[level-membership-for-pediatrics-category]
Chapter 604 Disorders of Neuromuscular Transmission and of Motor Neurons
604.1 Myasthenia Gravis
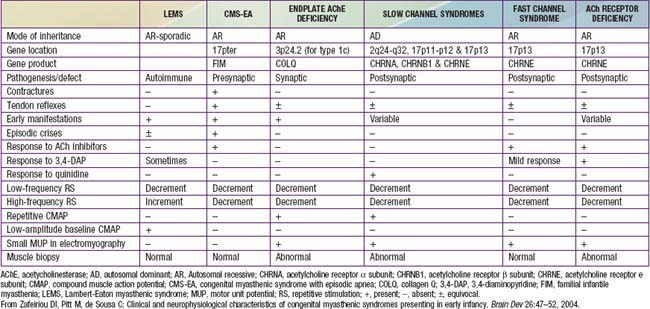
Myasthenia gravis is a chronic disease characterized by rapid fatigability of striated muscle. The most common cause is an immune-mediated neuromuscular blockade. The release of acetylcholine (ACh) into the synaptic cleft by the axonal terminal is normal, but the postsynaptic muscle membrane or motor endplate is less responsive than normal. A decreased number of available ACh receptors is due to circulating receptor-binding antibodies in most cases of acquired myasthenia. The disease is generally not hereditary and is an autoimmune disorder. A rare familial myasthenia gravis is probably an autosomal recessive trait and is not associated with plasma anti-ACh antibodies. One familial form is a deficiency of motor endplate acetylcholinesterase (AChE). Infants born to myasthenic mothers can have a transient neonatal myasthenic syndrome secondary to placentally transferred anti-ACh receptor antibodies, distinct from congenital myasthenia gravis (Table 604-1).
Table 604-1 CLINICAL, PATHOLOGIC, AND NEUROPHYSIOLOGIC CHARACTERISTICS OF VARIOUS CONGENITAL MYASTHENIC SYNDROMES
Clinical Manifestations
The syndrome of transient neonatal myasthenia gravis is to be distinguished from a rare and often hereditary congenital myasthenia gravis not related to maternal myasthenia that is nearly always a permanent disorder without spontaneous remission (see Table 604-1). Several distinct genetic forms are recognized, all with onset at birth or in early infancy with hypotonia, ophthalmoplegia, ptosis, dysphagia, weak cry, facial weakness, easy muscle fatigue generally, and sometimes respiratory insufficiency or failure, the last often precipitated by a minor respiratory infection. Cholinesterase inhibitors have a favorable effect in most, but in some forms the symptoms and signs are actually worsened. Most congenital myasthenic syndromes are transmitted as autosomal recessive traits, but the slow channel syndrome is autosomal dominant. Five defective postsynaptic molecules have been identified in the pathogenesis of congenital myasthenia gravis and account for 85% of cases; rapsyn may be the most common. Acetylcholine receptor deficiencies have >60 identified genetic mutations. Anti-AChR and anti-MuSK antibodies are absent in serum, unlike autoimmune forms of myasthenia gravis affecting older children and adults.
Laboratory Findings and Diagnosis
Recommendations on the Use of Cholinesterase Inhibitors as a Diagnostic Test for Myasthenia Gravis in Infants and Children
Children 2 Years and Older
For Children Younger than 2 Years
Treatment
Neonates with transient maternally transmitted myasthenia gravis require cholinesterase inhibitors for only a few days or occasionally for a few weeks, especially to allow feeding. No other treatment is usually necessary. In non–maternally transmitted congenital myasthenia gravis, identification of the specific molecular defect is important for treatment; specific therapies for each type are summarized in Table 604-2.
Table 604-2 POTENTIAL THERAPIES IN CONGENITAL MYASTHENIC SYNDROMES
| AChE |
Modified from Eyemard B, Hantai D, Estounet B: Congenital myasthenic syndromes. In Dulac O, Sarnat HB, Lassonde M, editors: Handbook of clinical neurology: paediatric neurology, vol 2, Philadelphia, Elsevier, in press.
Other Causes of Neuromuscular Blockade
Organophosphate chemicals, commonly used as insecticides, can cause a myasthenia-like syndrome in children exposed to these toxins (Chapter 58).
Botulism results from ingestion of food containing the toxin of Clostridium botulinum, a gram-positive, spore-bearing, anaerobic bacillus (Chapter 202). Honey is a common source of contamination. The incubation period is short, only a few hours, and symptoms begin with nausea, vomiting, and diarrhea. Cranial nerve involvement soon follows, with diplopia, dysphagia, weak suck, facial weakness, and absent gag reflex. Generalized hypotonia and weakness then develop and can progress to respiratory failure. Neuromuscular blockade is documented by EMG with repetitive nerve stimulation. Respiratory support may be required for days or weeks until the toxin is cleared from the body. No specific antitoxin is available. Guanidine, 35 mg/kg/24 hr, may be effective for extraocular and limb muscle weakness but not for respiratory muscle involvement.
Andrews PL. Autoimmune myasthenia gravis in childhood. Semin Neurol. 2004;24:101-110.
Barisic N, Müller JS, Paucic-Kirincic E, et al. Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. Eur J Paediatr Neurol. 2005;9:7-12.
Eyemard B, Hantai D, Estounet B: Congenital myasthenic syndromes. In Dulac O, Sarnat HB, Lassonde M, editors: Handbook of clinical neurology, vol 2, paediatric neurology, Philadelphia, Elsevier, in press.
Felice KJ, DiMario F, Conway SR. Postinfectious myasthenia gravis: report of 2 cases. J Child Neurol. 2005;20:441-444.
Harper CM. Congenital myasthenic syndromes. Semin Neurol. 2004;24:111-123.
Maselli RA, Kong DZ, Bowe CM, et al. Presynaptic congenital myasthenic syndrome due to quantal release deficiency. Neurology. 2001;57:279-289.
Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn. Neurology. 2009;73:228-235.
Muller JS, Herczegfalvi A, Vilchez JJ, et al. Phenotypical spectrum of DOK7 mutations in congenital myasthenic syndromes. Brain. 2007;130:1497-1506.
Scherer K, Bedlack RS, Simel DL. Does this patient have myasthenia gravis? JAMA. 2005;293:1906-1914.
Schmidt C, Abicht A, Krampfl K, et al. Congenital myasthenic syndrome due to a novel missense mutation in the gene encoding choline acetyltransferase. Neuromuscul Disord. 2003;13:245-251.
Vincent A, McConville J, Farrugia ME, et al. Seronegative myasthenia gravis. Semin Neurol. 2004;24:125-133.
Wylam ME, Anderson PM, Kuntz NL, Rodriguez V. Successful treatment of refractory myasthenia gravis using rituximab: a pediatric case report. J Pediatr. 2003;143:674-677.
Zafeiriou DI, Pitt M, de Sousa C. Clinical and neurophysiological characteristics of congenital myasthenic syndromes presenting in early infancy. Brain Dev. 2004;26:47-52.
Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis. Neurology. 2007;68:837-841.
604.2 Spinal Muscular Atrophies
SMA is classified into a severe infantile form, also known as Werdnig-Hoffmann disease or SMA type 1; a late infantile and more slowly progressive form, SMA type 2; and a more chronic or juvenile form, also called Kugelberg-Welander disease, or SMA type 3. A severe fetal form that is usually fatal in the perinatal period has been described as SMA type 0, with motor neuron degeneration demonstrated in the spinal cord as early as midgestation. These distinctions of types are clinical and are based on age at onset, severity of weakness, and clinical course; muscle biopsy does not distinguish types 1 and 2, although type 3 shows a more adult than perinatal pattern of denervation and reinnervation. Type 0 can show biopsy features more similar to myotubular myopathy because of maturational arrest; scattered myotubes and other immature fetal fibers also are demonstrated in the muscle biopsies of patients with types 1 and 2, but do not predominate. About 25% of patients have type 1, 50% type 2, and 25% type 3; type 0 is rare and accounts for <1%. Some patients are transitional between types 1 and 2 or between types 2 and 3 in terms of clinical function. A variant of SMA, Fazio-Londe disease, is a progressive bulbar palsy resulting from motor neuron degeneration more in the brainstem than the spinal cord. Other variants are noted in Table 604-3.
Table 604-3 SPINAL MUSCULAR ATROPHY VARIANTS: PROGRESSIVE OR SEVERE NEONATAL ANTERIOR HORN CELL DISEASE NOT LINKED TO SMN
| VARIANT | MAJOR FEATURES |
|---|---|
| SMA with respiratory distress type 1 (SMARD1) |
SMA, spinal muscular atrophy.
From Vole JJ: Neurology of the newborn, ed 5, Philadelphia, 2008, Saunders Elsevier, p 775.
Clinical Manifestations
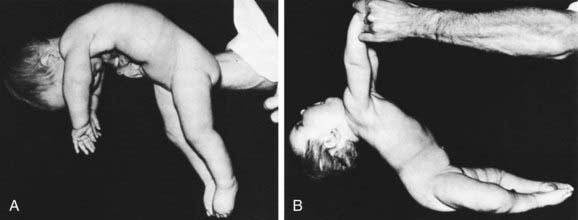
The cardinal features of SMA type 1 are severe hypotonia (Fig. 604-1); generalized weakness; thin muscle mass; absent tendon stretch reflexes; involvement of the tongue, face, and jaw muscles; and sparing of extraocular muscles and sphincters. Diaphragmatic involvement is late. Infants who are symptomatic at birth can have respiratory distress and are unable to feed. Congenital contractures, ranging from simple clubfoot to generalized arthrogryposis, occur in about 10% of severely involved neonates. Infants lie flaccid with little movement, unable to overcome gravity (see Fig. 599-1). They lack head control. More than 65% of children die by 2 yr of age, and many die early in infancy.
Diagnosis
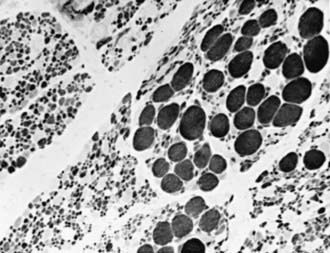
The simplest, most definitive diagnostic test is a molecular genetic marker in blood for the SMN gene. Muscle biopsy reveals a characteristic pattern of perinatal denervation that is unlike that of mature muscle. Groups of giant type I fibers are mixed with fascicles of severely atrophic fibers of both histochemical types (Fig. 604-2). Scattered immature myofibers resembling myotubes also are demonstrated. In juvenile SMA, the pattern may be more similar to adult muscle that has undergone many cycles of denervation and reinnervation. Neurogenic changes in muscle also may be demonstrated by EMG, but the results are less definitive than by muscle biopsy in infancy. Sural nerve biopsy sometimes shows mild sensory neuropathic changes, and sensory nerve conduction velocity may be slowed; hypertrophy of unmyelinated axons also is seen. At autopsy, mild degenerative changes are seen in sensory neurons of dorsal root ganglia and in somatosensory nuclei of the thalamus, but these alterations are not perceived clinically as sensory loss or paresthesias. The most pronounced neuropathologic lesions are the extensive neuronal degeneration and gliosis in the ventral horns of the spinal cord and brainstem motor nuclei, especially the hypoglossal nucleus.
Berger A, Mayr JA, Meierhofer D, et al. Severe depletion of mitochondrial DNA in spinal muscular atrophy. Acta Neuropathol. 2003;105:245-251.
Chung BHY, Wong VCN, Ip P. Spinal muscular atrophy: survival pattern and functional status. Pediatrics. 2004;114:e548-e553.
Grohmann K, Varon R, Stolz P, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann Neurol. 2003;54:719-724.
Hardart MKM, Truog RD. Spinal muscular atrophy-type 1. Arch Dis Child. 2003;88:848-850.
Kizilates SU, Talim B, Sel K, et al. Severe lethal spinal muscular atrophy variant with arthrogryposis. Pediatr Neurol. 2005;32:201-204.
Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120-2132.
Mercuri E, Messina S, Kinali M, et al. Congenital form of spinal muscular atophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromusc Disord. 2004;14:125-129.
Nadeau A, Anjou GD, Debray FG, et al. A newborn with spinal muscular atrophy type 0 presenting with a clinicopathological picture of centronuclear myopathy. Can J Neurol Sci. 2005;32(Suppl 1):S45.
Roper H, Quinlivan R. on Behalf of Workshop Participants: Implementation of “the consensus statement for the standard of care in spinal muscular atrophy” when applied to infants with severe type 1 SMA in the UK. Arch Dis Child. 2010;95:845-849.
Sarnat HB, Trevenen CL. Motor neuron degeneration in a 20-week male fetus: spinal muscular atrophy type 0. Can J Neurol Sci. 2007;34:215-220.
Souchon F, Simard LR, Lebrun S, et al. Clinical and genetic study of chronic (types II and III) childhood onset spinal muscular atrophy. Neuromuscul Disord. 1996;6:419-424.
Tachi N, Kikuchi S, Nozuka N, et al. A new mutation of IGHMBP2 gene in spinal muscular atrophy with respiratory distress type 1. Pediatr Neurol. 2005;32:288-290.
604.3 Other Motor Neuron Diseases
Motor neuron diseases other than SMA are rare in children. Poliomyelitis used to be a major cause of chronic disability, but since the routine use of polio vaccine, this viral infection is now rare (Chapter 241). Other enteroviruses, such as coxsackievirus and echovirus, or the live polio vaccine virus can also cause an acute infection of motor neurons with symptoms and signs similar to poliomyelitis, although usually milder. Specific polymerase chain reaction tests and viral cultures of cerebrospinal fluid are diagnostic. Motor neuron infection with the West Nile virus also occurs.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 604 Disorders of Neuromuscular Transmission and of Motor Neurons
604.1 Myasthenia Gravis
Myasthenia gravis is a chronic disease characterized by rapid fatigability of striated muscle. The most common cause is an immune-mediated neuromuscular blockade. The release of acetylcholine (ACh) into the synaptic cleft by the axonal terminal is normal, but the postsynaptic muscle membrane or motor endplate is less responsive than normal. A decreased number of available ACh receptors is due to circulating receptor-binding antibodies in most cases of acquired myasthenia. The disease is generally not hereditary and is an autoimmune disorder. A rare familial myasthenia gravis is probably an autosomal recessive trait and is not associated with plasma anti-ACh antibodies. One familial form is a deficiency of motor endplate acetylcholinesterase (AChE). Infants born to myasthenic mothers can have a transient neonatal myasthenic syndrome secondary to placentally transferred anti-ACh receptor antibodies, distinct from congenital myasthenia gravis (Table 604-1).
Table 604-1 CLINICAL, PATHOLOGIC, AND NEUROPHYSIOLOGIC CHARACTERISTICS OF VARIOUS CONGENITAL MYASTHENIC SYNDROMES
Clinical Manifestations
The syndrome of transient neonatal myasthenia gravis is to be distinguished from a rare and often hereditary congenital myasthenia gravis not related to maternal myasthenia that is nearly always a permanent disorder without spontaneous remission (see Table 604-1). Several distinct genetic forms are recognized, all with onset at birth or in early infancy with hypotonia, ophthalmoplegia, ptosis, dysphagia, weak cry, facial weakness, easy muscle fatigue generally, and sometimes respiratory insufficiency or failure, the last often precipitated by a minor respiratory infection. Cholinesterase inhibitors have a favorable effect in most, but in some forms the symptoms and signs are actually worsened. Most congenital myasthenic syndromes are transmitted as autosomal recessive traits, but the slow channel syndrome is autosomal dominant. Five defective postsynaptic molecules have been identified in the pathogenesis of congenital myasthenia gravis and account for 85% of cases; rapsyn may be the most common. Acetylcholine receptor deficiencies have >60 identified genetic mutations. Anti-AChR and anti-MuSK antibodies are absent in serum, unlike autoimmune forms of myasthenia gravis affecting older children and adults.
Laboratory Findings and Diagnosis
Recommendations on the Use of Cholinesterase Inhibitors as a Diagnostic Test for Myasthenia Gravis in Infants and Children
Children 2 Years and Older
For Children Younger than 2 Years
[/not-level-membership-for-pediatrics-category]
