-fold more often in girls than in boys and is usually sporadic. A high prevalence of idiopathic central precocious puberty has been reported in girls adopted from developing countries, with the limitation that the exact date of birth may be uncertain.
Although approximately 90% of girls have an idiopathic form, a structural central nervous system (CNS) abnormality can be demonstrated in up to 75% of boys with central precocious puberty. Beyond its etiology, which thus needs to be specifically addressed, central precocious puberty can affect linear growth and the child’s growth potential.
Clinical Manifestations
Sexual development can begin at any age and generally follows the sequence observed in normal puberty. In girls, early menstrual cycles may be more irregular than they are with normal puberty. The initial cycles are usually anovulatory, but pregnancy has been reported as early as 5.5 yr of age (Fig. 556-1). In boys, testicular biopsies have shown stimulation of all elements of the testes, and spermatogenesis has been observed as early as 5-6 yr of age. In affected girls and boys, height, weight, and osseous maturation are advanced. The increased rate of bone maturation results in early closure of the epiphyses, and the ultimate stature is less than it would have been otherwise. Without treatment, approximately 30% of girls and an even larger percentage of boys achieve a height less than the 5th percentile as adults. Mental development is usually compatible with chronological age. Emotional behavior and mood swings are common, but serious psychological problems are rare.
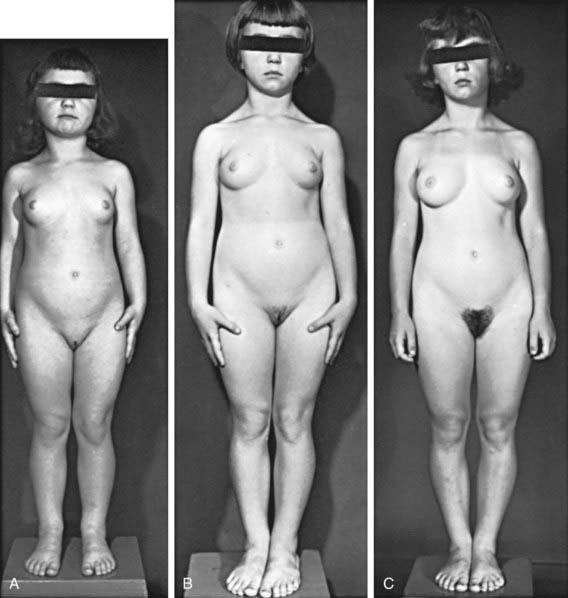
Figure 556-1 Natural course of idiopathic central precocious puberty. Patient (A) at 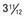 , (B) at
, (B) at 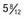 , and (C) at
, and (C) at 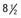 yr of age. Breast development and vaginal bleeding began at
yr of age. Breast development and vaginal bleeding began at 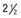 yr of age. Bone age was
yr of age. Bone age was 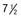 yr at
yr at 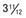 and 14 yr at 8 yr of age. Intelligence and dental age were normal for chronological age. Growth was completed at 10 yr; ultimate height was 142 cm (56 in). No effective therapy was available at the time this patient sought medical attention.
and 14 yr at 8 yr of age. Intelligence and dental age were normal for chronological age. Growth was completed at 10 yr; ultimate height was 142 cm (56 in). No effective therapy was available at the time this patient sought medical attention.
Although the clinical course is variable, 3 main patterns of pubertal progression can be identified. Most girls (particularly those <6 yr of age at the onset) and a large majority of boys have rapidly progressive puberty, characterized by rapid physical and osseous maturation, leading to a loss of height potential. Some girls (>6 yr of age at the onset with an idiopathic form) have a slowly progressive variant, characterized by parallel advancement of osseous maturation and linear growth, with preserved height potential. A small percentage of girls have spontaneously regressive or unsustained central precocious puberty. This variability in the natural course of sexual precocity underscores the need for longitudinal observation at the onset of sexual development, before treatment is considered.
Laboratory Findings
Sex hormone concentrations are usually appropriate for the stage of puberty in both sexes. Serum estradiol concentrations in girls are low or undetectable in the early phase of sexual precocity, as they are in normal puberty. In boys, serum testosterone levels are detectable or clearly elevated by the time the parents seek medical attention, particularly if an early morning blood sample is obtained.
Sensitive immunometric (including immunoradiometric, immunofluorometric, and chemiluminescent) assays for luteinizing hormone (LH) have replaced the traditional LH radioimmunoassays and offer greater diagnostic sensitivity using random blood samples. With sensitive assays, serum LH concentrations are undetectable in prepubertal children but become detectable in 50-75% of girls and a higher percentage of boys with central sexual precocity. Measurement of LH in serial blood samples obtained during sleep has greater diagnostic power than measurement in a single random sample, and it typically reveals a well-defined pulsatile secretion of LH.
Intravenous administration of gonadotropin-releasing hormone (GnRH stimulation test) or a GnRH agonist (leuprolide stimulation test) is a helpful diagnostic tool, particularly for boys, in whom a brisk LH response (LH peak >5-10 IU/L) with predominance of LH over follicle-stimulating hormone (FSH) tends to occur early in the course of precocious puberty. In girls with sexual precocity, however, the nocturnal LH secretion and the LH response to GnRH or GnRH agonist may be quite low at breast stages II to early III (immunometric LH peak <5IU/L), and the LH : FSH ratio can remain low until mid-puberty. In such girls with “low” LH response, the central nature of sexual precocity can be proved by detecting pubertal levels of estradiol (>50 pg/mL), 20-24 hr after stimulation with leuprolide.
Osseous maturation is variably advanced, often more than 2-3 standard deviations (SD). Pelvic ultrasonography in girls reveals progressive enlargement of the ovaries, followed by enlargement of the uterus to pubertal size. An MRI scan usually demonstrates physiologic enlargement of the pituitary gland, as seen in normal puberty; it may also reveal CNS pathology (Chapter 556.2).
Differential Diagnosis
Organic CNS causes of central sexual precocity should be ruled out by MRI scans. They are more likely in girls with rapid breast development, girls with estradiol >30 pg/mL, girls <6 yr of age, and in all boys, but specific criteria for ordering brain imaging are still lacking. Some authorities recommend MRI scans for all children with central precocious puberty.
Gonadotropin-independent causes of isosexual precocious puberty must be considered in the differential diagnosis (see Table 556-1). For girls, these include tumors of the ovaries, autonomously functioning ovarian cysts, feminizing adrenal tumors, McCune-Albright syndrome, and exogenous sources of estrogens. In boys, congenital adrenal hyperplasia, adrenal tumors, Leydig cell tumors, chorionic gonadotropin–producing tumors, and familial male precocious puberty should be considered.
Treatment
Virtually all boys and the large subgroup of girls with rapidly progressive precocious puberty are candidates for treatment. Girls with slowly progressive idiopathic central precocious puberty do not seem to benefit in terms of height prognosis from GnRH-agonist therapy. Children who were small for gestational age may be at greater risk of short stature as adults and can require more-aggressive treatment of precocious puberty, possibly in conjunction with human growth hormone (hGH) therapy. Certain patients require treatment solely for psychological or social reasons, including children with special needs and very young girls at risk of early menarche.
The observation that the pituitary gonadotropic cells require pulsatile, rather than continuous, stimulation by GnRH to maintain the ongoing release of gonadotropins provides the rationale for using GnRH agonists for treatment of central precocious puberty. By virtue of being more potent and having a longer duration of action than native GnRH, these GnRH agonists (after a brief period of stimulation) desensitize the gonadotropic cells of the pituitary to the stimulatory effect of endogenous GnRH and effectively halt the progression of central sexual precocity.
Long-acting formulations of GnRH agonists, which maintain fairly constant serum concentration of the drug for weeks or months, constitute the preparations of choice for treatment of central precocious puberty. In the USA, the most commonly used preparation is leuprolide acetate (Lupron Depot Ped), in a dose of 0.25-0.3 mg/kg (minimum, 7.5 mg) intramuscularly once every 4 wk. Other preparations (D-Trp6-GnRH [Decapeptyl], goserelin acetate [Zoladex]) are approved for treatment of precocious puberty in other countries. Recurrent sterile fluid collections at the sites of injections are the most troublesome local side effect and occur in <2-3% of treated patients. Alternatively, histrelin (Supprelin LA), a subcutaneous 50 mg implant with effects lasting 12 mo, is approved by the FDA for use in central precocious puberty. Other available treatment options include subcutaneous injections of aqueous leuprolide, given once or twice daily (total dose 60 µg/kg/24 hr), or intranasal administration of the GnRH agonist nafarelin (Synarel), 800 µg bid. The potential for irregular compliance with daily administration, as well as the variable absorption of the intranasal route for nafarelin, can limit the long-term benefit of the latter preparations on adult height. Preparations of depot-leuprolide with longer duration of action (90 days) are currently not FDA approved for treatment of central precocious puberty. GnRH antagonists are relatively new and have not been investigated sufficiently. Oral GnRH antagonists are also being investigated.
Treatment results in decrease of the growth rate, generally to age-appropriate values, and an even greater decrease of the rate of osseous maturation. Some children, particularly those with greatly advanced (pubertal) bone age, can show marked deceleration of their growth rate and a complete arrest in the rate of osseous maturation. Treatment results in enhancement of the predicted height, although the actual adult height of patients followed to epiphyseal closure is approximately 1 SD less than their mid-parental height.
In girls, breast development can regress in those with Tanner stage II-III development. Most commonly, the size of the breasts remains unchanged in girls with stage III-V development or even increases slightly because of progressive adipose tissue deposition. The amount of glandular tissue decreases. Pubic hair usually remains stable in girls or progresses slowly during treatment, reflecting the gradual increase in adrenal androgens. Menses, if present, cease. Pelvic sonography demonstrates a decrease of the ovarian and uterine size. In boys, there is decrease of testicular size, variable regression of pubic hair, and decrease in the frequency of erections.
Except for a reversible decrease in bone density (of uncertain clinical significance), no serious adverse effects of GnRH analogs have been reported in children treated for sexual precocity. If treatment is effective, the serum sex hormone concentrations decrease to prepubertal levels (testosterone, <10-20 ng/dL in boys; estradiol, <5-10 pg/mL in girls). The serum LH and FSH concentrations, as measured by sensitive immunometric assays, decrease to <1 IU/L in most patients, although almost never does the LH return to truly prepubertal levels (<0.1 IU/L). The incremental FSH and LH responses to GnRH stimulation decrease to <2-3 IU/L. Serum LH and sex hormone levels remain suppressed for as long as therapy is continued, but puberty resumes promptly when therapy is discontinued, typically at a “pubertal” chronological age. In girls, menarche and ovulatory cycles generally appear at an average of 18 mo (range 6-24 mo) of cessation of therapy. The addition of hGH to GnRH agonists has been used in children with precocious puberty, markedly advanced bone age, and prediction of short stature. The available data indicate that combined therapy can increase the adult height.
Antoniazzi F, Zamboni G, Bertoldo F. Bone development during GH and GnRH analog treatment. Eur J Endocrinol. 2004;151:S47-S54.
Carel JC, Eugster EA, Rogol A, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics. 2009;123:e752-e762.
Chalumeau M, Chemaitilly W, Trivin C, et al. Central precocious puberty in girls: an evidence based diagnosis tree to predict central nervous system abnormalities. Pediatrics. 2002;109:61-67.
Cromer B, Gordon CM. Early pubertal development in Chinese girls. Pediatrics. 2009;124:789-801.
Hirsch HJ, Gillis D, Strich D, et al. The histrelin implant: a novel treatment for central precocious puberty. Pediatrics. 2005;116:e798-e802.
Klein K, Barnes KM, Jones JV, et al. Increase in final height in precocious puberty after long-term treatment with LHRH agonists: the National Institutes of Health experience. J Clin Endocrinol Metab. 2001;86:4711-4716.
Ng SM, Kumar Y, Cody D, et al. Cranial MRI scans are indicated in all girls with central precocious puberty. Arch Dis Child. 2003;88:414-418.
Pucarelli I, Segni M, Ortore M, et al. Combined therapy with GnRH analog plus growth hormone in central precocious puberty. J Pediatr Endocrinol Metab. 2000;13:811-820.
Tanaka T, Niimi H, Matsuo N, et al. Results of long-term follow-up after treatment of central precocious puberty with leuprorelin acetate: evaluation of effectiveness of treatment and recovery of gonadal function. J Clin Endocrinol Metab. 2005;90:1371-1376.
Tuvemo T, Gustafsson J, Proos LA, et al. Suppression of puberty in girls with short acting intranasal versus subcutaneous depot GnRH agonist. Horm Res. 2002;57:27-31.
Tuvemo T, Jonsson B, Gustafsson J, et al. Final height after combined growth hormone and GnRH analogue treatment in adopted girls with early puberty. Acta Paediatr. 2004;93:1456-1462.
Van Beek JT, Sharafuddin MJ, Kao SC, et al. Prospective evaluation of pituitary size and shape on MR imaging after suppressive hormonal therapy in central precocious puberty. Pediatr Radiol. 2000;30:444-446.
556.2 Precocious Puberty Resulting from Organic Brain Lesions
Etiology
Hypothalamic hamartomas are the most common brain lesion causing central precocious puberty (Fig. 556-2). This congenital malformation consists of ectopically located neural tissue. Glial cells within the hamartoma have been shown to produce transforming growth factor β (TGF-β), which has the potential to activate the GnRH pulse generator. On MRI, it appears as a small pedunculated mass attached to the tuber cinereum or the floor of the third ventricle or, less often, as a sessile mass (Fig. 556-3) that remains static in size over years. Hypothalamic hamartomas, especially the sessile variant, can be associated with gelastic or psychomotor seizures.
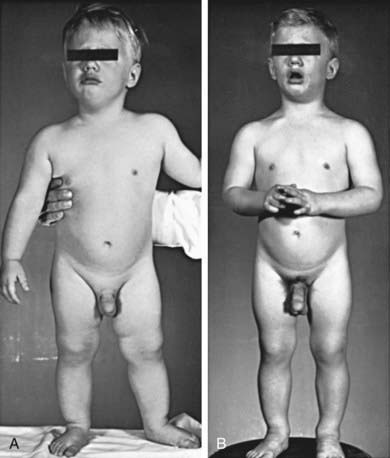
Figure 556-2 Natural course of precocious puberty with central nervous system lesion. Photographs at 1.5 (A) and 2.5 (B) yr of age. Accelerated growth, muscular development, osseous maturation, and testicular development were consistent with the degree of secondary sexual maturation. In early infancy, the patient began having frequent spells of rapid, purposeless motion; later in life, he had episodes of uncontrollable laughing with ocular movements. At 7 yr, he exhibited emotional lability, aggressive behavior, and destructive tendencies. Although a hypothalamic hamartoma had been suspected, it was not established until CT scanning became available when the patient was 23 yr of age. Epiphyses fused at 9 yr of age; final height was 142 cm (56 in). At 24 yr of age, he developed an embryonal cell carcinoma of the retroperitoneum.
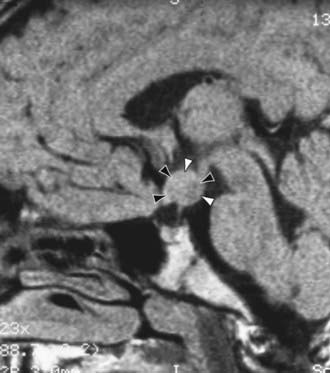
Figure 556-3 MRI of a central nervous system lesion in a child with central precocious puberty. A 6 yr old girl was referred for stage IV breast development and growth acceleration. Serum luteinizing hormone and estradiol concentrations were in the adult range. The midsagittal T1-weighted image shows an isointense hypothalamic mass (arrowheads), typical of a hamartoma.
(From Sharafuddin M, Luisiri A, Garibaldi LR, et al: MR imaging diagnosis of central precocious puberty: importance of changes in the shape and size of the pituitary gland, Am J Roentgenol 162:1167–1173, 1994.)
A wide variety of other CNS lesions or insults, usually involving the hypothalamus by scarring, invasion, or pressure, have been associated with gonadotropin-dependent sexual precocity. They include postencephalitic scars, tuberculous meningitis, tuberous sclerosis, severe head trauma, and hydrocephalus, either isolated or associated with myelomeningocele. Neoplasms causing precocious puberty include astrocytomas, ependymomas, and optic tract tumors. Tumors of the latter type (typically slowly progressive or indolent optic gliomas) are highly prevalent (15-20%) in children with neurofibromatosis type 1 (NF-1) and constitute the main etiologic factor for the central sexual precocity encountered in a small subset (approximately 3%) of children with NF-1.
About 50% of the tumors in the pineal region are germinomas or astrocytomas; the remainder consists of a wide variety of histologically distinct tumor types. In boys, pineal or hypothalamic germinomas can cause central precocious puberty by secreting human chorionic gonadotropin (hCG), which stimulates the LH receptors in the Leydig cells of the testes. These same tumors usually do not produce precocious puberty in girls, presumably because complete ovarian function cannot occur without FSH priming.
Clinical Manifestations
Some of these tumors or malformations (hypothalamic hamartomas) remain static in size or grow slowly, producing no signs other than precocious puberty. For lesions causing neurologic symptoms, the neuroendocrine manifestations may be present for 1-2 yr before the tumor can be detected radiologically. Hypothalamic signs or symptoms such as diabetes insipidus, adipsia, hyperthermia, unnatural crying or laughing (gelastic seizures), obesity, and cachexia should suggest the possibility of an intracranial lesion. Visual signs (proptosis, decreased visual acuity, visual field defects) may be the first manifestation of an optic glioma.
The sexual precocity is always isosexual, and the endocrine patterns are generally those found in children without demonstrable organic lesions. Rapidly progressive sexual precocity in very young children suggests the likelihood of a hypothalamic hamartoma. In conditions other than hypothalamic hamartoma, growth hormone deficiency can occur and may be masked by the growth-promoting effect of the increased sex hormone levels.
Treatment
Regardless of the cause, therapy with GnRH agonists is as effective in children with organic brain lesions causing central precocious puberty, and these analogs are the therapy of choice to halt premature sexual development. This includes patients with a hypothalamic hamartoma, if precocious puberty is its only manifestation. In patients with hypothalamic hamartoma and associated intractable gelastic or psychomotor seizures, however, stereotactic radiation therapy (gamma knife surgery) is effective and less risky than neurosurgical intervention. For other neurologic lesions, therapy depends on the nature and location of the pathologic process. Combined growth hormone therapy should be considered for patients with associated growth hormone deficiency.
Carel JC, Eugster EA, Rogol A, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics. 2009;123:e752-e762.
Virdis R, Street ME, Bandello MA, et al. Growth and pubertal disorders in neurofibromatosis type 1. J Pediatr Endocrinol Metab. 2003;16:289-292.
556.3 Precocious Puberty Following Irradiation of the Brain
Luigi Garibaldi and Wassim Chemaitilly
Radiation therapy, generally for leukemia or intracranial tumors, increases the risk of precocious puberty considerably, whether the irradiation is directed to the hypothalamic area or to areas of the brain anatomically distant from the hypothalamus. Low-dose radiation (18-24 Gy) hastens the onset of puberty almost exclusively in girls. High-dose radiation (25-47 Gy), conversely, appears to trigger precocious sexual development in both sexes, and the risk of sexual precocity is inversely proportional to the age of the child at the time radiation was given.
This type of sexual precocity is often associated with growth hormone deficiency and may also be combined with other conditions (spinal irradiation, hypothyroidism) adversely affecting the prognosis for a reasonable adult height. Unless careful attention is paid to early signs of pubertal development in these children, the combination of growth hormone deficiency and the growth-promoting effect of sex steroids often results in a “normal” growth rate at the expense of a rapidly advancing bone age and impaired adult height potential.
Treatment
GnRH analogs are effective in arresting pubertal progression in this patient population. However, concomitant growth hormone deficiency (and/or thyroid hormone deficiency) should be diagnosed and treated promptly to improve the adult height prognosis.
Paradoxically, hypopituitarism with gonadotropin deficiency can subsequently develop as a late effect of high-dose CNS irradiation in patients with or without a history of precocious puberty, and it can require substitution therapy with sex steroids.
556.4 Syndrome of Precocious Puberty and Hypothyroidism
Luigi Garibaldi and Wassim Chemaitilly
In children with untreated hypothyroidism, the onset of puberty is usually delayed until epiphyseal maturation reaches 12-13 yr of age. Precocious puberty in a child with untreated hypothyroidism and a prepubertal bone age presents a strikingly unphysiologic association, yet it is common and occurs in as many as 50% of children with severe hypothyroidism of long duration. These children have the usual manifestations of hypothyroidism, including retardation of growth and of osseous maturation (Chapter 559). The cause of the hypothyroidism is usually Hashimoto thyroiditis, which often goes undiagnosed, especially in children with special needs such as those with trisomy 21, in whom the symptoms of profound hypothyroidism may be more difficult to recognize.
Sexual development in girls consists primarily of breast enlargement and menstrual bleeding; the latter can occur even in girls with minimal breast enlargement. Pelvic sonography can reveal large, multicystic ovaries. Boys have testicular enlargement associated with modest or no penile enlargement and no pubic hair development. Enlargement of the sella, which is typical of long-standing primary hypothyroidism, may be demonstrated by skull film or MRI.
Plasma levels of thyroid-stimulating hormone (TSH) are markedly elevated, often >500 µU/mL, and plasma levels of prolactin are mildly elevated. Although serum FSH is low and LH is undetectable, when measured by specific assays, the massively elevated concentrations of TSH appear to interact with the FSH receptor (specificity spillover), thus inducing FSH-like effects in the absence of LH effects on the gonads. As a consequence, unlike in central precocious puberty, testicular enlargement occurs without substantial testosterone secretion in boys. Thus, the precocious puberty associated with hypothyroidism behaves as an incomplete form of gonadotropin-dependent puberty.
Treatment of the hypothyroidism results in rapid return to normal of the biochemical and clinical manifestations. Rapid bone age advancement and possible progression to central puberty could occur in the months following the initiation of thyroid hormone replacement, a complication that justifies delaying puberty with GnRH analogs. Macroorchidism (testicular volume >30 mL) can persist in men despite adequate thyroxine therapy.
556.5 Gonadotropin-Secreting Tumors
Luigi Garibaldi and Wassim Chemaitilly
Hepatic Tumors
Isosexual precocious puberty is uncommonly associated with hepatoblastoma. All reported patients have been male, with the age of onset varying from 4 mo to 8 yr (average 2 yr). An enlarged liver or mass in the upper quadrant should suggest the diagnosis. The tumor cells produce hCG, which stimulates the LH receptors in the Leydig cells of the testes. The testicles are only minimally enlarged, and the testicular histology reveals interstitial cell hyperplasia and absence of spermatogenesis. Plasma levels of hCG and α-fetoprotein are usually markedly elevated; they serve as useful markers for following the effects of therapy. Plasma levels of testosterone are elevated, and the FSH and LH levels, as measured by specific immunometric assays, are low; in the past, LH levels were falsely elevated because of cross reaction with hCG on radioimmunoassay.
Treatment for these tumors is the same as that for other carcinomas of the liver; prognosis for survival beyond 1-2 yr from the time of diagnosis is poor.
Other Tumors
Chorionic gonadotropin–secreting choriocarcinomas, teratocarcinomas, or teratomas (also called ectopic pinealomas or atypical teratomas), located in the CNS, mediastinum, gonads, or even adrenal glands, can cause precocious puberty, more commonly (10- to 20-fold) in boys than in girls. Affected patients often have marked elevations of hCG and α-fetoprotein. Mediastinal tumors, but not gonadal tumors, have been reported to cause precocious puberty in boys with Klinefelter syndrome.
Peripheral Precocious Puberty
The adrenal causes of pseudopuberty are discussed in Chapter 570, and the gonadal causes are discussed in Chapters 578 and 581.
556.6 McCune-Albright Syndrome (Precocious Puberty with Polyostotic Fibrous Dysplasia and Abnormal Pigmentation)
The McCune-Albright syndrome is associated with patchy cutaneous pigmentation and fibrous dysplasia of the skeletal system. It is a rare condition with a prevalence between 1/100,000 and 1/1,000,000. A classic cause of peripheral precocious puberty, it can also induce pituitary, thyroid, and adrenal aberrations. It is characterized by autonomous hyperfunction of many glands and is caused by a missense mutation in the gene encoding the α-subunit of GS, the G protein that stimulates cyclic adenosine monophosphate (cAMP) formation, resulting in the formation of the putative gsp oncoprotein. Activation of receptors (corticotropin [ACTH], TSH, FSH, and LH receptors) that operate via a cAMP-dependent mechanism, as well as cell proliferation, ensue. Because the mutation is somatic rather than genomic, it is expressed differently in different tissues, hence the variability of clinical expression.
Precocious puberty has been described predominantly in girls (Fig. 556-4). The average age at onset in affected girls is about 3 yr, but vaginal bleeding has occurred as early as 4 mo of age and secondary sex characters have occurred as early as 6 mo. Young girls have suppressed levels of LH and FSH, and there is no response to GnRH stimulation. Estradiol levels vary from normal to markedly elevated (>900 pg/mL), are often cyclic, and can correlate with the size of the cysts. In boys, precocious puberty is less common but has been reported in several instances. Unlike ovarian enlargement in girls, testicular enlargement in boys is fairly symmetric. It is followed by the appearance of phallic enlargement and pubic hair, as in normal puberty. Testicular histology has demonstrated large seminiferous tubules and no or minimal Leydig cell hyperplasia; these findings might simply reflect the fact that biopsy specimens were obtained at an early stage of pubertal development. In girls and boys, when the bone age reaches the usual pubertal age range, gonadotropin secretion begins, and the response to GnRH becomes pubertal. Central precocious puberty overrides the antecedent (gonadotropin-independent) precocious pseudopuberty. In girls, menses become more regular, but often not completely so, and fertility has been documented.
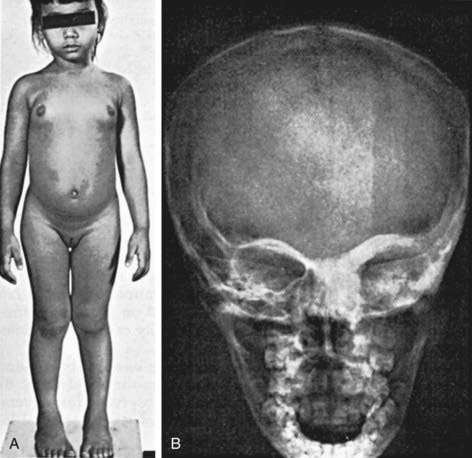
Figure 556-4 Precocious puberty associated with polyostotic fibrous dysplasia (McCune-Albright syndrome) in a girl 4.5 yr of age; at this time, her height age and bone age were normal. Menarche occurred at 4 yr of age. A, Note the bilateral breast development, hyperpigmented spots on the abdomen, and prominence of the left side of the face. B, Roentgenograms revealed fibrous dysplasia in the distal end of the left ulna and thickening of the bones about the left orbit and the maxillary portion of the frontal bones shown here.
Pubertal progression is variable in these patients. Functioning ovarian cysts often disappear spontaneously; aspiration or surgical excision of cysts is rarely indicated. For girls with persistent estradiol secretion, agents that interfere with the final step of estrogen biosynthesis, including aromatase inhibitors such as letrozole (1.25-2.5 mg/day orally) or antiestrogens (such as tamoxifen; fulvestrant is currently being investigated) limit, to a variable extent, the estrogen effects on pubertal and osseous maturation. The same compounds have also been used in boys, in combination with antiandrogens (such as spironolactone 50-100 mg bid, or flutamide 125-250 mg bid). These compounds are not approved by the FDA for this indication; tamoxifen and flutamide may be hepatotoxic, and high-dose spironolactone rarely causes hyperkalemia. Associated therapy with long-acting analogs of GnRH is indicated only for patients whose puberty has shifted from a gonadotropin-independent to a predominantly gonadotropin-dependent mechanism.
Extragonadal Manifestations
The hyperthyroidism that occurs in this condition differs from that characteristic of Graves disease. There is an equal distribution between male and female patients; the goiters are multinodular. Clinical hyperthyroidism is uncommon in children, but goiters, mildly elevated triiodothyronine levels, suppressed TSH levels, and abnormalities on ultrasound have been reported. Thyroidectomy is rarely necessary.
In patients with associated Cushing syndrome, bilateral nodular adrenocortical hyperplasia has occurred in early infancy, antedating the sexual precocity. ACTH levels are low, and adrenal function is not suppressed by large doses of dexamethasone. Treatment is bilateral adrenalectomy.
Increased secretion of growth hormone occurs uncommonly and is manifested clinically by gigantism or acromegaly or by increased rates of growth even in the absence of precocious puberty. Girls and boys are equally affected. Serum levels of growth hormone are elevated and increase during sleep; they are augmented by thyrotropin (TRH) and poorly inhibited by oral glucose. Serum levels of prolactin are increased in most patients, but less than  of the patients have a demonstrable pituitary tumor. Octreotide or lanreotide, long-acting somatostatin analogs, can be used to treat the hypersomatotropism. The prognosis is favorable for longevity, but deformities, repeated fractures, pain, and occasional cranial nerve compression can result from the bony lesions.
of the patients have a demonstrable pituitary tumor. Octreotide or lanreotide, long-acting somatostatin analogs, can be used to treat the hypersomatotropism. The prognosis is favorable for longevity, but deformities, repeated fractures, pain, and occasional cranial nerve compression can result from the bony lesions.
Of the extraglandular manifestations, phosphaturia, leading to rickets or osteomalacia, is the most common. Cardiovascular and hepatic involvement is rare but may be life threatening (severe neonatal cholestasis).
556.7 Familial Male Gonadotropin-Independent Precocious Puberty
Familial male gonadotropin-independent precocious puberty is a rare, autosomal dominant form of peripheral precocious puberty that is transmitted from affected male patients and unaffected female carriers of the gene to their male offspring. Signs of puberty appear by 2-3 yr of age. The testes are only slightly enlarged. Testicular biopsies show Leydig cell maturation and, sometimes, marked hyperplasia. Maturation of seminiferous tubules may be present. Testosterone levels are markedly elevated to the same range seen in boys with true precocious puberty; however, baseline levels of LH are prepubertal, pulsatile secretion of LH is absent, and LH does not respond to stimulation with GnRH or GnRH agonist. The cause for activation of Leydig cells independently of gonadotropin stimulation is a missense mutation of the LH receptor, leading to constitutive activation of cAMP production. Osseous maturation may be markedly advanced; when it reaches the pubertal age range, hypothalamic maturation shifts the mechanism of pubertal development to a gonadotropin-dependent one. This sequence of events is similar to that occurring in children with McCune-Albright syndrome (see earlier discussion) or in those with congenital adrenal hyperplasia (Chapter 570.1).
Gonadotropin-independent precocious puberty has been diagnosed in a few unrelated boys with type IA pseudohypoparathyroidism who had a single mutation of the Gsα protein. This mutation is inactivating at normal body temperature and causes pseudohypoparathyroidism, but in the cooler temperature of the testes, it is constitutionally activating, resulting in adenyl cyclase stimulation and production of testosterone. Although this mutation differs from the constitutive LH receptor mutation, which usually causes familial male gonadotropin-independent precocious puberty, the end result is the same.
Treatment
Young boys have been successfully treated with ketoconazole (600 mg/24 hr in divided doses every 8 hr), an antifungal drug that inhibits C-17,20-lyase and testosterone synthesis. Other investigators use a combination of antiandrogens (such as spironolactone 50-100 mg bid or flutamide 125-250 mg bid) and aromatase inhibitors (letrozole 2.5 mg/day, or anastrozole 1 mg/day), because estrogens derived from androgens stimulate bone maturation. These medications are unable to revert the serum testosterone to the normal (prepubertal) concentrations or completely offset the unfavorable effects of the elevated sex hormones. They slow down, but do not halt, the progression of puberty and might not improve the height prognosis. Boys whose GnRH pulse generator has matured require combined therapy with GnRH agonists.
556.8 Incomplete (Partial) Precocious Development
Isolated manifestations of precocity without development of other signs of puberty are not unusual; development of the breasts in girls and growth of sexual hair in both sexes are the two most common forms.
Premature Thelarche
This term applies to a transient condition of isolated breast development that most often appears in the first 2 yr of life. In some girls, breast development is present at birth and persists. It may be unilateral or asymmetric and often fluctuates in degree. Growth and osseous maturation are normal or slightly advanced. The genitalia show no evidence of estrogenic stimulation. The condition is usually sporadic. Breast development might regress after 2 yr, often persists for 3-5 yr, and is rarely progressive. Menarche occurs at the expected age, and reproduction is normal. Basal serum levels of FSH and the FSH response to GnRH stimulation may be greater than that seen in normal girls. Plasma levels of LH and estradiol are consistently less than the limits of detection. Ultrasound examination of the ovaries reveals normal size, but a few small (<9 mm) cysts are not uncommon.
In some girls, breast development is associated with definite evidence of systemic estrogen effects, such as growth acceleration or bone age advancement. Pelvic sonography might reveal enlarged ovaries or uterus. This condition, referred to as exaggerated or atypical thelarche, differs from central precocious puberty because it spontaneously regresses. Leuprolide or GnRH stimulation elicits a robust FSH response, a low LH response, and (after leuprolide only) a moderate estradiol increment at 24 hr (average 60-90 pg/mL).
The pathogenesis of typical and exaggerated forms of thelarche is unclear; activating mutations of the GNAS1 gene encoding the α-subunit of the GS protein have been described in some patients without other signs of McCune-Albright syndrome (Chapter 556.6). Premature thelarche is a benign condition but may be the first sign of true or peripheral precocious puberty, or it may be caused by exogenous exposure to estrogens. In addition to a detailed history, a bone age should be obtained. The serum concentrations of FSH, LH, and estradiol are generally low and not diagnostic. Pelvic ultrasound examination is rarely indicated. Continued observation is important because the condition cannot be readily distinguished from true precocious puberty. Regression and recurrence suggest functioning follicular cysts. Occurrence of thelarche in children older than 3 yr most often is caused by a condition other than benign premature thelarche.
Premature Pubarche (Adrenarche)
The term premature pubarche applies to the appearance of sexual hair before the age of 8 yr in girls or 9 yr in boys without other evidence of maturation. It is much more frequent in girls than in boys and might occur more commonly in African American girls than in others. Hair appears on the mons and labia majora in girls and in the perineal and scrotal area in boys; axillary hair generally appears later. Adult-type axillary odor is common. Affected children are slightly advanced in height and osseous maturation.
Premature adrenarche is an early maturational event of adrenal androgen production. This event coincides with precocious maturation of the zona reticularis, an associated decrease in 3β-hydroxysteroid dehydrogenase activity, and an increase in C-17,20-lyase activity. These enzymatic changes result in increased basal and ACTH-stimulated serum concentrations of the Δ5-steroids (17-hydroxypregnenolone and dehydroepiandrosterone [DHEA]) and, to a lesser extent, of the Δ4-steroids (particularly androstenedione) compared with age-matched control subjects. The levels of these steroids and of DHEA sulfate (DHEAS) are usually comparable to those of older children in the early stages of normal puberty.
Premature adrenarche is a slowly progressive condition that requires no therapy. However, a subset of patients with precocious pubarche have one or more features of systemic androgen effect, such as marked growth acceleration, clitoral (girls) or phallic (boys) enlargement, cystic acne, or advanced bone age (>2 SD above the mean for age). In these patients with atypical premature adrenarche, an ACTH stimulation test with measurement of steroid intermediates (mainly, serum 17-hydroxyprogesterone concentrations) is indicated to rule out nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Epidemiologic and molecular genetic studies have shown that the prevalence of nonclassic 21-hydroxylase deficiency is approximately 3-6% of unselected children with precocious pubarche; the prevalence of other enzyme defects (i.e., 3β-hydroxysteroid dehydrogenase or 11β-hydroxylase deficiencies) is extremely low.
Although idiopathic premature adrenarche has been considered a benign condition, longitudinal observations suggest that approximately 50% of girls with premature adrenarche are at high risk for hyperandrogenism and polycystic ovary syndrome, alone or more often in combination with other components of the metabolic syndrome (insulin resistance possibly progressing to type 2 diabetes mellitus, dyslipidemia, hypertension, increased abdominal fat) as adults. Whether the unfavorable progression to pubertal hyperandrogenism can be prevented by insulin-sensitizing agents (metformin 850-1000 mg/day) or lifestyle interventions (diet, exercise) remains to be proved in large studies. An increased risk of premature adrenarche and the metabolic syndrome has been documented in children born small for their gestational age. This appears to be associated with insulin resistance and decreased β-cell reserve, perhaps as a consequence of fetal undernutrition.
Premature Menarche
Premature menarche is rare, much less common than premature thelarche or premature adrenarche, and is a diagnosis of exclusion. In girls with isolated vaginal bleeding in the absence of other secondary sexual characters, more common causes such as vulvovaginitis, a foreign body, or sexual abuse, and uncommon causes such as urethral prolapse and sarcoma botryoides must be carefully excluded. The majority of girls with idiopathic premature menarche have only 1-3 episodes of bleeding; puberty occurs at the usual time, and menstrual cycles are normal. Plasma levels of gonadotropins are normal, but estradiol levels may be elevated, probably owing to episodic ovarian estrogen secretion. Occasional patients are found to have ovarian follicular cysts on ultrasound.
Armengaud JB, Charkaluk ML, Trivin C, et al. Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J Clin Endocrinol Metab. 2009;94:2835-2840.
deVries L, Guz-Mark A, Lazar L, et al. Premature thelarche: age at presentation affects clinical course but not clinical characteristics or risk to progress to precocious puberty. J Pediatr. 2010;156:466-471.
Ibanez L, Ferrer A, Ong K, et al. Insulin sensitization early after menarche prevents progression from precocious pubarche to polycystic ovary syndrome. J Pediatr. 2004;144:4-6.
Koulouri O, Conway GS. Management of hirsutism. BMJ. 2009;338:823-826.
Lappalainen S, Utrianen P, Kuulasmaa T, et al. ACTH receptor promoter polymorphism associates with severity of premature adrenarche and modulates hypothalamo-pituitary-adrenal axis in children. Pediatr Res. 2008;63:410-414.
Lumbroso S, Paris F, Sultan C. McCune-Albright syndrome: molecular genetics. J Pediatr Endocrinol Metab. 2002;15(Suppl 3):875-882.
Neville KA, Walker JL. Precocious pubarche is associated with SGA, prematurity, weight gain, and obesity. Arch Dis Child. 2005;90:258-261.
Noordam C, Dhir V, McNelis JC, et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N Engl J Med. 2009;360:2310-2318.
Roman R, Johnson MC, Codner E, et al. Activating GNAS1 gene mutations in patients with premature thelarche. J Pediatr. 2004;145:218-222.
556.9 Medicational Precocity
A variety of medicaments can induce the appearance of secondary sexual characters that may be confused with precocious puberty. A careful history focused on exploring the possibility of accidental exposure to or ingestion of sex hormones is important. Peripheral precocious puberty has occurred in boys and girls from the accidental ingestion of estrogens (including contraceptive pills) and from the administration of anabolic steroids. Estrogens in cosmetics, hair creams, and breast augmentation creams have caused breast development in girls and gynecomastia in boys; estrogens are readily absorbed through the skin. The high prevalence of premature thelarche and peripheral precocious puberty in Puerto Rico has been attributed to contamination of meats, particularly chicken, with estrogens used in animal husbandry, but this has not been proved. Exogenous estrogens can produce a darkening of the areola that is not usually seen in endogenous types of precocity. The precocious changes disappear after cessation of exposure to the hormones. The use of testosterone gels or creams, which are applied to the skin for treatment of male hypogonadism, has resulted in virilization of children and women following skin contact at, and systemic absorption from, the area where the gel or cream was applied by their family member.





