Chapter 124 Disorders of Phagocyte Function
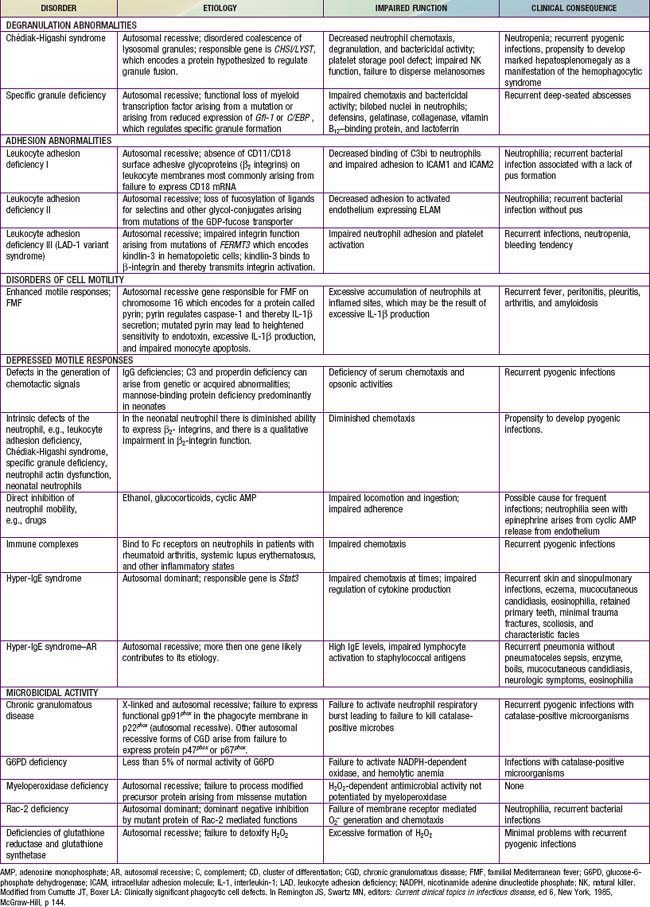
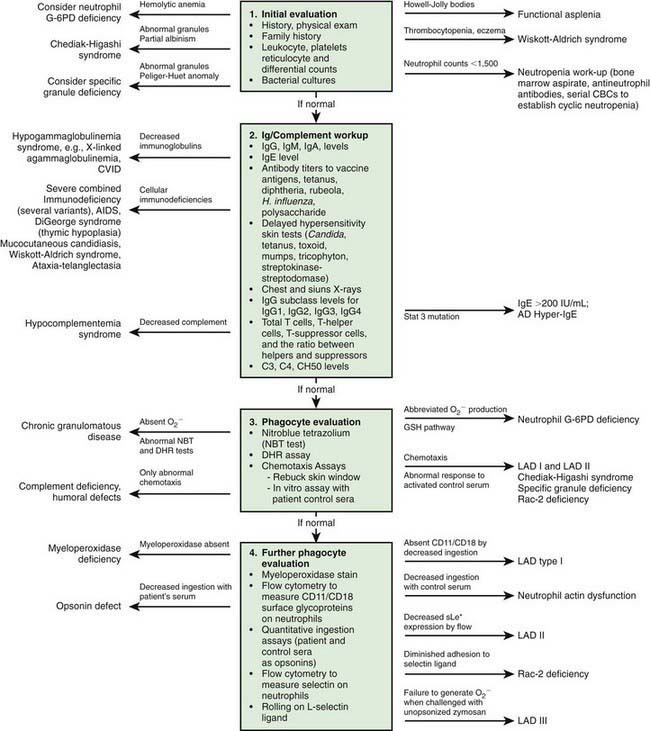
Immunologic evaluation of patients with suspected immunodeficiency (Chapter 116) should focus on disorders of phagocyte function (Table 124-1) in patients with recurrent or unusual bacterial infections (Fig. 124-1).
Chemotaxis, the direct migration of cells into sites of infection, involves a complex series of events (Chapter 121). Studies of defective in vitro chemotaxis of neutrophils obtained from children having various clinical conditions have not established whether frequent infections arise from a primary chemotactic abnormality or occur as secondary medical complications of the underlying disorder. For example, variable but at times severe abnormalities in neutrophil motility accompany the hyper-IgE syndrome, which is characterized by markedly elevated levels of IgE, chronic dermatitis, and recurrent sinopulmonary infections, as well as coarse facial features, retention of primary teeth, and a propensity for recurrent bone fractures (Chapter 123).
Leukocyte Adhesion Deficiency
Genetics and Pathogenesis
Mutations in the CD18 gene either impair gene expression or affect the structure of the synthesized CD18 peptide, leading to functionally abnormal CD11/CD18. Some mutations of CD11/CD18 allow a low level of assembly and activity of integrin molecules. These children retain some neutrophil integrin adhesion function and have a moderate phenotype. Failure of neutrophils to bear the β2-integrins leads to inability to migrate to sites of inflammation outside the blood vessel lumen because of their inability to adhere firmly to surfaces and undergo transendothelial migration. Failure of the CD11/CD18–deficient neutrophils to undergo transendothelial migration occurs because the β2-integrins bind to intercellular adhesion molecules 1 (ICAM-1) and 2 (ICAM-2) expressed on inflamed endothelial cells (Chapter 121). Neutrophils that do arrive at inflammatory sites by CD11/CD18–independent processes fail to recognize microorganisms opsonized with complement fragment iC3b, an important stable opsonin formed by the cleavage of C3b. Hence, other neutrophil functions such as degranulation and oxidative metabolism normally triggered by iC3b binding are also markedly compromised in LAD-1 neutrophils, resulting in impaired phagocytic function and high risk for serious and recurrent bacterial infections.
Clinical Manifestations
The pathogens infecting patients with LAD-1 are similar to those affecting patients with severe neutropenia (Chapter 125) and include Staphylococcus aureus and enteric gram-negative organisms such as Escherichia coli. These patients are also susceptible to opportunistic infection by fungi such as Candida and Aspergillus. Typical signs of inflammation such as swelling, erythema, and warmth may be absent. Pus does not form, and few neutrophils are identified microscopically in biopsy specimens of infected tissues. Despite the paucity of neutrophils within the affected tissue, the circulating neutrophil count during infection typically exceeds 30,000/µL and can surpass 100,000/µL. During intervals between infections, the peripheral blood neutrophil count may chronically exceed 12,000/µL. LAD-1 genotypes producing moderate amounts of functional integrins at the surface of the neutrophil significantly reduce the severity and frequency of infections compared to children with the severe form.
Chédiak-Higashi Syndrome
Chédiak-Higashi syndrome (CHS) is a rare autosomal recessive disorder characterized by increased susceptibility to infection due to defective degranulation of neutrophils, a mild bleeding diathesis, partial oculocutaneous albinism, progressive peripheral neuropathy, and a tendency to develop a life-threatening form of hemophagocytic lymphohistiocytosis (Chapter 501). CHS is a disorder of generalized cellular dysfunction caused by a fundamental defect in granule morphogenesis that results in abnormally large granules in multiple tissues. Pigmentary dilution involving the hair, skin, and ocular fundi results from pathologic aggregation of melanosomes. Neurologic deficits are associated with a failure of decussation of the optic and auditory nerves. Patients exhibit an increased susceptibility to infection that can be explained in part by defects in neutrophil chemotaxis, degranulation, and bactericidal activity. Giant granules in the neutrophils probably interfere with the cells’ ability to traverse the narrow passages between endothelial cells into tissue.
Myeloperoxidase Deficiency
Chronic Granulomatous Disease
Genetics and Pathogenesis
Activation of the phagocyte NADPH oxidase requires stimulation of the neutrophils and involves assembly from cytoplasmic and integral membrane subunits (see Fig. 121-3). Oxidase activation initiates with phosphorylation of a cationic cytoplasmic protein, p47phox (47-kd phagocyte oxidase protein). Phosphorylated p47phox, together with 2 other cytoplasmic components of the oxidase, p67phox and the low molecular weight GTPase Rac-2, translocates to the membrane where they combine with the cytoplasmic domains of the transmembrane flavocytochrome b558 to form the active oxidase complex (see Fig. 121-3). The flavocytochrome is a heterodimer composed of p22phox and highly glycosylated gp91phox. Current models predict that 3 transmembrane domains within the N-terminus of the flavocytochrome contain the histidines that coordinate heme binding. The p22phox component is required for stability of gp91phox and provides a docking site for the cytoplasmic subunits. The gp91phox glycoprotein catalyzes electron transport through its NADPH-binding, flavin-binding, and heme-binding domains. The cytoplasmic p47phox, p67phox, and Rac-2 components appear to serve as regulatory elements for activation of cytochrome b558. An addition protein, p40phox, stabilizes the preactivation cytoplasmic complex of p67phox and p47phox and protects p67phox from degradation. Defects in any of these NADPH oxidase components can lead to CGD.
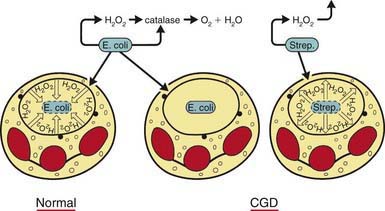
The metabolic deficiency of the CGD neutrophil predisposes the host to infection. The CGD phagocytic vacuoles lack microbicidal reactive oxygen species and remain acidic, so bacteria are not killed or digested properly (Fig. 124-2). Hematoxylin-eosin–stained sections from patients’ tissues show multiple granulomas that give CGD its descriptive name; the macrophages may contain a golden pigment that reflects an abnormal accumulation of ingested material and contributes to granuloma formation. The metabolic impairment in the CGD neutrophil leads to delayed neutrophil apoptosis and subsequent clearance of degenerating neutrophils by macrophages, which in turn leads to ongoing local tissue damage from release of proteases and other granule proteins.
Beauté J, Obenga G, Le Mignot L, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease. Pediatr Infect Dis J. 2011;30(1):57-62.
Bender JM, Rand TH, Ampofo K, et al. Family clusters of variant X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2009;28:529-533.
Dale DC, Boxer L, Liles W. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935.
Dinauer MC, Newburger PE. The phagocyte system and disorders of granulopoiesis and granulocyte function. In: Orkin SH, Ginsburg D, Nathan DG, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders/Elsevier; 2009:1109-1217.
Freeman AF, Holland SM. The hyper-IgE syndromes. Immunol Allergy Clin North Am. 2007;28:277-291.
Hansson M, Olsson I, Naufeef W. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445:214.
Kuhns DB, Alvord G, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600-2610.
Lee PPW, Chan KW, Jiang L, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease. Pediatr Infect Dis J. 2008;27:224-230.
Malech HL, Hickstein DD. Genetics, biology and clinical management of myeloid cell primary immune deficiencies: chronic granulomatous disease and leukocyte adhesion deficiency. Curr Opin Hematol. 2007;14:29.
Nussbaum C, Moser M, Sperandio M. Leukocyte adhesion deficiency-III: when leukocytes cannot stop. Pediatr Res. 2010;67:339.
Qasim W, Cavazzana-Calvo M, Davies EG, et al. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics. 2009;123:836-840.
Reichenbach J, Van de Velde H, De Rycke M, et al. First successful bone marrow transplantation for X-linked chronic granulomatous disease by using preimplantation female gender typing and HLA matching. Pediatrics. 2008;122:e778-e782.
Seger RA. Modern management of chronic granulomatous disease. Brit J Haematol. 2008;140:255.
Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198-203.
Svensson L, Howarth K, McDowall A, et al. Leukocyte adhesion deficiency-III is caused by mutations on KINDLIN3 affecting integrin activation. Nat Med. 2009;15:306-312.



